

Abstract
Antithrombin (AT) is an anticoagulant serpin that irreversibly inactivates the clotting proteinases factor Xa and thrombin by forming covalent complexes with them. Mutations in its critical domains, such as those that impair the conformational rearrangement required for proteinase inactivation, increase the risk of venous thrombosis. Águila et al. characterize for the first time the destabilizing effects of mutations in the region of AT that makes contact with the proteinase in the final acyl-enzyme complex. Their work adds new insight into the unique structural intricacies of the inhibitory mechanism.
Introduction
Inhibitory serpins (serine proteinase inhibitors) belong to a superfamily of proteins with a characteristic fold of three β sheets, eight or nine α helices, and a reactive center loop (RCL)2 that contains a specific proteinase cleavage site (1). Of the ∼1,500 identified serpin gene sequences, 36 are present in humans where they perform diverse functions. Antithrombin (AT), one of the 29 known human inhibitory serpins, is a major regulator of hemostasis, as inhibition of its targets thrombin and factor Xa (FXa) leads to a reduction in clotting. Inhibitory serpins are metastable: They undergo one of the largest known conformational changes upon forming covalent complexes with their proteinase targets (2). Whereas the importance of the RCL structure in this conformational change is well documented, much less is known about the structural requirements of residues in the base of the serpin, in particular the region that becomes the “landing place” for the inactivated proteinase in the covalent complex. Águila et al. (3) now present clinical and biochemical proof that this region in the serpin is critical for inhibitory function and may be considered a new regulatory domain.
Serpin action has been likened to that of a mousetrap. In the Michaelis complex, the proteinase is docked to the RCL of the native serpin; upon cleavage of the reactive site in the RCL, an acyl-enzyme intermediate is formed, and the N-terminal RCL fragment inserts as an extra strand into the principal β sheet of AT, rapidly translocating the attached proteinase 180° to the distal end of the serpin (Fig. 1, A and B). In this energetically favorable and stable complex, the proteinase active site is distorted, and the serpin acts as a suicide substrate (1, 2, 4, 5). Proteinase translocation, final docking, and distortion are steps in the inhibitory pathway of the branched serpin mechanism (Fig. 1C). Disturbing the serpin structure favors the alternative side of the branched mechanism, the substrate pathway, in which the acyl-enzyme intermediate completes hydrolysis to form a cleaved serpin and regenerated proteinase.
Figure 1.
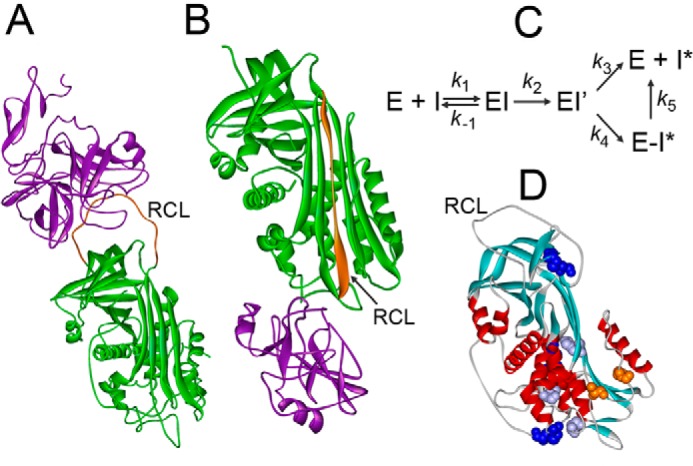
Mechanism of proteinase inactivation by serpins. A, Michaelis complex of S195A FXa and AT (Protein Data Bank code 2GD4). B, covalent complex of α1PI with trypsin (Protein Data Bank code 1EZX). Proteinases are shown in purple, serpins are shown in green, RCL are shown in orange. C, mechanism of enzyme (E) and serpin (I) forming a Michaelis complex (EI), acyl-enzyme (EI′), and partitioning between the substrate (k3) and inhibitory pathway (k4). Unstable E-I* complexes dissociate into free E and cleaved I* (k5). D, native AT with positions of Ser365 and Ile207 (orange); L340F, S349P, and H369Y (purple), and three disulfide bridges (blue) (Protein Data Bank code 1E05).
The elaborate structure of inhibitory serpins makes them particularly prone to dysfunction caused by mutations. Moderate and even mild AT deficiency, as observed in patients with heterozygous mutations, significantly increases the risk of deep vein thrombosis and pulmonary embolism. In type II AT deficiency, normal antigen levels are present but the inhibitory activity is reduced due to mutations in critical regions of the serpin. RCL mutations increase the contribution of the substrate pathway by affecting RCL insertion and proteinase translocation. Relative rates of kinetic trapping of the stable complex (k4) and the substrate reaction (k3) are altered, and the stoichiometry of inhibition (equal to (k3 + k4)/k4)) becomes larger than 1 (6). Dissociation of the final serpin-protease complex (k5) also increases this stoichiometry, and artificial perturbation of the serpin-proteinase interface has been shown to destabilize the final complex (7). Águila et al. (3) were the first to identify natural, destabilizing AT mutations at this interface in patients with AT type II deficiency (3) and to propose two potential mechanisms for impaired FXa and thrombin inactivation by these mutants.
In their new study, Águila et al. (3) consider five previously undescribed mutations outside key functional domains associated with type II AT deficiencies, I207T, L340F, S349P, S365L, and H369Y (Fig. 1D), and investigate two of these sites in depth. A patient with the S365P mutation was previously identified as type I-deficient, with poor AT expression. Moreover, Ser365 and Ile207 are highly conserved and are located in the area of contact with the protease in the covalent complex and near the RCL. Despite these features, it was not intuitively obvious how these residues would impact the inhibition mechanism. Studying these residues therefore provides an opportunity to assign functional roles to new regions of AT as well as to differentiate between mechanisms of increased substrate partitioning during RCL insertion versus instability of the final complex as possible causes for an increased inhibition stoichiometry.
To explore these mutations, the authors first measured AT activity and dimer formation in patient plasma. They also purified recombinant wild-type and mutant AT and characterized the proteins by kinetic and physical-chemical analysis. Unstable AT S365L behaved as a pure substrate for both FXa and thrombin. In contrast, AT I207T exhibited a modest increase in inhibition stoichiometry that was more predominant in heparin-catalyzed reactions. Heparin vastly accelerates the rates of thrombin and fXa inactivation by AT; however, it also moderately promotes the substrate pathway of the branched mechanism (8), and the findings of Águila et al. (3) are consistent with this. AT I207A, which was created for comparison, behaved largely as a substrate with both proteinases but with still measurable inhibitory activity. Multiplying the apparent second-order inactivation rate constant kapp by the stoichiometry of inhibition obtained from titrations, resulted in normalized kapp values that were similar to those measured for wild-type AT, except for reactions of AT I207A with FXa. In the latter reactions a 5–8-fold overcorrection was observed. Normalization to wild-type kapp values suggests that substrate partitioning results from decreasing the rates of the two-step process of proteinase translocation and stabilization of the final serpin-proteinase complex (9). In contrast, kapp overcorrections for AT I207A reacting with FXa suggested that additional cleaved AT was generated by partial dissociation of the final complex after its formation during end point titrations. No inhibition was seen with AT S365L, thus prohibiting the authors from distinguishing between these two different mechanisms underlying the effect of the mutation.
These important findings illustrate the intricate structural requirements of the serpin-proteinase interface for stable, covalent complex formation and warrants further studies on complex-stabilizing roles of the conserved residues Ser365 and Ile207 or their homologues in other serpins. For example, based on my initial scan of the structure, the propensity of the S365L but not I207T AT to form covalent dimers in plasma may be due to long-range destabilizing effects of the S365L mutation on the Cys21–Cys95 disulfide bond. As a simple proxy to evaluate whether stability might explain the authors' results, I used the Site-directed Mutator server (10) to probe the stability of the AT mutants reported by these authors. The resultant pseudoΔΔG stability scores I obtained did not point to inherent AT instability in the uncomplexed state for the S365L and S365P mutations (perhaps the impaired in vivo AT S365P expression in type I deficiency may be due to structurally altered mRNA?), whereas the I207T and I207A did seem to be less stable. I look forward to the authors' further mechanistic insights along these lines.
Águila et al. (3) have an extensive track record of defining functional effects of various AT mutations in elegant translational studies that connect clinical findings with altered biochemical properties of AT. The discovery of new AT mutations outside the RCL that promote proteinase recycling and AT consumption provides additional insight into the many subtleties of the AT mechanism and structure. Such insights could lead to new approaches to treating blood-clotting disorders.
This work was supported in part by National Institutes of Health Grant HL130018. The author declares that she has no conflicts of interest with the contents of this article. The content is solely the responsibility of the author and does not necessarily represent the official views of the National Institutes of Health.
- RCL
- reactive center loop
- AT
- antithrombin
- FXa
- factor Xa.
References
- 1. Olson S. T., and Gettins P. G. (2011) Regulation of proteases by protein inhibitors of the serpin superfamily. Prog. Mol. Biol. Transl. Sci. 99, 185–240 [DOI] [PubMed] [Google Scholar]
- 2. Stratikos E., and Gettins P. G. (1997) Major proteinase movement upon stable serpin-proteinase complex formation. Proc. Natl. Acad. Sci. U.S.A. 94, 453–458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Águila S., Izaguirre G., Martínez-Martínez I., Vicente V., Olson S. T., and Corral J. (2017) Disease-causing mutations in the serpin antithrombin reveal a key domain critical for inhibiting protease activities. J. Biol. Chem. 292, 16513–16520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Johnson D. J., Li W., Adams T. E., and Huntington J. A. (2006) Antithrombin-S195A factor Xa-heparin structure reveals the allosteric mechanism of antithrombin activation. EMBO J. 25, 2029–2037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Dementiev A., Dobó J., and Gettins P. G. (2006) Active site distortion is sufficient for proteinase inhibition by serpins: structure of the covalent complex of alpha1-proteinase inhibitor with porcine pancreatic elastase. J. Biol. Chem. 281, 3452–3457 [DOI] [PubMed] [Google Scholar]
- 6. Gettins P. G. (2002) Serpin structure, mechanism and function. Chem. Rev. 102, 4751–4804 [DOI] [PubMed] [Google Scholar]
- 7. Swanson R., Raghavendra M. P., Zhang W., Froelich C., Gettins P. G., and Olson S. T. (2007) Serine and cysteine proteases are translocated to similar extents upon formation of covalent complexes with serpins. Fluorescence perturbation and fluorescence resonance energy transfer mapping of the protease binding site in CrmA complexes with granzyme B and caspase-1. J. Biol. Chem. 282, 2305–2313 [DOI] [PubMed] [Google Scholar]
- 8. Izaguirre G., Aguila S., Qi L., Swanson R., Roth R., Rezaie A. R., Gettins P. G., and Olson S. T. (2014) Conformational activation of antithrombin by heparin involves an altered exosite interaction with protease. J. Biol. Chem. 289, 34049–34064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Maddur A. A., Swanson R., Izaguirre G., Gettins P. G., and Olson S. T. (2013) Kinetic intermediates en route to the final serpin-protease complex: studies of complexes of α1-protease inhibitor with trypsin. J. Biol. Chem. 288, 32020–32035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Worth C. L., Preissner R., and Blundell T. L. (2011) SDM–a server for predicting effects of mutations on protein stability and malfunction. Nucleic Acids Res. 39, W215–W222 [DOI] [PMC free article] [PubMed] [Google Scholar]