

Abstract
HIV fusion with the cell membrane can be inhibited by blocking coreceptor binding or by preventing fusion-inducing conformational changes in the Env protein. Logically, inhibitors that act by these two mechanisms should act synergistically, but previous studies have reported conflicting results. A new study by Ahn and Root reconciles these discordant reports by demonstrating that synergy emerges when Env engages multiple coreceptors prior to inducing fusion and when high-affinity inhibitory peptides are used, a condition that may not be satisfied in vivo.
Introduction
Glycoproteins of enveloped viruses, including the HIV envelope glycoprotein (Env),2 initiate infection by promoting membrane fusion. Env, arguably the most intensely studied viral fusion protein, is a trimer of gp120 and gp41 heterodimers that is activated upon sequential engagement of the receptor CD4 and the coreceptors, CCR5 or CXCR4, by the gp120 subunit (1). Binding to CD4 and the coreceptors loosens the interaction between gp120 and the transmembrane gp41 subunit, driving the transition to pre-hairpin intermediates characterized by the formation of the gp41 N-terminal coiled-coil and the C-terminal heptad repeat domains (Fig. 1) (2, 3). The gp41 pre-hairpin engages a target membrane through insertion of the hydrophobic N-terminal fusion peptide, thus coupling its subsequent folding into a stable trimer of hairpins to the merger of the viral and cellular membranes. The overwhelming majority of virions fail to undergo fusion, due in part to off-path conformational changes leading to irreversible functional inactivation of Env (Fig. 1, bottom).
Figure 1.
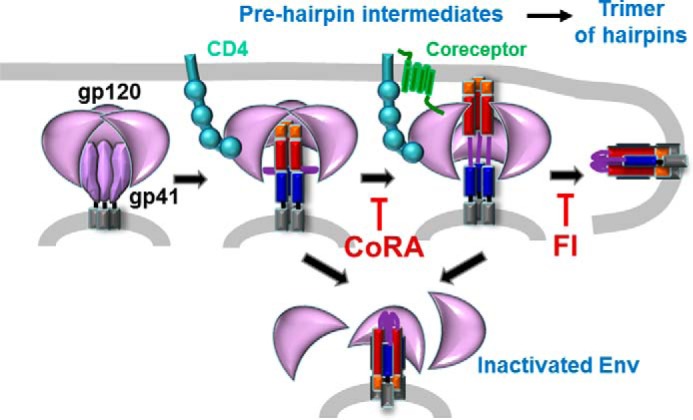
A model for HIV Env-mediated fusion. Fusion-promoting conformational changes in Env glycoprotein mediated by binding to CD4 and coreceptor are illustrated. Red and blue rectangles represent N- and C-terminal heptad repeat domains of gp41, respectively; orange boxes are the fusion peptides.
The kinetic competition between productive refolding and irreversible inactivation of Env can be studied by varying the experimental conditions (4–7). An alternative approach to delineate the interplay between these processes is introduced in this issue of JBC by Ahn and Root (8), who examined the interaction between fusion inhibitors targeting distinct steps of the fusion reaction. These inhibitors include the HIV coreceptor antagonists (CoRAs), which interrupt the interaction between Env and the host coreceptor, and fusion inhibitory peptides (FIs), which block the transition of gp41 pre-hairpins to the final hairpin structure. As CoRAs serve to prolong the exposure of pre-hairpin intermediates targeted by FIs, their activity should be synergistic, and indeed potentiation of FIs by CoRA, and by decreasing coreceptor density on target cells, has been previously demonstrated (e.g. Ref. 9). However, synergistic interaction between these inhibitors has not been reproducibly observed.
Ahn and Root (8) explore the basis for drug synergy by examining the viral and cellular factors affecting synergy between CoRAs and FIs. This group has previously shown that the potency of FIs that bind the gp41 pre-hairpins with very high affinity is kinetically limited by the relatively short lifetime of these intermediates (5, 6). By comparison, the potency of the weakly binding FIs is proportional to the binding affinity and is less affected by the fusion kinetics that define the time of pre-hairpin exposure. The potency of the commonly used medium-affinity enfuvirtide (also known as T20) (4) appears to be dependent on both the binding affinity and the pre-hairpin lifetime. In their efforts to elucidate the basis of CoRA/FI synergy, Ahn and Root (8) assembled a panel of FIs and Env mutants to progressively reduce the inhibitor-binding affinity and thereby render the potency of inhibitory peptides insensitive to the fusion kinetics. In addition, Env-coreceptor binding stoichiometry was varied by producing mixed wild-type/mutant trimers containing only 1 or 2 coreceptor-binding sites. Measurements of infectivity under these conditions revealed that synergy between the two inhibitors depended not only on the FI-binding affinity, but also on coreceptor density and the coreceptor-Env binding stoichiometry. In contrast to the high-affinity FIs, the low-affinity peptides did not synergize with CoRAs, consistent with the diminished dependence of these FIs on the pre-hairpin exposure time. Similarly, reducing the stoichiometry of coreceptor binding to Env or using target cells expressing low levels of coreceptor-abrogated synergy, irrespective of the FI-gp41 binding affinity.
These findings suggest that the minimal number of Env-bound coreceptor molecules required for HIV fusion (likely 1 coreceptor per trimer) is lower than the number that supports synergy between the two inhibitors. The synergy modulation results support the model proposed by Ahn and Root (see also Refs. 5 and 6) that binding of additional coreceptor(s) to Env accelerates the pre-hairpin to hairpin conversion and, at the same time, increases the inactivation rate of the FI-bound Env. Thus, in addition to offering these mechanistic insights into HIV fusion, this study helps reconcile the divergent findings related to CoRA/FI interactions by demonstrating a complex dependence of synergy on viral and cellular factors that affect the fusion and inactivation kinetics. The working model predicts that drug synergy in patients may be observed, depending on the requisite coreceptor stoichiometry of Env variants and on the coreceptor expression levels on host cells. One would anticipate that the CoRA maraviroc would exhibit stronger synergy with the high-affinity second generation T1249 peptide than with enfuvirtide, which binds gp41 with modest affinity (4).
Viral fusion in general, and HIV Env-mediated fusion in particular, is an asynchronous stochastic process that is not readily amenable to studies using conventional techniques. The Root laboratory has developed a unique set of experimental tools to dissect the fusion process, including inhibitory peptides and gp41 mutants with well-characterized binding affinities and fusion kinetics, as well as gp120 mutants that are deficient in CD4 or coreceptor binding. This combination of genetic and functional approaches enabled the researchers to gain new insights into the kinetic determinants of HIV fusion and inactivation and suggested potential ways to exploit its vulnerability. In addition to aspects of HIV fusion examined in this study, it would be interesting to elucidate the effects of CD4 density and CD4 binding affinity on CoRA/FI synergy. The Root laboratory has recently shown that engagement of a single CD4 triggers symmetric/synchronous Env refolding, whereas binding of multiple CD4 molecules results in asymmetric activation of monomers within the trimer (7). It is thus likely that CD4 binding stoichiometry can also modulate the magnitude of CoRA/FI synergy. Further methodological improvements should help refine the working model for HIV fusion and inactivation. For instance, because coreceptor density can affect the postfusion steps of HIV entry through cellular signaling, it might be worth implementing a direct virus-cell fusion assay for studies of drug synergy. The development and application of single-molecule techniques, such as single-molecule FRET (10), will also help assess the dynamics of single Env refolding and inactivation under fusion-permissive conditions and in the presence of inhibitors. Insights from these studies would lead to better understanding of fundamental principles of viral fusion and will help design effective preventative strategies.
Acknowledgments
Critical reading and helpful comments on the manuscript by the members of the Melikyan laboratory are greatly appreciated. The HIV fusion work in the Melikyan laboratory is supported by the National Institutes of Health Grant R01 GM054787.
The author declares that he has no conflicts of interest with the contents of this article. The content is solely the responsibility of the author and does not necessarily represent the official views of the National Institutes of Health.
- Env
- HIV-1 envelope glycoprotein
- CoRA
- chemokine receptor antagonist
- FI
- fusion inhibitor.
References
- 1. Berger E. A., Doms R. W., Fenyö E. M., Korber B. T., Littman D. R., Moore J. P., Sattentau Q. J., Schuitemaker H., Sodroski J., and Weiss R. A. (1998) A new classification for HIV-1. Nature 391, 240. [DOI] [PubMed] [Google Scholar]
- 2. Wilen C. B., Tilton J. C., and Doms R. W. (2012) Molecular mechanisms of HIV entry. Adv. Exp. Med. Biol. 726, 223–242 [DOI] [PubMed] [Google Scholar]
- 3. Melikyan G. B. (2008) Common principles and intermediates of viral protein-mediated fusion: The HIV-1 paradigm. Retrovirology 5, 111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Champagne K., Shishido A., and Root M. J. (2009) Interactions of HIV-1 inhibitory peptide T20 with the gp41 N-HR coiled coil. J. Biol. Chem. 284, 3619–3627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Kahle K. M., Steger H. K., and Root M. J. (2009) Asymmetric deactivation of HIV-1 gp41 following fusion inhibitor binding. PLoS Pathog. 5, e1000674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Steger H. K., and Root M. J. (2006) Kinetic dependence to HIV-1 entry inhibition. J. Biol. Chem. 281, 25813–25821 [DOI] [PubMed] [Google Scholar]
- 7. Khasnis M. D., Halkidis K., Bhardwaj A., and Root M. J. (2016) Receptor activation of HIV-1 Env leads to asymmetric exposure of the gp41 trimer. PLoS Pathog. 12, e1006098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ahn K. W., and Root M. J. (2017) Complex interplay of kinetic factors governs the synergistic properties of HIV-1 entry inhibitors. J. Biol. Chem. 292, 16498–16510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Reeves J. D., Gallo S. A., Ahmad N., Miamidian J. L., Harvey P. E., Sharron M., Pohlmann S., Sfakianos J. N., Derdeyn C. A., Blumenthal R., Hunter E., and Doms R. W. (2002) Sensitivity of HIV-1 to entry inhibitors correlates with envelope/coreceptor affinity, receptor density, and fusion kinetics. Proc. Natl. Acad. Sci. U.S.A. 99, 16249–16254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Munro J. B., Gorman J., Ma X., Zhou Z., Arthos J., Burton D. R., Koff W. C., Courter J. R., Smith A. B. 3rd, Kwong P. D., Blanchard S. C., and Mothes W. (2014) Conformational dynamics of single HIV-1 envelope trimers on the surface of native virions. Science 346, 759–763 [DOI] [PMC free article] [PubMed] [Google Scholar]