

Abstract
Mavacamten, formerly known as MYK-461 is a recently discovered novel small-molecule modulator of cardiac myosin that targets the underlying sarcomere hypercontractility of hypertrophic cardiomyopathy, one of the most prevalent heritable cardiovascular disorders. Studies on isolated cells and muscle fibers as well as intact animals have shown that mavacamten inhibits sarcomere force production, thereby reducing cardiac contractility. Initial mechanistic studies have suggested that mavacamten primarily reduces the steady-state ATPase activity by inhibiting the rate of phosphate release of β-cardiac myosin-S1, but the molecular mechanism of action of mavacamten has not been described. Here we used steady-state and presteady-state kinetic analyses to investigate the mechanism of action of mavacamten. Transient kinetic analyses revealed that mavacamten modulates multiple steps of the myosin chemomechanical cycle. In addition to decreasing the rate-limiting step of the cycle (phosphate release), mavacamten reduced the number of myosin-S1 heads that can interact with the actin thin filament during transition from the weakly to the strongly bound state without affecting the intrinsic rate. Mavacamten also decreased the rate of myosin binding to actin in the ADP-bound state and the ADP-release rate from myosin-S1 alone. We, therefore, conclude that mavacamten acts on multiple stages of the myosin chemomechanical cycle. Although the primary mechanism of mavacamten-mediated inhibition of cardiac myosin is the decrease of phosphate release from β-cardiac myosin-S1, a secondary mechanism decreases the number of actin-binding heads transitioning from the weakly to the strongly bound state, which occurs before phosphate release and may provide an additional method to modulate myosin function.
Keywords: cardiac hypertrophy, cardiomyopathy, myosin, presteady-state kinetics, small molecule, MYK-461, sarcomere
Introduction
Hypertrophic cardiomyopathy (HCM)2 is one of the most prevalent forms of heritable cardiovascular disorders, affecting approximately 1 in 500 individuals (1). Clinical pathology of HCM is characterized by thickening of the left ventricular wall, decreased left ventricular cavity volume, and diastolic dysfunction, with characteristically hyperdynamic contraction (2, 3). More than 70% of all HCM cases can be attributed to mutations in a variety of sarcomeric proteins (4). One of the most prevalent sites of mutation can be found on the gene encoding myosin heavy chain 7 (MYH7), which encodes β-cardiac myosin heavy chain, the major myosin isoform found in the adult human heart (5). Myosins are molecular motors responsible for converting the chemical energy of ATP hydrolysis into the mechanical force necessary for such processes as cell division and migration, vesicle trafficking, and muscle contraction (6).
There has been extensive research conducted over the years to determine the mechanistic consequences of the various mutations of β-cardiac myosin. Although a vast amount of data had been gathered, there are inconsistencies and disagreements with regard to the effects of each mutation from a biochemical and biophysical perspective. Some mutations, such as the R403Q mutation, have shown decreases in the enzymatic activity of myosin (7), which was later shown to be largely dependent on the isoform of myosin being studied, whereas other studies have reported increases in the enzymatic activity, tension development, and/or increased in vitro filament sliding velocities of myosin (8). It has been previously reported that a direct correlation exists between the ATPase activity of β-cardiac myosin and the rate of force generation in cardiac fibers (9). We believe that modulation of this increased ATPase activity can help to return the hyperdynamic unit back to normal levels and thus alleviate the pathological phenotype.
This increase in enzymatic activity led us to postulate that a molecule that decreases the ATPase activity of β-cardiac myosin would improve the overall contractile properties of a diseased HCM heart. It was previously shown that this can be achieved using the small molecule inhibitor mavacamten (Fig. 1) in mouse models harboring multiple HCM mutations (10). In this work Green et al. (10) showed that mavacamten reduced the steady-state ATPase activity of permeabilized mouse cardiac myofibrils and basal bovine cardiac myosin subfragment-1 (S1), thereby affecting both the α (primary isoform in rodent hearts) and β (primary isoform in large animal hearts) isoforms of cardiac myosin. It was further determined that mavacamten inhibited the phosphate release rate of bovine cardiac myosin subfragment-1 (S1) without affecting the ADP release rate in an actin-activated state (10).
Figure 1.
Chemical structure of mavacamten.
The purpose of this current work is to delve into the molecular mechanism of mavacamten and its effect on multiple types of myosin. This work will utilize systems of increasing complexity to minimal systems to identify the minimal catalytic unit on which mavacamten has an effect. Transient kinetic analysis was performed to determine the mechanism by which mavacamten acts. Green et al. (10) reported that the primary mechanism of mavacamten is inhibition of the phosphate release rate; our analysis suggests that another step in the chemomechanical cycle of myosin is also affected and may explain the mechanism in greater detail.
Results
Steady-state characterization of mavacamten
As previously reported (10), a small molecule screen was conducted against the bovine cardiac myofibril system to identify molecules that decreased the steady-state ATPase rate, thereby reducing the ensemble force generation of the sarcomere. Optimization of chemical matter from this screen led to the development of mavacamten. Myofibrils generated from human and bovine cardiac tissues as well as rabbit psoas (fast skeletal) were used to determine pCa50 values for each system. Concentration-dependent changes in myofibril ATPase at their respective pCa50 values show a similar IC50 for bovine and human systems, both of which have a 4-fold potency enhancement over fast skeletal myofibrils (Table 1). Mavacamten was found to have an IC50 value of 0.49 μm in the bovine system, 0.71 μm in the human system, and 2.14 μm in the rabbit system, indicating selectivity of >4-fold for cardiac myosin (Fig. 2). Analysis of the curve fit for these samples also shows a calculated Hill coefficient of ∼2 for both the rabbit skeletal and human cardiac myofibril systems. This deviation may be due to the heterogeneous nature of the protein preparation for each sample and potential variations in their respective preparations. A concentration-dependent response of mavacamten was also run in the in vitro motility (IVM) assay to assess its effect on actin filament sliding velocities. Cardiac heavy meromyosin (HMM) on a glass surface was used to propel actin filaments in the presence of ATP and compound or DMSO. It was found that mavacamten slowed the sliding velocity of actin filaments in a concentration-dependent manner with a calculated IC50 of 0.587 ± 0.149 μm, confirming the effect of mavacamten on a soluble form of double-headed myosin (Fig. 3). Notably, the movement of these filaments was more sporadic and “search-like” with mavacamten consistent with a mechanism that decreases the phosphate release rate of myosin and, therefore, entry into the strongly bound state measured by IVM.
Table 1.
Mavacamten IC50 and Hill coefficient values for multiple myofibril systems
| Myofibril type (pCa50) | IC50 value | Hill coefficient |
|---|---|---|
| μm | ||
| Bovine cardiac (6.25) | 0.490 ± 0.027 | 1.1 ± 0.02 |
| Rabbit fast skeletal (6.00) | 2.14 ± 0.27 | 2.14 ± 0.06 |
| Human cardiac (6.00) | 0.711 ± 0.099 | 2.04 ± 0.06 |
Figure 2.

Concentration-dependent response curve of mavacamten (MYK-461) in multiple myofibril-based systems. Myofibrils were assayed at pCa50 values of 6.25, 6.00, and 6.00 for bovine cardiac (1 mg/ml), rabbit skeletal (0.25 mg/ml), and human cardiac (1 mg/ml) respectively. Final assay conditions contained 2% DMSO with varying concentrations of mavacamten. Data were normalized to a DMSO control for each myofibril type assayed and analyzed using a four parameter fit (GraphPad Prism) to determine IC50 values of mavacamten. DMSO control rates of 0.097 ± 0.0002 μm/s, 0.158 ± 0.01 μm/s, and 0.104 ± 0.005 μm/s were measured for bovine cardiac, rabbit skeletal, and human cardiac myofibrils, respectively.
Figure 3.

Mavacamten (MYK-461) decreased in vitro motility-sliding velocities. The final in vitro motility buffer contained 2% DMSO with or without mavacamten at varying concentrations. Sliding velocities were normalized to DMSO only velocities. n = 3 for all data points. For each n, >500 filament trajectories were quantified, and the median velocity from each of the 3 data sets was averaged.
To further elucidate the effect of mavacamten and determine the minimal system in which mavacamten elicits a response, concentration-dependent response measurements were performed on a system utilizing myosin S1 generated from bovine cardiac, rabbit psoas, and chicken gizzard tissues as well as a recombinant form of human β-cardiac myosin and purified bovine cardiac actin (Fig. 4). Mavacamten was shown to inhibit all systems with potencies in the same range as the myofibril system, with the exception of smooth muscle myosin derived from chicken gizzard (Table 2). These data, along with concentration-dependent response measurements against basal myosin (data not shown), allows us to deduce that mavacamten exerts its effect via the myosin-S1 motor. It is noteworthy that there is increased specificity between the cardiac and skeletal isoforms (∼10×) compared with the myofibril system (∼4×), possibly due to the increased complexity of the myofibril system and that mavacamten shows no activity against the smooth muscle isoform of myosin-S1. To answer an open question of whether mavacamten would exert the same activity on mutant myosin-S1 as compared with wild-type bovine and human protein, concentration-dependent response experiments were performed against five mutations known to be associated with HCM. Fig. 5A shows the response curves for the mutant forms of recombinant human cardiac myosin, and their respective IC50 values are given in Table 2. Mavacamten inhibits each of these mutants to varying degrees, with the greatest potency conferred against the G741R mutant, 0.653 μm, and lower potency against the R719W mutant, 1.31 μm. Given that the potency of mavacamten against the wild-type form of human cardiac myosin-S1 is 0.727 μm, these values are remarkably similar (Fig. 5B).
Figure 4.

Concentration-dependent response of mavacamten (MYK-461) in purified actomyosin systems. Bovine cardiac (0.25 μm), rabbit skeletal (0.1 μm), chicken gizzard (0.5 μm), and human cardiac (0.25 μm) myosin-S1 were assayed with a constant concentration of actin (14 μm) at varying concentrations of mavacamten. ATPase rates were normalized to DMSO controls for each myosin type assayed, and EC50 values were calculated using a four-parameter-fit model in Prism (GraphPad). DMSO controls rates of 0.145 ± 0.002 μm/s, 0.121 ± 0.001 μm/s, 0.154 ± 0.001 μm/s, and 0.134 ± 0.002 μm/s were measured for bovine cardiac, rabbit skeletal, chicken gizzard, and human cardiac myosin-S1, respectively.
Table 2.
Mavacamten IC50 and Hill coefficient values for multiple purified actomyosin systems
| Actomyosin system (myosin concentration in μm) | IC50 values | Hill coefficient |
|---|---|---|
| μm | ||
| Bovine cardiac (0.25) | 0.473 ± 0.006 | 1.11 ± 0.01 |
| Rabbit fast skeletal (0.1) | 5.85 ± 0.65 | 0.867 ± 0.051 |
| Chicken gizzard smooth (0.5) | >50 | N/A |
| Human cardiac, wild type (0.25) | 0.727 ± 0.052 | 0.833 ± 0.029 |
| Human cardiac, R403Q (0.25) | 0.710 ± 0.015 | 0.968 ± 0.009 |
| Human cardiac, R453C (0.25) | 1.02 ± 0.053 | 0.947 ± 0.037 |
| Human cardiac, R719W (0.25) | 1.31 ± 0.072 | 0.898 ± 0.034 |
| Human cardiac, R723G (0.25) | 1.04 ± 0.062 | 0.984 ± 0.007 |
| Human cardiac, G741R (0.25) | 0.653 ± 0.033 | 0.912 ± 0.021 |
Figure 5.

Concentration-dependent response of mavacamten (MYK-461) in purified human actomyosin systems. A, human cardiac myosin-S1 (0.25 μm) and all mutants were assayed with a constant concentration of actin (14 μm) at varying concentrations of mavacamten. B, an expanded panel, showing the subtle differences in the potency of mavacamten against all mutant myosins assayed. ATPase rates were normalized to DMSO controls for each myosin type assayed, and EC50 values were calculated using a four-parameter-fit model in Prism (GraphPad). DMSO controls rates of 0.269 ± 0.002 μm/s, 0.142 ± 0.001 μm/s, 0.138 ± 0.002 μm/s, 0.184 ± 0.001 μm/s, 0.135 ± 0.001 μm/s, and 0.078 ± 0.001 μm/s were measured for wild-type, R403Q, R453C, R719W, R723G, and G741R human cardiac myosin-S1, respectively.
Transient kinetic analysis of mavacamten
To begin dissecting the mechanism by which mavacamten exerts its effect on cardiac myosin, we utilized presteady-state methods to interrogate multiple steps in the chemomechanical cycle of myosin (Fig. 6). As previously reported (10), mavacamten is known to inhibit the phosphate release rate (Fig. 6, step 5) of bovine cardiac myosin-S1 using the method of Brune et al. (11). We found that under single turnover conditions mavacamten inhibited the actin-associated phosphate release rate in a concentration-dependent manner with a maximal inhibition of 80% at the highest concentration tested (10 μm) and with an IC50 value of 1.85 μm (10). We have built upon this work to determine the effect of mavacamten on the recombinant human cardiac myosin-S1 construct. Mavacamten inhibited the human form of the enzyme in a concentration-dependent manner and to a similar extent as the bovine cardiac myosin-S1 system (Fig. 7). The IC50 value for the human construct was calculated to be 1.78 μm.
Figure 6.
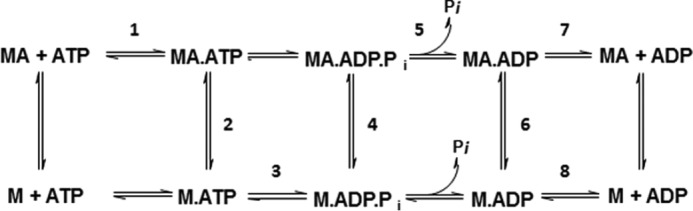
Generalized chemomechanical cycle of myosin. M is defined as myosin, A as actin, and Pi as inorganic phosphate.
Figure 7.

Phosphate release of myosin-S1 with varying concentrations of mavacamten (MYK-461). Myosin-S1 (0.5 μm final) was mixed with ATP (0.5 μm final) and aged for 2 s. This solution was then mixed with actin (14 μm final) and PBP (10 μm final), the fluorescence change upon binding of inorganic phosphate was recorded at an excitation wavelength of 430 nm, and emission was monitored through a 455-nm cutoff filter. Data were been normalized to DMSO controls. IC50 values of 1.85 ± 0.14 μm (bovine cardiac, A) and 1.78 ± 0.069 μm (human cardiac, B) were calculated using a four-parameter-fit model in Prism (GraphPad). A DMSO controls rate of 2.72 ± 0.16 s−1 and 0.257 ± 0.025 s−1 was measured for bovine cardiac and human cardiac myosin, respectively.
To further the mechanistic characterization of mavacamten, the ADP release rates were measured for the actin-associated (Fig. 6, step 7) and basal (myosin alone) (Fig. 6, step 8) states of bovine cardiac myosin-S1 (Fig. 8) using mant-ADP. Little difference was observed in the ADP release rate in an actomyosin system: DMSO and 20 μm mavacamten response rates of 128.9 ± 3.10 s−1 and 132.2 ± 3.05 s−1, respectively. Alternatively, ADP release rates in the basal system were found to be 0.2616 ± 0.0005 s−1 and 0.1402 ± 0.0007 s−1 for DMSO and 20 μm mavacamten, respectively, resulting in a 46% decrease in the overall rate.
Figure 8.
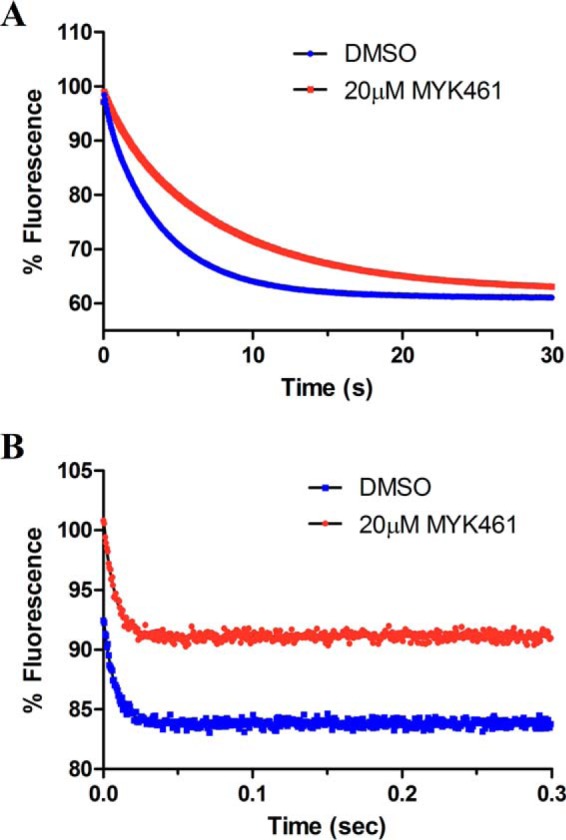
Mant-ADP release experiments were performed in both the basal (A)- and actin-associated (B) states of bovine cardiac myosin-S1, and representative traces are shown. In the basal (actin-free) state, bovine cardiac myosin-S1 (1.25 μm final) was incubated with mant-ADP (1 μm final) in the presence of 2% DMSO or 20 μm mavacamten (MYK-461) and rapidly mixed with 1 mm ATP. The fluorescence decrease of the mant-ADP was followed and fit to a single exponential, yielding mant-ADP release rates of 0.2616 ± 0.01455 s−1 and 0.1402 ± 0.010356 s−1 for DMSO and mavacamten, respectively. In the actin-associated state, bovine cardiac myosin-S1 (1.25 μm final), actin (4 μm final), and mant-ADP (1 μm final) were incubated and rapidly mixed with 1 mm ATP. The rate of actin-associated mant-ADP release was measured to be 128.9 ± 3.10 s−1 and 132.2 ± 3.05 s−1, for DMSO and mavacamten, respectively.
Previously it was reported that mavacamten did not affect the dissociation of myosin from actin in the ADP-bound state (10). To determine the effect of mavacamten on the rate of actin association in a strongly bound state (Fig. 6, step 6), a single mix stopped-flow experiment was performed in which myosin in the ADP-bound state of S1 was mixed with varying concentrations of pyrene actin in the presence and absence of 20 μm mavacamten (Fig. 9). The binding rate was measured via a decrease in the fluorescent transient of pyrene actin. The data in Fig. 9 were plotted and fit by linear regression, and the second order rate constant was calculated to decrease by ∼4-fold, with values of 63.48 ± 0.76 μm−1s−1 and 15.77 ± 0.75 μm−1s−1 for DMSO and 20 μm mavacamten, respectively.
Figure 9.

Actin association measured via pyrene actin in the ADP state. Bovine cardiac myosin-S1 (0.25 μm final) was rapidly mixed with pyrene actin (10 μm final) in an ADP-bound state (1.25 μm final). The pyrene actin fluorescence decrease was recorded and fit to a single exponential. This method was done over a range of pyrene actin concentrations in the presence and absence of mavacamten (MYK-461). It was found that the second order rate constant of myosin-S1 binding was reduced ∼4-fold in the presence of mavacamten.
To determine the effect of mavacamten on the weak to strong transition (Fig. 6, step 4) we used the method previously reported by De La Cruz et al. (12) for measuring the transition of myosin V. The rate of transition was measured with varying concentrations of mavacamten, and it was found that the compound had no effect on the transition rate, which was ∼25 s−1 (Fig. 10). The amplitudes were compared to determine if there was an effect on the number of myosin heads in the transition state that could bind to actin filaments. It was observed that increasing amounts of mavacamten decreased the amplitude of pyrene quenching by cardiac myosin-S1 in the transition state by 43% at the highest concentration.
Figure 10.
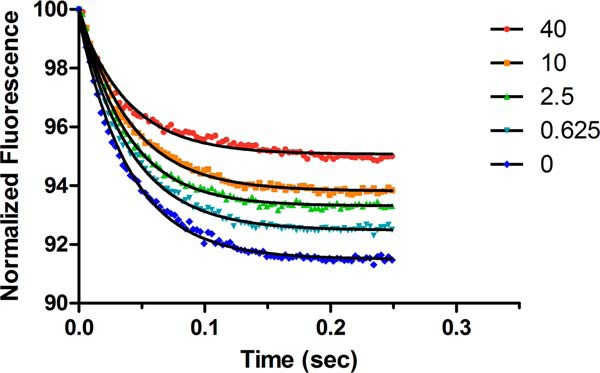
Weak to strong transition of myosin-ADP-Pi binding to pyrene actin in the presence of 1 mm ADP. Single turnover conditions in double mix mode are shown. Myosin-S1 (0.5 μm final) and ATP (0.5 μm final) were mixed and aged for 2 s and then mixed with 0.5 μm pyrene actin and 1 mm ADP. Representative traces show that increasing concentrations of mavacamten (MYK-461) reduce the amplitude of pyrene quenching caused by fewer myosin heads interacting with the actin filament.
Discussion
Because the discovery of mavacamten and the findings that showed encouraging effects in mice with a hypertrophic cardiomyopathy phenotype, we have begun experiments to deduce its mechanism of action on β-cardiac myosin. Steady-state characterization has taken us from discovery on the level of a complex myofibril to potency measurements on more purified systems, such as actomyosin and the myosin-S1 motor unit. Collectively, the data show that mavacamten reduced the ATPase rate of both the actin-associated and basal systems. Further findings showed that mavacamten is selective with respect to the β-cardiac isoform of myosin, with IC50 values of 0.473, 0.727, 5.852, and >50 μm in the bovine cardiac, human cardiac, rabbit fast skeletal, and chicken gizzard smooth muscle actomyosin systems respectively (Fig. 4). An in vitro motility assay was also performed and showed that mavacamten has a very similar IC50 compared with the purified actomyosin system.
Mavacamten showed similar IC50 values (a measure of potency) in both assays. The velocity with which actin filaments move across the HMM-covered surface in the IVM assay is largely thought to be rate limited by the ADP release rate of the myosin motor (15), whereas the steady-state ATPase assay is rate-limited by phosphate release (13, 14). However, it has been recently shown that sliding velocities in IVM may not be detachment-limited (gated by the ADP release rate) but also attachment-limited (gated by the entry into the strongly bound state or phosphate release) (16). The observation that mavacamten slowed steady-state ATPase kinetics as well as sliding velocities led us to perform stopped flow experiments to interrogate individual steps in the chemomechanical cycle of bovine cardiac myosin-S1 to determine the more precise mechanism of action of mavacamten.
Mavacamten reduces the phosphate release rate of bovine cardiac myosin-S1 in a concentration-dependent fashion, and we have also confirmed this effect on recombinant human β-cardiac myosin-S1, with an IC50 of 1.78 μm (Fig. 7). This finding coupled with the effect of mavacamten on the mutant myosins also tested allows us to conclude that the primary mechanism of action for this compound is the inhibition of the phosphate release step in the chemomechanical cycle. But this may not be the only step of the cycle that is affected by mavacamten. Looking at the inhibitory effect of mavacamten in the in vitro motility assay, an argument can be made that ADP release may also be affected by the compound. When looking at the kinetic transients of ADP release in Fig. 8, we see that the ADP release rate of bovine cardiac myosin-S1 in an actin-associated state was not affected by mavacamten (Fig. 8B), but the basal (myosin alone) rate of ADP release was inhibited by ∼50%. Furthermore, looking at the actin association rate of myosin-S1 in the ADP state, we saw a 4-fold reduction in the second-order rate constant for actin binding in the presence of mavacamten (Fig. 9). Taking these two parameters into account, we can see that decreased binding in a known strongly bound state can begin to describe the motility data. By decreasing the rate of ADP release to prime the myosin head to initiate another turnover, thereby another stroke, and decreasing the rate of binding in this strongly bound state, mavacamten may ultimately be taking heads out of the cycle and causing them to be “parked” into a state that is nonproductive in the chemomechanical cycle of myosin-S1.
This hypothesis was interrogated using single turnover kinetics and pyrene actin fluorescence to determine the weak to strong transition rate of myosin-S1 to actin. The data collected showed very little change in the rate of transition, 24–26 s−1, but a marked difference in the amplitudes of the reaction comparing DMSO and 40 μm mavacamten. The representative traces shown in Fig. 10 show a >40% reduction in fluorescence amplitude, which has been shown to represent the number of heads bound during this transition (12). This is the first piece of evidence to show a direct effect of mavacamten on the binding of myosin-S1 to actin in the transition state and gives some clarity as to its mechanism of action.
Experimental procedures
Protein preparation
Cardiac myofibrils were prepared as previously described (17). Bovine cardiac tissue was harvested, placed immediately on wet ice, and shipped overnight, and the left ventricle and septum were dissected, frozen in liquid nitrogen, and stored at −80 °C. Human tissue was procured from BioReclamations IVT (New York), and myofibrils were prepared on the day of receipt. Cardiac and skeletal myosin S1 was prepared using a chymotryptic digestion of full-length myosin prepared from bovine cardiac left ventricle and rabbit psoas muscle, respectively (18). Bovine cardiac HMM was prepared according to Margossian and Lowey (18). Human cardiac myosin subfragment-1 was expressed in differentiated murine C2C12 myotubes using an adenovirus infection method (19). The recombinant product utilized a 6×-histidine tag on the essential light chain for initial purification on Ni2+-resin with further purification by anion exchange and size exclusion chromatography. All myofibril and myosin-S1 preparations were brought to 10% sucrose, snap-frozen in liquid nitrogen, and stored at −80 °C. Actin was prepared from a bovine cardiac acetone powder (Pel Freez Biologicals) according to the method of Spudich and Watt (20). Pyrene actin was prepared according to the method of Criddle et al. (21).
Data analysis
All curve-fitting was conducted using GraphPad Prism 7.0. Data were processed or preprocessed using Microsoft Excel, SoftMax Pro (Molecular Devices), or KinetAsyst (TgK Scientific). All experiments were performed in triplicate, and the means and standard deviations were obtained from three independent experiments, with each performed in triplicate.
Steady-state characterization
ATPase measurements were conducted using a coupled enzyme system utilizing pyruvate kinase and lactate dehydrogenase. Unless otherwise stated, the buffer system used in all experiments was 12 mm Pipes, 2 mm MgCl2, 1 mm DTT at pH 6.8 (PM12 buffer). All steady-state experiments were carried out at 20 °C using a SpectraMax 384Plus plate reader, and rates were recorded using the SoftMax Pro software package. For all steady-state experiments, data were collected in triplicate and averaged, with n = 3. The value for n refers to the number of individual experiments performed. All data analysis of the steady-state systems were conducted using GraphPad Prism.
Transient kinetic characterization
Transient kinetic experiments were performed using a stopped-flow apparatus (Hi-Tech Scientific, SF-61 DX2) to determine the effects of mavacamten on myosin association and dissociation from actin filaments (21, 22), phosphate (Pi) release (11, 23), and 2′-(or-3′)-O-(N-methylanthraniloyl)-ADP (mant-ADP/ATP) release by myosin (23). For each data point, transient traces were collected in triplicate and averaged for each experiment, with n = 3. All transient experiments were performed with either varying amounts of mavacamten or single concentrations of mavacamten at varying substrate concentrations to determine a concentration-dependent change in each kinetic parameter, and control experiments were carried out with 2% DMSO final. For mant-ATP or mant-ADP experiments, fluorescence emission was measured through a 400-nm cutoff filter with excitation at 365 nm. The increase in fluorescence upon myosin binding mant-ATP or decrease in fluorescence after release of mant-ADP was monitored as previously described (24).
The rates of Pi release were measured using the bacterial phosphate-binding protein (PBP) modified with 7-diethyl-amino-3-[[[2-(maleimidyl)ethyl]amino]carbonyl] coumarin (MDCC) dye prepared according to Brune et al. (11). The stopped flow instrument was set up in double mix mode. In this configuration nucleotide free myosin-S1 was mixed with ATP at a 1:1 molar ratio and aged for 2 s to allow for complete hydrolysis. The myosin-nucleotide complex was then rapidly mixed with actin plus MDCC-PBP, and the fluorescence increase due to phosphate binding was measured through a 455-nm cutoff filter with excitation at 425 nm. This system was used to measure the effect of mavacamten by varying the concentration of compound in all syringes and comparing the data to a DMSO control. Before data collection, contaminating phosphate was removed from the system by soaking with a “Pi-mop,” which consisted of purine nucleoside phosphorylase and 7-methylguanosine at concentrations of 1 units/ml and 0.5 mm, respectively. This Pi-mop was also present in all solutions at concentrations of 0.1 units/ml purine nucleoside phosphorylase and 0.25 mm 7-methylguanosine to remove any residual phosphate.
Myosin-S1 association to pyrene-actin filaments was monitored by the quenching of pyrene fluorescence that occurs upon S1 binding to pyrene-actin (22, 25). The kinetics of ATP-induced acto-S1 dissociation was measured by monitoring the increase in pyrene fluorescence upon mixing pyrene-acto-S1 with increasing concentrations of ATP. Pyrene fluorescence was measured using a 400-nm cutoff filter with excitation at 360 nm. This interaction was also used to monitor the transition from the weakly to strongly bound state of myosin to actin. Briefly, bovine cardiac myosin-S1 and ATP were mixed under single turnover conditions and allowed to age for 2 s to hydrolyze the ATP to ADP-Pi. This mixture was then mixed with pyrene actin in a 1:1 ratio with myosin and 1 mm ADP to shift the equilibrium to the strongly bound state. The quenching of pyrene actin was monitored with varying concentrations of mavacamten, and the reaction amplitudes were analyzed.
In vitro motility assay
After thawing HMM, inactive HMM heads were removed before performing the motility assay. Actin was polymerized by adding 25 mm KCl and 2 mm MgCl2 and incubating for 30 min at 4 °C. HMM was combined with polymerized actin, and 2 mm ATP was then added to the actomyosin solution to release functional HMM heads from actin, and the sample was centrifuged at 350,000 × g for 15 min. The supernatant containing active HMM was used for experiments. Fluorescent actin filaments were made by combining 1 μm tetramethylrhodamine phalloidin with 1 μm actin in IVM assay buffer (25 mm Imidazole, pH 7.4, 25 mm KCl, 4 mm MgCl2, 1 mm EGTA, 1 mm DTT) containing 1 mm ATP. Tetramethylrhodamine-phalloidin-labeled filaments were stored in the dark at 4 °C for up to 1 week.
Movement of F-actin filaments over HMM-coated surfaces was achieved using a modification to the method of Uyeda et al. (26). When used, MyoKardia compounds were added to the final motility buffer with a final DMSO concentration of 2%, a concentration that does not significantly affect actin sliding velocities (data not shown). Images were acquired on a Nikon Eclipse Ti microscope at a frame rate of 2 Hz with a 100× oil immersion objective. Rigorous experimental design ensured that data sets contained >95% moving filaments. Moving filaments were defined as those with a velocity of >25 nm/s. Custom analysis software was created by VigeneTech, in which images were set to a threshold based on pixel intensity, filaments were identified, and trajectories were determined for each filament. Only filaments >2 μm were recorded. 3–4 movies, each with 100–1000 filament trajectories, from different surfaces were analyzed, and the median velocities of these were averaged for a single n. Each data point shown is a combination of 3–4 individual experiments (n = 3–4, each containing 500–3000 filament tracks).
Author contributions
R. F. K. conducted all steady-state and most transient kinetic experiments, analyzed the results, and wrote the manuscript. R. L. A. conducted the transient kinetic experiments and analyzed the results. S. R. B. I. conducted the IVM experiments, analyzed the data, and wrote the corresponding sections of the manuscript. Y. S. and A. S. S. synthesized mavacamten. H. M. R. provided guidance for the experiments needed to prepare the manuscript. All authors reviewed the results, provided comments, and approved the final version of the manuscript.
All authors are stockholders of MyoKardia, Inc.
- HCM
- hypertrophic cardiomyopathy
- IVM
- in vitro motility
- HMM
- heavy meromyosin
- S1
- subfragment-1
- PBP
- phosphate-binding protein
- MDCC
- 7-diethyl-amino-3-[[[2-(maleimidyl)ethyl]amino]carbonyl] coumarin
- mant-ADP
- 2′-(or-3′)-O-(N-methylanthraniloyl) ADP.
References
- 1. Maron B. J., Gardin J. M., Flack J. M., Gidding S. S., Kurosaki T. T., and Bild D. E. (1995) Prevalence of hypertrophic cardiomyopathy in a general population of young adults: echocardiographic analysis of 4111 subjects in the CARDIA study. coronary artery risk development in (young) adults. Circulation 92, 785–789 [DOI] [PubMed] [Google Scholar]
- 2. Wilson W. S., Criley J. M., and Ross R. S. (1967) Dynamics of left ventricular emptying in hypertrophic subaortic stenosis: A cinaengiographic and hemodynamic study. Am. Heart J. 73, 4–16 [DOI] [PubMed] [Google Scholar]
- 3. Stewart S., Mason D. T., and Braunwald E. (1968) Impared rate of left ventricular filling in idiopathic hypertrophic subaortic stenosis and valvular aortic stenosis. Circulation 37, 8–14 [DOI] [PubMed] [Google Scholar]
- 4. Ramaraj R. (2008) Hypertrophic cardiomyopathy: etiology, diagnosis, and treatment. Cardiol. Rev. 16, 172–180 [DOI] [PubMed] [Google Scholar]
- 5. Frey N., Luedde M., and Katus H. A. (2011) Mechanisms of disease: hypertrophic cardiomyopathy. Nat. Rev. Cardiol. 9, 91–100 [DOI] [PubMed] [Google Scholar]
- 6. Krendel M., and Mooseker M. S. (2005) Myosins: tails (and heads) of functional diversity. Physiology 20, 239–251 [DOI] [PubMed] [Google Scholar]
- 7. Cuda G., Fananapazir L., Epstein N. D., and Sellers J. R. (1997) The in vitro motility activity of β-cardiac myosin depends on the nature of the β-myosin heavy chain gene mutation in hypertrophic cardiomyopathy. J. Muscle Res. Cell Motil. 18, 275–283 [DOI] [PubMed] [Google Scholar]
- 8. Tyska M. J., Hayes E., Giewat M., Seidman C. E., Seidman J. G., and Warshaw D. M. (2000) Single-molecule mechanics of R403Q cardiac myosin isolated form the mouse model of familia hypertrophic cardiomyopathy. Circ. Res. 86, 737–744 [DOI] [PubMed] [Google Scholar]
- 9. Brenner B., and Eisenberg E. (1986) Rate of force generation in muscle: correlation with actomyosin ATPase activity in solution. Proc. Natl. Acad. Sci. U.S.A. 83, 3542–3546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Green E. M., Wakimoto H., Anderson R. L., Evanchik M. J., Gorham J. M., Harrison B. C., Henze M., Kawas R., Oslob J. D., Rodriguez H. M., Song Y., Wan W., Leinwand L. A., Spudich J. A., McDowell R. S., Seidman J. G., and Seidman C. E. (2016) A small-molecule inhibitor of sarcomere contractility suppresses hypertrophic cardiomyopathy in mice. Science 351, 617–621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Brune M., Hunter J. L., Corrie J. E., and Webb M. R. (1994) Direct, real-time measurement of rapid organic phosphate release using a novel fluorescent probe and its application to actomyosin subfragment 1 ATPase. Biochemistry 33, 8262–8271 [DOI] [PubMed] [Google Scholar]
- 12. De La Cruz E. M., Wells A. L., Rosenfeld S. S., Ostap E. M., and Sweeney H. L. (1999) The kinetic mechanism of myosin V. Proc. Natl. Acad. Sci. 96, 13726–13731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Lionne C., Iorga B., Candau R., Piroddi N., Webb M. R., Belus A., Travers F., and Barman T. (2002) Evidence that phosphate release is the rate-limiting step on the overall ATPase of psoas myofibrils prevented from shortening by chemical cross-linking. Biochemistry 41, 13297–13308 [DOI] [PubMed] [Google Scholar]
- 14. Iorga B., Candau R., Travers F., Barman T., and Lionne C. (2004) Does phosphate release limit the ATPases of soleus myofibrils? Evidence that (A)M·ADP·Pi states predominate on the cross-bridge cycle. J. Muscle Res. Cell Motil. 25, 367–378 [DOI] [PubMed] [Google Scholar]
- 15. Sato M. K., Ishihara T., Tanaka H., Ishijima A., and Inoue Y. (2012) Velocity-dependent actomyosin ATPase cycle revealed by in vitro motility assay with kinetic analysis. Biophys. J. 103, 711–718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Brizendine R. K., Alcala D. B., Carter M. S., Haldeman B. D., Facemyer K. C., Baker J. E., and Cremo C. R. (2015) Velocities of unloaded muscle filaments are not limited by drag forces imposed by myosin cross-bridges. Proc. Natl. Acad. Sci. 112, 11235–11240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Patel B. G., Wilder T., Solaro R. J. (2013) Novel control of cardiac myofilament response to calcium by S-glutathionylation at specific sites of myosin binding protein C. Front. Physiol. 4, 336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Margossian S. S., and Lowey S. (1982) Preparation of myosin and its subfragments from rabbit skeletal muscle. Methods Enzymol. 85, 55–71 [DOI] [PubMed] [Google Scholar]
- 19. Deacon J. C., Bloemink M. J., Rezavandi H., Geeves M. A., and Leinwand L. A. (2012) Identification of functional differences between recombinant human α and β cardiac myosin motors. Cell. Mol. Life Sci. 69, 2261–2277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Spudich J. A., and Watt S. (1971) The regulation of rabbit skeletal muscle contraction. I. Biochemiscal studies of the interaction of the tropomyosin-troponin complex with actin and the proteolytic fragments of myosin. J. Biol. Chem. 246, 4866–4871 [PubMed] [Google Scholar]
- 21. Criddle A. H., Geeves M. A., and Jeffries T. (1985) The use of actin labelled with N-(1-pyrenyl)iodoacetamide to study the interaction of actin with myosin subfragments and troponin/tropomyosin. Biochem. J. 232, 343–349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Cooper J. A., Walker S. B., and Pollard T. D. (1983) Pyrene actin: documentation of the validity of a sensitive assay for actin polymerization. J. Muscle Res. Cell Motil. 4, 253–262 [DOI] [PubMed] [Google Scholar]
- 23. Gilbert S. P., and Mackey A. T. (2000) Kinetics: a tool to study molecular motors. Methods 22, 337–354 [DOI] [PubMed] [Google Scholar]
- 24. Radke M. B., Taft M. H., Stapel B., Hilfiker-Kleiner D., Preller M., Manstein D. J. (2014) Small molecule-mediated refolding and activation of myosin motor function. eLife 3, e01603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kovács M., Tóth J., Hetényi C., Málnási-Csizmadia A., and Sellers J. R. (2004) Mechanism of blebbistatin inhibition of myosin II. J. Biol. Chem. 279, 35557–35563 [DOI] [PubMed] [Google Scholar]
- 26. Uyeda T. Q., Kron S. J., and Spudich J. A. (1990) Myosin step size: estimation from slow sliding movement of actin over low densities of heavy meromyosin. J. Mol. Biol. 214, 699–710 [DOI] [PubMed] [Google Scholar]