

Abstract
Ring-between-ring (RBR) E3 ligases have been implicated in autoimmune disorders and neurodegenerative diseases. The functions of many RBR E3s are poorly defined, and their regulation is complex, involving post-translational modifications and allosteric regulation with other protein partners. The functional complexity of RBRs, coupled with the complexity of the native ubiquitination reaction that requires ATP and E1 and E2 enzymes, makes it difficult to study these ligases for basic research and therapeutic purposes. To address this challenge, we developed novel chemical probes, ubiquitin C-terminal fluorescein thioesters UbMES and UbFluor, to qualitatively and quantitatively assess the activity of the RBR E3 ligase PARKIN in a simple experimental setup and in real time using fluorescence polarization. First, we confirmed that PARKIN does not require an E2 enzyme for substrate ubiquitination, lysine selection, and polyubiquitin chain formation. Second, we confirmed that UbFluor quantitatively detects naturally occurring activation states of PARKIN caused by Ser65 phosphorylation (pPARKIN) and phosphorylated ubiquitin (pUb). Third, we showed that both pUb and the ubiquitin-accepting substrate contribute to maximal pPARKIN ubiquitin conjugation turnover. pUb enhances the transthiolation step, whereas the substrate clears the pPARKIN∼Ub thioester intermediate. Finally, we established that UbFluor can quantify activation or inhibition of PARKIN by structural mutations. These results demonstrate the feasibility of using UbFluor for quantitative studies of the biochemistry of RBR E3s and for high-throughput screening of small-molecule activators or inhibitors of PARKIN and other RBR E3 ligases.
Keywords: enzyme, enzyme mechanism, parkin, Parkinson disease, ubiquitin, UbFluor
Introduction
Parkinson's disease is the second most common type of neurodegenerative disease, and it is characterized by defects in mitochondrial function. Ubiquitin ligase PARKIN and phosphatase and tensin homolog-induced putative Ser/Thr kinase 1 (PINK1) reconstitute a major pathway to eliminate dysfunctional mitochondria and are thought to protect dopaminergic neurons from oxidative stress (1). PINK1 acts upstream of PARKIN and is continuously imported to the mitochondria, where it is cleaved by PARL protease, exported back to the cytosol, and degraded by the ubiquitin proteasome system according to the N-end rule. Upon mitochondrial damage, PINK1 processing by PARL is inhibited, and PINK1 accumulates at the mitochondrial outer membrane (2–4). There PINK1 promotes the recruitment of cytoplasmic PARKIN and activation of its ubiquitin ligase activity (4). Active PARKIN causes extensive polyubiquitination of mitochondrial outer membrane proteins and triggers mitophagy (5). PARKIN ubiquitination is antagonized by deubiquitinases, such that a threshold amount of ubiquitination in a given time may be necessary to successfully signal mitophagy (6).
The mechanism of PARKIN activation is complex, but it is known that phosphorylation of Ser65 in the PARKIN Ubl domain and Ser65 in mono- and/or polyubiquitin chains by PINK1 promotes the mitochondrial translocation and activity of PARKIN, which in turn stimulate mitophagy (7–22). Dozens of mutations in both PINK1 and PARKIN have been found in patients with autosomal-recessive juvenile parkinsonism and have been extensively characterized in vitro and in cells (1). In the case of PARKIN, these mutations are characterized by diverse translocation and biochemical phenotypes (23–33). It is currently thought that pharmacological activators of PINK1 and/or PARKIN may provide potential therapy to treat Parkinson's disease. For example, it was shown that overexpression of PARKIN in rat and mouse Parkinson's disease models is neuroprotective, supporting this hypothesis (34–37). However, although pharmacological activators of PINK1 are known (38), activators of ubiquitin ligase PARKIN are not known in the literature, and attempts to develop them were not successful (39). To develop such probes, it is essential to understand Parkin enzymatic mechanisms.
PARKIN is a ring-between-ring (RBR)4 E3 ligase, which forms an obligatory RBR E3∼Ub thioester intermediate prior to the transfer of the ubiquitin onto the acceptor lysine (16, 40–42). PARKIN is composed of six distinct domains: an N-terminal ubiquitin-like domain (Ubl); a unique PARKIN-specific domain (UPD, also referred to as RING0); RING1; in-between-RING (IBR); repressor element (REP); and catalytic RING2 domains (supplemental Fig. S1E) (42–44). RING1 domain of PARKIN is thought to recruit E2∼Ub thioester (44), whereas the RING2 domain of PARKIN harbors the catalytic cysteine (40). Mutation of this catalytic cysteine (C431F) is found in some Parkinson's disease patients (45). Recently, it was shown that the RING2 domain of RBR E3 HHARI also harbors a weak Ub-binding site important for E2∼Ub/HHARI transthiolation, whereas the RING1–IBR–Ubl domains of PARKIN form another ubiquitin-binding surface important for PARKIN activity (46).
A shared feature of PARKIN and several other RBR ligases is autoinhibition, which is relieved by binding partners and/or by phosphorylation (47–51, 71). In autoinhibited PARKIN, the E2∼Ub-binding site on RING1 is blocked by the REP domain, whereas the catalytic cysteine on RING2 is blocked by the UPD domain (supplemental Fig. S1E) (16, 43, 44). Phosphorylation of PARKIN (pPARKIN) and pUb binding to PARKIN/pPARKIN disrupt these autoinhibitory conformations and activate PARKIN. Interestingly, although the activation roles of PINK1 and pUb have been investigated, the role of protein substrates in PARKIN catalytic turnover have escaped attention. PARKIN substrates can provide acceptor lysines that clear PARKIN∼Ub thioester, thus facilitating PARKIN catalytic turnover (52).
Most importantly, there is a lack of methods to a robust and simple quantifying of PARKIN and other RBR E3s activities, which is needed for both biochemical studies and small molecule screens. Typical in vitro ubiquitination assays use a reconstituted native cascade composed of at least five components and a substrate, if used: E1, E2, E3, ubiquitin, and ATP. In this setting, the assay is operationally complex and expensive, and there are many enzyme intermediates that make it difficult to conduct biochemical studies. Coupled with the complex autoregulatory mechanisms that govern the function of PARKIN and other RBR E3s, the complexity of native ubiquitination assays is a major bottleneck in assessing the activity of RBR E3 enzymes.
Recently developed electrophilic activity-based probes, such as UbVME and electrophilic E2∼Ub thioester mimics (53, 68), substantially reduce the complexity of PARKIN and other catalytic cysteine-containing E3 ligase assays. However, these probes are stoichiometric suicide inhibitors and therefore do not report on catalytic turnover, lack high throughput capabilities, and rely on Western blotting for quantitation. Absolute equantification-mass spectrometry (AQUA-MS) enables quantification of polyubiquitin chain formation by PARKIN over multiple rounds of Ub conjugation in vitro (8). Although this method offers exquisite sensitivity, it is not amenable to high-throughput screening, and it requires considerable expertise and instrumentation beyond typical laboratory operations.
To begin addressing these challenges, we previously demonstrated that a ubiquitin C-terminal thioester probe (ubiquitin mercaptoethanesulfonate, UbMES) and its fluorescent analogue UbFluor could bypass the need for E1, ATP, and E2 enzymes, thereby simplifying assessment of HECT E3 ligase activity in vitro (Fig. 1) (54–56). Because the resulting system bypasses the need for ATP and E1 and E2 enzymes, we called it bypassing system or ByS. We reasoned that the same system could be useful for RBRs, as RBR ligases also form an obligate E3∼Ub thioester prior to ligation. However, two unique features of RBR E3 ligases require consideration. First, unlike HECT ligases, RBRs are cysteine-rich. For example, human PARKIN has 20 surface-exposed cysteines that could potentially undergo non-specific transthiolation with Ub-MES (44). Second, PARKIN and other RBR ligases have complex, multistep activation mechanisms (69), and it was not clear at the outset of this work whether Ub-MES and its analogues could recapitulate these mechanisms. For example, it was not clear whether UbMES that lacks the E2 enzyme could sense activating mutations that disrupt REP/RING1 interface and open the E2 enzyme-binding site (44).
Figure 1.

Chemical activation of the C terminus of ubiquitin as a thioester (e.g. UbMES or UbFluor) can bypass the need for E1, E2, and ATP, downsizing the 5-component native cascade reaction (E1, E2, ATP, PARKIN, and Ub) to 2 components (PARKIN and ByS probes). Transthiolation with UbMES releases a mercaptoethanesulfonate group (MES), whereas transthiolation with UbFluor releases fluorescein thiol.
In this work, we report that E2-independent ByS probes can be directly and specifically charged to the catalytic cysteine of PARKIN or pPARKIN, resulting in native-like autoubiquitination, substrate ubiquitination, and polyUb chain formation.
We further adapted the E2-independent ByS to create UbFluor, a quantitative probe for assessment of PARKIN activity in vitro and for high-throughput screening to identify PARKIN activator compounds (Fig. 1 and Scheme 1). UbFluor is a fluorescent thioester that features a fluorescein thiol that is attached to the C terminus of the ubiquitin via a thioester bond. Effectively, UbFluor mimics an E2∼Ub thioester, except E2 enzyme is replaced with fluorescein. When UbFluor is charged to PARKIN's catalytic cysteine, it releases fluorescein thiol much like Ub∼E2 releases E2 enzyme upon transthiolation. The fluorescein release can be detected by fluorescence polarization (FP). Thus, fluorescence-based detection is built into the transthiolation reaction, enabling real-time monitoring of PARKIN catalytic turnover, while eliminating any need for extra reagent transfer steps or SDS-PAGE analysis. UbFluor can be used to analyze transthiolation under single turnover reaction conditions (excess PARKIN), similar to electrophilic Ub-VS/E2-Ub probes (53, 68) or an E2∼Ub discharge assay, or under multiple turnover reaction conditions (excess UbFluor), similar to the native ubiquitination assays. Using UbFluor, we verified PARKIN activation by Ser65 phosphorylation and by pUb binding, demonstrating that the ByS is sensitive to the native autoregulation mechanisms governing PARKIN activity. We also discovered that the PARKIN substrate Miro1 and pUb cooperatively increased PARKIN catalytic turnover. Thus, although most work in the area has focused on the roles of PINK1 and pUb on PARKIN activation, our results suggest that PARKIN substrates such as MIRO can also enhance PARKIN catalytic turnover.
Scheme 1.

Synthesis of UbFluor.
Finally, we demonstrated that UbFluor can detect unnatural activation or inactivation of PARKIN by point mutations that disrupt either the REP/RING1 domain or the UPD/RING2 domain interface or activation of PARKIN that lacks Ubl and UPD domains altogether, further validating the biochemical relevance of the UbFluor probe toward PARKIN mechanisms (44). These results validate UbFluor as a probe to study the biochemistry of PARKIN and other RBR E3 ligases and as a high-throughput screening tool to identify small molecules that target these enzymes.
Results
Chemically activated ubiquitin thioester recapitulates PARKIN function in a simplified two-component reaction
We first examined how well the E2-independent ByS reaction recapitulates PARKIN activity in the native cascade. To test whether the UbMES-based ByS recapitulates Ser65 phosphorylation-dependent activation, we prepared human PARKIN that was phosphorylated by Tribolium castaneum PINK1 (TcPINK1) on Ser65 (pPARKIN, supplemental Fig. S1A and B) (8) and ubiquitin mercaptoethanesulfonate thioester (UbMES) (57) as described previously (supplemental Fig. S1C). Miro1 (mitochondrial Rho1) is a calcium-sensitive regulator of mitochondrial dynamics on the outer mitochondrial membrane (58). Miro1 is a known substrate that is modified by pPARKIN both in cells and in in vitro assays (5, 14, 59). We prepared full-length human Miro1 (hMiro1) as well as an N-terminally truncated construct (MiroS) that are expressed more robustly and can be purified in larger amounts than hMiro1 (MiroS; hMiro1 residues 177–592 with a C-terminal His6 tag, 51.6 kDa, supplemental Fig. S1D) (60).
We examined pPARKIN autoubiquitination, MiroS substrate ubiquitination, and polyubiquitin chain formation using UbMES (Fig. 2). In the presence of UbMES, pPARKIN ubiquitinated both itself and MiroS (Fig. 2A, lanes 4 and 6, respectively). Despite having ∼28 surface lysines (supplemental Fig. S1D), MiroS was not modified by UbMES in the absence of pPARKIN (Fig. 2A, lane 5). To show that the observed ubiquitination requires UbMES thioester functionality, we conducted similar experiments with hydrolyzed UbMES (ubiquitin) indicated as Hyd (Fig. 2A). Importantly, both pPARKIN autoubiquitination and MiroS substrate ubiquitination by UbMES were strictly dependent on the catalytic cysteine (Cys431) of pPARKIN. Therefore, despite the fact that PARKIN is extremely cysteine-rich, UbMES is selective for the catalytic site cysteine (Fig. 2A, lane 7) (44, 60).
Figure 2.

PARKIN/ByS recapitulates several aspects of the native cascade. A, ByS reaction of pPARKIN, UbMES, and the MiroS substrate. Lane 4 shows pPARKIN autoubiquitination and chain formation, and lane 6 shows pPARKIN autoubiquitination, chain formation, and MiroS substrate ubiquitination. Notably, UbMES has no ubiquitination activity in the absence of pPARKIN or in the presence of catalytically inactive C431A pPARKIN (lanes 5 and 7). Hyd. indicates hydrolyzed UbMES before the reaction. B, pPARKIN undergoes autoubiquitination and MiroS ubiquitination using UbMES. Mutation of the primary phosphorylation site (S65A) or the catalytic cysteine (C431A) blocks this activity. P indicates PARKIN; M indicates MiroS, and * indicates impurities. PARKIN (S65A) was pretreated with TcPINK1 under the same reaction conditions as PARKIN, prior to its use in assay. C, ByS ubiquitination depends on the concentration of UbMES and requires pPARKIN. D, pPARKIN/ByS forms Lys48- and Lys63-linked chains, similar to the native cascade. The reactions with pPARKIN, MiroS, and the corresponding UbMES were analyzed by Western blot. A K48R mutation in UbMES blocks formation of Lys48-linked chains but not Lys63-linked chains (lane 3), whereas a K63R mutation in UbMES blocks formation of Lys63-linked chains but not Lys48-linked chains (lane 4). E, MALDI-TOF analysis of a slice from the stacking gel of a native cascade reaction and the ByS reaction from D, lane 2, confirming Lys48 and Lys63 chain formation. F, MiroS ubiquitination by pPARKIN using the ByS (1 μm pPARKIN and 30 μm UbMES) proceeds on a similar time scale to common native reaction conditions (100 nm UBE1, 1 μm UbcH7, 1 μm pPARKIN, 30 μm Ub, and 4 mm ATP). P indicates PARKIN, and M indicates MiroS.
Next, we demonstrate that PARKIN ubiquitination using the ByS is still subject to its native phosphoregulation mechanism (Fig. 2B). Although both autoubiquitination and MiroS substrate ubiquitination by wild-type PARKIN were strongly activated by TcPINK1 phosphorylation (pPARKIN), mutation of Ser65 to alanine (PARKINS65A) abolished this activation. Both non-phosphorylated wild-type PARKIN and PARKINS65A treated with TcPINK1 showed only a minimal amount of MiroS ubiquitination with UbMES (Fig. 2B, lanes 1 and 2 and 5 and 6, respectively). Mutation of the catalytic cysteine Cys431 to alanine also abolished PARKIN activity even after TcPINK1 phosphorylation (pPARKINC431A, Fig. 2B, lanes 7 and 8). pPARKIN ubiquitination of MiroS increased with increasing concentrations of UbMES, but without pPARKIN, and MiroS-Ub adduct formation was negligible (Fig. 2C). Only after incubating for a 3-fold longer reaction time (120 min) at 80–120 μm UbMES was a faint band corresponding to the molecular weight of MiroS-Ub adduct observed (lanes 12–14, Fig. 2C). This result shows that our standard reaction conditions (30 μm UbMES and 40 min reaction time at room temperature) do not cause non-specific modification of PARKIN, ubiquitin, or the MiroS substrate.
We next examined ubiquitin chain formation by pPARKIN under the ByS reaction. Previous work demonstrated that pPARKIN can form Lys48-, Lys63-, Lys6-, and Lys11-linked chains in vitro (8). We compared PARKIN ubiquitination using UbMES, Ub(K48R)MES, and Ub(K63R)MES in the ByS reaction. For all of these Ub variants, similar amounts of ubiquitin adducts are formed (Fig. 2D). Antibodies against either Lys48 or Lys63 linkages detected the corresponding chains, which were absent when Ub(K48R)-MES or Ub(63R)-MES, respectively, was used. MALDI analysis confirmed the presence of both Lys63 and Lys48 linkages, similar to the native cascade reaction (Fig. 2E). Importantly, this experiment directly shows that the E2 enzyme is dispensable for the formation of polyubiquitin chains by PARKIN under the ByS, consistent with our previous report (54).
We next compared the sites within full-length hMiro1 that are ubiquitinated by pPARKIN either under ByS or native reaction conditions, using tandem mass spectrometry analysis. We limited hMiro1 modification to ∼2–3 ubiquitin molecules in both the ByS and in the native cascade, and we found that Lys153, Lys330, and Lys572 within hMiro1 were modified in both reactions (supplemental Fig. S2). These lysines have been previously reported as ubiquitination sites within hMiro1 (5, 6, 8, 14). Because the ByS reaction recapitulates the activity of pPARKIN toward target lysine residues within hMiro1, we can conclude that pPARKIN alone can select target substrate lysines without the help of an E2 enzyme in vitro. Testing a larger panel of different substrates could further confirm this finding. Taken together, our results suggest that the E2-independent ByS reaction recapitulates several aspects of pPARKIN's native function as follows: ubiquitin ligation; polyubiquitin chain formation; polyubiquitin chain-type specificity; and selection of substrate lysines.
Finally, we showed that the reaction conditions of the ByS can be adjusted to approach the time scale of the native ubiquitination reaction, such that similar amounts of the reaction products are formed on a similar time scale (Fig. 2F). For these experiments, we used the same amount of ubiquitin species, PARKIN enzyme, and MiroS substrate in each reaction. The native cascade reaction contained 100 nm UBE1, 1 μm UbcH7, 1 μm pPARKIN, 30 μm Ub, 4 mm ATP, and 1 μm MiroS. The ByS reaction contained 1 μm PARKIN, 30 μm UbMES, and 1 μm MiroS. The similar time scales of MiroS ubiquitination in the ByS using 30 μm UbMES and in a native cascade reaction with a maximum of 1 μm UbcH7∼Ub certainly do not indicate that UbMES is as good a PARKIN substrate as UbcH7∼Ub. However, this result demonstrates that ByS reactions proceed on a very reasonable time scale, providing an experimentally feasible and simplified ubiquitination reaction. Furthermore, the developed ByS system can be used to prepare site-specific mono- or diubiquitinated PARKIN substrates using PARKIN and UbMES (Fig. 2F, lane 3). This could allow us to bypass challenges using regular protein synthesis approaches. A slower time scale of the ByS system and its simplicity (i.e. just two components E3 and UbMES) make it easier to adjust the stoichiometry of reagents.
UbFluor enables real-time monitoring of PARKIN activity by FP
Encouraged that UbMES specifically recognized the PARKIN catalytic cysteine and recapitulated several essential aspects of PARKIN activity, we designed and prepared UbFluor, a probe that enables quantitative analysis of PARKIN activity using the ByS. UbFluor preparation is described under the supplemental Material and methods and in previously published procedures (Scheme 1, supplemental Fig. S3) (55, 56). When UbFluor is charged to PARKIN's catalytic cysteine, it releases fluorescein thiol, and this release can be monitored in real time using FP (Fluor-SH, Fig. 3A).
Figure 3.

UbFluor reports pPARKIN activity in real time by fluorescence polarization. A, design of the ByS reaction using UbFluor and PARKIN. B, BME (lane 2) or pPARKIN (lane 4) can release free fluorescein (SFluor) from UbFluor but not pPARKINC431A (lane 6). C, gel-based analysis of UbFluor/pPARKIN reaction. At the indicated time points, the reaction was stopped by addition of non-reducing Laemmli buffer and analyzed by SDS-PAGE. A time-dependent decrease in the intensity of the UbFluor band and an increase in the free fluorescein band were observed. D, FP-based analysis of UbFluor (0.5 μm) consumption by the indicated amount of pPARKIN (0–5 μm). Mean ± S.E. of three measurements for each concentration of pPARKIN are shown. E, plot of the initial rates (0–5 min) from D showing that the rate of UbFluor consumption increases linearly with pPARKIN concentration (R2 = 0.996).
First, we tested whether UbFluor releases free Fluor-SH in a pPARKIN-dependent manner. When UbFluor (0.5 μm) was mixed with β-mercaptoethanol (BME; Fig. 3B, lane 2), cysteamine (supplemental Fig. S4, lane 2), or pPARKIN (5 μm, Fig. 3B, lane 4), the fluorescent band corresponding to UbFluor (∼9 kDa) diminished in intensity, whereas Fluor-SH intensity increased, in a time-dependent manner (Fig. 3C and supplemental Fig. S5). pPARKIN harboring a C431A mutation failed to release fluorescein from UbFluor after 60 min, demonstrating that the release of Fluor-SH from UbFluor requires the catalytic cysteine of PARKIN (Fig. 3B, lane 6).
Next, we tested whether the consumption of UbFluor can be measured in real-time using FP (Fig. 3, D and E). As expected, increasing the amount of pPARKIN in the UbFluor reaction accelerated the rate of FP decrease under excess enzyme conditions (Fig. 3, D and E). Early time point data (1–5 min) were fitted to obtain initial rates of FP decrease at an effectively constant concentration of UbFluor (supplemental Fig. S6). The initial rate of UbFluor consumption increased linearly with pPARKIN concentration, as expected for a bimolecular reaction. Because UbFluor lacks the E2 enzyme, high Km values can be expected, similar to HECT E3s for which Km values were ∼800 μm (55). These data demonstrate that UbFluor provides a robust FP-based assay for real-time monitoring of PARKIN activity.
UbFluor confirms the native activation mechanism of PARKIN by Ser65 phosphorylation and pUb
Changing the ratio of UbFluor to E3 ligase in ByS reactions enables examination of different aspects of the E3 ligase mechanism. In a single turnover (ST) reaction with excess PARKIN (10-fold excess of PARKIN over UbFluor), the rate of FP decay only reflects the rate of transthiolation reaction between UbFluor and PARKIN. However, under multiple turnover (MT) conditions (10-fold excess of UbFluor over PARKIN), the UbFluor consumption rate (FP decay) should reflect both transthiolation and isopeptide ligation rates. Under normal MT conditions, one molecule of PARKIN can consume >1 molecule of UbFluor. However, if there is a defect in the isopeptide ligation step, PARKIN can only consume one molecule of UbFluor, because the resulting inactive PARKIN∼Ub thioester will not be able to react with the second molecule of UbFluor.
We used both ST and MT reaction conditions to examine PARKIN activation by Ser65 phosphorylation and by pUb to test whether the UbFluor assay can detect these known PARKIN activation mechanisms, similar to the native reaction. We prepared pUb according to previously established methods (supplemental Fig. S7) (8). ST experiments were performed at 5 μm PARKIN and 0.5 μm UbFluor, whereas the MT experiments were performed at 2 μm PARKIN and 20 μm UbFluor. To measure initial velocities during which <20% of available UbFluor was consumed, ST reactions were carried out for 5 min, and MT reactions were carried out for 15 min.
We titrated 0–25 μm pUb into ST (Fig. 4A) and MT (Fig. 4B) UbFluor reactions using both PARKIN and pPARKIN. Titrations are written as PARKIN/pUb or pPARKIN/pUb, with the titrated reaction component after the slash, throughout this work. Without PARKIN or pPARKIN present, the small amount of background UbFluor consumption is not affected by pUb concentrations up to 25 μm (No Enz/pUb, Fig. 4, A and B). However, addition of pUb increases the UbFluor consumption rate of both PARKIN and pPARKIN under both ST and MT conditions (Fig. 4, A and B). The pUb-dependent increase in UbFluor consumption by PARKIN under ST conditions was small but significant (∼1.5-fold increase) (supplemental Fig. S8, A–C). This slight activation of PARKIN by pUb was not previously detected using Ub-VS (8). As expected based on previous results, the pUb-dependent increase in UbFluor consumption by pPARKIN was much larger for both ST and MT conditions (∼7-fold for ST conditions) (pPARKIN/pUb, Fig. 4, A and B) (7, 8). Non-phosphorylated ubiquitin did not increase UbFluor consumption by pPARKIN (pPARKIN/Ub, Fig. 4, A and B), emphasizing the importance of Ser65 phosphorylation of ubiquitin for activating PARKIN. Together, these experiments confirm that the UbFluor assay can detect the natural Ser65 phosphorylation and pUb-dependent activation mechanisms of PARKIN.
Figure 4.
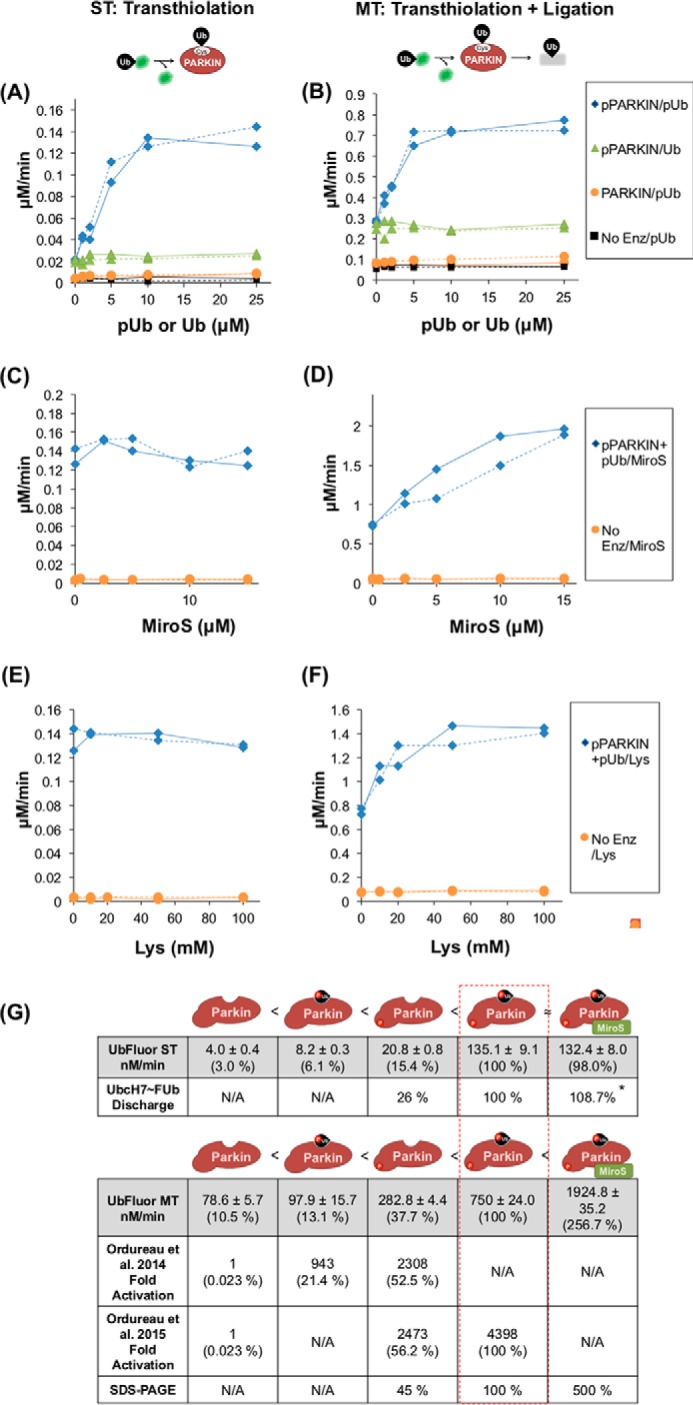
UbFluor confirms multiple activation states of PARKIN. Titrations in the left panels (A, C, and E) were performed under ST conditions (5 μm PARKIN, 0.5 μm UbFluor), and the right panels (B, D, and F) were performed under MT conditions (2 μm PARKIN, 20 μm UbFluor). Each titration was repeated twice, and all data are shown. Solid lines join the rates determined from one titration experiment, and dashed lines join rates determined from a second titration. A and B, Ub or pUb titration into a UbFluor reaction with PARKIN or pPARKIN. C and D, MiroS titration into pPARKIN + 25 μm pUb. E and F, lysine titration into pPARKIN + 25 μm pUb. G, comparison of PARKIN activation measured using UbFluor with previous data. UbFluor measurements under ST and MT conditions and SDS-PAGE-based quantification are reported in this work. AQUA-MS data were reported in by Ordureau et al. (7, 8). For comparison of UbFluor to AQUA-MS, the maximal rate corresponding to each state is determined as a percentage of pPARKIN + pUb activity. Schematics depict the relative activity of each PARKIN species, with unmodified PARKIN having the lowest activity and pPARKIN + pUb + MiroS having the highest. N/A, not applicable. * rate was too fast to accurately resolve.
pUb and substrate enhance pPARKIN catalytic turnover rate by different mechanisms
Next, we examined how the inclusion of substrate affects the catalytic turnover rate of pPARKIN in the presence of a saturating concentration of pUb (pPARKIN + pUb; 25 μm pUb in our assays), which is the maximally activated state of pPARKIN in the native reaction that has been previously described (7). We titrated increasing concentrations of MiroS into pPARKIN+ pUb. Although 15 μm MiroS did not increase the ST rate of pPARKIN + pUb, it did increase the MT rate 2.6-fold (Fig. 4, C, D, and G). In contrast to MiroS addition, adding an additional 15 μm pUb to a total concentration of 40 μm did not further increase either the ST or the MT rate of UbFluor turnover by pPARKIN (supplemental Fig. S9). The simplest interpretation of these results is that the presence of the MiroS substrate does not significantly increase the transthiolation rate but does enhance the ligation rate of pPARKIN + pUb. To determine whether the MiroS-dependent increase in pPARKIN + pUb activity under MT conditions that we observed could be due to lysine-dependent clearance of the Ub thioester, we titrated free lysine into the UbFluor reaction with pPARKIN + pUb in place of MiroS. We note that UbFluor is stable at 100 mm free lysine (Fig. 4, E and F, and supplemental Fig. S10). Similar to MiroS, addition of free lysine increased the MT rate of pPARKIN + pUb (∼2-fold), but not the ST rate (Fig. 4, E and F). Based on these results it is reasonable to conclude that the MiroS-dependent increase in the MT rate may be due to the increased ligation rate that facilitates the clearance of pPARKIN∼Ub thioester, regenerating the free enzyme able to consume another equivalent of UbFluor. Furthermore, the increase in UbFluor consumption of pPARKIN + pUb upon lysine or MiroS titration suggests that pUb may be a poor substrate for the isopeptide ligation step compared with MiroS or lysine.
We further confirmed that pUb activates transthiolation of pPARKIN but is a poor substrate for ligation using the native reaction with fluorescent ubiquitin (FUb). FUb is different from UbFluor, as it features a ubiquitin that has a permanent fluorophore at its N terminus. This fluorophore is for gel imaging purposes only and is not transferred or released from FUb at any point. FUb can be activated by E1 and then be transferred onto E2 and then E3 enzyme. We evaluated the effects of pUb on both E2-discharge (ST) and total ubiquitination (MT) activity of pPARKIN (supplemental Fig. S11). In an E2-discharge assay, the E2∼FUb thioester adduct (UbcH7∼FUb in our assays) is separately prepared and chased by excess pPARKIN with the indicated amount of pUb, MiroS, or both (supplemental Fig. S11A). The appearance of a fluorescent band at ∼55 kDa (pPARKIN∼FUb and Miro-FUb, supplemental Fig. S11B) and the reduction in the 25-kDa UbcH7∼Ub band (supplemental Fig. S11C) corresponded to transthiolation activity. The total ubiquitination activity of pPARKIN under native cascade conditions was measured after adding pUb, MiroS, or both (supplemental Fig. S11D). The appearance of fluorescent bands above 10 kDa (supplemental Fig. S11E) and the reduction in the FUb band at 10 kDa (supplemental Fig. S11F) were quantified to reveal total Ub turnover. We confirmed that pUb enhances the transthiolation of pPARKIN at least ∼4-fold, whereas MiroS has no effect under these end-point assay conditions (supplemental Fig. S11A, lanes 3 and 4). In contrast, we observed that the total ubiquitin consumption for pPARKIN + pUb was ∼2-fold lower than pPARKIN + MiroS (supplemental Fig. S11D, lanes 3 and 4). These results indicate that pUb is a strong allosteric activator of pPARKIN transthiolation but is not a good ubiquitin acceptor compared with MiroS.
We were unable to conclusively determine whether or not MiroS addition further enhanced the pPARKIN + pUb transthiolation rate in the native reaction, because transthiolation reactions using pPARKIN + pUb were too fast to quantify in the discharge assay (supplemental Fig. S11A, lanes 3 and 5). Similar to the UbFluor MT reaction, pPARKIN shows the highest ubiquitin conjugation efficiency when both pUb and MiroS are present in a native cascade assay (supplemental Fig. S11D, lane 5).
Together, all of these data suggest that pUb activates pPARKIN by increasing its transthiolation rate in both the E2-independent UbFluor reaction and in the native cascade. However, substrate is required for maximal pPARKIN ubiquitination turnover even in the presence of pUb, because pUb is a poor Ub acceptor for pPARKIN. These results also confirm that UbFluor ST and MT assays can conveniently reveal different aspects of PARKIN ubiquitination mechanisms; ST assays detect transthiolation, whereas MT assays detect both transthiolation and ligation. Furthermore, our results reveal a similar trend between the UbFluor assay and the native reaction under both ST and MT conditions.
Comparison of UbFluor quantitation of PARKIN activity with previous measurements using the native reaction
Fig. 4G compares the maximal rates of UbFluor consumption that we observed in this work to previous AQUA-MS-based measurements, which have provided a quantitative measure of PARKIN activity using the native reaction (7, 8). pPARKIN + pUb gave the highest PARKIN activity in these experiments; therefore, we set the activity of pPARKIN + pUb as a standard value (100%) for comparison.
The apparent activity of non-phosphorylated PARKIN is much higher in the UbFluor assay than in the native reaction experiments. PARKIN activity is ∼10% of pPARKIN + pUb activity in the UbFluor assay, whereas it is only 0.02% of pPARKIN + pUb activity in the native reaction. This difference may arise because UbFluor detects catalytic site activation apart from E2 binding, unlike the native reaction. This catalytic site activation may reflect the following: 1) physical opening of the catalytic site, exposing the catalytic cysteine (Cys431) on RING2 that is otherwise occluded by the UPD, and 2) chemical activation of Cys431 by neighboring residues for transthiolation or ligation reactions, or both. In contrast, binding of the E2∼Ub complex is strictly required to observe any activation in the native reaction. Therefore, the larger difference between minimal and maximal activity in the native reaction may be due to multiple regulation sites, the E2∼Ub interaction site and the catalytic site (supplemental Fig. S1E).
Aside from higher basal PARKIN activity in the UbFluor assay, the agreement between these two measurements is remarkable. Both measurements find that activation of PARKIN by pUb is less than activation by Ser65 phosphorylation, whereas the greatest activation occurs with both pUb and pPARKIN. This set of findings poses an interesting question. How can UbFluor reproduce the activity trend of PARKIN observed in the native reaction, despite using a distinct transthiolation mechanism? It may be that although E2 binding clearly does not happen in the ByS, the E2-independent UbFluor transthiolation reaction is still relevant to the mechanism of Ub binding during the native transthiolation.
Consistent with this conclusion, Kumar et al. (64) showed that UbcH7-Ub binds to pPARKIN 20-fold tighter than UbcH7 alone, suggesting that ubiquitin binding itself has an important role in native transthiolation (8). Furthermore, recent work showed that RING2 and RING1 domains of RBR E3s harbor a ubiquitin-binding site (44, 46). RING2 ubiquitin-binding site is particularly interesting because it was shown that it recruits E2∼Ub thioester for transthiolation reaction. In this respect, the RING2 domain is functionally similar to the C-lobe of HECT E3 ligases that also has a Ub-binding site near the catalytic cysteine. The latter serves to recruit E2∼Ub thioester for transthiolation reaction and subsequently retains Ub in HECT E3∼Ub thioester for isopeptide ligation step (61–63).
S65E is a poor mimetic of pPARKIN
Several studies have used overexpressed PARKINS65E in cells as a phosphomimetic for pPARKIN, but it is unclear whether PARKINS65E is activated similar to pPARKIN. PARKINS65E failed to show increased free ubiquitin chain formation compared with PARKIN in a Western blot assay (7). In apparent contrast, Kumar et al. (64) demonstrated enhanced polyubiquitin chain formation by PARKINS65E compared with PARKIN. We used the UbFluor-based ByS to quantitatively examine whether pUb activates PARKINS65E as it does for pPARKIN. We performed titration experiments using PARKINS65E under the same ST and MT conditions as defined for pPARKIN in Fig. 4 (Fig. 5, A and B). pUb slightly increases the UbFluor consumption rate of PARKINS65E under both ST and MT conditions. This slight, <1.5-fold change under ST conditions is similar to what we observed for pUb activation of PARKIN under ST conditions (supplemental Fig. S8A), but it is significantly smaller than the ∼7- and 3-fold activation of pPARKIN by pUb under ST and MT conditions, respectively (Fig. 5, A and B). These results indicate that S65E does not mimic Ser65 phosphorylation of pPARKIN in terms of activation by pUb binding.
Figure 5.

S65E is a poor mimetic of pPARKIN. A and B, ST (A) and MT (B) experiments titrating Ub or pUb into a UbFluor reaction with PARKINS65E (green triangles) were compared with PARKIN (orange circles) and pPARKIN (blue diamonds) titrations from Fig. 4. Conditions were as described in Fig. 4. Each titration was repeated twice, and all data are shown. Solid lines join the rates determined from one titration experiment, and dashed lines join rates determined from a second titration. C and D, ubiquitination of MiroS by pPARKIN, pPARKINC431A, PARKIN, or PARKINS65E were performed by the UbMES-based ByS reaction (C) or the native reaction (D). Conditions for these experiments were identical to those used in Fig. 2F. * indicates ubiquitinated MiroS. P indicates PARKIN and M indicates MiroS.
We further compared the MiroS substrate ubiquitination efficiency of PARKINS65E and pPARKIN using both the UbMES-based ByS and the native reaction (Fig. 5, C and D). In both reactions, pPARKIN robustly ubiquitinated MiroS, whereas PARKINS65E had only slight activity (Fig. 5, C and D, lanes 2 and 8). As we described previously for the UbFluor assay, unphosphorylated PARKIN showed higher background activity in the UbMES-based ByS reaction than in the native reaction (Fig. 5C, lane 6). The PARKINS65E mutation did not increase Miro ubiquitination significantly above this background in the ByS. This result supports our conclusion that S65E is not a good mimetic of pPARKIN. While this work was in progress, Ordureau et al. (7) published similar findings.
UbFluor can detect PARKIN activation by point mutations
PARKIN is autoregulated both by mechanisms that block E2∼Ub binding and by mechanisms that occlude the catalytic site. Because the UbFluor-based ByS reaction does not include an E2, we may expect it to only detect activation of the catalytic site (supplemental Fig. S1E). In fact, when used with truncated HECT E3s, the ByS detects mutations that alter activity near the Ub-binding site on the C-lobe and near the catalytic cysteine but not those that alter E2 binding (55). However, for the RBR ligase PARKIN, reports have postulated that the REP/RING1 interface and the RING2/UPD interface are allosterically coupled (11, 20). For example, it was shown that disrupting the REP/RING1 interface with the W403A mutation mimics phosphorylated PARKIN (70). This result suggests that changes in regulation of the E2∼Ub-binding site may translate to changes in the catalytic site, such that these changes may be observable using the ByS.
To determine the extent to which UbFluor can detect different mechanisms of PARKIN activation, we prepared five constructs as follows: four harboring point mutations that are known to activate PARKIN and one harboring a mutation that abolishes ubiquitination by fully active PARKIN. We assume that the activating mutations represent potential modes of activation by small molecules. Activating mutations disrupt the REP/RING1 interface (A398T and W403A) (43, 44) or the UPD/RING2 interface (F463Y and ΔUPD; the ΔUPD construct consists of PARKIN residues 219–465) (44). The inactivating mutation C431A ablates the catalytic cysteine residue (supplemental Fig. S1E) (40, 42). Importantly, A398T is a Parkinson's disease mutation (1). To enable a direct comparison with past data evaluating the effects of these mutations on PARKIN activity, PARKIN point mutations were prepared using a base construct of rat PARKIN(141–465) with an N-terminal GST tag (ΔUbl, supplemental Fig. S1E). The ΔUbl construct lacks the ubiquitin-like domain and linker (residues 1–140), and therefore it represents a minimally autoregulated construct that still harbors a low level of ubiquitination activity (44).
We first confirmed that the E2-independent ByS recapitulates the known properties of Ubl, ΔUPD, and ΔUPD (C431A) using UbMES (Fig. 6A). As reported previously from native assays (44), Ubl exhibited a small amount of activity; UPD had robust activity, and the C431A mutation completely abolished the activity of ΔUPD, demonstrating that the activity of UPD in the ByS assay requires the catalytic cysteine (Fig. 6A, lanes 1–3, 4–6 and 7–9 respectively). Next, we examined three activating point mutations in the ΔUbl background using UbMES. As expected, the F463Y mutation that disrupts the UPD/RING2 interaction showed obvious activation (compare Fig. 6A, lanes 16–18 to lanes 1–3). We observed modest activation by the REP/RING1 disrupting mutation A398T and more robust activation by W403A (compare Fig. 6A, lanes 10–12 and 13–15 to lanes 1–3). Although there is no E2∼Ub-bound structure of PARKIN, a simple explanation for these results is that the W403A mutation may relieve autoinhibition of the Ub-binding site in PARKIN, which is occupied by UbMES/UbFluor to a larger extent than A398T, or to expose the catalytic cysteine of RING2 domain to a larger extent. Regardless of the unknown structural mechanisms by which these mutations activate PARKIN, our W403A result demonstrates that the E2-independent ByS can detect the PARKIN catalytic site activation by significant disruption of the REP/RING1 interface, where E2 enzyme presumably binds, and that is ∼50 Å away from the catalytic cysteine of PARKIN.
Figure 6.

UbFluor can quantify PARKIN activation or inactivation by known point mutations. A, UbMES assay on ΔUbl, ΔUPD, and ΔUPD (C431A) and activating PARKIN mutations in the ΔUbl background (A398T, W403A, and F463Y). Experiments with each mutant were performed using 30 μm UbMES and 1.0 μm PARKIN for the time indicated. W403A and F463Y were run on a separate gel. B, bar graph showing bimolecular reaction rates of UbFluor consumption under MT conditions by PARKIN mutants as described above using the UbFluor assay. Errors are based on linear fits of data from two repeats of UbFluor consumption data at four different concentrations of UbFluor (eight total measurements).
We next investigated whether the same unnatural activation mechanisms could be detected using the UbFluor assay. To generate a quantitative comparison between mutants, we determined the apparent bimolecular rate of UbFluor consumption (kobs) for each mutant. First, we performed UbFluor assays using four different concentrations of UbFluor under either ST conditions (1 μm rat PARKIN with 0.25, 0.5, 0.75, and 1 μm UbFluor) or MT conditions (5 μm rat PARKIN with 10, 12.5, 15, and 20 μm UbFluor). Each condition was repeated twice for a total of eight measurements. Next, we obtained the initial UbFluor consumption rate by performing a linear fit of the raw FP decrease at early time points (1–5 min for ST; 1–15 min for MT; supplemental Fig. S12A). We then performed a linear fit of the initial UbFluor consumption rate versus the UbFluor concentration (supplemental Fig. S12B). Finally, the slope of this linear fit was divided by the PARKIN concentration to obtain kobs (Fig. 6B for MT and supplemental Fig. S13 for ST).
Using this bimolecular rate analysis, we found that UbFluor detected activation of the ΔUPD construct, as well as activation of the ΔUbl construct by F463Y and W403A mutations under both ST and MT conditions, similar to the UbMES-based ByS (Fig. 6B and supplemental Fig. S13, respectively). The weak activation by A398T observed in the UbMES assay was not significant enough to be detected in the UbFluor assay, as the activation was smaller than the error in the kobs measurement. We applied the same method to compare kobs of S65E and pPARKIN, and we found that in contrast to phosphorylation, the S65E mutation did not significantly increase PARKIN catalytic turnover under either ST or MT conditions (supplemental Fig. S14).
Together, these results demonstrate that UbFluor can recapitulate the majority of the effects of PARKIN mutations observed using UbMES. In addition, these results confirmed that UbFluor assays are robust to changes in the UbFluor concentration. We determined kobs values using initial rates obtained from four different UbFluor concentrations under both ST and MT conditions, and we found that the rate of each mutant relative to the wild-type ΔUbl construct is conserved at all UbFluor concentrations used (supplemental Fig. S15) (65). The robustness of the UbFluor assay to different concentrations of probe validates its use for single-concentration and end-point high-throughput screening assays.
Finally, after determining that the UbFluor-based ByS can quantitatively detect both natural and unnatural activation mechanisms of the RBR E3 ligase PARKIN, we performed an initial test of the dynamic range of the UbFluor assay for high-throughput screening to identify small molecules that affect RBR E3 ligase activity. We monitored UbFluor consumption by pPARKIN and GST-tagged HHARI RBR E3 ligases in a 384-well plate using a microplate reader (Synergy 4, supplemental Fig. S16). Each E3 ligase (1.0 μm) was incubated with either DMSO (0.2%; negative control) or iodoacetamide (1 mm; positive control) for 1 h. UbFluor (5 μm) was then added, and end-point FP readings were recorded after 1 and 2 h of reaction. The 2–3-fold difference between the end-point values of these positive and negative controls further demonstrates the feasibility of using UbFluor for high-throughput screening of bioactive molecules for RBR E3 ligases under MT conditions.
Discussion
We began this work by applying our previously developed UbMES-based ByS assay to study the RBR E3 ligase PARKIN (55, 56). Despite the fact that PARKIN, like other RBRs, is very cysteine-rich (44), we found that the reaction with UbMES was highly specific for the catalytic cysteine and that the E2-independent bypassing system recapitulates many aspects of biochemical mechanisms of PARKIN. Our results demonstrate that in the absence of any E2 enzyme, pPARKIN can build polyUb chains with specific linkages and select target lysine residues within substrates in vitro.
After confirming that the E2-independent ByS reaction recapitulated the native activity of PARKIN, we used the novel probe UbFluor to quantitatively evaluate PARKIN activation by both natural mechanisms (pUb and substrate) and unnatural mechanisms (PARKIN mutations that disrupt the REP/RING1 and UPD/RING2 interface). Simple titration experiments using UbFluor enabled us to quantitatively compare and rank the native activation mechanisms acting on PARKIN, Ser65 phosphorylation and pUb. Our titration experiments also provided new insight on pPARKIN + pUb activity. We found that although pUb strongly activates the transthiolation step of PARKIN and E2∼Ub/UbFluor, maximal ubiquitination turnover by pPARKIN is only observed when a substrate or free lysine is present, suggesting that pUb itself is not a good ubiquitin acceptor for pPARKIN. Importantly, most efforts on studying the mechanisms of PARKIN activation have been focused on the activating role of PINK1 and pUb. Here, we show that substrates that provide acceptor lysines can also enhance PARKIN catalytic turnover. Thus, the local concentration of the substrate can affect Parkin activity as evident from ByS and native ubiquitination systems (Fig. 4G and supplemental Fig. S11).
We would like to highlight the remarkable simplicity of the assay: only two reagents are needed, and by changing the ratio of UbFluor and enzyme we can quantify transthiolation and isopeptide ligation steps. Furthermore, we can easily test the effect of other protein cofactors, such as pUb and substrates such as Miro on PARKIN activity, and investigate their roles on the enzyme mechanism (i.e. activation of transthiolation step versus isopeptide ligation step). We envision that our findings will expand the use of the UbFluor probe for a robust quantification of the enzymatic activity of HECT, RBR, and bacterial HECT-like and NEL E3 ligases, upon structural mutations, in the presence of protein partners such as pUb or substrates, or in the presence of small molecules.
Importantly, our data demonstrated that UbFluor can detect changes in the ligation efficiency of PARKIN under MT conditions, despite the fact that the signal change from FP occurs during transthiolation (Fig. 4 and supplemental Fig. S11). This is an essential property of the UbFluor probe for high-throughput screening, as our goal is to identify molecules that increase ligation efficiency as well as the transthiolation step.
Finally, we demonstrated several properties of the UbFluor assay that indicate its suitability for high-throughput screening to identify small molecules affecting RBR E3 ligase activity. We found that the E2-independent UbFluor reaction can, at least in some cases, detect significant activation at the REP/RING1 site of PARKIN. In addition, the UbFluor-based end-point assay detected activity changes of both PARKIN and HHARI caused by iodoacetamide with a strong signal change in a 384-well plate format. Because UbFluor is stable at 100 mm lysine, direct consumption of UbFluor by small molecules containing amines is not expected. However, because high concentrations of free thiols (such as BME and cysteamine) release Fluor-SH, small molecules with free thiols should be avoided. Overall, these results provide a very encouraging demonstration that UbFluor can be used in a highly simplified high-throughput screening assay to detect allosteric activators or inhibitors of PARKIN and perhaps other RBR E3 ligases. Further use of UbFluor in application to HECT/RBR/HECT-like bacterial/NEL E3s will be reported in the future.
Experimental procedures
Synthesis of compound I
We followed a previously described method (66). We mixed cysteamine hydrochloride (1 g, 8.8 mmol) and trifluoroacetic acid (1.3 ml, 17.0 mmol) in CH2Cl2 (30 ml), then added trityl chloride (2.5 g, 8.8 mmol), and stirred the reaction mixture for 16 h at room temperature. We added 1 m NaOH solution (20 ml) to quench the reaction, then diluted the organic phase with methylene chloride (50 ml), washed with brine (20 ml), and dried over magnesium sulfate. Diethyl ether/n-pentane (1:4) precipitation yielded white crystalline compound I (455 mg, 1.42 mmol, 16% yield). 1H NMR (400 MHz, CDCl3), δ = 7.43–7.42 (d, 6H), 7.29–7.26 (t, 3H), 7.21–7.19 (t, 3H), 7.16–7.11 (m, 3H), 2.62 (t, 2H), 2.23 (t, 2H); 13C NMR (125 MHz, CDCl3), δ = 144.4, 129.8, 128.6, 127.4, 67.7, 36.9, 29.4.
Synthesis of compound II
We followed a previously described method (66). We mixed compound I (330 mg, 1.0 mmol) and diisopropylethylamine (350 μl, 2.0 mmol) in dimethylformamide (5 ml), then added FITC (400 mg, 1.0 mmol), and stirred the reaction mixture for 16 h at room temperature. N,N-Dimethylformamide was evaporated down to ∼1–2 ml by blowing dry nitrogen over the mixture. We poured the reaction into 48 ml of 50 mm HEPES, pH 6.5, buffer in a 50-ml Falcon tube and precipitated the product by centrifugation at 4000 × g for 10 min. The precipitate was dissolved in 2–3 ml of MeOH and passed through a silica plug with 50–60 ml of ethyl acetate. It was further washed with ethyl acetate (8% MeOH) until all fluorescent material had eluted. The eluted material was concentrated under reduced pressure to yield an orange powder (650 mg, 90% yield). 1H NMR (400 MHz, MeOD), δ = 8.17 (d, 1H), 7.74 (dd, 1H), 7.44–7.41 (m, 6H), 7.32–7.21 (d, 10H), 7.12 (d, 1 H),_6.69 (m, 3H), 6.53 (dd, 2H), 3.53 (t, 2H), 2.56 (t, 2H); 13C NMR (125 MHz, MEOD), δ = 31.0, 42.6, 66.4, 102.1, 110.0, 112.2, 116.7, 124.2, 126.4, 127.6, 128.7, 128.9, 129.4, 130.5, 140.8, 144.8, 152.7, 159.9.
Synthesis of Fluor-SH
Compound II (30 mg, 0.042 mmol) was added to the flask containing 1 ml of TFA solution (2.5% triethylsilane and 2.5% water), and the reaction mixture was for 1 h at room temperature, followed by the addition of 50 ml of cold diethyl ether. Fluor-SH formed a cloudy precipitate that was pelleted in a centrifuge (4000 × g, 10 min) to yield an orange pellet (16.3 mg, 0.035 mmol, 83% yield). 1H NMR (400 MHz, DMSO), δ = 8.22 (s, 1H), 7.74 (d, 1H), 7.19 (dd, 1H), 6.67–6.55 (m, 6H), 2.73 (t, 2H), 2.47 (t, 2H); 13C NMR (125 MHz, DMSO), δ = 23.2, 47.3, 102.7, 110.2, 113.1, 121.1, 124.5, 129.5, 141.5, 147.7, 152.3, 159.9, 168.9.
Preparation of UbFluor
E1-mediated Ub-MES preparation
We followed a previously described method (57). Briefly, we mixed Ub (100 μm), E1 (250 nm), and MESNa (100 mm) in Ub-MES reaction buffer and then added 10 mm ATP. We incubated the reaction for 5 h at 37 °C and then purified UbMES by FPLC using an S75 column with Ub-MES FPLC buffer.
For the Ub-MES reaction buffer, we used 50 mm sodium phosphate buffer, pH 8, 10 mm MgCl2. For Ub-MES FPLC buffer, we used 12.5 mm HEPES, pH 6.7, 25 mm NaCl.
Transthiolation reaction
We prepared Fluor-SH stock solution in water (50% DMSO, 50 mm) and then added an aqueous solution of saturated NaHCO3 until the color changed to dark red and a thiol odor arose. Our final reaction mixture contained 110 μl of HEPES (1 m, pH 7.5), 100 μl of TCEP (100 mm), 300 μl of Fluor-SH stock solution (50 mm), 300 μl of guanidine HCl (6 m), and 300 μl of Ub-MES (0.8–0.9 mm), and then we vortexed the mixture for 1 h at room temperature.
Purification
After a 2 × 2 h dialysis using a Slide-A-Lyzer dialysis cassette in 2 liters of UbFluor Storage Buffer to remove remaining free Fluor-SH, the reaction was concentrated and purified by HiLoad Superdex 75 FPLC column equilibrated with UbFluor Storage Buffer. The pooled eluate was concentrated, snap-frozen, and stored at −80 °C. UbFluor can be stored at −80 °C indefinitely.
For UbFluor storage buffer, we used 250 mm NaCl, 12.5 mm HEPES, pH 6.0.
Measurement of concentration
We prepared a standard curve using a series of Fluor-SH dilutions in characterization buffer (1× PBS, 10 mm BME) and measured absorption at 498 nm using a BiospecNano (Shimadzu Corp., Columbia, MD). We diluted UbFluor 10-fold in characterization buffer and measured absorption at 498 nm to confirm the concentration using the FITC standard curve.
Analysis
MS/MS analysis of phosphorylation and ubiquitination sites
Ubiquitinated or phosphorylated proteins were denatured with 8 m urea by incubating at room temperature for 60 min. After denaturation, proteins were reduced in 10 mm DTT after incubating at 50 °C for 15 min. After reduction, proteins were alkylated in 100 mm iodoacetamide and incubated in the dark at room temperature for 15 min. Subsequently, urea was diluted to 1 m by adding 100 mm ammonium bicarbonate, followed by proteolytic digestion by sequencing grade trypsin. The sample was digested at 37 °C overnight. The digested samples were desalted using reverse phase C18 spin columns (Thermo Fisher Scientific, Rockford, IL) and concentrated to dryness in vacuo. The samples were loaded directly onto a 10-cm long × 75-μm reversed phase capillary column (PicoChip, packed with ProntoSIL C18aq, 300 Å, 3 μm particle size, New Objective, Woburn MA) on a Thermo Easy n-LC II (Thermo Scientific, San Jose, CA). The data were searched using Mascot (version 2.5, Matrix Science, Boston, MA) and reported using Scaffold (Proteome Software, Portland OR).
MALDI-TOF analysis
The SDS-polyacrylamide gels were stained using Coomassie dye. Polyubiquitinated bands within the stacking gel were excised and in-gel digested with trypsin for 12–15 h. The digested peptides were purified and concentrated by zip-tip (Millipore, ZTC04S008) and analyzed by MALDI-TOF (Bruker Autoflex III MALDI, reflectron positive mode, α-cyano-4-hydroxycinnamic acid as a matrix). Masses corresponding to Lys48- and Lys63-linkage peptides were predicted using Swiss-Prot software.
UbcH7∼FUb discharge assay
UbcH7∼FUb was prepared by incubating Ube1 (0.5 μm), UbcH7 (10 μm), fluorescent ubiquitin (FUb, LifeSensors, 15 μm), and 2 mm ATP in 300 mm NaCl, 8 mm MgCl2, 50 mm Tris, pH 7.6, for 30 min at room temperature. The reaction (30 μl, total) was diluted with Quench buffer to 130 μl. The solution was further desalted into the same buffer with a Zeba spin column (Thermo Fisher Scientific, Grand Island, NY). These reactions containing ∼0.5 μm UbcH7∼FUb were then chased by adding pPARKIN (2 μm) in 300 mm NaCl, 50 mm Tris, pH 7.6, on ice, and quenched after 5 s with 3× non-reducing Laemmli buffer. For Quench buffer, we used 25 mm HEPES, pH 7.5, 100 mm NaCl, and 25 mm EDTA.
Fluorescence polarization measurement
Fluorescence polarization was measured using a Synergy4 (BioTek) fluorescence plate reader running Gen5 software (BioTek). Reactions were run in a 384-well plate and had a total volume of 25 μl. All the reactions contained 150 mm NaCl, 6 μm Tween 20, 0.5 mm TCEP, 50 mm HEPES, pH 7.5. All components for a given reaction were added to a 1.5-ml microcentrifuge tube; the appropriate volume of PARKIN was placed on the wall of the tube, and then the reaction was initiated by centrifuging the tube at 10,000 × g for 7 s. The reaction solution was mixed by pipetting five times and then loaded (25 μl) in the 384-well plate (Corning 3820). Once all the samples were loaded to the plate, the plate was spun at 1500 × g for 8 s and then added to the plate reader to record fluorescence polarization. The dead time between spinning PARKIN into solution and FP measurement was ∼2 min. Typically, four to eight reactions were analyzed in parallel. For single turnover assays, four concentrations of UbFluor (0.25, 0.50, 0.75, and 1.0 μm) with 5 μm PARKIN were analyzed every 5–15 s. For multiple turnover assays, four concentrations of UbFluor (10, 12.5, 15, and 20 μm) with 2 μm PARKIN were analyzed every 5–20 s. For conversion of FP measurements (mP values) to the actual concentration of UbFluor remaining during reactions, we referenced three measurements recorded at the beginning of our study: the FP value before the reaction started and after the reaction completed. These two points correspond to the initial concentration of UbFluor in the assay and 0 μm UbFluor, respectively. These measurements were used to set the reference points for both ST and MT reactions, as shown in supplemental Table S1. All FP experiments were repeated at least three times, except for the titration experiments of Fig. 4 (repeated twice).
The following antibodies were used: ubiquitin antibody (Cell Signaling, catalogue no. 3933); Lys48-linkage-specific antibody (Cell Signaling, catalogue no. 4289); Lys63-linkage-specific antibody (Millipore, catalogue no. 05-1308); PARKIN antibody (Santa Cruz Biotechnology, catalogue no. sc-32282); and anti-RHOT2 antibody (Abcam, catalogue no. ab154946).
Protein expression
For T. castaneum PINK1 TcPINK1) preparation, the plasmid for TcPINK1 was a generous gift from Professor Miratul Muqit (Medical Research Council Protein Phosphorylation and Ubiquitination Unit, Dundee, Scotland, UK). The expression protocol follows his laboratory's previous report (22). TcPINK1 was expressed as an MBP-TcPINK1 fusion in BL21 (DE3) Rosetta cells. The overnight culture was used to inoculate 2 liters of LB media containing 50 μg/ml carbenicillin. The culture was grown to OD ∼0.3 at 37 °C, and the temperature was lowered to 16 °C. When the cells grew to OD ∼0.6, the culture was induced by 250 μl of 1 m IPTG and incubated overnight at 16 °C. Lysis was performed with TcPINK1 lysis buffer (described below). Amylose resin (2 ml) was washed with TcPINK1 lysis buffer (two times, 10 ml). The lysate was incubated with beads at 4 °C for 30 min. After washing with TcPINK1 lysis buffer (three times, 10 ml), protein was completely eluted using 10 ml of TcPINK1 elution buffer. ∼10 ml of eluate was concentrated to a total volume under 3 ml using an Amicon filter (MWCO 30,000, EMD Millipore, Billerica, MA) and then dialyzed into TcPINK1 storage buffer (below) overnight at 4 °C. Glycerol was added to 10%, and TcPINK1 was aliquoted and stored at −80 °C.
For TcPINK1 lysis buffer, 1 × PBS, 15 mm imidazole, 1 mm EDTA, 1 mm EGTA, 1% (v/v) Triton X-100, 0.1% (v/v) 2-mercaptoethanol, and 1 mm benzamidine were used. For TcPINK1 elution buffer, 1× PBS, 250 mm imidazole, 0.1 mm EGTA, 0.25% (v/v) Triton X-100, 0.1% (v/v) 2-mercaptoethanol, 1 mm benzamidine, and 0.1 mm PMSF were used. For TcPINK1 storage buffer, 50 mm Tris-HCl, pH 7.6, 150 mm NaCl, 0.1 mm EGTA, 0.25% (v/v) Triton X-100, 0.1% (v/v) 2-mercaptoethanol, and 10% glycerol were used.
PARKIN preparation
The plasmid for His6-SUMO-PARKIN was a generous gift from Professor Miratul Muqit. The expression protocol follows his laboratory's previous report (22). The His6-SUMO-PARKIN plasmid was transformed into BL21-CodonPlus(DE3)-RIL Escherichia coli cells. An overnight culture was grown from a single colony in 20 ml of LB medium (50 μg/ml carbenicillin), and 10 ml was used to inoculate 1 liter of the same medium at 37 °C. Cells were grown at 37 °C until the OD600 was 0.2–0.3, and the temperature was dropped to 15 °C. At OD600, 0.6–0.7 (45–60 min later), expression was induced with 10 μm IPTG (1 m) and 100 μl of ZnCl2 (2.5 m). The cells were grown overnight at 15 °C. Cells were harvested and lysed in 20 ml of PARKIN lysis buffer (below). After sonication and removal of insoluble material at 18,000 × g for 30 min, His6-SUMO-Parkin was purified via Ni2+-NTA-agarose (GE Healthcare). Ni2+-NTA-agarose beads (1.5 ml of slurry for 1 liter of culture) were washed into PARKIN lysis buffer and incubated with the lysate for ∼30 min at 4 °C. Beads were washed with 50 ml of PARKIN lysis buffer. Protein was eluted using PARKIN elution buffer (below). Eluted protein was concentrated using an Amicon filter (MWCO 30,000) to a final volume of 2 ml. The concentrate was purified using an S75 sizing column (GE Healthcare) on an FPLC and was eluted in PARKIN storage buffer (see below). The pooled eluate was mixed with SUMO1/sentrin specific peptidase 1 (SENP1) (PARKIN/SENP1 = 10:1) expressed as described (67) and incubated at room temperature for 2 h. Ni2+-NTA-agarose beads (0.5 ml of slurry, washed into PARKIN storage buffer) were added to the mixture to remove cleaved His6-SUMO, SENP1, and uncleaved His6-SUMO-Parkin. The solution was concentrated to 2 ml using Amicon filter and further purified using the S75 column into PARKIN storage buffer. The eluate was concentrated to ∼1 ml using an Amicon filter, and glycerol was added (10% glycerol). Aliquots were snap-frozen in liquid nitrogen and stored at −80 °C.
To prepare pPARKIN, the pooled His6-SUMO-Parkin (10 ml, 15 μm) in PARKIN storage buffer was treated with 4 μm TcPINK1, 5 mm ATP, and 1 ml of 10× TcPINK1 phosphorylation buffer and incubated at 30 °C for 4 h. The mixture was further treated with SENP1 (PARKIN/SENP1 = 10:1) and incubated at room temperature for 1 h. Ni2+-NTA-agarose beads (0.5 ml slurry, washed into PARKIN storage buffer) were added to the mixture to remove cleaved His6-SUMO, SENP1, and uncleaved His6-SUMO-pPARKIN. The reaction was concentrated using an Amicon filter (MWCO 30,000) and purified by FPLC using an S75 sizing column in PARKIN storage buffer (below). The pooled eluate was concentrated to ∼1 ml using an Amicon filter (MWCO 30,000). 10% glycerol was added to the concentrate, and it was aliquoted and snap-frozen in liquid nitrogen.
For PARKIN lysis buffer, we used 75 mm Tris, pH 7.5, 200 mm NaCl, 0.2% Triton X-100, 25 mm imidazole, 0.5 mm TCEP, 1 mm Pefabloc, and 10 μg/ml leupeptin. For PARKIN elution buffer, we used 400 mm imidazole in 25 mm Tris, pH 8.0, 200 mm NaCl, and 0.5 mm TCEP. For PARKIN storage buffer, we used 25 mm HEPES, pH 8.0, 200 mm NaCl, and 0.5 mm TCEP. For 10× TcPINK1 phosphorylation buffer, we used 500 mm Tris-HCl (pH 7.6), 100 mm MgCl2, 1 mm EGTA, and 20 mm DTT.
Phosphorylation of ubiquitin
Ubiquitin phosphorylation was performed as described in a previous report (8) Ubiquitin solution (100 μm) in TcPINK1 phosphorylation buffer was treated with 10 μm TcPINK1 and 5 mm ATP and incubated at 30 °C overnight. The 10-ml phosphorylation reaction was concentrated using an Amicon filter (MWCO 3,000) to ∼0.5 ml. 9.5 ml of deionized (DI) water was added, and the sample was re-concentrated. This process was repeated for a total of three times to exchange the sample into DI water. The exchanged solution was loaded onto a high trap Q (GE Healthcare, 1 ml) and eluted using a gradient (0% was DI water, 100% was 50 mm Tris-HCl, pH 7.6; gradient was 0–100% in 100 min). The product eluted at ∼30%. 10% glycerol and 0.5 mm TCEP were added to the eluate, which was aliquoted and snap-frozen in liquid nitrogen.
For the TcPINK1 phosphorylation buffer, we used 50 mm Tris-HCl (pH 7.6), 10 mm MgCl2, 0.1 mm EGTA, and 2 mm DTT.
Author contributions
S. P., S. E. R., and A. V. S. designed the study. S. P., P. K. F., and D. T. K. conducted experiments and collected the data. S. P., P. K. F., D. T. K., S. E. R., and A. V. S. analyzed the data. The manuscript was written by S. E. R., S. P., and A. V. S. All authors participated in discussions about the manuscript and approved the final version.
Supplementary Material
This work was supported in part by National Institutes of Health Awards R01GM072656 (to S. E. R.) and R01GM115632 (to A. V. S.) from NIGMS, Northwestern University, and CLP Cornew Innovation Award. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains supplemental Table S1, Figs. S1–S16, and Refs. 1–8.
- RBR
- ring-between-ring
- UPD
- unique PARKIN-specific domain
- Ub
- ubiquitin
- pUb
- Ser65 phosphorylated ubiquitin
- REP
- repressor element
- Ubl
- ubiquitin-like domain
- MT
- multiple turnover
- ST
- single turnover
- FP
- fluorescence polarization
- FUb
- fluorescent ubiquitin
- IBR
- in-between RING
- UbMES
- ubiquitin mercaptoethanesulfonate
- AQUA-MS
- absolute equantification-mass spectrometry
- BME
- β-mercaptoethanol
- h
- human
- IPTG
- isopropyl 1-thio-β-d-galactopyranoside
- Ni2+-NTA
- nickel-nitrilotriacetic acid
- TCEP
- tris(2-carboxyethyl)phosphine
- Ub-VS
- Ub-vinylsulfone
- DI
- deionized.
References
- 1. Pickrell A. M., and Youle R. J. (2015) The roles of PINK1, Parkin, and mitochondrial fidelity in Parkinson's disease. Neuron 85, 257–273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Jin S. M., Lazarou M., Wang C., Kane L. A., Narendra D. P., and Youle R. J. (2010) Mitochondrial membrane potential regulates PINK1 import and proteolytic destabilization by PARL. J. Cell Biol. 191, 933–942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Meissner C., Lorenz H., Weihofen A., Selkoe D. J., and Lemberg M. K. (2011) The mitochondrial intramembrane protease PARL cleaves human Pink1 to regulate Pink1 trafficking. J. Neurochem. 117, 856–867 [DOI] [PubMed] [Google Scholar]
- 4. Narendra D. P., Jin S. M., Tanaka A., Suen D. F., Gautier C. A., Shen J., Cookson M. R., and Youle R. J. (2010) PINK1 is selectively stabilized on impaired mitochondria to activate Parkin. PLoS Biol. 8, e1000298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Sarraf S. A., Raman M., Guarani-Pereira V., Sowa M. E., Huttlin E. L., Gygi S. P., and Harper J. W. (2013) Landscape of the PARKIN-dependent ubiquitylome in response to mitochondrial depolarization. Nature 496, 372–376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bingol B., Tea J. S., Phu L., Reichelt M., Bakalarski C. E., Song Q., Foreman O., Kirkpatrick D. S., and Sheng M. (2014) The mitochondrial deubiquitinase USP30 opposes parkin-mediated mitophagy. Nature 510, 370–375 [DOI] [PubMed] [Google Scholar]
- 7. Ordureau A., Heo J. M., Duda D. M., Paulo J. A., Olszewski J. L., Yanishevski D., Rinehart J., Schulman B. A., and Harper J. W. (2015) Defining roles of PARKIN and ubiquitin phosphorylation by PINK1 in mitochondrial quality control using a ubiquitin replacement strategy. Proc. Natl. Acad. Sci. U.S.A. 112, 6637–6642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ordureau A., Sarraf S. A., Duda D. M., Heo J. M., Jedrychowski M. P., Sviderskiy V. O., Olszewski J. L., Koerber J. T., Xie T., Beausoleil S. A., Wells J. A., Gygi S. P., Schulman B. A., and Harper J. W. (2014) Quantitative proteomics reveal a feedforward mechanism for mitochondrial PARKIN translocation and ubiquitin chain synthesis. Mol. Cell 56, 360–375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Heo J. M., Ordureau A., Paulo J. A., Rinehart J., and Harper J. W. (2015) The PINK1-PARKIN mitochondrial ubiquitylation pathway drives a program of OPTN/NDP52 recruitment and TBK1 activation to promote mitophagy. Mol. Cell 60, 7–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Yamano K., Queliconi B. B., Koyano F., Saeki Y., Hirokawa T., Tanaka K., and Matsuda N. (2015) Site-specific interaction mapping of phosphorylated ubiquitin to uncover Parkin activation. J. Biol. Chem. 290, 25199–25211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Caulfield T. R., Fiesel F. C., Moussaud-Lamodière E. L., Dourado D. F., Flores S. C., and Springer W. (2014) Phosphorylation by PINK1 releases the UBL domain and initializes the conformational opening of the E3 ubiquitin ligase Parkin. PLoS Comput. Biol. 10, e1003935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Shiba-Fukushima K., Imai Y., Yoshida S., Ishihama Y., Kanao T., Sato S., and Hattori N. (2012) PINK1-mediated phosphorylation of the Parkin ubiquitin-like domain primes mitochondrial translocation of Parkin and regulates mitophagy. Sci. Rep. 2, 1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Wauer T., Swatek K. N., Wagstaff J. L., Gladkova C., Pruneda J. N., Michel M. A., Gersch M., Johnson C. M., Freund S. M., and Komander D. (2015) Ubiquitin Ser65 phosphorylation affects ubiquitin structure, chain assembly and hydrolysis. EMBO J. 34, 307–325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kazlauskaite A., Kelly V., Johnson C., Baillie C., Hastie C. J., Peggie M., Macartney T., Woodroof H. I., Alessi D. R., Pedrioli P. G., and Muqit M. M. (2014) Phosphorylation of Parkin at serine 65 is essential for activation: elaboration of a Miro1 substrate-based assay of Parkin E3 ligase activity. Open Biol. 4, 130213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kondapalli C., Kazlauskaite A., Zhang N., Woodroof H. I., Campbell D. G., Gourlay R., Burchell L., Walden H., Macartney T. J., Deak M., Knebel A., Alessi D. R., and Muqit M. M. (2012) PINK1 is activated by mitochondrial membrane potential depolarization and stimulates Parkin E3 ligase activity by phosphorylating serine 65. Open Biol. 2, 120080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Zheng X., and Hunter T. (2013) Parkin mitochondrial translocation is achieved through a novel catalytic activity coupled mechanism. Cell Res. 23, 886–897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Iguchi M., Kujuro Y., Okatsu K., Koyano F., Kosako H., Kimura M., Suzuki N., Uchiyama S., Tanaka K., and Matsuda N. (2013) Parkin-catalyzed ubiquitin-ester transfer is triggered by PINK1-dependent phosphorylation. J. Biol. Chem. 288, 22019–22032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Koyano F., Okatsu K., Kosako H., Tamura Y., Go E., Kimura M., Kimura Y., Tsuchiya H., Yoshihara H., Hirokawa T., Endo T., Fon E. A., Trempe J. F., Saeki Y., Tanaka K., and Matsuda N. (2014) Ubiquitin is phosphorylated by PINK1 to activate parkin. Nature 510, 162–166 [DOI] [PubMed] [Google Scholar]
- 19. Kane L. A., Lazarou M., Fogel A. I., Li Y., Yamano K., Sarraf S. A., Banerjee S., and Youle R. J. (2014) PINK1 phosphorylates ubiquitin to activate Parkin E3 ubiquitin ligase activity. J. Cell Biol. 205, 143–153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Aguirre J. D., Dunkerley K. M., Mercier P., and Shaw G. S. (2017) Structure of phosphorylated UBL domain and insights into PINK1-orchestrated parkin activation. Proc. Natl. Acad. Sci. U.S.A. 114, 298–303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wauer T., Simicek M., Schubert A., and Komander D. (2015) Mechanism of phospho-ubiquitin-induced PARKIN activation. Nature 524, 370–374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kazlauskaite A., Kondapalli C., Gourlay R., Campbell D. G., Ritorto M. S., Hofmann K., Alessi D. R., Knebel A., Trost M., and Muqit M. M. (2014) Parkin is activated by PINK1-dependent phosphorylation of ubiquitin at Ser(65). Biochem. J. 460, 127–139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kitada T., Asakawa S., Hattori N., Matsumine H., Yamamura Y., Minoshima S., Yokochi M., Mizuno Y., and Shimizu N. (1998) Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature 392, 605–608 [DOI] [PubMed] [Google Scholar]
- 24. Lücking C. B., Dürr A., Bonifati V., Vaughan J., De Michele G., Gasser T., Harhangi B. S., Meco G., Denèfle P., Wood N. W., Agid Y., Brice A., French Parkinson's Disease Genetics Study Group, and European Consortium on Genetic Susceptibility in Parkinson's Disease (2000) Association between early-onset Parkinson's disease and mutations in the parkin gene. New Engl. J. Med. 342, 1560–1567 [DOI] [PubMed] [Google Scholar]
- 25. Valente E. M., Abou-Sleiman P. M., Caputo V., Muqit M. M., Harvey K., Gispert S., Ali Z., Del Turco D., Bentivoglio A. R., Healy D. G., Albanese A., Nussbaum R., González-Maldonado R., Deller T., Salvi S., et al. (2004) Hereditary early-onset Parkinson's disease caused by mutations in PINK1. Science 304, 1158–1160 [DOI] [PubMed] [Google Scholar]
- 26. Fiesel F. C., Caulfield T. R., Moussaud-Lamodière E. L., Ogaki K., Dourado D. F., Flores S. C., Ross O. A., and Springer W. (2015) Structural and functional impact of Parkinson disease-associated mutations in the E3 ubiquitin ligase Parkin. Hum. Mutat. 36, 774–786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Safadi S. S., and Shaw G. S. (2007) A disease state mutation unfolds the parkin ubiquitin-like domain. Biochemistry 46, 14162–14169 [DOI] [PubMed] [Google Scholar]
- 28. Sriram S. R., Li X., Ko H. S., Chung K. K., Wong E., Lim K. L., Dawson V. L., and Dawson T. M. (2005) Familial-associated mutations differentially disrupt the solubility, localization, binding and ubiquitination properties of Parkin. Hum. Mol. Genet. 14, 2571–2586 [DOI] [PubMed] [Google Scholar]
- 29. Hampe C., Ardila-Osorio H., Fournier M., Brice A., and Corti O. (2006) Biochemical analysis of Parkinson's disease-causing variants of Parkin, an E3 ubiquitin-protein ligase with monoubiquitylation capacity. Hum. Mol. Genet. 15, 2059–2075 [DOI] [PubMed] [Google Scholar]
- 30. Henn I. H., Gostner J. M., Lackner P., Tatzelt J., and Winklhofer K. F. (2005) Pathogenic mutations inactivate parkin by distinct mechanisms. J. Neurochem. 92, 114–122 [DOI] [PubMed] [Google Scholar]
- 31. Matsuda N., Kitami T., Suzuki T., Mizuno Y., Hattori N., and Tanaka K. (2006) Diverse effects of pathogenic mutations of parkin that catalyze multiple monoubiquitylation in vitro. J. Biol. Chem. 281, 3204–3209 [DOI] [PubMed] [Google Scholar]
- 32. Spratt D. E., Martinez-Torres R. J., Noh Y. J., Mercier P., Manczyk N., Barber K. R., Aguirre J. D., Burchell L., Purkiss A., Walden H., and Shaw G. S. (2013) A molecular explanation for the recessive nature of parkin-linked Parkinson's disease. Nat. Commun. 4, 1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kazlauskaite A., Martínez-Torres R. J., Wilkie S., Kumar A., Peltier J., Gonzalez A., Johnson C., Zhang J., Hope A. G., Peggie M., Trost M., van Aalten D. M., Alessi D. R., Prescott A. R., Knebel A., Walden H., and Muqit M. M. (2015) Binding to serine 65-phosphorylated ubiquitin primes Parkin for optimal PINK1-dependent phosphorylation and activation. EMBO Rep. 16, 939–954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Clark I. E., Dodson M. W., Jiang C., Cao J. H., Huh J. R., Seol J. H., Yoo S. J., Hay B. A., and Guo M. (2006) Drosophila pink1 is required for mitochondrial function and interacts genetically with parkin. Nature 441, 1162–1166 [DOI] [PubMed] [Google Scholar]
- 35. Park J., Lee S. B., Lee S., Kim Y., Song S., Kim S., Bae E., Kim J., Shong M., Kim J. M., and Chung J. (2006) Mitochondrial dysfunction in Drosophila PINK1 mutants is complemented by parkin. Nature 441, 1157–1161 [DOI] [PubMed] [Google Scholar]
- 36. Paterna J. C., Leng A., Weber E., Feldon J., and Büeler H. (2007) DJ-1 and Parkin modulate dopamine-dependent behavior and inhibit MPTP-induced nigral dopamine neuron loss in mice. Mol. Ther. 15, 698–704 [DOI] [PubMed] [Google Scholar]
- 37. Bian M., Liu J., Hong X., Yu M., Huang Y., Sheng Z., Fei J., and Huang F. (2012) Overexpression of Parkin ameliorates dopaminergic neurodegeneration induced by 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine in mice. PLoS One 7, e39953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Hertz N. T., Berthet A., Sos M. L., Thorn K. S., Burlingame A. L., Nakamura K., and Shokat K. M. (2013) A neo-substrate that amplifies catalytic activity of Parkinson's-disease-related kinase PINK1. Cell 154, 737–747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Regnström K., Yan J., Nguyen L., Callaway K., Yang Y., Diep L., Xing W., Adhikari A., Beroza P., Hom R. K., Riley B., Rudolph D., Jobling M. F., Baker J., Johnston J., Konradi A., Bova M. P., Artis D. R., and Artis R. D. (2013) Label free fragment screening using surface plasmon resonance as a tool for fragment finding-analyzing Parkin, a difficult CNS target. PLoS One 8, e66879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Wenzel D. M., Lissounov A., Brzovic P. S., and Klevit R. E. (2011) UBCH7 reactivity profile reveals parkin and HHARI to be RING/HECT hybrids. Nature 474, 105–138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Lazarou M., Narendra D. P., Jin S. M., Tekle E., Banerjee S., and Youle R. J. (2013) PINK1 drives Parkin self-association and HECT-like E3 activity upstream of mitochondrial binding. J. Cell Biol. 200, 163–172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Riley B. E., Lougheed J. C., Callaway K., Velasquez M., Brecht E., Nguyen L., Shaler T., Walker D., Yang Y., Regnstrom K., Diep L., Zhang Z., Chiou S., Bova M., Artis D. R., et al. (2013) Structure and function of Parkin E3 ubiquitin ligase reveals aspects of RING and HECT ligases. Nat. Commun. 4, 1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Wauer T., and Komander D. (2013) Structure of the human Parkin ligase domain in an autoinhibited state. EMBO J. 32, 2099–2112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Trempe J. F., Sauvé V., Grenier K., Seirafi M., Tang M. Y., Ménade M., Al-Abdul-Wahid S., Krett J., Wong K., Kozlov G., Nagar B., Fon E. A., and Gehring K. (2013) Structure of Parkin reveals mechanisms for ubiquitin ligase activation. Science 340, 1451–1455 [DOI] [PubMed] [Google Scholar]
- 45. Joselin A. P., Hewitt S. J., Callaghan S. M., Kim R. H., Chung Y. H., Mak T. W., Shen J., Slack R. S., and Park D. S. (2012) ROS-dependent regulation of Parkin and DJ-1 localization during oxidative stress in neurons. Hum. Mol. Genet. 21, 4888–4903 [DOI] [PubMed] [Google Scholar]
- 46. Dove K. K., Stieglitz B., Duncan E. D., Rittinger K., and Klevit R. E. (2016) Molecular insights into RBR E3 ligase ubiquitin transfer mechanisms. EMBO Rep. 17, 1221–1235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Duda D. M., Olszewski J. L., Schuermann J. P., Kurinov I., Miller D. J., Nourse A., Alpi A. F., and Schulman B. A. (2013) Structure of HHARI, a RING-IBR-RING ubiquitin ligase: autoinhibition of an ariadne-family E3 and insights into ligation mechanism. Structure 21, 1030–1041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Stieglitz B., Rana R. R., Koliopoulos M. G., Morris-Davies A. C., Schaeffer V., Christodoulou E., Howell S., Brown N. R., Dikic I., and Rittinger K. (2013) Structural basis for ligase-specific conjugation of linear ubiquitin chains by HOIP. Nature 503, 422–426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Kelsall I. R., Duda D. M., Olszewski J. L., Hofmann K., Knebel A., Langevin F., Wood N., Wightman M., Schulman B. A., and Alpi A. F. (2013) TRIAD1 and HHARI bind to and are activated by distinct neddylated Cullin-RING ligase complexes. EMBO J. 32, 2848–2860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Lechtenberg B. C., Rajput A., Sanishvili R., Dobaczewska M. K., Ware C. F., Mace P. D., and Riedl S. J. (2016) Structure of a HOIP/E2 similar to ubiquitin complex reveals RBR E3 ligase mechanism and regulation. Nature 529, 546–550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Ho S. R., Lee Y. J., and Lin W. C. (2015) Regulation of RNF144A E3 ubiquitin ligase activity by self-association through its transmembrane domain. J. Biol. Chem. 290, 23026–23038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Bender M. L., Kezdy F. J., and Wedler F. C. (1967) α-Chymotrypsin–enzyme concentration and kinetics. J. Chem. Educ. 44, 84. [DOI] [PubMed] [Google Scholar]
- 53. Pao K. C., Stanley M., Han C., Lai Y. C., Murphy P., Balk K., Wood N. T., Corti O., Corvol J. C., Muqit M. M., and Virdee S. (2016) Probes of ubiquitin E3 ligases enable systematic dissection of parkin activation. Nat. Chem. Biol. 12, 324–331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Park S., Krist D. T., and Statsyuk A. V. (2015) Protein ubiquitination and formation of polyubiquitin chains without ATP, E1 and E2 enzymes. Chem. Sci. 6, 1770–1779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Krist D. T., Park S., Boneh G. H., Rice S. E., and Statsyuk A. V. (2016) UbFluor: a mechanism-based probe for HECT E3 ligases. Chem. Sci. 7, 5587–5595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Krist D. T., Foote P. K., and Statsyuk A. V. (2017) UbFluor: a fluorescent thioester to monitor HECT E3 ligase catalysis. Curr. Protoc. Chem. Biol. 9, 11–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. El Oualid F., Merkx R., Ekkebus R., Hameed D. S., Smit J. J., de Jong A., Hilkmann H., Sixma T. K., and Ovaa H. (2010) Chemical synthesis of ubiquitin, ubiquitin-based probes, and diubiquitin. Angew. Chem. Int. Ed. Engl. 49, 10149–10153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Birsa N., Norkett R., Higgs N., Lopez-Domenech G., and Kittler J. T. (2013) Mitochondrial trafficking in neurons and the role of the Miro family of GTPase proteins. Biochem. Soc. Trans. 41, 1525–1531 [DOI] [PubMed] [Google Scholar]
- 59. Wang X., Winter D., Ashrafi G., Schlehe J., Wong Y. L., Selkoe D., Rice S., Steen J., LaVoie M. J., and Schwarz T. L. (2011) PINK1 and Parkin target miro for phosphorylation and degradation to arrest mitochondrial motility. Cell 147, 893–906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Klosowiak J. L., Focia P. J., Chakravarthy S., Landahl E. C., Freymann D. M., and Rice S. E. (2013) Structural coupling of the EF hand and C-terminal GTPase domains in the mitochondrial protein Miro. EMBO Rep. 14, 968–974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Kamadurai H. B., Souphron J., Scott D. C., Duda D. M., Miller D. J., Stringer D., Piper R. C., and Schulman B. A. (2009) Insights into ubiquitin transfer cascades from a structure of a UbcH5B similar to ubiquitin-HECT(NEDD4L) complex. Mol. Cell 36, 1095–1102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Maspero E., Valentini E., Mari S., Cecatiello V., Soffientini P., Pasqualato S., and Polo S. (2013) Structure of a ubiquitin-loaded HECT ligase reveals the molecular basis for catalytic priming. Nat. Struct. Mol. Biol. 20, 696–701 [DOI] [PubMed] [Google Scholar]
- 63. Kamadurai H. B., Qiu Y., Deng A., Harrison J. S., Macdonald C., Actis M., Rodrigues P., Miller D. J., Souphron J., Lewis S. M., Kurinov I., Fujii N., Hammel M., Piper R., Kuhlman B., and Schulman B. A. (2013) Mechanism of ubiquitin ligation and lysine prioritization by a HECT E3. Elife 2, e00828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Kumar A., Aguirre J. D., Condos T. E., Martinez-Torres R. J., Chaugule V. K., Toth R., Sundaramoorthy R., Mercier P., Knebel A., Spratt D. E., Barber K. R., Shaw G. S., and Walden H. (2015) Disruption of the autoinhibited state primes the E3 ligase Parkin for activation and catalysis. EMBO J. 34, 2506–2521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Eisenthal R., Danson M. J., and Hough D. W. (2007) Catalytic efficiency and kcat/KM: a useful comparator? Trends Biotechnol. 25, 247–249 [DOI] [PubMed] [Google Scholar]
- 66. Baumann S., Schoof S., Bolten M., Haering C., Takagi M., Shin-ya K., Arndt H. D. (2010) Molecular determinants of microbial resistance to thiopeptide antibiotics. J. Am. Chem. Soc. 132, 6973–6981 [DOI] [PubMed] [Google Scholar]
- 67. Mikolajczyk J., Drag M., Békés M., Cao J. T., Ronai Z., and Salvesen G. S. (2007) Small ubiquitin-related modifier (SUMO)-specific proteases: profiling the specificities and activities of human SENPs. J. Biol. Chem. 282, 26217–26224 [DOI] [PubMed] [Google Scholar]
- 68. Borodovsky A., Ovaa H., Kolli N., Gan-Erdene T., Wilkinson K. D., Ploegh H. L., and Kessler B. M. (2002) Chemistry-based functional proteomics reveals novel members of the deubiquitinating enzyme family. Chem. Biol. 9, 1149–1159 [DOI] [PubMed] [Google Scholar]
- 69. Dove K. K., and Klevit R. E. (2017) RING-Between-RING E3 ligases: emerging themes amid the variations. J. Mol. Biol. 10.1016/j.jmb.2017.08.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Tang M. Y., Vranas M., Krahn A. I., Pundlik S., Trempe J. F., and Fon E. A. (2017) Structure-guided mutagenesis reveals a hierarchical mechanism of Parkin activation. Nat. Commun. 8, 14697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Chaugule V. K., Burchell L., Barber K. R., Sidhu A., Leslie S. J., Shaw G. S., and Walden H. (2011) Autoregulation of Parkin activity through its ubiquitin-like domain. EMBO J. 30, 2853–2867 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.