

Abstract
Salivary gland inflammation is a hallmark of Sjögren's syndrome (SS), a common autoimmune disease characterized by lymphocytic infiltration of the salivary gland and loss of saliva secretion, predominantly in women. The P2X7 receptor (P2X7R) is an ATP-gated nonselective cation channel that induces inflammatory responses in cells and tissues, including salivary gland epithelium. In immune cells, P2X7R activation induces the production of proinflammatory cytokines, including IL-1β and IL-18, by inducing the oligomerization of the multiprotein complex NLRP3-type inflammasome. Here, our results show that in primary mouse submandibular gland (SMG) epithelial cells, P2X7R activation also induces the assembly of the NLRP3 inflammasome and the maturation and release of IL-1β, a response that is absent in SMG cells isolated from mice deficient in P2X7Rs (P2X7R−/−). P2X7R-mediated IL-1β release in SMG epithelial cells is dependent on transmembrane Na+ and/or K+ flux and the activation of heat shock protein 90 (HSP90), a protein required for the activation and stabilization of the NLRP3 inflammasome. Also, using the reactive oxygen species (ROS) scavengers N-acetyl cysteine and Mito-TEMPO, we determined that mitochondrial reactive oxygen species are required for P2X7R-mediated IL-1β release. Lastly, in vivo administration of the P2X7R antagonist A438079 in the CD28−/−, IFNγ−/−, NOD.H-2h4 mouse model of salivary gland exocrinopathy ameliorated salivary gland inflammation and enhanced carbachol-induced saliva secretion. These findings demonstrate that P2X7R antagonism in vivo represents a promising therapeutic strategy to limit salivary gland inflammation and improve secretory function.
Keywords: ATP, autoimmune disease, inflammasome, inflammation, P2X7, P2X7 receptor, purinergic receptor, IL-1β, Sjögren's syndrome, salivary gland
Introduction
Salivary gland inflammation and dysfunction are serious clinical problems that affect millions of people whose quality of life is severely impacted because of dry mouth, periodontitis, oral bacterial and yeast infections, and speech problems (1). Major causes of salivary gland degeneration leading to hyposalivation in humans are the autoimmune inflammatory disease Sjögren's syndrome (SS)2 and the unintended side effects of radiotherapies for head and neck cancers (1). A hallmark of salivary gland inflammation in SS is mononuclear cell infiltration, mainly T and B lymphocytes, dendritic cells and macrophages, which produce proinflammatory cytokines and chemokines and ultimately promote salivary gland degeneration in addition to extraglandular manifestations including skin vasculitis, glomerulonephritis, peripheral neuropathy, and malignant lymphomas (2, 3). There is increasing evidence that salivary gland epithelial cells contribute to immunological responses associated with salivary gland inflammation (4, 5). Hence, understanding the mechanism of initiation of inflammatory events in salivary epithelium is crucial to developing more effective and localized treatments for salivary gland disorders than are currently available. To date, relatively ineffective therapies for salivary hypofunction include stimulation of salivary flow in residual salivary acinar cells of SS patients using muscarinic receptor agonists (e.g. pilocarpine or cevimeline), oral administration of anti-inflammatory agents (e.g. interferon-α), and utilization of artificial saliva (6).
In addition to its role as a source of energy in intracellular metabolism and signaling, ATP is well-established as an extracellular signaling molecule that regulates various physiological processes, including immune cell recruitment, neurotransmission, and pain perception (7–10). Under normal conditions, the levels of extracellular ATP (eATP) are tightly regulated by ecto-ATPases, however, in the microenvironment of injured, inflamed, or stressed cells, eATP is known to be present at high concentrations (11). Extracellular ATP is the natural agonist for most members of the purinergic receptor family, which includes the ionotropic P2X (P2X1–7) and the G protein–coupled P2Y (P2Y1,2,4,6,11–14) receptors (12). The P2X7 receptor (P2X7R) is of particular interest as its activation is linked to a number of inflammatory diseases, e.g. glomerulonephritis, neuroinflammation, and rheumatoid arthritis (13–16). P2X7R is widely expressed in hematopoietic cells, neurons, glial cells, osteoblasts, endothelial cells, and epithelial cells (12). Brief stimulation of the ionotropic P2X7R with ATP causes plasma membrane depolarization because of Na+, Ca2+, and K+ flux down their electrochemical gradients (12, 17). Sustained activation of the P2X7R by high ATP concentrations in some cell types, including macrophages, astrocytes, and epithelial cells, can lead to the increased permeability of hydrophilic molecules up to 900 Da (18), membrane blebbing (19, 20), reactive oxygen species (ROS) production, and intracellular nucleotide release resulting in cell death (12). P2X7R activation plays a key role in the maturation and release of the leaderless proinflammatory cytokine IL-1β in immune cells by inducing the oligomerization of the multiprotein complex NLRP3-type inflammasome (21, 22). Notably, IL-1β and the components of the NLRP3 inflammasome, including a scaffold NOD-like receptor (NLR) protein, NLR family pyrin domain containing protein 3 (NLRP3), apoptosis-associated specklike protein containing a CARD (ASC), and pro-caspase-1 (22), along with P2X7Rs, are up-regulated in salivary gland biopsies from SS patients, as compared with normal controls (23). Although widely studied in immune cells, P2X7R-mediated NLRP3 inflammasome activation in salivary epithelial cells has not been investigated.
P2X7Rs are highly expressed in salivary epithelium (19, 24) and have been implicated in the regulation of ATP-induced saliva secretion, probably through calcium influx (25), but their role in salivary gland inflammation is not well-understood. Our previous studies showed that P2X7R activation by ATP induces inflammatory responses, e.g. increases in caspase-1 activity and membrane blebbing, in freshly prepared mouse submandibular gland (SMG) cells from wild-type mice, but not from P2X7R-deficient (P2X7R−/−) mice (19). In addition, duct ligation after ATP perfusion in wild-type mouse SMG increases lymphocytic infiltration, a response that is absent in P2X7R−/− mouse SMG (19). Studies have shown that P2X7R−/− mice exhibit reduced lung inflammation and neuropathic pain as compared with wild-type mice (26, 27) and systemic administration of selective P2X7R antagonists, e.g. A438079, attenuates inflammatory responses in rodent models of lung inflammation, nephritis, nociception, and intracerebral hemorrhage (28–31). Taken together, the P2X7R represents a novel therapeutic target for reducing inflammation (15, 32) as further indicated by previous phase I and II clinical trials with P2X7R antagonists for treatment of rheumatoid arthritis and spinal cord injury (33, 34). To date, there have been no clinical studies investigating the P2X7R as a therapeutic target to prevent salivary gland inflammation.
Here, we demonstrate for the first time in salivary gland epithelial cells that P2X7R activation promotes NLRP3 inflammasome activation and assembly and IL-1β release through a mechanism involving transmembrane Na+ and K+ flux, ROS production, and heat shock protein 90 (HSP90) function. Moreover, our in vivo results show that administration of the P2X7R antagonist, A438079, in the CD28−/−, IFNγ−/−, NOD.H-2h4 mouse model of autoimmune exocrinopathy (35), reduced salivary gland lymphocytic infiltrates and enhanced saliva secretion, suggesting that antagonism of the P2X7R is a promising approach to prevent salivary gland inflammation and associated hyposalivation in the autoimmune disease SS.
Results
P2X7R activation induces the release of IL-1β in primary SMG epithelial cell aggregates from wild-type, but not P2X7R−/−, mice
Activation of the P2X7R in immune cells is well-known to play a role in the maturation and release of the proinflammatory cytokine IL-1β (21, 36, 37). We have previously shown that P2X7R activation in primary mouse SMG epithelial cell aggregates increases the activity of caspase-1, the protease responsible for IL-1β processing (19); however, it is unclear how P2X7R activation in salivary glands regulates IL-1β maturation and release. Here, we show that IL-1β release is significantly increased in wild-type primary SMG cell aggregates (97–99% epithelial cells) (supplemental Fig. S1A) stimulated with ATP (3 mm), as compared with untreated controls (Fig. 1A). This response is abolished by pretreating aggregates with the P2X7R antagonist A438079 (25 μm), whereas IL-1β release induced by the K+ ionophore nigericin (10 μm) is unaffected (Fig. 1A). Moreover, ATP-induced IL-1β release is absent in SMG cell aggregates from P2X7R−/− mice (Fig. 1B). To further validate that SMG epithelial cells are the source of IL-1β, rather than immune cells contaminating the SMG cell preparations, epithelial cell purity was enriched using the magnetic bead-based EasySep Mouse Epithelial Cell Enrichment Kit to remove hematopoietic and endothelial cells and fibroblasts. ATP-induced IL-1β release was equally robust in the enriched SMG epithelial cells (supplemental Fig. S1B) and the standard SMG cell preparations (Fig. 1A), suggesting that SMG epithelial cells are the primary source of ATP-induced IL-1β release in our in vitro experiments.
Figure 1.

P2X7R activation induces the release of IL-1β in primary mouse SMG cell aggregates. A, dispersed SMG cell aggregates from wild-type mice were cultured in the presence or absence of A438079 (25 μm) for 30 min and then incubated with ATP (3 mm) or nigericin (10 μm) for 90 min (n = 4). B, dispersed SMG cell aggregates isolated from P2X7R−/− mice were incubated with ATP (3 mm) or nigericin (10 μm) for 90 min (n = 3). Cells were collected by centrifugation and IL-1β was quantified in the supernatant using the IL-1β Quantikine ELISA kit. Data represent means ± S.E., where **, p < 0.01 indicates a significant increase over basal levels, and ##, p < 0.01 indicates a significant decrease compared with ATP-treated cells.
NLRP3 inflammasome activation is required for P2X7R-mediated IL-1β release in primary SMG epithelial cell aggregates
Activation of the multiprotein complex NLRP3 inflammasome is responsible for the maturation and release of the proinflammatory cytokine IL-1β in a variety of cell types, including intestinal epithelial cells, monocytes, and macrophages (22, 38, 39). To investigate whether NLRP3 inflammasome activation is required for P2X7R-mediated IL-1β release in salivary epithelium, primary SMG cell aggregates from wild-type mice were pretreated with the selective NLRP3 inflammasome inhibitors MCC-950 (10 μm) (40) or Bay 11-7082 (15 m) (41) and then stimulated with ATP (3 mm). Results show that inhibition of NLRP3 inflammasome activity abolishes ATP-induced IL-1β release (Fig. 2A). Furthermore, immunoprecipitation of NLRP3 was used to demonstrate that ATP (3 mm) potentiates coprecipitation of ASC and pro-caspase-1 proteins, components of the NLRP3 inflammasome, in SMG cell aggregates from wild-type but not P2X7R−/− mice (Fig. 2B). These results indicate that P2X7R-mediated IL-1β release in salivary epithelial cells is mediated by activation and assembly of the NLRP3 inflammasome.
Figure 2.

The NLRP3 inflammasome mediates P2X7R-induced IL-1β release. A, dispersed SMG cell aggregates from wild-type mice were cultured in the presence or absence of Bay 11–7082 (15 μm) for 1 h or MCC-950 (10 μm) for 30 min and then stimulated with ATP (3 mm) for 90 min (n = 3). Cells were collected by centrifugation and IL-1β was quantified in the supernatant using the IL-1β Quantikine ELISA kit. Data represent means ± S.E., where **, p < 0.01 and ***, p < 0.001 indicate significant decreases from ATP-treated only. B, upper panel, cell lysates from dispersed wild-type or P2X7R−/− SMG cell aggregates treated with or without ATP (3 mm) for 15 min were subjected to immunoprecipitation (IP) with anti-NLRP3 antibody, followed by immunoblotting (IB) with anti-NLRP3, anti-ASC, or anti–pro-caspase-1 antibodies. Images represent results from three independent experiments. Lower panel, quantification of the coimmunoprecipitated proteins in wild-type SMG cells (n = 3). Data represent means ± S.E., where *, p < 0.05 and **, p < 0.01 indicate significant increases in relative band intensities as compared with basal conditions.
Ionic dependence of P2X7R-mediated IL-1β release in primary SMG epithelial cell aggregates
P2X7R is an ATP-gated nonselective cation channel whose stimulation induces the efflux of K+ and influx of Na+ and Ca2+ ions (12, 17). K+ efflux and Na+ influx are necessary for the activation of the NLRP3 inflammasome complex in immune cells (42, 43). Therefore, we sought to evaluate the role of transmembrane ion flux in ATP-induced IL-1β release in salivary epithelial cells. K+ efflux is a necessary response in immune cells to various NLRP3 inflammasome activators, including ATP, monosodium urate crystals, and bacterial lipopolysaccharide (43–45). As mentioned above, the H+/K+ ionophore nigericin induces IL-1β release in primary SMG cell aggregates from wild-type and P2X7R−/− mice (Fig. 1), suggesting that K+ efflux is required for inflammasome activation in salivary epithelium. To determine whether ATP treatment perturbs intracellular K+ homeostasis in salivary epithelial cells, primary SMG cell aggregates from wild-type and P2X7R−/− mice were treated with ATP (3 mm) and the intracellular [K+] was quantified with an inductively coupled plasma optical emission spectrometer (ICP-OES). ATP induced a significant decrease in the intracellular [K+] after 10 and 30 min, a response that was absent in P2X7R−/− SMG cell aggregates (Fig. 3A). To determine whether the transmembrane [Na+] and [K+] gradients are required for NLRP3 inflammasome activation, primary SMG cell aggregates were assayed for IL-1β release in a buffer containing the physiological extracellular [Na+] and [K+] (145 mm and 5 mm, respectively) or in a buffer where the NaCl (145 mm) was substituted with KCl (145 mm) or choline chloride (145 mm). Replacement of extracellular NaCl with either KCl or choline chloride inhibited ATP-induced IL-1β release (Fig. 3B), indicating that P2X7R-mediated IL-1β release in salivary epithelial cells is dependent on physiological transmembrane Na+ and K+ gradients.
Figure 3.

K+ efflux, ROS, and HSP90 are required for P2X7R-mediated IL-1β release. A, dispersed SMG cell aggregates from wild-type and P2X7R−/− mice were stimulated with ATP (3 mm) for 0, 10, or 30 min. Then, the intracellular [K+] was quantified using ICP-OES and expressed as a percentage of the intracellular [K+] at 0 min of ATP treatment (n = 3). Data represent means ± S.E., where *, p < 0.05 indicates a significant decrease from ATP-treated cells at 0 min. B, dispersed SMG cell aggregates from wild-type mice were stimulated with ATP (3 mm) for 90 min in the presence of the specified concentrations of NaCl, KCl, or choline chloride (n = 4). C–E, dispersed SMG cell aggregates from wild-type mice also were cultured in presence or absence of (C) the ROS inhibitor NAC (25 mm), (D) the mitochondrial ROS inhibitor Mito-Tempo (1 mm), or (E) the HSP90 inhibitor geldanamycin (GA) (5 μm) for 1 h and then stimulated with ATP (3 mm) for 90 min (n = 3). Cells were collected by centrifugation, and IL-1β was quantified in the supernatant using the IL-1β Quantikine ELISA kit. Data represent means ± S.E., where *, p < 0.05, **, p < 0.01, and ***, p < 0.001 indicate significant decreases from (B) ATP-treated cells in 145 mm Na+ or (C–E) ATP-treated only. F, upper panel, cell lysates from dispersed wild-type SMG cell aggregates were treated with or without GA (5 μm) for 1 h and subjected to immunoblotting with anti-NLRP3 or anti-β-tubulin antibodies. Images represent results from three independent experiments. Lower panel, quantification of NLRP3 expression in wild-type SMG cells (n = 3). Data represent means ± S.E., where *, p < 0.05 indicates a significant decrease in relative band intensity as compared with basal conditions.
Inhibition of ROS production significantly reduces P2X7R-mediated IL-1β release in primary SMG epithelial cell aggregates
ROS are generated by all known inflammasome activators at times of inflammation and infection, where they play crucial roles in innate immune responses, including wound healing (46), and act as antimicrobial agents (47). ROS scavengers, e.g. N-acetyl cysteine (NAC), are known to block inflammasome activity, caspase-1 activation, and IL-1β release in immune cells (48). To examine whether generation of cytosolic ROS is required for ATP-induced IL-1β maturation and subsequent release in salivary epithelial cells, primary SMG cell aggregates were pretreated with the ROS scavenger NAC (25 mm) and then stimulated with ATP (3 mm). NAC significantly inhibited P2X7R-mediated IL-1β release (Fig. 3C). Because the mitochondria are one of the major sources of cellular ROS under stress conditions (49, 50), the contribution of mitochondrial ROS to P2X7R-mediated IL-1β release was evaluated by pretreating primary SMG cell aggregates with the mitochondrial-targeted antioxidant Mito-TEMPO (1 mm), before stimulation with ATP (3 mm). Mito-TEMPO partially blocked ATP-induced IL-1β release, demonstrating that ROS generated in the mitochondria are partially required for P2X7R-mediated IL-1β release in mouse SMG epithelial cells (Fig. 3D) and that other sources of ROS (e.g. NADPH oxidase) contribute to inflammasome activation.
Functional HSP90 is required for P2X7R-mediated IL-1β release in primary SMG epithelial cell aggregates
HSP90 is a molecular chaperone whose function is to maintain the proper folding of proteins required for their stability and activity, including proteins of the NLRP3 inflammasome (51, 52). In monocytes, HSP90 is a member of the P2X7R multiprotein complex and is required for NLRP3 inflammasome activity (52). To determine whether HSP90 contributes to P2X7R-mediated IL-1β release in salivary epithelial cells, primary SMG cell aggregates were pretreated with the HSP90 inhibitor geldanamycin (GA) (5 μm), then stimulated with ATP (3 mm). GA abolished ATP-induced IL-1β release in salivary epithelial cells (Fig. 3E), indicating that HSP90 is required for P2X7R-mediated IL-1β release. Furthermore, GA (5 μm) pretreatment significantly reduced the endogenous expression of NLRP3 protein, suggesting that functional HSP90 contributes to inflammasome activity in salivary epithelial cells by maintaining the stability of the NLRP3 protein (Fig. 3F).
In vivo administration of the P2X7R antagonist A438079 reduces salivary gland inflammation and improves saliva flow in the CD28−/−, IFNγ−/−, NOD.H-2h4 mouse model of salivary gland exocrinopathy
P2X7R antagonists, including A438079, have been shown to reduce inflammation in a number of inflammatory diseases, including glomerulonephritis, pulmonary fibrosis, and rheumatoid arthritis (33, 34). Hence, we sought to determine whether in vivo antagonism of the P2X7R in a mouse model of salivary gland exocrinopathy would reduce salivary gland inflammation. Recently, we showed that the CD28−/−, IFNγ−/−, NOD.H-2h4 mouse exhibits extensive lymphocytic infiltrates in salivary glands that correlate with diminished saliva flow by 4–6 months of age, as compared with the NOD.H-2h4 mouse control (35). Therefore, the P2X7R antagonist A438079 (34.2 mg/kg/day) or saline was injected intraperitoneally for 10 days in CD28−/−, IFNγ−/−, NOD.H-2h4 mice. Histochemical analysis by hematoxylin and eosin (H&E) staining revealed a decrease in lymphocytic infiltrates in SMGs from A438079-injected CD28−/−, IFNγ−/−, NOD.H-2h4 mice, as compared with saline-injected control mice (Fig. 4A). This result was further confirmed by RT-PCR analysis, in which the expression of the pan-immune cell antigen CD45 was significantly reduced in response to A438079 treatment (Fig. 4B), as compared with mice injected with saline alone.
Figure 4.

P2X7R antagonism ameliorates salivary gland inflammation and reduces expression of IL-1β and immunoactive molecules in CD28−/−, IFNγ−/−, NOD.H-2h4 mice. A, H&E staining of SMG sections isolated from CD28−/−, IFNγ−/−, NOD.H-2h4 mice (5 months old) injected intraperitoneally with saline alone or the P2X7R antagonist A438079 (34.2 mg/kg/day) for 10 days. Images are representative of n = 10 mice per each group. B–E, cDNA was prepared from SMGs of NOD.H-2h4 and CD28−/−, IFNγ−/−, NOD.H-2h4 mice injected with saline alone or A438079, as above, then analyzed by RT-PCR using specific primers for (B) the pan-immune cell marker CD45 (n = 5 for NOD.H-2h4 mice; n = 9 for saline-injected; and n = 10 for A438079-injected CD28−/−, IFNγ−/−, NOD.H-2h4 mice); (C) IL-1β; (D) ICAM-1, VCAM, or E-Selectin; or (E) CD80 or CD86 (n = 4 for NOD.H-2h4 mice; n = 9 for saline-injected; and n = 10 for A438079-injected CD28−/−, IFNγ−/−, NOD.H-2h4 mice). Data represent means ± S.E., where *, p < 0.05 and **, p < 0.01 indicate significant differences from saline-injected CD28−/−, IFNγ−/−, NOD.H-2h4 mice.
Because ATP-induced inflammasome activation is responsible for the maturation and release of the leaderless proinflammatory cytokine IL-1β in mouse SMG cells (Figs. 1 and 2), we evaluated the role of the P2X7R on the expression of IL-1β in whole SMGs isolated from CD28−/−, IFNγ−/−, NOD.H-2h4 mice injected with the P2X7R antagonist A438079 or saline alone. Results indicated that A438079 significantly decreased the expression of IL-1β in CD28−/−, IFNγ−/−, NOD.H-2h4 mice, as compared with saline alone (Fig. 4C).
During inflammation, as in SS, salivary gland epithelial cells exhibit aberrant expression of immunoactive molecules (4, 53) that mediate innate immune responses and induce chronic inflammation known as “autoimmune epithelitis” (53). These immunoactive molecules include costimulatory molecules (e.g. CD80 and CD86) (4) that are required for T-cell activation (54) and adhesion molecules (e.g. E-selectin, ICAM-1, and VCAM) that are required for immune cell homing (4). Using RT-PCR analysis, the expression of these molecules was assessed in whole SMGs isolated from CD28−/−, IFNγ−/−, NOD.H-2h4 mice following treatment for 10 days with the P2X7R antagonist A438079 or saline alone. The results show that expression levels of the adhesion molecules E-selectin, ICAM-1, and VCAM (Fig. 4D) and the costimulatory molecules CD80 and CD86 (Fig. 4E) were significantly reduced in CD28−/−, IFNγ−/−, NOD.H-2h4 mice treated with A438079, as compared with saline alone.
Hyposalivation is associated with SS and although its cause is not fully understood, inflammation is likely a contributing factor (55). Because the CD28−/−, IFNγ−/−, NOD.H-2h4 mouse model of autoimmune exocrinopathy exhibits diminished saliva flow as compared with the NOD.H-2h4 control (35), the effects of a 10-day treatment with A438079 or saline alone on carbachol-induced saliva flow were compared in CD28−/−, IFNγ−/−, NOD.H-2h4 mice. Importantly, there was a significant improvement in saliva flow in A438079-injected CD28−/−, IFNγ−/−, NOD.H-2h4 mice (Fig. 5), as compared with saline-injected controls.
Figure 5.

P2X7R antagonism improves saliva flow rate in CD28−/−, IFNγ−/−, NOD. H-2h4 mice. Carbachol-induced saliva flow rate in CD28−/−, IFNγ−/−, NOD.H-2h4 mice (5 months old) injected with saline alone or the P2X7R antagonist A438079 (34.2 mg/kg/day) for 10 days (n = 9 for saline-injected and n = 10 for A438079-injected CD28−/−, IFNγ−/−, NOD.H-2h4 mice). Data represent means ± S.E., where **, p < 0.01 indicates a significant difference from saline-injected CD28−/−, IFNγ−/−, NOD.H-2h4 mice.
Discussion
The lack of an understanding of inflammatory processes that predispose the salivary gland to degeneration and dysfunction is a major obstacle hindering the development of effective therapies to prevent or reverse hyposalivation caused by Sjögren's syndrome or inadvertent radiation-induced damage from head and neck cancer treatments. Epithelial cells have emerged as active players in immune responses that underlie inflammation, in addition to their well-accepted role in the formation of a physical barrier to the outside environment in a wide variety of tissues. Inflammatory disorders of the epithelium in addition to SS include inflammatory bowel disease (56), primary biliary cirrhosis (57), pulmonary fibrosis (58), and psoriasis (59). In these diseases, changes in the morphology and ionic and molecular permeabilities of the plasma membrane of epithelial cells initiate interactions with immune cells, resulting in tissue inflammation (5). In SS, salivary epithelial cells participate in inflammatory responses by releasing proinflammatory cytokines and chemokines (5) and expressing immunoactive molecules that modulate early innate immune responses preceding hyposalivation or autoimmune exocrinopathy (4, 53). Although there are multiple reports of inflammatory responses in immune cells that are associated with SS, very few studies have focused on salivary epithelial cells in the initiation of inflammatory disease despite the critical role played by the epithelium in homeostasis required for normal gland functions (60). In this study, we show that P2X7R activation by eATP in freshly isolated primary mouse SMG epithelial cells increases the activity of the multiprotein NLRP3 inflammasome required for the subsequent release of the proinflammatory cytokine IL-1β (Figs. 1 and 2). These data are consistent with reports that the P2X7R stimulates inflammasome activity in intestinal epithelial cells (61) and immune cells, e.g. monocytes and macrophages (38, 39).
Mechanisms have been proposed in immune cells to define how eATP enhances NLRP3 inflammasome activity, including by alteration of the ionic composition of the inflammasome's microenvironment through transmembrane Na+ influx and K+ efflux (42–44). Besides eATP, other activators of the NLRP3 inflammasome in immune cells promote K+ efflux, including monosodium ureate, peptidoglycan, and silica, leading to the secretion of mature IL-1β (43–45). It was reported that in neutrophils following Streptococcus pneumoniae infection of the cornea, a low intracellular [K+] enhanced P2X7R-mediated inflammasome activation and IL-1β maturation and release (36). Also, pro-caspase-1 was not recruited to the inflammasome complex in monocytes in the presence of a high extracellular [K+] (43). However, media devoid of Na+ also impaired inflammasome activation in human monocytes (42, 44). Here, we show that the H+/K+ ionophore nigericin induced IL-1β release in SMG cell aggregates prepared from either wild-type or P2X7R−/− mice (Fig. 1). Stimulation of the P2X7R with ATP for 10 or 30 min in wild-type salivary epithelial cells resulted in a significant reduction in the intracellular [K+] that did not occur with SMG cells from P2X7R−/− mice (Fig. 3A). Moreover, P2X7R-mediated IL-1β release was diminished when cells were incubated in a high extracellular [K+] or in absence of extracellular Na+ (Fig. 3B). These results suggest that disruption of the normal transmembrane Na+ and K+ gradients inhibits P2X7R-dependent NLRP3 inflammasome activation and the release of IL-1β.
In addition to ionic dependence, the generation of ROS is a common response to various inflammasome activators, including ATP (48, 62), and ROS have been shown to enhance inflammasome activity (43, 48). Mitochondria are a major source of ROS, hence, abnormal mitochondria and uncontrolled ROS production are known to contribute to inflammatory diseases, including neurodegenerative diseases and sepsis (63, 64). Mitochondrial ROS are generated by the electron transport chain in complex I and the Q cycle in complex III (65, 66). Accumulation of mitochondrial ROS, as a consequence of mitochondrial dysfunction, is a trigger for the activation of the NLRP3 inflammasome in murine and human macrophages upon ATP or nigericin treatment, a response that was abolished in the presence of the mitochondrial-specific ROS scavenger Mito-Tempo (49, 67). In bronchial epithelial cells, mitochondrial ROS induce the colocalization of NLRP3 and ASC inflammasome proteins in the mitochondria and are responsible for airway inflammation in bronchial asthma (68). Consistent with these data, our results show that in salivary epithelial cells NAC or Mito-Tempo reduce P2X7R-mediated inflammasome activation indicated by a significant decrease in the ATP-induced release of mature IL-1β (Fig. 3, C and D). The molecular chaperone HSP90 is also required for NLRP3 inflammasome stability in monocytes, because it prevents the degradation of the NLRP3 protein in the absence of inflammation (52), a homeostatic mechanism that likely prepares cells to respond to inflammasome activators, including ATP. We found that inhibition of HSP90 activity in mouse SMG epithelial cells abolishes P2X7R-mediated IL-1β release (Fig. 3E) and significantly reduces the endogenous levels of NLRP3 protein (Fig. 3F), consistent with a role for this chaperone in inflammasome stability in salivary epithelium. Taken together, these findings demonstrate for the first time that P2X7R activation in salivary gland epithelial cells induces the assembly of an active NLRP3 inflammasome that regulates the maturation and release of the proinflammatory cytokine IL-1β, a pathway that requires physological transmembrane Na+ and K+ gradients, the production of mitochondrial ROS, and the presence of a functional HSP90 protein.
P2X7R antagonists have been explored as inhibitors of inflammation in several mouse models of inflammatory diseases, including lung inflammation, nephritis, nociception and intracerebral hemorrhage (28–31, 33, 61, 69), and P2X7R antagonists have shown promise in phase I and phase II clinical trials for the treatment of inflammatory bowel syndrome, spinal cord injury, and rheumatoid arthritis (33, 34, 70). In this study, the selective P2X7R antagonist A438079 was administered in vivo to CD28−/−, IFNγ−/−, NOD.H-2h4 mice that we recently demonstrated develop extensive immune cell infiltrates of salivary glands and hyposalivation (35). The results revealed that P2X7R antagonism dramatically reduced infiltration of CD45-expressing lymphocytes in the SMG of this mouse model of inflammation (Fig. 4, A and B). In addition, A438079 reduced the expression of the proinflammatory cytokine IL-1β in SMG (Fig. 4C). The role of IL-1β in promoting tissue inflammation and recruiting systemic immune cells is well-established in several inflammatory diseases, including myocardial infarction, gout, and rheumatoid arthritis (71). Also, IL-1β is known to be up-regulated in salivary glands isolated from SS patients, as compared with normal controls (72). IL-1β activates the NF-κB transcription factor in cells, thereby inducing the expression of several proinflammatory genes, including cyclooxygenase-2 and inducible nitric oxide synthase (73), which in turn increase the production of the proinflammatory factors prostaglandin E2 and nitric oxide that are responsible for vasodilation and the recruitment of immune cells to sites of inflammation (73–75). In addition, IL-1β enhances the expression of cell adhesion molecules, including ICAM-1 and VCAM, to promote immune cell homing to inflamed tissue (73). Notably, our previous studies showed that IL-1β increases NF-κB-dependent expression of the G protein–coupled P2Y2 receptor (P2Y2R) (76, 77), a member of the purinergic receptor family. The P2Y2R is up-regulated in the epithelium of inflamed and/or damaged salivary glands (78–82) and upon activation mediates integrin-dependent cell migration and enhances acinar formation (81). Additional studies have shown that P2Y2Rs regulate localized immune responses, induce immune cell infiltration (83), and up-regulate VCAM-1 expression (80). Thus, P2X7R and P2Y2R may partner together in the initiation of an inflammatory response and in epithelial tissue repair during inflammation. Nonetheless, the reduced expression of IL-1β seen in SMGs of CD28−/−, IFNγ−/−, NOD.H-2h4 mice treated with the P2X7R antagonist A438079 (Fig. 4C) could explain, at least in part, the diminution of lymphocytic infiltrates in the SMGs of this mouse model of salivary gland inflammation upon A438079 administration (Fig. 4, A and B).
Salivary glands of SS patients exhibit immune cell infiltrates, primarily comprised of B- and T-cells (55), and increased expression of immunoactive proteins, including the costimulatory molecules CD80 and CD86 and the cell adhesion molecules VCAM, ICAM-1, and E-selectin (4). The increased expression of immunoactive proteins propagates inflammatory responses and enhances the interaction between immune and salivary epithelial cells. Here, we show that P2X7R antagonism in CD28−/−, IFNγ−/−, NOD.H-2h4 mice induces a dramatic decrease in the expression of the adhesion molecules VCAM, ICAM-1, and E-selectin (Fig. 4D) and the costimulatory molecules CD80 and CD86 (Fig. 4E), suggesting that P2X7R antagonism could be used to prevent chronic inflammation that ultimately results in the loss of salivary gland function (1).
Current treatments for hyposalivation, including muscarinic receptor agonists (1), do not address the underlying salivary gland inflammation, which is a well-appreciated factor in salivary gland dysfunction (55), particularly in the autoimmune disease SS. In the NOD mouse model of SS, increased expression of proinflammatory cytokines has been shown to precede a decrease in saliva flow (84). In addition, a recent proteomic analysis of saliva from SS patients demonstrated that hyposalivation is associated with increased expression and activity of the IL-1 family of proinflammatory cytokines (i.e. IL-1β and IL-18) (55) and adiponectin, a metabolic regulatory protein that promotes IL-1 cytokine interaction with downstream targets (85). Some anti-inflammatory drugs, such as interferon-α, have undergone clinical trials for treatment of salivary gland dysfunction (1). Similarly, our results encourage attempts to use P2X7R antagonists to treat hyposalivation in humans, based on the ability of A438079 to enhance the saliva flow rate in the CD28−/−, IFNγ−/−, NOD.H-2h4 mouse model of salivary gland inflammation (Fig. 5).
In conclusion, we propose a role for the P2X7R in salivary gland inflammation, in which stimulation of the P2X7R with eATP in salivary epithelial cells induces the assembly and activation of the NLRP3 inflammasome and consequently the maturation and release of the proinflammatory cytokine IL-1β. This process requires a transmembrane Na+ and/or K+ flux, mitochondrial ROS production, and a functional HSP90 protein (Fig. 6). In addition, P2X7R activation is associated with increased immune cell infiltration of the salivary gland that eventually results in tissue degeneration (Fig. 6). Hence, this study suggests that P2X7R antagonism represents a novel therapeutic strategy to prevent chronic inflammation in human salivary gland inflammatory disorders.
Figure 6.
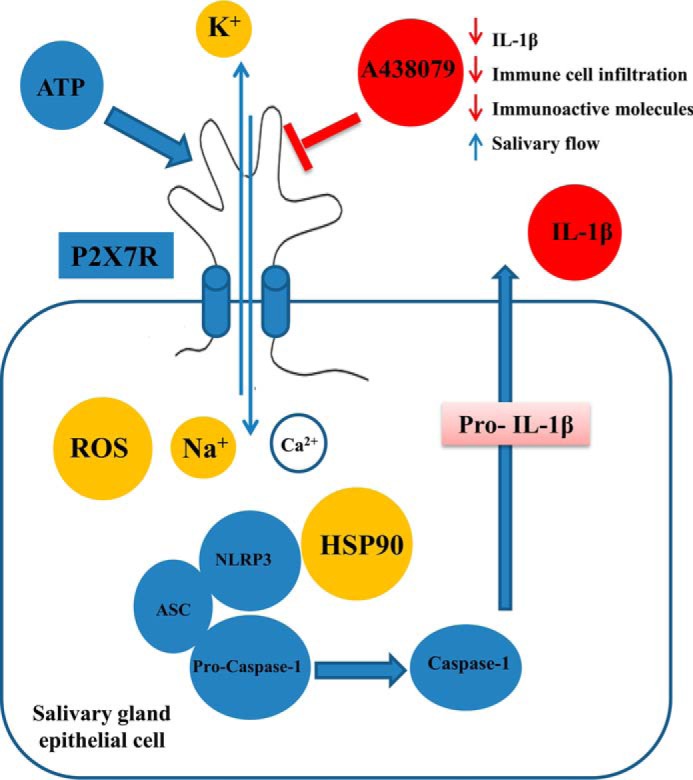
Schematic diagram illustrating the proposed role of the P2X7R in salivary gland inflammation. In salivary epithelial cells, P2X7R activation by eATP induces the assembly and activation of the NLRP3 inflammasome and the subsequent release of mature IL-1β. This process involves transmembrane Na+ and/or K+ flux, production of ROS, and the presence of an active HSP90 protein. P2X7R antagonism reduces immune cell infiltration and salivary gland expression of IL-1β, ICAM-1, VCAM, E-Selectin, CD80, and CD86 and enhances carbachol-induced saliva secretion in the CD28−/−, IFNγ−/−, NOD.H-2h4 mouse model of salivary gland inflammation.
Experimental procedures
Materials
DMEM and Ham's Nutrient Mixture F-12 (DMEM/F12) and penicillin-streptomycin 100× solution were obtained from Life Technologies. All other reagents were purchased from Sigma-Aldrich, unless stated otherwise.
Mice
Male C57/BL6 (stock no. 000664) and P2X7R−/− (stock no. 005576) mice (6–8 weeks old) were purchased from The Jackson Laboratory (Bar Harbor, ME), whereas the P2X7R−/− mice (stock no. 005576) were originally donated by Pfizer Pharmaceuticals. The NOD.H-2h4 and CD28−/−, IFNγ−/−, NOD.H-2h4 mice were provided by Dr. Helen Braley-Mullen, Department of Molecular Microbiology and Immunology, University of Missouri. The mice were bred in the Christopher S. Bond Life Sciences Center Animal Facility of the University of Missouri and housed in vented cages with 12-h light-dark cycles and free access to standard laboratory diet and water. For P2X7R antagonism, A438079 (34.2 mg/kg/day) or saline was injected intraperitoneally for 10 days in CD28−/−, IFNγ−/−, NOD.H-2h4 mice. The mice were treated under the guidelines and approval of the University of Missouri Institutional Animal Care and Use Committee (IACUC).
Preparation of dispersed cell aggregates from mouse SMG
Dispersed SMG cell aggregates were prepared as previously described (19). Briefly, mice were anesthetized with isoflurane in a chamber and euthanized by cervical dislocation. SMGs were isolated, minced, and incubated at 37 °C in a shaking water bath for 1 h in dispersion media (1:1 DMEM/F12 containing 50 units/ml collagenase (Worthington Biochemical), 400 units/ml hyaluronidase, 1% (w/v) BSA, and 0.2 mm CaCl2) in an atmosphere of 5% CO2 and 95% O2. For further dispersion, the minced SMGs were passed through a 10 ml pipette after 20, 30, and 40 min of incubation. Finally, the dispersed cell aggregates were filtered through a nylon mesh and incubated for an additional hour at 37 °C in 1:1 DMEM/F12 containing 100 units/ml penicillin and 100 μg/ml streptomycin in an atmosphere of 5% CO2 and 95% O2, before further use. For further SMG epithelial cell purification, the EasySep Mouse Epithelial Cell Enrichment Kit (STEMCELL Technologies, Cambridge, MA) was used according to the manufacturer's protocol.
IL-1β enzyme-linked immunosorbent assay
Dispersed SMG cells in fresh 1:1 DMEM/F12 containing 100 units/ml penicillin and 100 μg/ml streptomycin were incubated with ATP (pH 7.4), nigericin (Tocris, Minneapolis, MN), A438079 (Tocris), Bay11–7082 (Tocris), MCC-950 (catalog no. AG-CR1–3615, AdipoGen Life Sciences, San Diego, CA), geldanamycin (Tocris, catalog no. 1368), or the ROS scavengers, N-acetyl cysteine (NAC) or Mito-TEMPO for the indicated time periods. Cell supernatants were collected and concentrated using Amicon Ultra-0.5 ml Centrifugal Filters. IL-1β was measured in the supernatant using the IL-1β Quantikine ELISA Kit (R&D Systems, Minneapolis, MN), according to the manufacturer's protocol. To measure the effect of ionic gradient alterations, dispersed SMG cells were incubated for 90 min with ATP (3 mm) in a buffer with physiological [Na+] and [K+] (145 mm NaCl, 5 mm KH2PO4, 10 mm HEPES, 1 mm MgCl2, 1 mm CaC12, and 1% (w/v) BSA), pH adjusted to 7.4 with NaOH, a buffer with high [K+] (145 mm KCl, 5 mm NaH2PO4, 10 mm HEPES, 1 mm MgCl2, 1 mm CaC12, and 1% (w/v) BSA), pH adjusted to 7.4 with KOH, or a buffer with low [Na+] in which NaCl is substituted with choline chloride (145 mm choline chloride, 5 mm KH2PO4, 10 mm HEPES, 1 mm MgCl2, 1 mm CaC12, and 1% (w/v) BSA), pH adjusted to 7.4 with KOH.
NLRP3 inflammasome immunoprecipitation
Dispersed SMG cells from wild-type or P2X7R−/− mice were incubated with ATP (3 mm) for 15 min. The cells were then collected by centrifugation and incubated in T-PER lysis buffer (Thermo Fisher Scientific) with 1× SIGMAFAST serine, cysteine, and metalloprotease inhibitors (2 mm AEBSF, 1 mm EDTA, 130 μm bestatin, 14 μm E-64, 1 μm leupeptin, 0.3 μm aprotinin). NLRP3 in SMG cell lysates (500–1,000 μg protein/ml) was immunoprecipitated with protein A MagBeads (GenScript, Piscataway, NJ) precoated with anti-mouse NLRP3 antibody (1:200 dilution; Cryo-2) (catalog no. AG-20B-0014-C100, Adipogen) by incubation for 1 h at room temperature. Immunoprecipitated proteins were solubilized using 40 μl of 1× SDS sample buffer (62.5 mm Tris-HCl at pH 6.8, 2% (w/v) SDS, 10% (v/v) glycerol, 50 mm dl-DTT, and 0.01% (w/v) bromphenol blue) followed by heating at 95 °C for 5 min. The samples were then subjected to SDS-PAGE on a 12% (w/v) gel (GenScript) and then transferred to nitrocellulose membranes for Western blot analysis.
Western blot analysis
Nitrocellulose membranes were blocked for 1 h with 5% (w/v) nonfat dry milk in TBS containing 0.1% (v/v) Tween 20 (TBST) and incubated overnight at 4 °C with anti-mouse NLRP3 antibody (1:1,000 dilution in 5% (w/v) nonfat dry milk in TBST, Cryo-2) (catalog no. AG-20B-0014-C100, Adipogen), rabbit anti-mouse ASC antibody (1:1,000 dilution in 5% (w/v) nonfat dry milk in TBST) (catalog no. AG-25B-0006, Adipogen), rabbit anti-mouse pro-caspase-1 antibody (1:1,000 dilution in 5% (w/v) nonfat dry milk in TBST) (catalog no. ab108362, Abcam, Cambridge, MA) or mouse anti-mouse β-tubulin antibody (1:5,000 dilution in 5% (w/v) nonfat dry milk in TBST). Membranes were washed three times in TBST and incubated with horseradish peroxidase-conjugated goat anti-rabbit IgG or goat anti-mouse IgG antibody (1:1,000 dilution in 5% (w/v) nonfat dry milk in TBST) (Santa Cruz, Dallas, TX) at room temperature for 1 h. Then, blots were washed three times in TBST and incubated in enhanced chemiluminescence reagent (Thermo Fisher Scientific) for 1 min. Protein bands were detected on X-ray film and quantified using Image Studio Lite software (LI-COR Biosciences, Lincoln, NE).
Intracellular [K+] quantification
Dispersed SMG cells from wild-type or P2X7R−/− mice were incubated with ATP (3 mm) for 0, 10, or 30 min. Cells were then collected by centrifugation and extracted with 10% (v/v) ultrapure HNO3. Intracellular [K+] was then quantified using an ICP-OES (PerkinElmer, Optima 8000) at wavelength 769.9 nm using manganese as the internal standard.
Immunohistochemistry
Whole SMGs were isolated from CD28−/−, IFNγ−/−, NOD.H-2h4 mice, placed in 4% (v/v) paraformaldehyde in PBS at 4 °C for 24 h, followed by 70% (v/v) ethanol for 24 h at 4 °C. Samples were then sent to IDEXX RADIL (Columbia, MO) where glands were embedded in paraffin, cut into 5-μm sections, and subjected to H&E staining.
RT-PCR
Whole SMGs were excised, homogenized in TRIzol reagent (Life Technologies), and incubated for 5 min at room temperature, followed by incubation with chloroform (0.2 ml/ml TRIzol) for 5 min at room temperature. Following centrifugation at 12,000 g for 15 min at 4 °C, the resulting aqueous phase containing RNA was collected and DNA-free RNA was isolated using the RNeasy Plus Mini Kit (Qiagen, Valencia, CA). cDNA was prepared from purified RNA using RNA to cDNA EcoDry Premix (Clontech, Mountain View, CA). TaqMan probes for CD45 (also known as protein tyrosine phosphatase receptor type C, a pan-immune cell marker), IL-1β, CD80, CD86, ICAM-1, VCAM, and E-selectin were obtained from Applied Biosystems (Foster City, CA) and used for RT-PCR with an Applied Biosystems 7500 real-time PCR machine. The mRNA expression of target genes was normalized to 18S ribosomal RNA as an internal control and data were analyzed using Applied Biosystems software.
Saliva collection
Mice were anesthetized with tribromoethanol (Avertin) (0.75 mg/g mouse weight) and an endotracheal tube (PE50 polyethylene tubing) was inserted through a 2-cm midventral incision to prevent aspiration. Saliva secretion was induced by intraperitoneal injection of 0.25 mg/kg carbachol. Saliva was collected from the oral cavity for 15 min using a pipette tip and placed in a preweighed Eppendorf tube. Results are presented as microliters of saliva produced per 15 min.
Statistical analysis
The statistical analyses were performed using Student's t test on GraphPad Prism software where significant differences were considered to be p < 0.05.
Author contributions
M. G. K. contributed to the experimental design, performed most of the experiments, and drafted the manuscript. L. T. W. performed the immunohistochemistry experiment, provided technical assistance, and participated in the critical review of the manuscript. J. M. C. supervised the experiments and performed the mouse studies. A. A. K. measured the [K+] using ICP-OES and participated in the critical review of the manuscript. K. H. L., M. J. P., and L. E. contributed to the experimental design and the critical review of the results and manuscript. G. A. W. designed and supervised the project and edited the manuscript.
Supplementary Material
This work was supported by National Institute of Dental and Craniofacial Research, National Institutes of Health Grant DE023342. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains supplemental Fig. S1.
- SS
- Sjögren's syndrome
- eATP
- extracellular ATP
- ROS
- reactive oxygen species
- SMG
- submandibular gland
- NOD
- nucleotide-binding oligomerization domain
- NLR
- NOD-like receptor
- NLRP3
- NLR family pyrin domain containing protein 3
- ASC
- apoptosis-associated specklike protein containing a CARD
- NAC
- N-acetyl cysteine
- ICP-OES
- inductively coupled plasma optical emission spectrometer
- GA
- geldanamycin
- ICAM-1
- intracellular cell adhesion molecule
- VCAM
- vascular cell adhesion molecule
- NOD.H-2h4 mouse
- non-obese diabetic H-2h4 mouse.
References
- 1. Atkinson J. C., Grisius M., and Massey W. (2005) Salivary hypofunction and xerostomia: Diagnosis and treatment. Dent. Clin. North Am. 49, 309–326 [DOI] [PubMed] [Google Scholar]
- 2. Delaleu N., Jonsson R., and Koller M. M. (2005) Sjögren's syndrome. Eur. J. Oral Sci. 113, 101–113 [DOI] [PubMed] [Google Scholar]
- 3. Tincani A., Andreoli L., Cavazzana I., Doria A., Favero M., Fenini M.-G., Franceschini F., Lojacono A., Nascimbeni G., Santoro A., Semeraro F., Toniati P., and Shoenfeld Y. (2013) Novel aspects of Sjögren's syndrome in 2012. BMC Med. 11, 93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Tsunawaki S., Nakamura S., Ohyama Y., Sasaki M., Ikebe-Hiroki A., Hiraki A., Kadena T., Kawamura E., Kumamaru W., Shinohara M., and Shirasuna K. (2002) Possible function of salivary gland epithelial cells as nonprofessional antigen-presenting cells in the development of Sjögren's syndrome. J. Rheumatol. 29, 1884–1896 [PubMed] [Google Scholar]
- 5. Manoussakis M. N., and Kapsogeorgou E. K. (2007) The role of epithelial cells in the pathogenesis of Sjögren's syndrome. Clin. Rev. Allergy Immunol. 32, 225–230 [DOI] [PubMed] [Google Scholar]
- 6. Fox P. C. (2004) Salivary enhancement therapies. Caries Res. 38, 241–246 [DOI] [PubMed] [Google Scholar]
- 7. Trautmann A. (2009) Extracellular ATP in the immune system: more than just a “danger signal.” Sci. Signal. 2, pe6. [DOI] [PubMed] [Google Scholar]
- 8. Tsuda M., Tozaki-Saitoh H., and Inoue K. (2010) Pain and purinergic signaling. Brain Res. Rev. 63, 222–232 [DOI] [PubMed] [Google Scholar]
- 9. Di Virgilio F. (1998) ATP as a death factor. Biofactors 8, 301–303 [DOI] [PubMed] [Google Scholar]
- 10. Riteau N., Gasse P., Fauconnier L., Gombault A., Couegnat M., Fick L., Kanellopoulos J., Quesniaux V. F., Marchand-Adam S., Crestani B., Ryffel B., and Couillin I. (2010) Extracellular ATP is a danger signal activating P2X7 receptor in lung inflammation and fibrosis. Am. J. Respir. Crit. Care Med. 182, 774–783 [DOI] [PubMed] [Google Scholar]
- 11. Deaglio S., and Robson S. C. (2011) Ectonucleotidases as regulators of purinergic signaling in thrombosis, inflammation, and immunity. Adv. Pharmacol. 61, 301–332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. North R. A. (2002) Molecular physiology of P2X receptors. Physiol. Rev. 82, 1013–1067 [DOI] [PubMed] [Google Scholar]
- 13. Deplano S., Cook H. T., Russell R., Franchi L., Schneiter S., Bhangal G., Unwin R. J., Pusey C. D., Tam F. W., and Behmoaras J. (2013) P2X7 receptor-mediated Nlrp3-inflammasome activation is a genetic determinant of macrophage-dependent crescentic glomerulonephritis. J. Leukocyte Biol. 93, 127–134 [DOI] [PubMed] [Google Scholar]
- 14. Monif M., Reid C. A., Powell K. L., Smart M. L., and Williams D. A. (2009) The P2X7 receptor drives microglial activation and proliferation: a trophic role for P2X7R pore. J. Neurosci. 29, 3781–3791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Di Virgilio F. (2015) P2X receptors and inflammation. Curr. Med. Chem. 22, 866–877 [DOI] [PubMed] [Google Scholar]
- 16. Lopez-Castejon G., Theaker J., Pelegrin P., Clifton A. D., Braddock M., and Surprenant A. (2010) P2X7 receptor-mediated release of cathepsins from macrophages is a cytokine-independent mechanism potentially involved in joint diseases. J. Immunol. 185, 2611–2619 [DOI] [PubMed] [Google Scholar]
- 17. Weisman G. A., De B. K., Friedberg I., Pritchard R. S., and Heppel L. A. (1984) Cellular responses to external ATP which precede an increase in nucleotide permeability in transformed cells. J. Cell Physiol. 119, 211–219 [DOI] [PubMed] [Google Scholar]
- 18. Surprenant A., Rassendren F., Kawashima E., North R. A., and Buell G. (1996) The cytolytic P2Z receptor for extracellular ATP identified as a P2X receptor (P2X7). Science 272, 735–738 [DOI] [PubMed] [Google Scholar]
- 19. Woods L. T., Camden J. M., Batek J. M., Petris M. J., Erb L., and Weisman G. A. (2012) P2X7 receptor activation induces inflammatory responses in salivary gland epithelium. Am. J. Physiol. Cell Physiol. 303, C790–C801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Wilson H. L., Wilson S. A., Surprenant A., and North R. A. (2002) Epithelial membrane proteins induce membrane blebbing and interact with the P2X7 receptor C terminus. J. Biol. Chem. 277, 34017–34023 [DOI] [PubMed] [Google Scholar]
- 21. Lister M. F., Sharkey J., Sawatzky D. A., Hodgkiss J. P., Davidson D. J., Rossi A. G., and Finlayson K. (2007) The role of the purinergic P2X7 receptor in inflammation. J. Inflamm. (Lond.) 4, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Rathinam V. A., and Fitzgerald K. A. (2016) Inflammasome complexes: Emerging mechanisms and effector functions. Cell 165, 792–800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Baldini C., Rossi C., Ferro F., Santini E., Seccia V., Donati V., and Solini A. (2013) The P2X7 receptor–inflammasome complex has a role in modulating the inflammatory response in primary Sjögren's syndrome. J. Intern. Med. 274, 480–489 [DOI] [PubMed] [Google Scholar]
- 24. Hwang S. M., Koo N. Y., Choi S. Y., Chun G. S., Kim J. S., and Park K. (2009) P2X7 receptor-mediated membrane blebbing in salivary epithelial cells. Korean J. Physiol. Pharmacol. 13, 175–179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Nakamoto T., Brown D. A., Catalán M. A., Gonzalez-Begne M., Romanenko V. G., and Melvin J. E. (2009) Purinergic P2X7 receptors mediate ATP-induced saliva secretion by the mouse submandibular gland. J. Biol. Chem. 284, 4815–4822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Lucattelli M., Cicko S., Müller T., Lommatzsch M., De Cunto G., Cardini S., Sundas W., Grimm M., Zeiser R., Dürk T., Zissel G., Sorichter S., Ferrari D., Di Virgilio F., Virchow J.C., Lungarella G., and Idzko M. (2011) P2X7 receptor signaling in the pathogenesis of smoke-induced lung inflammation and emphysema. Am. J. Respir. Cell Mol. Biol. 44, 423–429 [DOI] [PubMed] [Google Scholar]
- 27. Chessell I. P., Hatcher J. P., Bountra C., Michel A. D., Hughes J. P., Green P., Egerton J., Murfin M., Richardson J., Peck W. L., Grahames C. B., Casula M. A., Yiangou Y., Birch R., Anand P., and Buell G. N. (2005) Disruption of the P2X7 purinoceptor gene abolishes chronic inflammatory and neuropathic pain. Pain 114, 386–396 [DOI] [PubMed] [Google Scholar]
- 28. Müller T, Vieira R. P., Grimm M., Dürk T., Cicko S., Zeiser R., Jakob T., Martin S. F., Blumenthal B., Sorichter S., Ferrari D., Di Virgilio F., and Idzko M. (2011) A potential role for P2X7R in allergic airway inflammation in mice and humans. Am. J. Respir. Cell Mol. Biol. 44, 456–464 [DOI] [PubMed] [Google Scholar]
- 29. Taylor S. R., Turner C. M., Elliott J. I., McDaid J., Hewitt R., Smith J., Pickering M. C., Whitehouse D. L., Cook H. T., Burnstock G., Pusey C. D., Unwin R. J., and Tam F. W. (2009) P2X7 deficiency attenuates renal injury in experimental glomerulonephritis. J. Am. Soc. Nephrol. 20, 1275–1281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. McGaraughty S., Chu K. L., Namovic M. T., Donnelly-Roberts D. L., Harris R. R., Zhang X. F., Shieh C. C., Wismer C. T., Zhu C. Z., Gauvin D.M., Fabiyi A. C., Honore P., Gregg R. J., Kort M. E., Nelson D. W., Carroll W. A., Marsh K., Faltynek C. R., and Jarvis M. F. (2007) P2X7-related modulation of pathological nociception in rats. Neuroscience 146, 1817–1828 [DOI] [PubMed] [Google Scholar]
- 31. Feng L., Chen Y., Ding R., Fu Z., Yang S., Deng X., and Zeng J. (2015) P2X7R blockade prevents NLRP3 inflammasome activation and brain injury in a rat model of intracerebral hemorrhage: Involvement of peroxynitrite. J. Neuroinflammation 12, 190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Alves L. A., Bezerra R. J. S., Faria R. X., Ferreira L. G. B., and da Silva Frutuoso V. (2013) Physiological roles and potential therapeutic applications of the P2X7 receptor in inflammation and pain. Molecules 18, 10953–10972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Arulkumaran N., Unwin R. J., and Tam F. W. (2011) A potential therapeutic role for P2X7 receptor (P2X7R) antagonists in the treatment of inflammatory diseases. Expert Opin. Investig. Drugs 20, 897–915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Bartlett R., Stokes L., and Sluyter R. (2014) The P2X7 receptor channel: Recent developments and the use of P2X7 antagonists in models of disease. Pharmacol. Rev. 66, 638–675 [DOI] [PubMed] [Google Scholar]
- 35. Kayes T. D., Weisman G. A., Camden J. M., Woods L. T., Bredehoeft C., Downey E. F., Cole J., and Braley-Mullen H. (2016) New murine model of early onset autoimmune thyroid disease/hypothyroidism and autoimmune exocrinopathy of the salivary gland. J. Immunol. 197, 2119–2130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Karmakar M., Katsnelson M. A., Dubyak G. R., and Pearlman E. (2016) Neutrophil P2X7 receptors mediate NLRP3 inflammasome-dependent IL-1β secretion in response to ATP. Nat. Commun. 7, 10555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Gombault A., Baron L., and Couillin I. (2012) ATP release and purinergic signaling in NLRP3 inflammasome activation. Front. Immunol. 3, 414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kahlenberg J. M., and Dubyak G. R. (2004) Mechanisms of caspase-1 activation by P2X7 receptor-mediated K+ release. Am. J. Physiol. Cell Physiol. 286, C1100–C1108 [DOI] [PubMed] [Google Scholar]
- 39. Kanneganti T. D., Lamkanfi M., Kim Y. G., Chen G., Park J. H., Franchi L., Vandenabeele P., and Núñez G. (2007) Pannexin-1-mediated recognition of bacterial molecules activates the cryopyrin inflammasome independent of Toll-like receptor signaling. Immunity 26, 433–443 [DOI] [PubMed] [Google Scholar]
- 40. Coll R. C., Robertson A. A., Chae J. J., Higgins S. C., Muñoz-Planillo R., Inserra M. C., Vetter I., Dungan L. S., Monks B. G., Stutz A., Croker D. E., Butler M. S., Haneklaus M., Sutton C. E., Núñez G., et al. (2015) A small-molecule inhibitor of the NLRP3 inflammasome for the treatment of inflammatory diseases. Nat. Med. 21, 248–255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Juliana C., Fernandes-Alnemri T., Wu J., Datta P., Solorzano L., Yu J. W., Meng R., Quong A. A., Latz E., Scott C. P., and Alnemri E. S. (2010) Anti-inflammatory compounds parthenolide and Bay 11–7082 are direct inhibitors of the inflammasome. J. Biol. Chem. 285, 9792–9802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Perregaux D. G., and Gabel C. A. (1998) Human monocyte stimulus-coupled IL-1β posttranslational processing: Modulation via monovalent cations. Am. J. Physiol. Cell Physiol. 275, C1538–C1547 [DOI] [PubMed] [Google Scholar]
- 43. Pétrilli V., Papin S., Dostert C., Mayor A., Martinon F., and Tschopp J. (2007) Activation of the NALP3 inflammasome is triggered by low intracellular potassium concentration. Cell Death Differ. 14, 1583–1589 [DOI] [PubMed] [Google Scholar]
- 44. Muñoz-Planillo R., Kuffa P., Martínez-Colón G., Smith B. L., Rajendiran T. M., and Núñez G. (2013) K+ efflux is the common trigger of NLRP3 inflammasome activation by bacterial toxins and particulate matter. Immunity 38, 1142–1153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Dostert C., Pétrilli V., Van Bruggen R., Steele C., Mossman B. T., and Tschopp J. (2008) Innate immune activation through Nalp3 inflammasome sensing of asbestos and silica. Science 320, 674–677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Niethammer P., Grabher C., Look A. T., and Mitchison T. J. (2009) A tissue-scale gradient of hydrogen peroxide mediates rapid wound detection in zebrafish. Nature 459, 996–999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Bolwell G. P. (1999) Role of active oxygen species and NO in plant defense responses. Curr. Opin. Plant Biol. 2, 287–294 [DOI] [PubMed] [Google Scholar]
- 48. Cruz C. M., Rinna A., Forman H. J., Ventura A. L., Persechini P. M., and Ojcius D. M. (2007) ATP activates a reactive oxygen species–dependent oxidative stress response and secretion of proinflammatory cytokines in macrophages. J. Biol. Chem. 282, 2871–2879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Zhou R., Yazdi A. S., Menu P., and Tschopp J. (2011) A role for mitochondria in NLRP3 inflammasome activation. Nature 469, 221–225 [DOI] [PubMed] [Google Scholar]
- 50. Cloonan S. M., and Choi A. M. (2012) Mitochondria: Commanders of innate immunity and disease? Curr. Opin. Immunol. 24, 32–40 [DOI] [PubMed] [Google Scholar]
- 51. Scroggins B. T., and Neckers L. (2007) Post-translational modification of heat-shock protein 90: Impact on chaperone function. Expert Opin. Drug Discov. 2, 1403–1414 [DOI] [PubMed] [Google Scholar]
- 52. Mayor A., Martinon F., De Smedt T., Pétrilli V., and Tschopp J. (2007) A crucial function of SGT1 and HSP90 in inflammasome activity links mammalian and plant innate immune responses. Nat. Immunol. 8, 497–503 [DOI] [PubMed] [Google Scholar]
- 53. Moutsopoulos H. (1994) Sjögren's syndrome: Autoimmune epithelitis. Clin. Immunol. Immunopathol. 72, 162–165 [DOI] [PubMed] [Google Scholar]
- 54. Thiel M., Wolfs M. J., Bauer S., Wenning A. S., Burckhart T., Schwarz E. C., Scott A. M., Renner C., and Hoth M. (2010) Efficiency of T-cell costimulation by CD80 and CD86 cross-linking correlates with calcium entry. Immunology 129, 28–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Delaleu N., Mydel P., Brun J. G., Jonsson M. V., Alimonti A., and Jonsson R. (2016) Sjögren's syndrome patients with ectopic germinal centers present with a distinct salivary proteome. Rheumatology 55, 1127–1137 [DOI] [PubMed] [Google Scholar]
- 56. Coskun M. (2014) Intestinal epithelium in inflammatory bowel disease. Front. Med. 1, 1–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Shimoda S., Harada K., Niiro H., Yoshizumi T., Soejima Y., Taketomi A., Maehara Y., Tsuneyama K., Nakamura M., Komori A., Migita K., Nakanuma Y., Ishibashi H., Selmi C., and Gershwin M. E. (2008) Biliary epithelial cells and primary biliary cirrhosis: The role of liver-infiltrating mononuclear cells. Hepatology 47, 958–965 [DOI] [PubMed] [Google Scholar]
- 58. Bläsche R., Ebeling G., Perike S., Weinhold K., Kasper M., and Barth K. (2012) Activation of P2X7R and downstream effects in bleomycin treated lung epithelial cells. Int. J. Biochem. Cell Biol. 44, 514–524 [DOI] [PubMed] [Google Scholar]
- 59. Witte E., Kokolakis G., Witte K., Warszawska K., Friedrich M., Christou D., Kirsch S., Sterry W., Volk H. D., Sabat R., and Wolk K. (2016) Interleukin-29 induces epithelial production of CXCR3A ligands and T-cell infiltration. J. Mol. Med. 94, 391–400 [DOI] [PubMed] [Google Scholar]
- 60. Barrera M. J., Bahamondes V., Sepúlveda D., Quest A. F., Castro I., Cortés J., Aguilera S., Urzúa U., Molina C., Pérez P., Ewert P., Alliende C., Hermoso M. A., González S., Leyton C., and González M. J. (2013) Sjögren's syndrome and the epithelial target: A comprehensive review. J. Autoimmun. 42, 7–18 [DOI] [PubMed] [Google Scholar]
- 61. Cesaro A., Brest P., Hofman V., Hébuterne X., Wildman S., Ferrua B., Marchetti S., Doglio A., Vouret-Craviari V., Galland F., Naquet P., Mograbi B., Unwin R., and Hofman P. (2010) Amplification loop of the inflammatory process is induced by P2X7R activation in intestinal epithelial cells in response to neutrophil transepithelial migration. Am. J. Physiol. Gastrointest. Liver Physiol. 299, G32–G42 [DOI] [PubMed] [Google Scholar]
- 62. Abais J. M., Xia M., Zhang Y., Boini K. M., and Li P. L. (2015) Redox regulation of NLRP3 inflammasomes: ROS as trigger or effector? Antioxid. Redox Signal. 22, 1111–1129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Akundi R. S., Huang Z., Eason J., Pandya J. D., Zhi L., Cass W. A., Sullivan P. G., and Büeler H. (2011) Increased mitochondrial calcium sensitivity and abnormal expression of innate immunity genes precede dopaminergic defects in Pink1-deficient mice. PLoS One 6, 10.1371/journal.pone.0016038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. d'Avila J. C. P., Santiago A. P. S. A., Amâncio R. T., Galina A., Oliveira M. F., and Bozza F. A. (2008) Sepsis induces brain mitochondrial dysfunction. Crit. Care Med. 36, 1925–1932 [DOI] [PubMed] [Google Scholar]
- 65. Muller F. L., Roberts A. G., Bowman M. K., and Kramer D. M. (2003) Architecture of the Qo site of the cytochrome bc1 complex probed by superoxide production. Biochemistry 42, 6493–6499 [DOI] [PubMed] [Google Scholar]
- 66. Turrens J. F. (2003) Mitochondrial formation of reactive oxygen species. J. Physiol. 552, 335–344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Heid M. E., Keyel P. A., Kamga C., Shiva S., Watkins S. C., and Salter R. D. (2013) Mitochondrial reactive oxygen species induces NLRP3-dependent lysosomal damage and inflammasome activation. J. Immunol. 191, 5230–5238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Kim S. R., Kim D. I., Kim S. H., Lee H., Lee K. S., Cho S. H., and Lee Y. C. (2014) NLRP3 inflammasome activation by mitochondrial ROS in bronchial epithelial cells is required for allergic inflammation. Cell Death Dis. 5, e1498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Friedle S. A., Curet M. A., and Watters J. J. (2010) Recent patents on novel P2X7 receptor antagonists and their potential for reducing central nervous system inflammation. Recent Pat CNS Drug Discov. 5, 35–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Keystone E. C., Wang M. M., Layton M., Hollis S., and McInnes I. B. (2012) Clinical evaluation of the efficacy of the P2X7 purinergic receptor antagonist AZD9056 on the signs and symptoms of rheumatoid arthritis in patients with active disease despite treatment with methotrexate or sulphasalazine. Ann. Rheum. Dis. 71, 1630–1635 [DOI] [PubMed] [Google Scholar]
- 71. Dinarello C. A. (2011) Interleukin-1 in the pathogenesis and treatment of inflammatory diseases. Blood 117, 3720–3732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Roescher N., Tak P. P., and Illei G. G. (2010) Cytokines in Sjögren's syndrome: Potential therapeutic targets. Ann. Rheum. Dis. 69, 945–948 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Dinarello C. A. (2009) Immunological and inflammatory functions of the interleukin-1 family. Annu. Rev. Immunol. 27, 519–550 [DOI] [PubMed] [Google Scholar]
- 74. Kalinski P. (2012) Regulation of immune responses by prostaglandin E2. J. Immunol. 188, 21–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Sharma J. N., Al-Omran A., and Parvathy S. S. (2007) Role of nitric oxide in inflammatory diseases. Inflammopharmacology 15, 252–259 [DOI] [PubMed] [Google Scholar]
- 76. Degagné E., Grbic D. M., Dupuis A. A., Lavoie E. G., Langlois C., Jain N., Weisman G. A., Sévigny J., and Gendron F.-P. (2009) P2Y2 receptor transcription is increased by NF-κB and stimulates cyclooxygenase-2 expression and PGE2 released by intestinal epithelial cells. J. Immunol. 183, 4521–4529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Peterson T. S., Thebeau C. N., Ajit D., Camden J. M., Woods L. T., Wood W. G., Petris M. J., Sun G. Y., Erb L., and Weisman G. A. (2013) Up-regulation and activation of the P2Y2 nucleotide receptor mediate neurite extension in IL-1β-treated mouse primary cortical neurons. J. Neurochem. 125, 885–896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Turner J. T., Weisman G. A., and Camden J. M. (1997) Up-regulation of P2Y2 nucleotide receptors in rat salivary gland cells during short-term culture. Am. J. Physiol. Cell Physiol. 273, C1100–C1107 [DOI] [PubMed] [Google Scholar]
- 79. Schrader A. M., Camden J. M., and Weisman G. A. (2005) P2Y2 nucleotide receptor up-regulation in submandibular gland cells from the NOD.B10 mouse model of Sjögren's syndrome. Arch. Oral Biol. 50, 533–540 [DOI] [PubMed] [Google Scholar]
- 80. Baker O. J., Camden J. M., Rome D. E., Seye C. I., and Weisman G. A. (2008) P2Y2 nucleotide receptor activation up-regulates vascular cell adhesion molecular-1 expression and enhances lymphocyte adherence to a human submandibular gland cell line. Mol. Immunol. 45, 65–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. El-Sayed F. G., Camden J. M., Woods L. T., Khalafalla M. G., Petris M. J., Erb L., and Weisman G. A. (2014) P2Y2 nucleotide receptor activation enhances the aggregation and self-organization of dispersed salivary epithelial cells. Am. J. Physiol. Cell Physiol. 307, C83–C96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Ahn J. S., Camden J. M., Schrader A. M., Redman R. S., and Turner J. T. (2000) Reversible regulation of P2Y2 nucleotide receptor expression in the duct-ligated rat submandibular gland. Am. J. Physiol. Cell Physiol. 279, C286–C294 [DOI] [PubMed] [Google Scholar]
- 83. Ayata C. K., Ganal S. C., Hockenjos B., Willim K., Vieira R. P., Grimm M., Robaye B., Boeynaems J. M., Di Virgilio F., Pellegatti P., Diefenbach A., Idzko M., and Hasselblatt P. (2012) Purinergic P2Y2 receptors promote neutrophil infiltration and hepatocyte death in mice with acute liver injury. Gastroenterology 143, 1620–1629 [DOI] [PubMed] [Google Scholar]
- 84. Jonsson M. V., Delaleu N., Brokstad K. A., Berggreen E., and Skarstein K. (2006) Impaired salivary gland function in NOD mice: association with changes in cytokine profile but not with histopathologic changes in the salivary gland. Arthritis Rheum. 54, 2300–2305 [DOI] [PubMed] [Google Scholar]
- 85. Lee Y. A., Choi H. M., Lee S. H., Yang H. I., Yoo M. C., Hong S. J., and Kim K. S. (2012) Synergy between adiponectin and interleukin-1β on the expression of interleukin-6, interleukin-8, and cyclooxygenase-2 in fibroblast-like synoviocytes. Exp. Mol. Med. 44, 440–447 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.