

Abstract
Radical S-adenosylmethionine (RS) enzymology has emerged as a major biochemical strategy for the homolytic cleavage of unactivated C–H bonds. At the same time, the post-translational modification of ribosomally synthesized peptides is a rapidly expanding area of investigation. We discuss the functional cross-section of these two disciplines, highlighting the recently uncovered importance of protein–protein interactions, especially between the peptide substrate and its chaperone, which functions either as a stand-alone protein or as an N-terminal fusion to the respective RS enzyme. The need for further work on this class of enzymes is emphasized, given the poorly understood roles performed by multiple, auxiliary iron–sulfur clusters and the paucity of protein X-ray structural data.
Keywords: iron–sulfur protein, oxidation–reduction (redox), peptide biosynthesis, post-translational modification (PTM), radical, RiPPs, SPASM, radical SAM
Introduction
Modern biochemistry has seen the exponential growth of two exciting areas of research that focus individually on the post-translational modification of ribosomally synthesized and post-translationally modified peptides (RiPPs)2 (1) and on the structure and mechanism of free radical conversions catalyzed by radical S-adenosylmethionine (RS) enzymes (2, 3). These separate fields have recently converged within a growing family of enzymes that bring about free radical-based, post-translational modifications on ribosomally produced peptide substrates. RS enzymes function by cleavage of a [4Fe-4S] cluster-bound S-adenosylmethionine to form methionine and a deoxyadenosyl radical. This radical then initiates the reaction by abstracting a hydrogen atom from the substrate. The [4Fe-4S] cluster that accomplishes this reaction has a characteristic CX3CXϕC motif, with each of the cysteines coordinated to a single iron, leaving an open coordination sphere where SAM binds and is cleaved. A subset of RS enzymes belongs to a family that contains an additional conserved structural motif annotated as a SPASM domain and referred to as RS-SPASM proteins. Although the exact function of the SPASM domain is unknown, it has been shown that it houses generally two additional iron–sulfur clusters that are critical in RS-SPASM chemistry. Using the perspective of the pathway for production of the bacterial cofactor pyrroloquinoline quinone (PQQ), we compare and highlight some of the unique properties among these RS-SPASM-dependent RiPP systems. Bioinformatics analyses suggest that the family members will extend far beyond the examples presented herein.
PQQ biosynthesis: Demonstration of a PqqE–PqqD complex and its requirement for PqqA cross-linking
PQQ is a bacterial cofactor that confers a growth advantage to selective bacteria via the introduction of new electron transfer pathways for the generation of cellular ATP. It is synthesized from the RiPP precursor PqqA by the enzymes PqqB, PqqC, PqqD, and PqqE (Fig. 1). A comparative bioinformatics analysis of the pqq operon among a large number of sequenced prokaryotic genomes provided one of the early intimations of a role for protein–protein complexes in the PQQ biosynthetic pathway (4). This study revealed a fully conserved ordering of each of five ORFs, pqqA–E. Although the retention of an ORF within a given operon is reflective of its essential function, the order in which each gene is located will normally undergo random drift. A second informative observation was the occasional addition of pqqD to either the 3′-end of pqqC or to the 5′-end of pqqE. Furthermore, among the annotated PqqE proteins across several genomes, members of the family Methylocystaceae show an open reading frame predicted to encode an N-terminal fusion of PqqD onto PqqE. The fusion of orthologous PqqD domains to RS proteins is also conserved among other RS-dependent RIPP pathways; however, until recently, little was known about the purpose for their co-occurrence.
Figure 1.

PQQ biosynthetic pathway. The ribosomally-produced peptide PqqA, containing a leader sequence and the to-be-modified Glu and Tyr, is recognized and bound by the peptide chaperone PqqD (1). The complex associates with PqqE, along with one equivalent of SAM (2). The radical SAM Fe-S cluster is reduced, and this electron is then transferred to reductively cleave SAM (3 and 4). The newly-produced deoxyadenosyl radical abstracts hydrogen from the glutamate γ-carbon, leading to formation of a carbon–carbon bond between two residues and a radical on the tyrosine (5). Oxidation of the tyrosyl radical leads to the cross-linked peptide product, which is later hydrolyzed and oxidized to give AHQQ, the substrate for PqqC, which converts AHQQ to PQQ in an oxygen-dependent manner.
RS enzymes contain, at a minimum, a single [4Fe-4S] cluster that when reduced, e.g. by sodium dithionite, shows a well-characterized EPR signal (3). Consistent with the prediction of protein–protein complexes, examination of the EPR pattern for PqqE from Klebsiella pneumoniae indicated detectable changes in the presence of PqqD (5). This was followed by measurements of binding constants among PqqA, PqqE, and PqqD from a more tractable source (Methylobacterium extorquens), using surface plasmon resonance spectroscopy and isothermal titration calorimetry (6). The interaction between PqqE and PqqD occurs in a 1:1 manner with a micromolar KD, whereas PqqA and PqqD interact more tightly, KD ∼200 nm. The formation of a ternary complex involving all three components, PqqA, PqqD, and PqqE, could be detected via native mass spectrometry, with little impact of pre-forming a complex of PqqA–PqqD on its binding to PqqE, KD ∼5 μm.
This series of observations has led to the proposal of a unique and formerly unrecognized role for PqqD as a peptide chaperone that binds and directs PqqA toward the first catalyst in the pathway, PqqE (Fig. 1) (6, 7). The finding of a peptide-binding role for PqqD immediately “opened a window” into our understanding of the control of the post-translational modification of peptides. First, after many unsuccessful efforts to demonstrate activity of PqqE toward PqqA, the addition of PqqD was shown to lead to de novo C–C bond formation within PqqA (7). Second, bioinformatics analyses have identified the presence of a large (SPASM) subfamily of RS enzymes, many of which are predicted to contain PqqD homologues (6). The SPASM designation of Haft and Basu (8) derives from the four inaugural families shown to contain multiple iron/sulfur centers and to act on either protein or peptide substrates: subtilosin; pyrroloquinoline quinone; anaerobic sulfatase-maturating enzyme (AnSME), and mycofactocin, discussed in greater detail below. Finally, Mitchell and co-workers (9) recognized the similarity between the PqqD structural motif and the N termini of enzymes that target peptides via non-free radical lantibiotic pathways, thereby greatly extending the commonality of peptide-binding motifs within the highly diverse family of RiPPs.
Three-dimensional structure of AnSME as a model for SPASM domain-containing RS enzymes that modify RiPPs
Prior to writing this review, the only structure of an RS-SPASM protein available, out of the >18,000 sequences annotated in the Interpro Database,3 was that of AnSME. AnSME is responsible for the post-translational modification of sulfatases, a widely distributed and physiologically significant class of enzymes (10, 11). The hydrolase activity of sulfatases requires the post-translational conversion of an active-site cysteine (or in some cases, serine) to a Cα-formylglycine (FGly). Under anaerobic conditions, the formation of FGly is catalyzed by AnSME (10, 11). Moreover, it has been proposed that AnSME functions cotranslationally, acting on the unfolded sulfatase protein (12). Indeed, in vitro AnSME peptide modification assays have been carried out on a truncated sulfatase peptide sequence containing the Cys or Ser of interest and not the full-length folded protein. However, it should be noted that unlike PqqE, AnSME does not require a PqqD domain to function. This observation signifies an outlier enzyme with regard to the proposal that RS-SPASM proteins characterized to date will require a PqqD-like domain or partner. This is likely the result of AnSME acting natively on full-length proteins (unfolded or partially folded) rather than peptides; nonetheless, AnSME shares significant sequence motifs with other RS-SPASM orthologues and has been employed as the primary structural surrogate for all RS-SPASM proteins.
When the X-ray crystal structure for Clostridium perfringens AnSME (Fig. 2, PDB codes 4K36, 4K37, 4K38, and 4K39) was published (13), it provided immediate insight to the nascent field of RS-SPASM-dependent RiPP modification pathways. First, the structure demonstrates that AnSME contains a triose-phosphate isomerase (TIM) barrel fold as an N-terminal domain (Fig. 2). The TIM barrel domain, consisting of a (β/α)6, is structurally conserved among most RS proteins and contains the CX3CXϕC sequence motif responsible for binding the RS-dependent [4Fe-4S] cluster that has been thoroughly described (14–16). Notably, the crystal structure shows that AnSME contains an elongated C-terminal (SPASM) domain consisting of ∼80 amino acids that form a two-stranded β-sheet, followed by an α-helix and several looped regions. Eight conserved cysteines are identified within this region, fully ligated to two auxiliary [4Fe-4S] clusters, denoted as Aux I and Aux II. Although their role in catalysis has yet to be verified, the distances between the Aux I cluster and substrate (10 Å) and the Aux I cluster to the Aux II cluster (13 Å) are consistent with the auxiliary clusters shuttling electrons between the active site and the surface of the protein (17). We note that prior to the availability of a three-dimensional structure for AnSME, a fruitful combination of sequence analyses, site-specific mutagenesis and extensive EPR characterizations had, in fact, predicted a role for two auxiliary [4Fe-4S] centers as a prerequisite for peptide modification as well as the uncoupled cleavage of the cofactor S-adenosylmethionine to 5′-deoxyadenosine (18).
Figure 2.

A, X-ray structure of the canonical AnSME from C. perfringens (PDB code 4K36) indicates the presence of a SPASM domain (red) in addition to the conserved TIM barrel (gray). The TIM barrel contains the RS [4Fe-4S] cluster (black asterisk), ligated by three cysteines and SAM (green), whereas the SPASM domain contains an auxiliary I cluster (blue asterisk) and an auxiliary II cluster (red asterisk), both ligated by four cysteines. B, chemical reaction catalyzed by AnSME.
While this paper was under review, the second crystal structure of an RS-SPASM protein, CteB, was published (19). CteB is a newly characterized thioether bond-forming enzyme responsible for the biosynthesis of a sactipeptide found in Clostridium thermocellum. The structure of CteB is the first to show an RS-SPASM/peptide chaperone fusion protein. The core RS-SPASM region of CteB shares many features with AnSME. In addition, the structure of CteB provides the first glimpse of the position of a peptide chaperone domain in relation to an RS-SPASM protein, indicating its proximity to the α6′-helix portion SPASM domain and the importance of hydrophobic interactions. The timely addition of the CteB structure to the growing RS-SPASM field is welcomed. Not only will the structure of CteB provide new insights into the RS-SPASM field, it provides an additional structural surrogate for most of the RS-SPASM proteins studied to date.
The chemistry behind RS-SPASM proteins
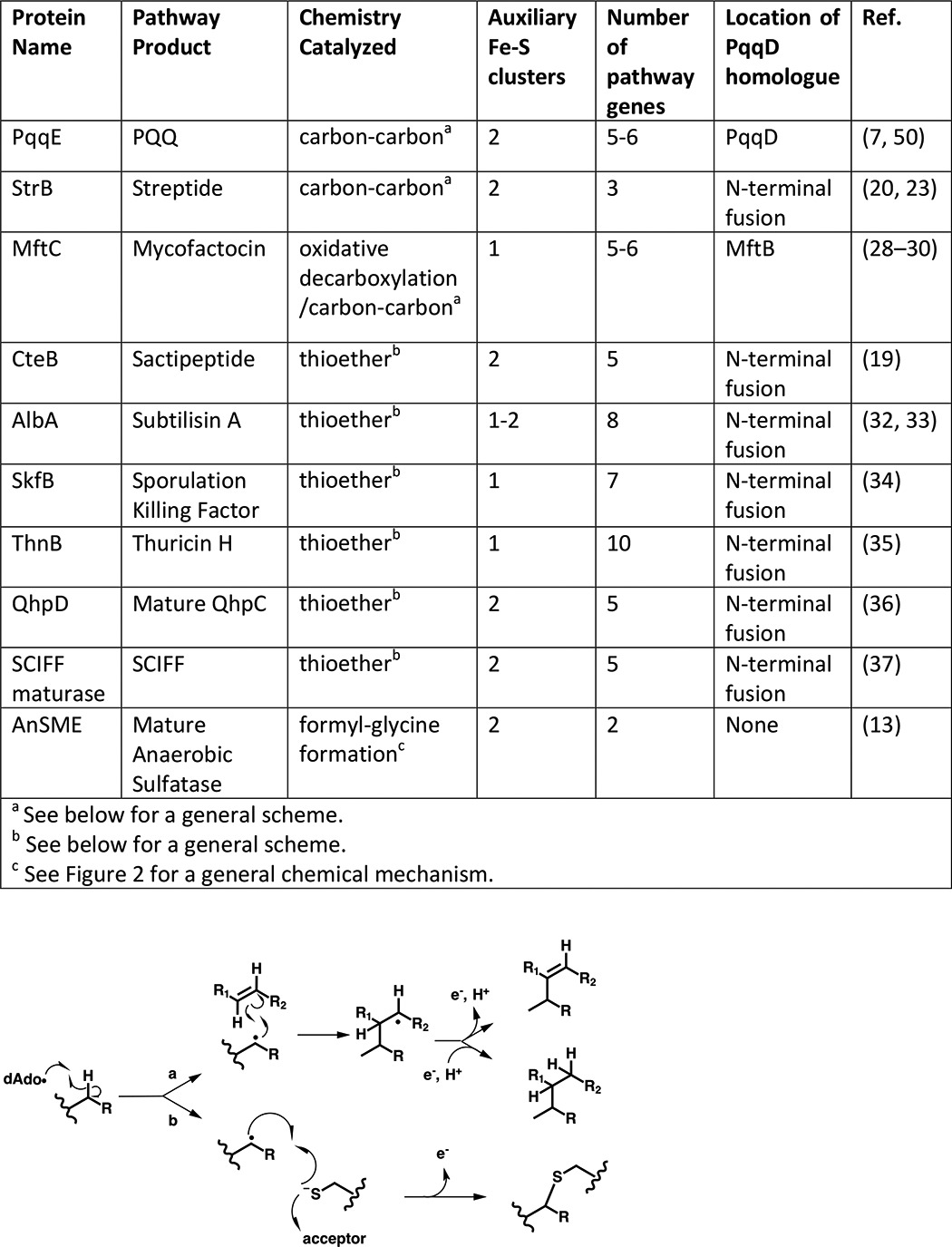
This section briefly describes the chemical mechanism for RS-SPASM proteins whose biochemical activities have been successfully reconstituted in vitro. Apart from AnSME, these RS-SPASM proteins are seen to act uniformly on peptide substrates and to utilize similar chemical strategies that result in two generic groups of chemical modifications. The first group of modifications generates a new carbon–carbon bond, and the second group leads to thioether bond formation. The common part of the chemical mechanism is the initial formation of an alkyl radical on the atom of the amino acid side chain that will participate in new bond formation. This radical subsequently reacts with an electron-rich center (e.g. at sulfur, oxygen, or a π-bonded carbon). An intermolecular transfer of electrons is also an essential part of this process, beginning with the reductive cleavage of SAM via the involvement of an exogenous donor as well as possible downstream removal of electrons from the substrate-derived free radical intermediates; a role for the auxiliary RS clusters in these electron transfer processes is inferred although currently unproven.
Carbon–carbon bond creation: PqqE, StrB, and MftC
To date, three RS-SPASM enzymes have been shown to catalyze the formation of intramolecular carbon–carbon bonds. First, PqqE is the least mechanistically understood in this class and is responsible for catalyzing a new carbon–carbon bond between the PqqA-containing glutamate and tyrosine side chains residing in the conserved sequence EX3Y (7). The position of the new carbon–carbon bond has been inferred from the structure of the final product (PQQ), as arising between the γ-carbon of glutamate and the C5 of the tyrosine ring. The detailed mechanism by which PqqE catalyzes this cross-linking is unknown and is under ongoing investigation.
The second demonstrated carbon–carbon bond formation pathway is found in the streptide system (20). Streptide is a macrocyclic peptide pheromone involved in bacterial communication (20–23). The first step of the biosynthesis of streptide is accomplished through the formation of an intramolecular carbon–carbon bond on the precursor peptide StrA by the RS-SPASM protein StrB. Like PqqE, StrB cross-links the two side chain residues, lysine and tryptophan, found in the conserved motif KGDGW (20). A potential unifying mechanism involves initial hydrogen atom abstraction from Glu (PqqE) or Lys (StrB) via a pre-formed 5′-deoxyadenosine radical, followed by de novo carbon–carbon bond formation on tyrosine (PqqE) or tryptophan (StrB) and subsequent re-aromatization of the aromatic ring; the latter is likely an oxidative process that may involve one or more of the auxiliary RS clusters. Experimental support for the first part of this mechanism comes from Schramma et al. (20), who demonstrated that the β-carbon hydrogen of lysine on the peptide substrate is abstracted by the 5′-deoxyadenosine radical generated by StrB. Recent work shows that StrB most likely contains two auxiliary [4Fe-4S] clusters and that an N-terminal region of StrA is necessary for the reaction (23). Other RS enzymes known to create carbon–carbon bonds include MoaA and NikJ, which modify nucleosides. Mechanistic investigations suggest that these function by abstraction of a hydrogen from an sp3 hybridized carbon and formation of a bond with a carbon that is initially sp2 hybridized, as is proposed for PqqE and StrB. However, in these cases, there is no re-aromatization through oxidation. Instead, the radical is quenched through reduction (24, 25).
The third example in this category comes from the newly characterized member of the RS-SPASM family, MftC. MftC belongs to the mycofactocin biosynthetic cluster composed of the genes mftABCDEF (26). Mycofactocin is predicted to be a redox cofactor used by a niche set of dehydrogenases found largely in the Mycobacterium genera (26, 27). Initially, it was thought that MftC catalyzed the oxidative decarboxylation of the C-terminal tyrosine found on the peptide MftA, resulting in an αβ-unsaturated bond (28, 29). However, a more detailed mechanistic study demonstrated that the decarboxylated peptide is only an intermediate of a two-step reaction (30). In the first step, the 5′-deoxyadenosine radical generated by MftC abstracts a β-carbon hydrogen from the C-terminal tyrosine to form a Cβ radical. The loss of an electron and proton leads to the formation of a benzenone. The collapse of the C-terminal carboxylate electrons to the acyl carbon results in decarboxylation and the formation of an αβ-unsaturated bond. In the second RS-dependent step, an alkyl radical is formed on the Cβ of the penultimate valine. This radical attacks the αβ-unsaturated bond resulting in the formation of a carbon–carbon bond between Cβ of valine and the Cα of tyrosine. Upon the injection of an additional electron and proton, the remaining radical on the Cβ of tyrosine becomes quenched, and the resulting product is a peptide containing a new five-membered ring between the penultimate valine and the C-terminal tyrosine (30). Although this mechanism requires the abstraction and insertion of electrons and protons, it is unclear which structural features of MftC serve these functions.
Thioether bond formation: AlbA, SkfB, ThnB, QhpD, CteB, and SCIFF maturase
By far the most diverse of the enzymes in the RS-RiPPs class are those responsible for the formation of thioether bonds. The bonds formed are between a cysteine sulfur and the α-, β-, or γ-carbon of another residue. The reaction has been shown to be initiated by abstraction of a hydrogen from the carbon atom that will form the thioether (31, 32). The resulting alkyl radical forms a bond with a thiol of a nearby cysteine resulting in a cyclized peptide. Interestingly, most members of this group (AlbA, QhpD, ThnB, CteB, and SkfB) have been found to carry out multiple modifications on the same peptide, distinguishing them from the rest of the enzymes in Table 1, which modify only a single site in their substrates (33–37). In addition, the diverse physiological functions for the products of these RS enzymes, which include antibiotics (e.g. subtilosin A and thuricin H), a growth regulator (sporulation killing factor), and an enzyme subunit (QhpC), have no obvious similarities. It is likely that RS-SPASM proteins have been recruited to generate thioether bonds on peptide substrates due to their proficiency in activating C–H bonds through the use of free radical chemistry.
Table 1.
Biochemically verified pathways with RS-SPASM activities
Outlier modifications
The remaining RS-SPASM enzyme that has been mechanistically characterized is AnSME. As mentioned above, AnSME catalyzes the anaerobic transformation of cysteine (or serine) to FGly, the active component in sulfatases (18, 38). A brief summary of the AnSME mechanism has been presented in Fig. 2B, showing the three-step process of hydrogen atom abstraction at the β-position of an active cysteine residue of the sulfatase to form an alkyl radical, an extraction of the alkyl radical electron that leads to collapse of the thiolate to form a thiocarbonyl, and its subsequent hydrolysis to produce hydrogen sulfide and the catalytic formyl glycine, FGly (18).
Auxiliary cluster content and function
The proteins summarized in Table 1 are annotated as containing, in addition to the canonical RS [4Fe-4S] cluster, most generally two but sometimes only one auxiliary Fe-S cluster. Establishing the exact number of such auxiliary clusters has been found to be technically challenging and frequently ambiguous, given the possibility of cofactor deterioration during protein isolation. For example, the original paper characterizing AlbA reported a single auxiliary [4Fe-4S] cluster (33); however, mutagenesis has suggested recently that AlbA contains two auxiliary clusters (32). PqqE was originally thought to have a single auxiliary cluster as well (39), but subsequent work has demonstrated that two auxiliary clusters are present (7). Likewise for StrB, an early paper suggested the protein contains a single auxiliary cluster; however, a subsequent report indicated that there are likely two auxiliary clusters present (20, 23).
Although a determination of redox potentials for the separate Fe-S clusters in RS-SPASM proteins has not yet been achieved, their ability to function may be affected differently by the choice of reductant used in in vitro assays. For example, PqqE was found to be unable to modify its substrate in the presence of the low potential reductant dithionite, whereas flavodoxin and flavodoxin reductase led to peptide cross-linking (7). In contrast, the remaining entries in Table 1 are able to catalyze both SAM cleavage and subsequent peptide modification using dithionite as the electron source. The dependence of successful peptide modifications on a reducing agent suggests that differences in redox potentials, particularly among the auxiliary clusters, will impact their precise mechanistic role(s). Even less clear is the detailed mechanistic role played by each of these clusters; as noted, proposed mechanisms for the carbon–carbon bond and thioether formation often include the oxidation of a substrate-derived intermediate as a final chemical step, which arguably could be catalyzed by auxiliary clusters. However, experimental validation of such a mechanistic feature remains inadequate. Furthermore, there is currently no clear relationship between the number of reported auxiliary clusters and the type of chemistry being carried out. For instance, QhpD and SCIFF maturase have two auxiliary clusters, and SkfB and ThnB have been reported to have only one each, yet all four form thioether bonds. Given the large number of unanswered and challenging questions, this aspect of RS-SPASM function is ripe for further investigation.
Toward a structural basis of protein–protein interactions within the RS-RiPP family
The crystal structure of Xanothomonas campestris (Fig. 3A) PqqD was made available nearly a decade ago (40). From this structure, it was postulated that PqqD could serve as a PQQ-releasing mechanism, a scaffold for protein complexes, or as a PQQ carrier (4, 40). These hypotheses were fed, in part, by the dimeric state of PqqD found in the crystal structure (Fig. 3A). Although it was ultimately shown that PqqD serves as a peptide chaperone by interacting with PqqE and PqqA (7), it was the publication of small-angle X-ray scattering, size-exclusion chromatography, and native mass spectrometry data that indicated the solution structure of PqqD is, in fact, a monomer (6). While the oligomeric state of PqqD may seem nuanced, the monomeric protein provided new opportunities to study the interactions of PqqD with PqqA and PqqE by solution NMR.
Figure 3.

A, X. campestris PqqD crystal structure (PDB code 3G2B) determined the oligomeric state to be a dimer. B, solution NMR 1H,15N-HSQC experiments on the monomeric M. extorquens AM1 PqqD (PDB code 5SXY) identify residues with significant chemical shifts when bound to PqqA (violet residues) and PqqE (green residues). C, homology-based modeling predicts that the putative peptide chaperone domains of MftB (light blue), QhpD (blue), AlbA (cyan), ThnB (violet), StrB (partial, light gray), and SCIFF maturase (magenta) all have similar structures to that of PqqD (dark gray). D, overlay of the pathway peptides from NisB (light blue) and LynD (dark blue) onto the structure for PqqD, where purple represents residues that are altered in the PqqD–PqqA complex, and green represents positions altered in the ternary complex consisting of PqqD, PqqA, and PqqE. E, complex of MccB with its cognate substrate where the peptide is colored purple.
Recently, the NMR assignments were published for 1H-, 13C-, and 15N-labeled PqqD from M. extorquens, both in the presence and absence of PqqA (41). Notably, more than half of the ∼90 residues on PqqD accrued a chemical shift. In fact, the study implied that most residues in PqqD are affected by the binding of PqqA, suggesting that either PqqA “wraps around” PqqD or that PqqD contributes a large surface for binding of PqqA. In a follow-up study with PqqD, the solution NMR structure has been solved, a first for peptide chaperones (42). The core structure is shown to be nearly identical to the crystal structure (root mean square deviation ∼1.9 Å) except for one notable feature; the solution structure is a monomer and therefore the β1 and β2 strands are not domain-swapped (Fig. 3B). In addition to solving the solution structure for PqqD, Evans et al. (42) conducted 1H,15N-HSQC-binding experiments with PqqA and PqqE. These inaugural experiments provide extraordinary information regarding protein–peptide and protein–protein interactions found in the PQQ biosynthetic pathway. First, Evans et al. (42) identified PqqD residues involved, directly or indirectly, in binding PqqA. Strikingly, these residues (Fig. 3B, violet) lie at the interface between the α-helical bundle and the β-sheet and implicate a hot spot that enters into both H-bonds and hydrophobic interactions with PqqA. Moreover, in similar HSQC experiments in the presence of PqqE, new residues were identified for the binding of the PqqD–PqqA binary complex to PqqE (Fig. 3B, green); a total of seven such side chains are implicated, among which there are three surface residues, two of which are near the C and N termini of the protein. These surface residues, consisting of Ser, Asp, and Arg, suggest a role for salt bridges or hydrogen bonds at one binding interface of PqqD with PqqE. There is an additional network of four hydrophobic residues on the opposite face of PqqD that may either contact PqqE (cf. 19) or undergo a conformational change upon ternary complex formation.
Although PqqD is currently the most characterized RS-SPASM peptide chaperone system, bioinformatic evidence has been presented implicating the usage of peptide chaperones in a large number of RS-SPASM-dependent RiPP pathways (6). This evidence has been substantiated by two independent studies in the mycofactocin and thurincin H biosynthetic pathways. In the mycofactocin biosynthetic pathway, the peptide chaperone is a stand-alone protein (MftB), and in the thurincin H biosynthetic pathway, the peptide chaperone is fused to the N terminus of the RS protein (ThnB). Although morphologically different, it was shown in both systems that the peptide chaperone protein/domain is required for catalytic turnover by the RS protein (28, 29, 35). These findings led us to the hypothesis that the PqqD domain is ubiquitous among the remaining characterized RS-SPASM proteins (excluding AnSME). To test this hypothesis, we turned to the sequences of Uniprot annotated PqqD domains (AlbA, ThnB, PqqD, and MftB) for homology-based structural modeling (43, 44). For RS-SPASM proteins without annotated PqqD domains (SkfB, SCIFF maturase, StrB, and QhpD), we used the N-terminal 80–100 amino acids leading up to the annotated RS domain for homology-based structural modeling. When the modeled structures are overlain with the structure of PqqD (Fig. 3C), it becomes clear that the N-terminal domains of ThnB, AlbA, SCIFF maturase, StrB, and QhpD are likely peptide chaperones (neither RaptorX nor Phyre2.0 provided a structure for SkfB). All available models show similar structural arrangements consisting of an α-helical bundle flanked on one side by two or three β-strands. Even though peptide chaperones are not ubiquitous across all RS pathways (e.g. AnSME and YydG, an RS-peptide epimerase (45)), there is a strong connection between N-terminal fusion peptide chaperones and RS-SPASM pathways that utilize a peptide as a substrate. The timely publication of the structure for CteB provides support for this proposal (19).
Peptide chaperones are not unique to RS-SPASM-dependent pathways. As mentioned previously, Mitchell and co-workers (9) used bioinformatics to demonstrate that peptide chaperones (defined as “RRE domains”) are utilized in over half of all known RiPP classes. Their study further characterized a handful of RRE domains by measuring dissociation constants with their respective peptide substrate; most were determined to be sub-micromolar. Strengthening these findings, crystal structures of several RRE domains found in RiPP pathways are available. Examples of the available structures are NisB (PDB code 4WD9) and LynD (nisin biosynthesis, PDB code 4V1T), both of which have been co-crystallized with their respective peptides (46, 47). Significantly, the RRE domains of NisB and LynD are structurally homologous to PqqD, and they allow us to visualize a possible binding mode for the interaction of PqqA with PqqD (Fig. 3D). However, the orientation of bound peptides in MccB (microcin C7 biosynthesis, PDB code 3H9G) is distinctive. As shown in Fig. 3E, the peptide–MccB interaction is between the RRE domain's β-sheet and the adenylase domain (gray), with interactions that are less evident or non-existent in the other characterized systems. This divergence of binding orientations and domain interactions furthers our knowledge gap about the structure–function relationship within this class of proteins and cautions against premature generalizations.
Future directions
Although the RS-SPASM field has become an exciting area of research, difficult biochemical questions must be answered if accurate functional prediction of the >18,000 RS-SPASM enzymes3 is to be realized. For instance, which enzyme structural features dictate thioether bond formation over carbon–carbon bond formation or is this encoded in the peptide substrate itself? To answer this question, the community will need to solve significantly more RS-SPASM protein structures. In addition, for all RS-SPASM proteins that have been characterized to date, the general reaction mechanism requires a process of gaining or losing an electron and proton. Which structural features lie behind these processes? Although site-directed mutagenesis data can support a role for the auxiliary clusters in long-range electron transfer, little additional experimental evidence has been provided. The latter question may best be answered using electrochemical techniques that are able to define the redox potentials for the individual auxiliary clusters, analogous to what has been accomplished in the closely related RS-TWITCH proteins (48, 49). In terms of proton transfer, adequate structural coverage of the RS-SPASM family will be required to fully evaluate which, if any, residues are able to function in this regard.
How prolific are the peptide chaperones and why are they required? Despite the difficulty of annotating a peptide substrate, we previously postulated that homologues of PqqD will be important for function in, at a minimum, ∼50% of all RS-SPASM proteins (6). Although only PqqD, MftB, and now CteB have been validated in this regard, using predictive structural homology we anticipate a peptide chaperone function for N-terminal fusions in virtually all RS-SPASM proteins studied to date (Table 1, with the exception of AnSME). Other than CteB (19), it remains unclear where the peptide chaperone associates with its RS-SPASM protein partner and the extent to which the SPASM domains containing the auxiliary iron–sulfur centers will participate in this binding process. Among a sea of unanswered questions, establishing a structural and functional link of SPASM domains to RS proteins that act on RiPPs constitutes one of the least understood and potentially fascinating future directions.
Since the inaugural study of AnSME nearly a decade ago, the RS-SPASM field has undergone a renaissance period of discovery. Currently, four distinct chemistries can be attributed to RS-SPASM proteins: thioether and carbon–carbon bond formation; formyl glycine formation, and oxidative decarboxylation. However, the diversity of their products, including a redox cofactor, antibiotics, quorum-sensing molecules, growth regulators, and mature proteins, is notable, presaging the emergence of fascinating new chemistries, mechanisms, and natural products.
This work was supported by National Institutes of Health Grants GM118117-02 (to J. P. K.) and GM124002-01 (to J. A. L.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
To acquire this value, we used the Interpro Database accession number IPR023885 for SPASM proteins and summed the number of proteins in the top three domain architectures that consisted of an RS domain (IPR007197) and a SPASM domain.
- RiPP
- ribosomally synthesized and post-translationally modified peptide
- SAM
- S-adenosylmethionine
- PQQ
- pyrroloquinoline quinone
- AnSME
- anaerobic sulfatase-maturating enzyme
- FGly
- Cα-formylglycine
- HSQC
- heteronuclear single quantum coherence
- PDB
- Protein Data Bank
- TIM
- triose-phosphate isomerase.
References
- 1. Arnison P. G., Bibb M. J., Bierbaum G., Bowers A. A., Bugni T. S., Bulaj G., Camarero J. A., Campopiano D. J., Challis G. L., Clardy J., Cotter P. D., Craik D. J., Dawson M., Dittmann E., Donadio S., et al. (2013) Ribosomally synthesized and post-translationally modified peptide natural products: overview and recommendations for a universal nomenclature. Nat. Prod. Rep. 30, 108–160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Booker S. J. (2012) Radical SAM enzymes and radical enzymology. Biochim. Biophys. Acta. 1824, 1151–1153 [DOI] [PubMed] [Google Scholar]
- 3. Broderick J. B., Duffus B. R., Duschene K. S., and Shepard E. M. (2014) Radical S-adenosylmethionine enzymes. Chem. Rev. 114, 4229–4317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Shen Y.-Q., Bonnot F., Imsand E. M., RoseFigura J. M., Sjölander K., and Klinman J. P. (2012) Distribution and properties of the genes encoding the biosynthesis of the bacterial cofactor, pyrroloquinoline quinone. Biochemistry 51, 2265–2275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Wecksler S. R., Stoll S., Iavarone A. T., Imsand E. M., Tran H., Britt R. D., and Klinman J. P. (2010) Interaction of PqqE and PqqD in the pyrroloquinoline quinone (PQQ) biosynthetic pathway links PqqD to the radical SAM superfamily. Chem. Commun. 46, 7031–7033 [DOI] [PubMed] [Google Scholar]
- 6. Latham J. A., Iavarone A. T., Barr I., Juthani P. V., and Klinman J. P. (2015) PqqD is a novel peptide chaperone that forms a ternary complex with the radical S-adenosylmethionine protein PqqE in the pyrroloquinoline quinone biosynthetic pathway. J. Biol. Chem. 290, 12908–12918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Barr I., Latham J. A., Iavarone A. T., Chantarojsiri T., Hwang J. D., and Klinman J. P. (2016) The pyrroloquinoline quinone (PQQ) biosynthetic pathway: demonstration of de novo carbon–carbon cross-linking within the peptide substrate (PqqA) in the presence of the radical SAM enzyme (PqqE) and its peptide chaperone (PqqD). J. Biol. Chem. 291, 8877–8884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Haft D. H., and Basu M. K. (2011) Biological systems discovery in silico: radical S-adenosylmethionine protein families and their target peptides for post-translational modification. J. Bacteriol. 193, 2745–2755 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Burkhart B. J., Hudson G. A., Dunbar K. L., and Mitchell D. A. (2015) A prevalent peptide-binding domain guides ribosomal natural product biosynthesis. Nat. Chem. Biol. 11, 564–570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Benjdia A., Subramanian S., Leprince J., Vaudry H., Johnson M. K., and Berteau O. (2008) Anaerobic sulfatase-maturating enzymes, first dual substrate radical S-adenosylmethionine enzymes. J. Biol. Chem. 283, 17815–17826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Grove T. L., Lee K. H., St Clair J., Krebs C., and Booker S. J. (2008) In vitro characterization of AtsB, a radical SAM formylglycine-generating enzyme that contains three [4Fe-4S] clusters. Biochemistry 47, 7523–7538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Fang Q., Peng J., and Dierks T. (2004) Post-translational formylglycine modification of bacterial sulfatases by the radical S-adenosylmethionine protein AtsB. J. Biol. Chem. 279, 14570–14578 [DOI] [PubMed] [Google Scholar]
- 13. Goldman P. J., Grove T. L., Sites L. A., McLaughlin M. I., Booker S. J., and Drennan C. L. (2013) X-ray structure of an AdoMet radical activase reveals an anaerobic solution for formylglycine post-translational modification. Proc. Natl. Acad. Sci. U.S.A. 110, 8519–8524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Frey P. A., Hegeman A. D., and Ruzicka F. J. (2008) The radical SAM superfamily. Crit. Rev. Biochem. Mol. Biol. 43, 63–88 [DOI] [PubMed] [Google Scholar]
- 15. Dowling D. P., Vey J. L., Croft A. K., and Drennan C. L. (2012) Structural diversity in the AdoMet radical enzyme superfamily. Biochim. Biophys. Acta 1824, 1178–1195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Shisler K. A., and Broderick J. B. (2012) Emerging themes in radical SAM chemistry. Curr. Opin. Struct. Biol. 22, 701–710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Grell T. A., Goldman P. J., and Drennan C. L. (2015) SPASM and twitch domains in AdoMet radical enzyme structures. J. Biol. Chem. 290, 3964–3971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Benjdia A., Subramanian S., Leprince J., Vaudry H., Johnson M. K., and Berteau O. (2010) Anaerobic sulfatase-maturating enzyme–a mechanistic link with glycyl radical-activating enzymes? FEBS J. 277, 1906–1920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Grove T. L., Himes P. M., Hwang S., Yumerefendi H., Bonanno J. B., Kuhlman B., Almo S. C., and Bowers A. A. (2017) Structural insights into thioether bond formation in the biosynthesis of sactipeptides. J. Am. Chem. Soc. 10.1021/jacs.7b01283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Schramma K. R., Bushin L. B., and Seyedsayamdost M. R. (2015) Structure and biosynthesis of a macrocyclic peptide containing an unprecedented lysine-to-tryptophan crosslink. Nat. Chem. 7, 431–437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ibrahim M., Guillot A., Wessner F., Algaron F., Besset C., Courtin P., Gardan R., and Monnet V. (2007) Control of the transcription of a short gene encoding a cyclic peptide in Streptococcus thermophilus: a new quorum-sensing system? J. Bacteriol. 189, 8844–8854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Gardan R., Besset C., Guillot A., Gitton C., and Monnet V. (2009) The oligopeptide transport system is essential for the development of natural competence in Streptococcus thermophilus strain LMD-9. J. Bacteriol. 191, 4647–4655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Benjdia A., Decamps L., Guillot A., Kubiak X., Ruffié P., Sandström C., and Berteau O. (2017) Insights into catalysis of lysine-tryptophan bond in bacterial peptides by a SPASM-domain radical SAM peptide cyclase. J. Biol. Chem. 292, 10835–10844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lilla E. A., and Yokoyama K. (2016) Carbon extension in peptidylnucleoside biosynthesis by radical-SAM enzymes. Nat. Chem. Biol. 12, 905–907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Hover B. M., Tonthat N. K., Schumacher M. A., and Yokoyama K. (2015) Mechanism of pyranopterin ring formation in molybdenum cofactor biosynthesis. Proc. Natl. Acad. Sci. U.S.A. 112, 6347–6352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Haft D. H. (2011) Bioinformatic evidence for a widely distributed, ribosomally produced electron carrier precursor, its maturation proteins, and its nicotinoprotein redox partners. BMC Genomics 12, 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Haft D. H., Pierce P. G., Mayclin S. J., Sullivan A., Gardberg A. S., Abendroth J., Begley D. W., Phan I. Q., Staker B. L., Myler P. J., Marathias V. M., Lorimer D. D., and Edwards T. E. (2017) Mycofactocin-associated mycobacterial dehydrogenases with non-exchangeable NAD cofactors. Sci. Rep. 7, 41074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Khaliullin B., Aggarwal P., Bubas M., Eaton G. R., Eaton S. S., and Latham J. A. (2016) Mycofactocin biosynthesis: modification of the peptide MftA by the radical S-adenosylmethionine protein MftC. FEBS Lett. 590, 2538–2548 [DOI] [PubMed] [Google Scholar]
- 29. Bruender N. A., and Bandarian V. (2016) The radical S-adenosyl-l-methionine enzyme MftC catalyzes an oxidative decarboxylation of the C-terminus of the MftA peptide. Biochemistry 55, 2813–2816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Khaliullin B., Ayikpoe R., Tuttle M., and Latham J. A. (2017) Mechanistic elucidation of the mycofactocin-biosynthetic radical-S-adenosylmethionine protein, MftC. J. Biol. Chem. 292, 13022–13033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Bruender N. A., and Bandarian V. (2016) SkfB abstracts a hydrogen atom from Cα on SkfA to initiate thioether crosslink formation. Biochemistry 55, 4131–4134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Benjdia A., Guillot A., Lefranc B., Vaudry H., Leprince J., and Berteau O. (2016) Thioether bond formation by SPASM domain radical SAM enzymes: Cα H-atom abstraction in subtilosin A biosynthesis. Chem. Commun. 52, 6249–6252 [DOI] [PubMed] [Google Scholar]
- 33. Flühe L., Knappe T. A., Gattner M. J., Schäfer A., Burghaus O., Linne U., and Marahiel M. A. (2012) The radical SAM enzyme AlbA catalyzes thioether bond formation in subtilosin A. Nat. Chem. Biol. 8, 350–357 [DOI] [PubMed] [Google Scholar]
- 34. Flühe L., Burghaus O., Wieckowski B. M., Giessen T. W., Linne U., and Marahiel M. A. (2013) Two [4Fe-4S] clusters containing radical SAM enzyme SkfB catalyze thioether bond formation during the maturation of the sporulation killing factor. J. Am. Chem. Soc. 135, 959–962 [DOI] [PubMed] [Google Scholar]
- 35. Wieckowski B. M., Hegemann J. D., Mielcarek A., Boss L., Burghaus O., and Marahiel M. A. (2015) The PqqD homologous domain of the radical SAM enzyme ThnB is required for thioether bond formation during thurincin H maturation. FEBS Lett. 589, 1802–1806 [DOI] [PubMed] [Google Scholar]
- 36. Nakai T., Ito H., Kobayashi K., Takahashi Y., Hori H., Tsubaki M., Tanizawa K., and Okajima T. (2015) The radical S-adenosyl-l-methionine enzyme QhpD catalyzes sequential formation of intra-protein sulfur-to-methylene carbon thioether bonds. J. Biol. Chem. 290, 11144–11166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Bruender N. A., Wilcoxen J., Britt R. D., and Bandarian V. (2016) Biochemical and spectroscopic characterization of a radical SAM enzyme involved in the formation of a peptide thioether crosslink. Biochemistry 55, 2122–2134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Schmidt B., Selmer T., Ingendoh A., and von Figura K. (1995) A novel amino acid modification in sulfatases that is defective in multiple sulfatase deficiency. Cell 82, 271–278 [DOI] [PubMed] [Google Scholar]
- 39. Wecksler S. R., Stoll S., Tran H., Magnusson O. T., Wu S.-P., King D., Britt R. D., and Klinman J. P. (2009) Pyrroloquinoline quinone biogenesis: demonstration that PqqE from Klebsiella pneumoniae is a radical S-adenosyl-l-methionine enzyme. Biochemistry 48, 10151–10161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Tsai T.-Y., Yang C.-Y., Shih H.-L., Wang A. H., and Chou S.-H. (2009) Xanthomonas campestris PqqD in the pyrroloquinoline quinone biosynthesis operon adopts a novel saddle-like fold that possibly serves as a PQQ carrier. Proteins 76, 1042–1048 [DOI] [PubMed] [Google Scholar]
- 41. Evans R. L. 3rd., Latham J. A., Klinman J. P., Wilmot C. M., and Xia Y. (2016) 1H, 13C, and 15N resonance assignments and secondary structure information for Methylobacterium extorquens PqqD and the complex of PqqD with PqqA. Biomol. NMR Assign. 10, 385–389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Evans R. L. 3rd., Latham J. A., Xia Y., Klinman J. P., and Wilmot C. M. (2017) NMR structural and binding studies of PqqD, a chaperone required in the biosynthesis of the bacterial dehydrogenase cofactor pyrroloquinoline quinone. Biochemistry 56, 2735–2746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Peng J., and Xu J. (2011) Raptorx: Exploiting structure information for protein alignment by statistical inference. Proteins 79, 161–171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Kelley L. A., and Sternberg M. J. (2009) Protein structure prediction on the Web: a case study using the Phyre server. Nat. Protoc. 4, 363–371 [DOI] [PubMed] [Google Scholar]
- 45. Benjdia A., Guillot A., Ruffié P., Leprince J., and Berteau O. (2017) synthesized peptides by a radical SAM epimerase in Bacillus subtilis. Nat. Chem. 9, 698–707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Ortega M. A., Hao Y., Zhang Q., Walker M. C., van der Donk W. A., and Nair S. K. (2015) Structure and mechanism of the tRNA-dependent lantibiotic dehydratase NisB. Nature 517, 509–512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Koehnke J., Mann G., Bent A. F., Ludewig H., Shirran S., Botting C., Lebl T., Houssen W., Jaspars M., and Naismith J. H. (2015) Structural analysis of leader peptide binding enables leader-free cyanobactin processing. Nat. Chem. Biol. 11, 558–563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Blaszczyk A. J., Silakov A., Zhang B., Maiocco S. J., Lanz N. D., Kelly W. L., Elliott S. J., Krebs C., and Booker S. J. (2016) Spectroscopic and electrochemical characterization of the iron–sulfur and cobalamin cofactors of TsrM, an unusual radical S-adenosylmethionine methylase. J. Am. Chem. Soc. 138, 3416–3426 [DOI] [PubMed] [Google Scholar]
- 49. Maiocco S. J., Grove T. L., Booker S. J., and Elliott S. J. (2015) Electrochemical resolution of the [4Fe-4S] centers of the AdoMet radical enzyme BtrN: evidence of proton coupling and an unusual, low-potential auxiliary cluster. J. Am. Chem. Soc. 137, 8664–8667 [DOI] [PubMed] [Google Scholar]