

Abstract
Nerve growth factor-β (NGF) is essential for the correct development of the nervous system. NGF exists in both a mature form and a pro-form (proNGF). The two forms have opposing effects on neurons: NGF induces proliferation, whereas proNGF induces apoptosis via binding to a receptor complex of the common neurotrophin receptor (p75NTR) and sortilin. The overexpression of both proNGF and sortilin has been associated with several neurodegenerative diseases. Insights into the conformational differences between proNGF and NGF are central to a better understanding of the opposing mechanisms of action of NGF and proNGF on neurons. However, whereas the structure of NGF has been determined by X-ray crystallography, the structural details for proNGF remain elusive. Here, using a sensitive MS-based analytical method to measure the hydrogen/deuterium exchange of proteins in solution, we analyzed the conformational properties of proNGF and NGF. We detected the presence of a localized higher-order structure motif in the pro-part of proNGF. Furthermore, by comparing the hydrogen/deuterium exchange in the mature part of NGF and proNGF, we found that the presence of the pro-part in proNGF causes a structural stabilization of three loop regions in the mature part, possibly through a direct molecular interaction. Moreover, using tandem MS analyses, we identified two N-linked and two O-linked glycosylations in the pro-part of proNGF. These results advance our knowledge of the conformational properties of proNGF and NGF and help provide a rationale for the diverse biological effects of NGF and proNGF at the molecular level.
Keywords: glycoprotein, hydrogen exchange mass spectrometry, hydrogen/deuterium exchange, intrinsically disordered protein, neurodegeneration, neurodegenerative disease, neurotrophin, protein conformation, protein structure
Introduction
Neurotrophins are a family of proteins critical for the correct development and maintenance of both the central and the peripheral nervous system (1). This family of proteins has a wide range of biological effects on neurons including the regulation of differentiation, promotion of survival, modulation of plasticity, and induction of apoptosis (2). The most abundant of the neurotrophins is nerve growth factor-β (NGF).2 Its neurotrophic effect is mediated via binding to both tropomyosin receptor kinase A (TrkA) and the common neurotrophin receptor p75NTR (1, 3).
NGF is expressed in a pro-form (proNGF), which is proteolytically cleaved to the mature form by several different proteases (e.g. furin) (4–6). For many years the pro-part was described only as a chaperone-like entity facilitating the folding of the mature part of the protein. Recently, several research groups have reported that proNGF inherently exhibits biological activity (2, 4, 7, 8). Interestingly, proNGF shows the opposite biological effect on neurons as compared with NGF, as proNGF induces apoptosis by activating a receptor complex of p75NTR and sortilin (8, 9). Furthermore, increased levels of proNGF and expression of sortilin have been associated with diverse pathological conditions in which apoptosis of neurons are prevalent, e.g. ischemic stroke, seizure, spinal cord injury, Alzheimer's disease, and multiple sclerosis (2, 10–13). The proNGF-sortilin-p75NTR interaction has been described as a new drug target in the treatment of the above-mentioned conditions, but the actual molecular mechanism behind the biological effect of proNGF is still inadequately understood. Simultaneous expression of p75NTR and sortilin appears to be necessary for the induction of apoptosis (8), and it has been hypothesized that the pro-part of proNGF interacts with sortilin, whereas the mature part binds to p75NTR (8, 9).
To be able to rationalize the diverse biological effect of proNGF and NGF and develop drugs targeting the proNGF-sortilin interaction, it is crucial to characterize the proteins involved at the molecular level. NGF has been described extensively in the literature and has been crystallized both alone and in complex with the TrkA and p75NTR receptors (14–17). It is a dimer under native conditions, and its structure is dominated by three anti-parallel β-strands and three intersecting disulfide bonds forming a so-called cysteine knot that tightly binds the protein together (Fig. 1) (18, 19).
Figure 1.

Structure of NGF. A, crystal structure of the NGF dimer (PDB ID: 1SG1). The cysteine knot is highlighted in yellow. B, schematic representation of the translation product of the NGF gene. Residues in the signal and the pro-part of proNGF are by convention assigned negative numbers (21). The connectivity of the three cysteine bonds in the mature part is shown. White bar, signal peptide of NGF; light gray bar, pro-part of NGF; dark gray bar, mature NGF.
The difference between the primary structure of NGF and proNGF is the presence of a 103-residue N-terminal pro-part in proNGF (Fig. 1B) (20). Unfortunately, the structural characterization of proNGF by traditional biophysical methods has thus far been complicated by the highly dynamic nature of the pro-part and its sensitivity to proteolysis (5, 9, 18, 21, 22). It has been shown that proNGF is also a homodimer under native conditions, and the structure of proNGF in complex with p75NTR has been solved by X-ray crystallography, but unfortunately clear electron density was only obtained for the mature part (9, 22, 23). Additionally, proNGF has been characterized by solution-based biophysical methods such as nuclear magnetic resonance (5), fluorescence (18), circular dichroism spectroscopy (5, 23), Fourier transform infrared spectroscopy (5), small-angle X-ray scattering (SAXS), (22) and hydrogen/deuterium exchange measured by mass spectrometry (HDX-MS) (21). These studies have described proNGF mainly at the global level, wherein the mature part of proNGF has been described as highly identical to NGF and the pro-part as highly flexible and dynamic, possibly with minor elements of structure. Finally, the pro-part of proNGF is known to be N-linked glycosylated, but the exact glycosylation sites and pattern have never been investigated thoroughly (4, 20).
HDX-MS is a sensitive analytical tool, which can be used in investigating the conformational dynamics and interactions of proteins under native solution-phase conditions (24, 25). During HDX-MS the exchange rate of backbone amide hydrogens is measured; this is defined by their engagement in hydrogen bonds (26–29). The exchange rate can vary by 7 orders of magnitude for an amide hydrogen in disordered proteins (non-hydrogen-bonded) compared with an amide hydrogen in a structured protein (hydrogen-bonded). The usefulness of HDX-MS in investigating highly-disordered parts of proteins, such as the pro-part of proNGF, has been indicated by the mapping of structured and disordered regions of intrinsically disordered proteins (30). Furthermore, HDX-MS can be used to investigate subtle changes in the protein dynamics caused by protein-protein interactions and might thereby be used to detect any conformational impact of the molecular interactions between the pro-part and the mature part of proNGF (25, 31, 32).
In this work, we used HDX-MS to study the conformation of proNGF and NGF. We show that the pro-part contains several O- and N-linked glycosylations and that the pro-part exhibits protection from HDX near the O-linked glycosites, identifying the presence of a transient and localized secondary/tertiary structure motif (e.g. a higher-order structure in which backbone amide hydrogens participate in hydrogen bonds). In conjunction, we show that the HDX of the mature part of the protein is directly impacted by the pro-part, in that several loops of NGF show reduced HDX when the pro-part remains attached to NGF. Based on these new findings, we have proposed a model for the conformational interplay between the ordered and disordered parts of the proNGF dimer regarding how the pro-part reduces the binding affinity of proNGF toward both TrkA and p75NTR compared with NGF.
Results
Expression and purification of human proNGF
Full-length, glycosylated proNGF is inherently difficult to express and purify in eukaryotic host cells due to the presence of three protease (e.g. furin) cleavage sites in the pro-part (9). To circumvent this challenge, most studies of proNGF thus far have been performed on nonglycosylated protein refolded following expression in Escherichia coli (5, 18, 21, 22). However, by the introduction of double mutations in the three protease cleavage sites, mouse proNGF has been expressed successfully in high and homogenous yield in insect cells (9, 33). The expressed mouse proNGF was shown to be biologically active and able to bind its natural receptors, sortilin and p75NTR. In the present study, we used this triple mutant approach and expressed human proNGF in Chinese hamster ovarian (CHO) cells. To our knowledge this is the first time that full-length, glycosylated, human proNGF has been successfully expressed in a mammalian host organism and purified in yields suitable for detailed structural characterization.
Identification of N- and O-linked glycosylations on human proNGF
To investigate the glycosylation status of proNGF, an intact mass analysis of nontreated and PNGase F- and sialidase-treated proNGF was performed by electrospray ionization (ESI) mass spectrometry (Fig. 2, A and B). The observed mass shift in the mass spectrum of proNGF after treatment with PNGase F and sialidase corresponds to the removal of two biantennary complex glycans, clearly showing that proNGF contains two N-linked glycans. The most abundant form of the N-linked glycans is core-fucosylated and end-capped with a single sialic acid moity at each antenna (Fig. 2, A and B). Of the four peaks in the PNGase F- and sialidase-treated mass spectrum (Fig. 2B), only the peak with the lowest mass (peak 1: 25,762.7 Da) corresponds to the theoretical mass of proNGF including two Asn-to-Asp modifications (34).
Figure 2.

Identification and mapping of N- and O-linked glycans in the pro-part of proNGF. Intact mass analysis of nontreated (A), PNGase- and sialidase-treated (B), and PNGase F-, sialidase-, and O-glycosidase-treated (C) proNGF. Treatment with both PNGase F and O-glycosidase caused a mass shift corresponding to known glycan structures, showing that proNGF is both N- and O-linked glycosylated. The ETD fragmentation spectra of glycopeptide Val−35–Arg−7 with either two (E) or one (D) O-linked glycan show that proNGF is O-linked glycosylated at Ser−32 and Thr−31. The mass error of all identified peaks is below 23 ppm. Yellow square, N-acetylgalactosamine; blue square, N-acetylglucosamine; yellow circle, galactose; green circle, mannose; purple diamond, sialic acid; red triangle, fucose.
Thus, proNGF exists as four different species even after enzymatic removal of the two N-linked glycans. Interestingly, the mass difference between these species of proNGF correlated with sugar moieties, indicating the presence of further glycosylations not cleavable by PNGase F and sialidase. The heterogeneity of proNGF disappeared after further O-glycosidase treatment, confirming the presence of O-linked glycans (Fig. 2, B and C) (35). From the intact mass analysis of proNGF, it can be estimated that 12% of proNGF is not O-linked glycosylated, whereas the rest of the protein population (88%) contains from one up to three O-linked glycosylations (Fig. 2B). The O-linked glycans most likely consist of one N-acetylgalactosamine, one galactose, and either one or two sialic acid entities (supplemental Fig. S1), as the biosynthesis of O-glycans containing multiple sugar moieties is most commonly initiated by the attachment of an α-linked N-acetylgalactosamine followed by the attachment of a galactose entity (36, 37). Furthermore, the identified types of O-glycosylations are similar to the ones reported previously for CHO cells (38).
Mapping of N- and O-linked glycosylation sites
To map the N-glycosylation sites on proNGF, a tryptic digestion of untreated and PNGase F-treated proNGF was performed, and the resulting peptides were analyzed by reverse-phase ultra-high-performance liquid chromatography (UPLC) ESI tandem mass spectrometry (MS/MS). A conventional peptide search algorithm (not able to identify glycopeptides) was used to identify tryptic peptides. The resulting sequence coverage map from the nontreated sample contained two regions without sequence coverage in the pro-part of proNGF: Val−59–Arg−47 and Val−35–Arg−4 (supplemental Fig. S2). These two regions, containing an N-linked consensus motif at Asn−53 and Asn−8, were covered by peptides in the PNGase F-treated sample (taking into account a single Asn–Asp conversion in each peptide) indicating the presence of an N-linked glycan at both Asn−53 and Asn−8 (34, 39). This indication was supported by manual identification of glycopeptide Asn−53–Arg−47 and glycopeptide Val−35–Arg−4, confirming the identity of N-linked glycans at both Asn−53 and Asn−8 (supplemental Fig. S3). The most prominent glycan form on both sites was a complex biantennary glycan that was fucosylated and end-capped with a single sialic acid at each antenna, as also observed in the intact mass analysis.
The mapping of O-linked glycosylation sites is more challenging than for N-linked glycosylation sites due to the absence of sequence consensus motifs (40). However, glycopeptides can be differentiated from non-glycopeptides by their gas-phase fragmentation pattern in collision-induced dissociation, as the attached glycan results in specific high-intensity fragments in the low m/z range (e.g. oxonium ions) (41–43). By exploiting this property, several O-linked glycopeptides covering the region Val−35–Arg−4 were detected. The Val−35–Arg−4 region of the pro-part contains three possible O-linked glycosylation sites (Ser−32, Thr−31, and Thr−22), and each peptide detected contained either one or two O-linked glycosylation sites. To pinpoint the individual O-linked glycosylation sites in proNGF, MS/MS experiments using electron transfer dissociation (ETD) were performed on four O-linked tryptic glycopeptides. ETD is a non-ergodic gas-phase fragmentation technique that gives rise to backbone fragmentation at the N–Cα bond (c and z ions) while retaining labile post-translational modifications (such as O-linked glycosylations) on the backbone fragments (44, 45).
The ETD spectrum of glycopeptide Val−35–Arg−7 containing one O-linked glycan shows abundant fragmentation of the backbone (Fig. 2D). The presence of the unmodified c4, z24, and z21 ions and glycosylated c5 and z25 ions allows an unambiguous assignment of the O-linked glycan to Thr−31. No evidence was found for a glycosylated c4 or z24 ion or for the presence of a chromatographically separated m/z isomer of the precursor mass, indicating that the O-linked glycosylation at Thr−31 probably precedes additional O-glycosylations of proNGF.
The ETD spectrum of glycopeptide Val−35–Arg−7 containing two O-linked glycans also displayed abundant fragmentation of the backbone (Fig. 2E). On the basis of the identified fragment ions and the specific identification of an unmodified z16 ion, single glycosylated c4 and z25 ions, and a doubly glycosylated z26 ion, the second O-linked glycosylation site on proNGF could be unambiguously assigned to Ser−32. The occupancy of the O-glycosylation sites at Ser−32 and Thr−31 was 49 and 88%, respectively (Fig. 2B). It was not possible to map the third O-linked glycosylation site because of the low occupancy (8%) at this site.
HDX-MS analysis of proNGF and NGF
Our initial experiments showed proNGF and NGF very challenging to investigate via a conventional “bottom-up” HDX-MS workflow, as the cysteine knot was completely resistant to reduction by the addition of tris(2-caboxyethyl)phosphine (TCEP) to the quench buffer. This resulted in low sequence coverage in major parts of NGF and the mature part of proNGF, e.g. total sequence coverage below 50% (46). To meet this challenge, a HDX-MS–compatible workflow employing online electrochemical reduction was developed, allowing us to successfully reduce the cysteine knot under quenched conditions (e.g. low pH and temperature). By employing this workflow, an effective sequence coverage, 100 and 87% of NGF and proNGF, respectively, was achieved (Fig. 3). The missing sequence coverage of proNGF from residues Glu−16–Cys15 could be ascribed to the heterogeneity of the N-linked glycan at Asn−8 diminishing the signal from the single glycopeptide.
Figure 3.

HDX-MS coverage map of proNGF and NGF. A, peptic peptides from which HDX data could be obtained are shown as: blue bars, data for both proNGF and NGF; gray bars, data only for proNGF; and black bars, data for NGF only. The bent arrow marks position 0, i.e. the beginning of the mature part of NGF. Glycosylations in the pro-part of proNGF are marked by white (N-linked) and gray (O-linked) hexagons. The alanine mutations at positions −73, −72, −43, −42, −2, and −1 are highlighted by a bold font, and the His6 tag is highlighted by an italic font. The graphs show absolute deuterium incorporation, plotted as a function of time, of a disordered region from the pro-part (B) and a structured region in the mature part (C) of proNGF. Blue-filled circles, deuterium incorporation of equilibrium labeled (90%) samples. S.D. is plotted as error bars (only slightly visible) (n = 3 for the 15-s, 1-min, and 60-min time points, and n = 2 for the 16-h time point and the equilibrated control sample).
Identification of local higher-order structure in the pro-part of proNGF
The structural dynamics of the pro-part of proNGF were investigated by HDX-MS. Backbone amide hydrogens in the disordered regions of proteins undergo rapid exchange compared with regions containing hydrogen bonding (higher-order structure) and are completely labeled within a few seconds after dilution into a deuterated buffer (Fig. 3, B and C) (30, 47). Deuterium incorporation following 15 s of peptides originating from the pro-part was compared with deuterium incorporation of an equilibrium-labeled (90%) sample (Fig. 4A and supplemental Fig. S4). Four peptides spanning residues Phe−45 to Glu−26 showed a significant protection from exchange (p < 0.01), and the protection could be sublocalized to residues Arg−42–Phe−33 based on the following three observations: (i) no protection was observed for peptide Ser−32–Phe−17; (ii) the absolute protection in peptide Ala−43–Phe−33 was not significantly different from any of the three other peptides where protection was observed; and (iii) the first amide (i.e. Ala−42 in peptide Ala−43–Phe−33) in a peptide normally back-exchanges too rapidly to be measured in a classic bottom-up HDX-MS experiment (47, 48). The use of overlapping peptides to sublocalize the protection is valid only if the back-exchange of the compared peptides is similar (less than 11 percentage points) (49, 50). No significant difference in the deuterium incorporation level was observed for longer peptides that also spanned this area (e.g. Phe−45–Phe−17), as the deuterium incorporation of larger peptides tends to have a higher standard deviation.
Figure 4.

Detection of local higher-order structure in the pro-part of proNGF. A, deuterium incorporation after 15 s (red bars) was compared with deuterium incorporation of equilibrium labeled (90%) samples (blue bars). All peptides shown originated from the pro-part of proNGF. A significantly lesser incorporation of deuterium was seen from residue Arg−41–Phe−33. Glycopeptides was grouped by themselves, as a possible protection could be assigned to both the peptide backbone and the attached glycans (72). Asterisks, indicate significant differences in exchange (p < 0.01). S.D. is plotted as error bars. a, peptide contains one O-linked glycan; b, peptide contains two O-linked glycans. (n = 3 for the 15-s time point, and n = 2 for the equilibrated control sample). B, structure prediction of the pro-part of proNGF performed by several algorithms. DISOPRED2 predicts whether a residue is natively disordered or structured, whereas PSIPRED v3.3 and JPRED 3 predict the presence of secondary structure elements, e.g. random coil, α-helix, and β-strand. The lack of disorder predicted for residues Arg−47–Ser−32 aligns well with the observed protection from exchange in the region of residues Arg−41–Phe−33. Gray bars, predicted disordered regions; green bars, predicted α-helix structure; yellow bars, predicted β-sheet structure. AA, double alanine mutation site; white hexagon, N-linked glycosylation site; gray hexagon, O-linked glycosylation site.
The observed protection from exchange in this region shows that at least one or several of the backbone amide hydrogens in this region engage in hydrogen bonds of moderate stability either with hydrogen bond acceptor sites in the pro-part or residues in the mature part of proNGF. In other words, this region of the pro-part is not fully disordered in solution and contains a localized element of higher-order structure.
The detected protection from exchange corresponds well with structural prediction algorithms (Fig. 4B) (51, 52). The protection was observed in the only region of the pro-part where disorder is not predicted and secondary structure elements are predicted at the same time. The α-helix structure is predicted slightly before the observed protection, which can be ascribed to the fact that the first four backbone amide hydrogens in the N terminus of an α-helix do not engage in hydrogen bonding and thereby are unprotected (53, 54).
The pro-part changes the structural dynamics of loop regions in the mature part of NGF
proNGF have been inherently difficult to investigate by traditional biophysical methods, and little is known about the conformational effect (if any) on the mature part of proNGF caused by the presence of the pro-part. To investigate this effect, a comparative HDX analysis of the mature part of proNGF and NGF was performed (Fig. 5 and supplemental Fig. S4). Interestingly, the presence of the pro-part provided significant protection from exchange of backbone amide hydrogens in three segments comprising loops I, II, and IV of the mature part of proNGF. In the peptides spanning loop I (residues 23–35) and loop II (residues 40–49) a difference in deuterium incorporation was seen at the first two time points, whereas the difference in deuterium incorporation for peptides spanning loop IV (residues 91–97) was most pronounced at the second time point. The three loop regions form a continuous surface in the top and along the side of the NGF dimer. Notably, the surface patch is situated in the opposite end of the molecule to the pro-part attachment site and the cysteine knot, for both of which no difference in exchange was observed (Fig. 5).
Figure 5.

The pro-part of proNGF perturbs the conformation of the mature part of proNGF. Absolute deuterium incorporation is plotted as a function of time for NGF (gray lines) and the mature part of proNGF (red lines). Equilibrium-labeled (90%) proNGF (control samples) are plotted as blue-filled circles at the 16-h time point. Regions in the mature part of proNGF where a significant protection of exchange was observed in the presence of the pro-part for at least two consecutive time points are colored red on a crystal structure of NGF (PDB ID: 1SG1). S.D. is plotted as error bars (most are too small to be visible). Asterisk, denotes a significant protection from exchange when comparing deuterium incorporation in NGF with the mature part of proNGF (p < 0.01). (n = 3 for the 15-s, 1-min, and 60-min time points, and n = 2 for the 16-h time point and the equilibrated control sample).
Discussion
Glycan analysis
Human proNGF has for a long time been suggested to be N-linked glycosylated at Asn−53 and Asn−8 (4, 9, 20). The results of the glycan analysis of proNGF presented in this study confirm this proposition. The further identification of two O-linked glycosylation sites at Ser−32 and Thr−31 has, to our knowledge, not been described previously in the literature. The presence of both N-linked and O-linked glycans on the pro-part of proNGF is intriguing, as glycosylations are known to modulate protein structure, stability, function, and in vivo activity (55–57). Furthermore, the glycosylations can be important in the interaction of proNGF with sortilin, which is specifically thought to be mediated by the pro-part (8). It is important to note that the identified glycoforms of proNGF expressed in CHO cells are likely not identical to the exact glycoforms of proNGF found in human brain tissue (58–60). Especially O-glycosylation sites have been shown to differ between recombinant protein and naturally derived protein material (61), and the identified O-glycosylations sites in the pro-part of proNGF can only be interpreted as an indication of the presence of O-linked glycosylations in human proNGF.
Interestingly, the identification of O-linked glycosylations sites close to one of the mutated furin cleavage sites, however, could be biologically relevant, as O-linked glycosylation have been shown to be protective toward pro-protein convertases such as furin (62, 63). Furthermore, the presences of the O-linked glycosylation sites (Ser−32 and Thr−31) are in very close proximity to residues Arg−41–Phe−33, where protection from exchange and thereby secondary structure was identified. It is thus possible that the O-linked glycans could potentially serve a structural role by providing a scaffold or nucleation point for the localized higher-order structure in the pro-part of proNGF.
The precise function of the identified O-glycosylations will require further experiments to clarify.
Identification of local higher-order structure in the pro-part of proNGF
The pro-part of proNGF has been studied by diverse biophysical methods and has previously been indicated as highly flexible with minor elements of higher-order structure (5, 18, 21). The SAXS data from proNGF support these findings and suggest that the relative hydrophobic pro-part does not fluctuate freely in solution but collapses onto the mature part of the protein (22). An interaction with the mature part of NGF is in excellent accordance with the finding of local secondary higher-order structure in the present study and the observed surface patch of mature NGF stabilized by the pro-part. We note that the proNGF construct used in this study contains three double Ala mutations in the pro-part. One of these is in immediate proximity to the segment where hydrogen-bonded amides were identified. Ala is known to be the greatest α-helix stabilizer of all the amino acids (64), but if the Ala mutations induce α-helix formation, they would likely be directly involved and thereby participate in hydrogen bonding. This is not the case, considering that the observed protection can be ascribed mainly to residues Arg−41–Phe−33.
The localization of the local higher-order structure in the pro-part of proNGF expressed in E. coli has been attempted earlier by HDX-MS (21). Here, a protection from exchange was detected in two peptides spanning residues Trp−83 to Ala−63, a region where no protection was observed in the present study. Furthermore, no protection of exchange was reported in the region from residues Arg−41 to Phe−33, where protection was identified in the present study. These diverging results can be explained by use of different protein material from different host organisms (i.e. E. coli is not able to glycosylate proteins). Furthermore, several questions can unfortunately be raised concerning the data quality and interpretation presented in the earlier study (21). First, the peptic peptides were assigned to a given sequence based only on their intact mass without supportive fragmentation (MS/MS) data. Secondly, the deuterium incorporation was compared with a calculated maximum level of incorporation, corrected for back-exchange by using only two control peptides at a length of 12 and 21 residues. This can be problematic, as back-exchange rates are dependent on the primary amino acid sequence and thereby are highly peptide-dependent (47, 50, 65). Finally, the reproducibility of the reported deuterium incorporation is remarkably low (48). For one of the peptides, where a protection was observed, a standard deviation of ±7.7 deuteriums was reported. In the present study, we addressed all these concerns by performing the HDX experiments in accordance with present guidelines (25, 48). The identification of peptides was supported by fragmentation data, an equilibrium-labeled sample was used as control to correct for differences in back-exchange from peptide to peptide, and a markedly higher reproducibility of deuterium incorporation was obtained (supplemental Fig. S4). We are convinced that the clear discrepancies between the results from our present study and that of Kliemannel et al. (21) should be explained by the above differences in experimental approach. We believe that our HDX-MS measurements show convincingly that structural elements are present in the pro-part, and that these can be mapped to the Arg−41–Phe−33 region of proNGF.
The pro-part changes the structural dynamics of loop regions in the mature part of NGF
proNGF has until now been elusive to characterize by conventional biophysical methods, and thus far only a single crystal structure of proNGF has been published (9). proNGF was crystallized in complex with p75NTR, but unfortunately clear and well-defined electron density was obtained only for the mature part of proNGF. In this structure, the mature part of proNGF is highly similar to NGF in complex with p75NTR, and only loop II adopts a slightly altered conformation (9, 16). In the proNGF-p75NTR complex, loop II is found in an open conformation, where the two loops in the dimer of proNGF bend away from each other and the dimer interface. This is opposite to the NGF-p75NTR complex, where the two loops bend toward each other and the dimer interface, in a closed conformation. Furthermore, unassigned electron density is situated between the two loops of loop II in the proNGF-p75NTR complex, which have been suggested to be a segment of the pro-part (9).
The conformational effect of the presence of the pro-part on the mature part of proNGF has also been investigated by fluorescence spectroscopy (21). This study suggests that Trp21 takes part in a binding surface between the pro-part and the mature part of proNGF. However, Trp21 adopts the same conformation in either crystal structures of NGF and proNGF in complex with p75NTR, and the Trp side chain engages in a hydrogen bond with p75NTR in both complexes (9, 16). Several simulations and models of the structure of proNGF have been presented, which have tried to incorporate the experimental data from the SAXS, HDX-MS, and fluorescence spectroscopy studies discussed above (5, 21, 22). These models are an ensemble of structures in which the pro-part is suggested to pack mainly in proximity to the bottom of the mature part of the protein, forming a “crab-like” structure where the relative hydrophobic pro-part collapses onto the bottom of the mature part and only seems to interact directly with loop I of the loop regions in the mature part of proNGF (5, 22).
In contrast, the presented solution-state HDX-MS data of proNGF and NGF show no significant differences in exchange in the bottom of the mature part in the presence of the pro-part (Fig. 5), which is tightly bound together by the cysteine knot. On the other hand, a conformational perturbation of loops I, II, and IV was observed in the presence of the pro-part. The change in conformation of loop II corresponds well with the crystal structure of proNGF in complex with p75NTR (9). The further change in structural dynamics observed in loops I and IV of our solution-phase experiments cannot be rationalized on the background of the crystal structure. Our data are, however, supported by binding studies of proNGF to NGF antibodies, in which NGF antibodies targeting loops I, II, and IV have reduced affinity toward proNGF compared with NGF (22). Thus, the pro-part either sterically shields the antibody epitopes or perturbs the epitope recognized by the antibodies.
A refined model for the mechanism of action of the pro-part of proNGF
Based on the present experimental study of proNGF in solution, we propose a model whereby the structural stabilization of the three loops in the upper part of the mature part of proNGF is caused by a direct molecular interaction between the pro-part and the mature part (Fig. 6A). The segment of the pro-part responsible for interaction with the mature part of NGF (residues Arg−41–Phe−33) does so by forming a localized higher-order structure scaffold in the otherwise disordered pro-part that optimizes binding to the continuous surface formed by loops I, II, and IV of the mature part of proNGF.
Figure 6.
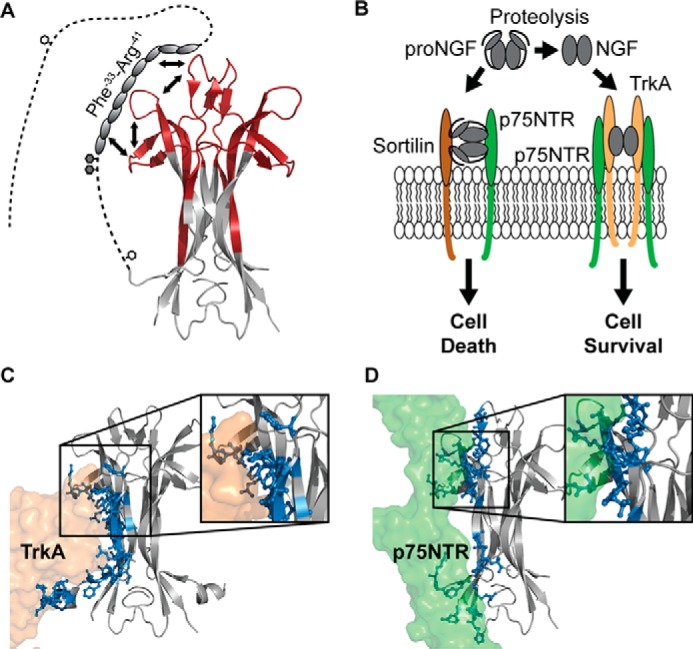
A refined structural model of proNGF. A, schematic representation of the findings presented in the current study. We propose that the part of the pro-part where local higher-order structure was identified interacts with parts of loops I, II, and IV via a combined intra- and intermolecular mechanism. The white hexagons mark N-linked glycosylation sites, and the gray hexagons mark O-linked glycosylation sites. To decrease the complexity, the pro-part was included on only one of the NGF monomers (PDB ID: 1SG1). B, biological effect of proNGF and NGF on neurons. NGF induces the growth of neurons by binding to the TrkA and the p75NTR receptors, whereas proNGF mediates apoptosis of neurons by binding to a receptor complex of sortilin and p75NTR. Figure adapted from (8). C and D, crystal structures of NGF (gray) in complex with TrkA (orange) and p75NTR (green) (PDB ID: 2IFG and 1SG1). Residues in NGF that form hydrogen bonds with the receptors are highlighted as sticks and colored blue. Inserts, zoom image of the binding surface implicating loops I, II, and IV of NGF. To simplify this figure, the second receptor molecule of TrkA and p75NTR was omitted.
It is in principle not possible to discern, from the HDX data alone, whether the interaction between the pro-part and the mature part is solely of an intramolecular nature (i.e. pro-part interacting solely with loops of its own mature part) or the pro-part adopts the more direct alignment in which it interacts with the NGF loops in closest proximity to the pro-part attachment site (i.e. from both molecules of the dimer) (Fig. 6A and supplemental Fig. S5). Such a “combined intra/intermolecular interaction” mode between the pro-part and the mature part would impart significant additional stability to the proNGF dimer compared with an exclusively intramolecular interaction mode. In good apparent correlation, it has been shown that the proNGF dimer is significantly more thermodynamically stable than the NGF dimer (5). In either case, the interaction mode and conformational alignment of each pro-part in the proNGF dimer appears to be identical, as no evidence of conformational heterogeneity (i.e. bimodal exchange patterns) was found in our HDX data for all regions of proNGF.
The opposing biological effects of proNGF and NGF can be ascribed to two features of proNGF; its specific interaction with sortilin and its reduced affinity towards both TrkA and p75NTR (Fig. 6B) (8).
The proposed model provides a structural rationale for the reduced binding of proNGF to both TrkA and p75NTR. The binding interface in the NGF-TrkA complex from the NGF point of view consists of two surfaces located spatially remote from each other, with which TrkA interacts in a bi-dendate fashion (Fig. 6C) (14, 15). Binding surface A is formed by the N-terminal residues, close to the site of pro-part attachment. Binding surface B consists of the residues in loop I and the residues in the top of the surface of the four β-strands forming the dimer interface (Fig. 6C). No sequence coverage was obtained for the N-terminal of the mature part of proNGF (Ser2–Ser13) in the current study, but the N terminus of NGF is known from other studies to be highly flexible and dynamic in solution (supplemental Fig. S4) (17). Upon complexation of NGF and TrkA, the N terminus in NGF forms a short α-helix, which is highly important for the binding and specificity of NGF toward TrkA (14). The presence of the pro-part, extending from the immediate vicinity, might sterically interfere with the formation of this helix and the correct alignment of TrkA for binding to binding surface A on NGF. More importantly, our experimental results show, for the first time, that a surface defined by loops I, II, and IV of NGF, which overlaps partially with binding surface B, becomes stabilized in solution in the presence of the pro-part (Fig. 5) (9, 21). Perturbation of the conformation of loops I, II, and IV of the pro-part by a direct binding model provides a rationale for how the pro-part directly impairs the ability of TrkA to access binding surface B on the mature part of proNGF. Hence, the detected local conformational stabilization of the mature part by the pro-part may directly explain the decreased affinity of proNGF for TrkA compared with NGF (8). In support of this concept, mutational studies have shown that both loop II and loop IV are important for the biological activity of NGF (66), and these two loops have also been suggested to associate with the membrane linker of TrkA (14, 15).
The binding interface between NGF and p75NTR can also be divided into two different binding sites (Fig. 6D). The first binding site constitutes residues in the upper part of the β-strands that form the dimer interface and loops I, II, and IV (overlapping with binding surface B, described above for the TrkA-NGF interaction) (16). As mentioned, the conformational stabilization of this region was observed in the presence of the pro-part (Fig. 5) and thus also explains the decrease in affinity of proNGF toward p75NTR compared with NGF. The second binding site is situated in the bottom part of NGF where protruding apolar side chains form a zipper-like interface between NGF and p75NTR (16). In this region, no conformational changes were detected in the presence of the pro-part (Fig. 5 and supplemental Fig. S4). As the conformation of this part of the mature part of NGF was not affected by the presence of the pro-part, as indicated by our HDX measurements, we propose that this region of NGF is still able to engage in binding to p75NTR. Our model suggests that the ability of proNGF to retain interactions with p75NTR, in turn, might be important for the correct alignment of the pro-part for interaction with the sortilin receptor in the context of the proNGF-p75NTR-sortilin complex (Fig. 6A) (8). Future experimental studies of the complexes formed between proNGF and NGF and their cognate receptors are needed to provide additional molecular insights into how neuronal development is regulated by the pro- and mature forms of NGF.
Experimental procedures
Materials
All reagents were purchased from Sigma-Aldrich in analytical grade except for the following: immobilized pepsin beads (Thermo Scientific), recombinant NGF (amino acid 1–117) (Sino Biological Inc.) and O-glycosidase (QA Bio). Sialidase was expressed and purified in-house as described in the literature (67). PNGase F was expressed and purified in-house using a similar vector and expression system as described above for sialidase.
Expression of proNGF
Human proNGF (residues Asp−103–Ala120) was modified by three double alanine mutations (K−73A, K−72A, K−43A, R−42A, and K−2A, R−1A) to increase expression yield and prevent proteolysis, and a C-terminal His6 tag was added at the C terminus (IEGRHHHHHH) (Fig. 3A) (9, 33). The coding sequence for proNGF-His6 was cloned into the pTT5 plasmid for transient gene expression in mammalian cells (GeneArt Gene Synthesis, Thermo Fisher Scientific). Transient gene expression was performed in CHO 3E7 cells using PEIpro (Polyplus-transfection SA) as a transfection reagent. The CHO 3E7 expression system, including the pTT5 vector, is licensed from the National Research Council of Canada. Cell culture supernatant was harvested by centrifugation followed by sterile filtration. Subsequently the medium was applied to a HisTrap column (GE Healthcare Life Sciences) in 20 mm sodium phosphate, pH 7.4, 1 m NaCl, and proNGF was eluted in a linear gradient to 0.5 m imidazole over 15 column volumes in the same buffer. Fractions were analyzed by SDS-PAGE and pooled according to the purity and concentration of proNGF determined by bicinchoninic acid assay (BCA, Pierce Biotechnology). The pooled fractions were finally dialyzed for 18 h against a PBS solution of 2.67 mm KCl, 1.47 mm KH2PO4, 137.93 mm NaCl, and 8.06 mm Na2HPO4-7 H2O, pH 7.4 (Gibco, Thermo Fisher Scientific).
Glycan analysis
PNGase F treatment of proNGF
proNGF was mixed at 4:1 (v/v) with PNGase F and incubated for 16 h at 37 °C in PBS buffer, pH 7.4.
PNGase F and sialidase treatment of proNGF
proNGF was mixed at 100:25:1 (v/v/v) with PNGase F and sialidase and incubated for 16 h at 37 °C in PBS buffer, pH 7.4.
O-Glycosidase treatment of proNGF
PNGase F- and sialidase-treated proNGF were treated with O-glycosidase according to instructions provided by QA Bio.
Tryptic digest of nontreated and glycosidase-treated proNGF
proNGF (either untreated or glycosidase-treated) was denaturated (3 m guanidinium chloride for 30 min at 60 °C), reduced (15 mm dithiothreitol for 120 min at 60 °C), alkylated in the dark (20 mm iodoacetamide for 30 min at 25 °C), and finally digested by trypsin (1:20 (w/w) for 16 h at 37 °C).
Mass analysis of intact proNGF
150–300 pmol of proNGF was loaded onto a reverse-phase UPLC system (nanoAquity, Waters Inc.). The protein was trapped unto a C4 trap column (ACQUITY UPLC BEH C4 1.7 μm VanGuard column, Waters Inc.), desalted for 3 min at 100 μl/min, eluted from the trap column by conventional reverse-phase chromatography, and ionized by positive ESI into a quadrupole time-of-flight (Q-TOF) mass spectrometer (Synapt G2, Waters Inc.).
Mapping of glycosylation sites
The tryptic peptide mixture (50 pmol) was loaded onto a reverse-phase UPLC system (nanoAquity, Waters Inc.), trapped on a C18 trap column (ACQUITY UPLC BEH C18 1.7 μm VanGuard column, Waters Inc.), and desalted for 6 min at 200 μl/min. The trap column was subsequently put in-line with a C18 analytical column (ACQUITY UPLC BEH C18 1.7 μm, 1 × 100 mm column, Waters Inc.), where the tryptic peptides were separated by reverse-phase chromatography and ionized by positive ESI into a Q-TOF mass spectrometer (Synapt G2, Waters Inc.). The tryptic peptides were analyzed by MS/MS with collision-induced dissociation. The mass spectrometer was operated in data-dependent acquisition mode. ETD of O-linked glycopeptides was performed in the trap traveling wave ion guide using 1,4-dicyanobenzene as the ETD reagent, as described elsewhere (68). Supplemental activation (10 eV of collision energy) was applied in the transfer traveling wave ion guide to increase ETD fragment ion yields (69).
Data analysis
Intact proteins
The acquired spectra were lock mass–corrected against Glu1-fibrinopeptide B and deconvoluted in the MassLynx software using the MaxEnt1 algorithm.
Mapping of glycosylation sites
The acquired mass spectra were lock mass–corrected against Glu1-fibrinopeptide B and analyzed using PLGS 2.5 software, which matched precursor and fragment ions to a local protein database. All positive hits were validated manually. Oxonium fragment ions of N-acetylglucosamine/N-acetylgalactosamine, sialic acid subtracted water, and N-acetylglycosamine/N-acetylgalactosamine connected with galactose (204.0875, 274.1732, and 366.14 m/z, respectively) were used as reporter ions to manually identify N- and O-linked glycopeptides (41–43).
HDX-MS analysis of proNGF and NGF
Exchange reaction
NGF and proNGF were buffer-exchanged into a 20 mm Tris buffer, pH 7.4, by spin-filtering (Vivaspin, 5,000 or 10,000 molecular weight cut-off, Sartorius Stedim). The exchange reaction was started by diluting proNGF or NGF samples (30pmol) 1:9 with 99% D2O, 20 mm Tris buffer (pDread 7.6) at 25 °C. After various time points (15 s, 1 min, 60 min, and 16 h) the exchange reaction was quenched by the addition of an ice-cold 2% formic acid solution in a 1:1 relationship, thereby decreasing the pH to 2.20. The quenched samples were immediately frozen to −80 °C until analysis. Equilibrium-deuterated control samples were prepared by incubating samples for 5 days at 25 °C. The early time points (15 s, 1 min, and 60 min) were performed in triplicate, and the final time point and the equilibrated control sample were performed in duplicate.
UPLC-HDX-MS setup
The quenched samples were thawed and injected onto a cooled (0 °C) reverse-phase UPLC-HDX system (Waters Inc.) with an integrated electrochemical reduction cell (high-pressure μ-prep cell, internal volume of 11 μl, Antec) (46, 70, 71) and a home-packed pepsin column (internal volume of 60 μl). The deuterated protein samples were subjected to online electrochemical reduction and subsequent pepsin digestion at 20 °C (46). The resulting peptic peptides were trapped unto a C18 trap column (ACQUITY UPLC BEH C18 1.7 μm VanGuard column, Waters Inc.) and desalted for 6 min at 50 μl/min with 0.23% formic acid. The trap column was put in-line with a C18 analytical column (ACQUITY UPLC BEH C18 1.7 μm, 1 × 100 mm column, Waters Inc.), and the peptic peptides were separated by conventional reverse-phase chromatography (solvent A, 0.23% formic acid in water; solvent B, 0.23% formic acid in acetonitrile) and ionized by positive ESI into a Q-TOF mass spectrometer (Synapt G2, Waters Inc.). Identification of peptides was performed on fully reduced and non-deuterated samples by MS/MS using a combination of data-independent acquisition (MSe) and data-dependent acquisition.
Data analysis
Identification of peptides
The acquired mass spectra were lock mass–corrected against Glu1-fibrinopeptide B and analyzed in PLGS 2.5, which matched precursor and fragment ions to a local protein database. The N- and O-linked peptic glycopeptides were identified manually.
Determination of deuterium incorporation
The acquired mass spectra were lock mass–corrected against Glu1-fibrinopeptide B, and the software DynamX 3.0 (Waters Inc.) was used to determine the deuterium uptake for all peptides.
Statistical analysis
All statistical analyses were performed using Excel software (Microsoft). All comparisons were performed with either a homoscedastic or a heteroscedastic Student's t test with the α-value set to 0.01. The choice to use either a homoscedastic or a heteroscedastic Student's t test was determined by using an F-test with the α-level set to 0.05. The F-test compares the variance of deuterium uptake of a single peptide at a single time point from two different states. In the comparative HDX analysis of NGF and the mature part of proNGF, a peptide was only considered to have a significant difference in HDX if two consecutive time points showed a significant difference in deuterium incorporation (p < 0.01).
Author contributions
S. C. and K. D. R. conceived and coordinated the study. S. C. and F. K. designed and constructed the vector for expression of proNGF and performed the expression and purification. E. T. performed all mass spectrometry experiments. S. C., K. D. R., and E. T. analyzed the data and wrote the paper. All authors approved the final version of the manuscript.
Supplementary Material
Acknowledgments
We thank Dr. Laurent Rieux (Antec, Zoeterwoude, Netherlands) for valuable technical advice and assistance and H. Lundbeck A/S for providing protein material.
This work was supported by Grant 1355-00165 from the Innovation Fund Denmark, Grant PCIG09-GA-2011-294214 from the Marie Curie Actions Programme of the European Union, and Steno Grant 11-104058 from the Danish Council for Independent Research, Natural Sciences. The authors declare that they have no conflicts of interest with the contents of this article.
This article contains supplemental Figs. S1–S5.
- NGF
- nerve growth factor-β
- TrkA
- tropomyosin receptor kinase A
- proNGF
- pro-form of nerve growth factor-β
- SAXS
- small-angle X-ray scattering
- HDX-MS
- hydrogen/deuterium exchange measured by mass spectrometry
- UPLC
- ultra-high performance liquid chromatography
- ESI
- electrospray ionization
- MS/MS
- tandem mass spectrometry
- ETD
- electron transfer dissociation
- Q-TOF
- quadrupole time-of-flight
- PDB
- Protein Data Bank.
References
- 1. Chao M. V. (2003) Neurotrophins and their receptors: A convergence point for many signalling pathways. Nat. Rev. Neurosci. 4, 299–309 [DOI] [PubMed] [Google Scholar]
- 2. Jansen P., Giehl K., Nyengaard J. R., Teng K., Lioubinski O., Sjoegaard S. S., Breiderhoff T., Gotthardt M., Lin F., Eilers A., Petersen C. M., Lewin G. R., Hempstead B. L., Willnow T. E., and Nykjaer A. (2007) Roles for the pro-neurotrophin receptor sortilin in neuronal development, aging and brain injury. Nat. Neurosci. 10, 1449–1457 [DOI] [PubMed] [Google Scholar]
- 3. Nykjaer A., Willnow T. E., and Petersen C. M. (2005) p75NTR: Live or let die. Curr. Opin. Neurobiol. 15, 49–57 [DOI] [PubMed] [Google Scholar]
- 4. Seidah N. G., Benjannet S., Pareek S., Savaria D., Hamelin J., Goulet B., Laliberte J., Lazure C., Chrétien M., and Murphy R. A. (1996) Cellular processing of the nerve growth factor precursor by the mammalian pro-protein convertases. Biochem. J. 314, 951–960 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Paoletti F., Malerba F., Kelly G., Noinville S., Lamba D., Cattaneo A., and Pastore A. (2011) Conformational plasticity of proNGF. PLoS ONE 6, e22615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bruno M. A., and Cuello A. C. (2006) Activity-dependent release of precursor nerve growth factor, conversion to mature nerve growth factor, and its degradation by a protease cascade. Proc. Natl. Acad. Sci. U.S.A. 103, 6735–6740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Clewes O., Fahey M. S., Tyler S. J., Watson J. J., Seok H., Catania C., Cho K., Dawbarn D., and Allen S. J. (2008) Human ProNGF: Biological effects and binding profiles at TrkA, P75NTR, and sortilin. J. Neurochem. 107, 1124–1135 [DOI] [PubMed] [Google Scholar]
- 8. Nykjaer A., Lee R., Teng K. K., Jansen P., Madsen P., Nielsen M. S., Jacobsen C., Kliemannel M., Schwarz E., Willnow T. E., Hempstead B. L., and Petersen C. M. (2004) Sortilin is essential for proNGF-induced neuronal cell death. Nature 427, 843–848 [DOI] [PubMed] [Google Scholar]
- 9. Feng D., Kim T., Ozkan E., Light M., Torkin R., Teng K. K., Hempstead B. L., and Garcia K. C. (2010) Molecular and structural insight into proNGF engagement of p75NTR and sortilin. J. Mol. Biol. 396, 967–984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Fahnestock M., Michalski B., Xu B., and Coughlin M. D. (2001) The precursor pro-nerve growth factor is the predominant form of nerve growth factor in brain and is increased in Alzheimer's disease. Mol. Cell. Neurosci. 18, 210–220 [DOI] [PubMed] [Google Scholar]
- 11. Beattie M. S., Harrington A. W., Lee R., Kim J. Y., Boyce S. L., Longo F. M., Bresnahan J. C., Hempstead B. L., and Yoon S. O. (2002) ProNGF induces p75-mediated death of oligodendrocytes following spinal cord injury. Neuron. 36, 375–386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Lowry K. S., Murray S. S., McLean C. A., Talman P., Mathers S., Lopes E. C., and Cheema S. S. (2001) A potential role for the p75 low-affinity neurotrophin receptor in spinal motor neuron degeneration in murine and human amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. Other Motor Neuron Disord. 2, 127–134 [DOI] [PubMed] [Google Scholar]
- 13. Perez S. E., He B., Nadeem M., Wuu J., Scheff S. W., Abrahamson E. E., Ikonomovic M. D., and Mufson E. J. (2015) Resilience of precuneus neurotrophic signaling pathways despite amyloid pathology in prodromal Alzheimer's disease. Biol. Psychiatry 77, 693–703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Wiesmann C., Ultsch M. H., Bass S. H., and de Vos A. M. (1999) Crystal structure of nerve growth factor in complex with the ligand-binding domain of the TrkA receptor. Nature 401, 184–188 [DOI] [PubMed] [Google Scholar]
- 15. Wehrman T., He X., Raab B., Dukipatti A., Blau H., and Garcia K. C. (2007) Structural and mechanistic insights into nerve growth factor interactions with the TrkA and p75 receptors. Neuron 53, 25–38 [DOI] [PubMed] [Google Scholar]
- 16. He X. L., and Garcia K. C. (2004) Structure of nerve growth factor complexed with the shared neurotrophin receptor p75. Science 304, 870–875 [DOI] [PubMed] [Google Scholar]
- 17. McDonald N. Q., Lapatto R., Murray-Rust J., Gunning J., Wlodawer A., and Blundell T. L. (1991) New protein fold revealed by a 2.3-A resolution crystal structure of nerve growth factor. Nature 354, 411–414 [DOI] [PubMed] [Google Scholar]
- 18. Kliemannel M., Rattenholl A., Golbik R., Balbach J., Lilie H., Rudolph R., and Schwarz E. (2004) The mature part of proNGF induces the structure of its pro-peptide. FEBS Lett. 566, 207–212 [DOI] [PubMed] [Google Scholar]
- 19. Kolmar H. (2008) Alternative binding proteins: Biological activity and therapeutic potential of cystine-knot miniproteins. FEBS J. 275, 2684–2690 [DOI] [PubMed] [Google Scholar]
- 20. Edwards R. H., Selby M. J., Mobley W. C., Weinrich S. L., Hruby D. E., and Rutter W. J. (1988) Processing and secretion of nerve growth factor: Expression in mammalian cells with a vaccinia virus vector. Mol. Cell. Biol. 8, 2456–2464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kliemannel M., Golbik R., Rudolph R., Schwarz E., and Lilie H. (2007) The pro-peptide of proNGF: Structure formation and intramolecular association with NGF. Protein Sci. 16, 411–419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Paoletti F., Covaceuszach S., Konarev P. V., Gonfloni S., Malerba F., Schwarz E., Svergun D. I., Cattaneo A., and Lamba D. (2009) Intrinsic structural disorder of mouse proNGF. Proteins 75, 990–1009 [DOI] [PubMed] [Google Scholar]
- 23. Rattenholl A., Lilie H., Grossmann A., Stern A., Schwarz E., and Rudolph R. (2001) The pro-sequence facilitates folding of human nerve growth factor from Escherichia coli inclusion bodies. Eur. J. Biochem. 268, 3296–3303 [DOI] [PubMed] [Google Scholar]
- 24. Zhang Z., and Smith D. L. (1993) Determination of amide hydrogen exchange by mass spectrometry: A new tool for protein structure elucidation. Protein Sci. 2, 522–531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Wales T. E., and Engen J. R. (2006) Hydrogen exchange mass spectrometry for the analysis of protein dynamics. Mass Spectrom. Rev. 25, 158–170 [DOI] [PubMed] [Google Scholar]
- 26. Skinner J. J., Lim W. K., Bédard S., Black B. E., and Englander S. W. (2012) Protein dynamics viewed by hydrogen exchange. Protein Sci. 21, 996–1005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Skinner J. J., Lim W. K., Bédard S., Black B. E., and Englander S. W. (2012) Protein hydrogen exchange: Testing current models. Protein Sci. 21, 987–995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. McAllister R. G., and Konermann L. (2015) Challenges in the interpretation of protein H/D exchange data: A molecular dynamics simulation perspective. Biochemistry 54, 2683–2692 [DOI] [PubMed] [Google Scholar]
- 29. Persson F., and Halle B. (2015) How amide hydrogens exchange in native proteins. Proc. Natl. Acad. Sci. U.S.A. 112, 10383–10388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Keppel T. R., Howard B. A., and Weis D. D. (2011) Mapping unstructured regions and synergistic folding in intrinsically disordered proteins with amide H/D exchange mass spectrometry. Biochemistry 50, 8722–87332 [DOI] [PubMed] [Google Scholar]
- 31. Jensen P. F., Larraillet V., Schlothauer T., Kettenberger H., Hilger M., and Rand K. D. (2015) Investigating the interaction between the neonatal Fc receptor and monoclonal antibody variants by hydrogen/deuterium exchange mass spectrometry. Mol. Cell. Proteomics 14, 148–161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kim M., Sun Z. Y., Rand K. D., Shi X., Song L., Cheng Y., Fahmy A. F., Majumdar S., Ofek G., Yang Y., Kwong P. D., Wang J. H., Engen J. R., Wagner G., and Reinherz E. L. (2011) Antibody mechanics on a membrane-bound HIV segment essential for GP41-targeted viral neutralization. Nat. Struct. Mol. Biol. 18, 1235–1243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Pagadala P. C., Dvorak L. A., and Neet K. E. (2006) Construction of a mutated pro-nerve growth factor resistant to degradation and suitable for biophysical and cellular utilization. Proc. Natl. Acad. Sci. U.S.A. 103, 17939–17943 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Maley F., Trimble R. B., Tarentino A. L., and Plummer T. H. (1989) Characterization of glycoproteins and their associated oligosaccharides through the use of endoglycosidases. Anal. Biochem. 180, 195–204 [DOI] [PubMed] [Google Scholar]
- 35. Iwase H., and Hotta K. (1993) Release of O-linked glycoprotein glycans by endo-α-N-acetylgalactosaminidase. Methods Mol. Biol. 14, 151–159 [DOI] [PubMed] [Google Scholar]
- 36. Gill D. J., Clausen H., and Bard F. (2011) Location, location, location: new insights into O-GalNAc protein glycosylation. Trends Cell Biol. 21, 149–158 [DOI] [PubMed] [Google Scholar]
- 37. Brockhausen I., and Schachter H. S. P. (2009) O-GalNAc glycans, in Essentials of Glycobiology (Varki A., Cummings R., and Esko J., eds) 2nd Ed., Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY: [PubMed] [Google Scholar]
- 38. North S. J., Huang H. H., Sundaram S., Jang-Lee J., Etienne A. T., Trollope A., Chalabi S., Dell A., Stanley P., and Haslam S. M. (2010) Glycomics profiling of Chinese hamster ovary cell glycosylation mutants reveals N-glycans of a novel size and complexity. J. Biol. Chem. 285, 5759–5775 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Bause E. (1983) Structural requirements of N-glycosylation of proteins: Studies with proline peptides as conformational probes. Biochem. J. 209, 331–336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Julenius K., Mølgaard A., Gupta R., and Brunak S. (2005) Prediction, conservation analysis, and structural characterization of mammalian mucin-type O-glycosylation sites. Glycobiology 15, 153–164 [DOI] [PubMed] [Google Scholar]
- 41. Cao L., Tolić N., Qu Y., Meng D., Zhao R., Zhang Q., Moore R. J., Zink E. M., Lipton M. S., Paša-Tolić L., and Wu S. (2014) Characterization of intact N- and O-linked glycopeptides using higher energy collisional dissociation. Anal. Biochem. 452, 96–102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Pompach P., Chandler K. B., Lan R., Edwards N., and Goldman R. (2012) Semi-automated identification of N-glycopeptides by hydrophilic interaction chromatography, nano-reverse-phase LC-MS/MS, and glycan database search. J. Proteome Res. 11, 1728–1740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Mayampurath A. M., Wu Y., Segu Z. M., Mechref Y., and Tang H. (2011) Improving confidence in detection and characterization of protein N-glycosylation sites and microheterogeneity. Rapid Commun. Mass Spectrom. 25, 2007–2019 [DOI] [PubMed] [Google Scholar]
- 44. Perdivara I., Petrovich R., Allinquant B., Deterding L. J., Tomer K. B., and Przybylski M. (2009) Elucidation of O-Glycosylation structures of the β-amyloid precursor protein by liquid chromatography-mass spectrometry using electron transfer dissociation and collision induced dissociation. J. Proteome Res. 8, 631–642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Syka J. E., Coon J. J., Schroeder M. J., Shabanowitz J., and Hunt D. F. (2004) Peptide and protein sequence analysis by electron transfer dissociation mass spectrometry. Proc. Natl. Acad. Sci. U.S.A. 101, 9528–9533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Trabjerg E., Jakobsen R. U., Mysling S., Christensen S., Jørgensen T. J., and Rand K. D. (2015) Conformational analysis of large and highly disulfide-stabilized proteins by integrating online electrochemical reduction into an optimized H/D exchange mass spectrometry workflow. Anal. Chem. 87, 8880–8888 [DOI] [PubMed] [Google Scholar]
- 47. Bai Y., Milne J. S., Mayne L., and Englander S. W. (1993) Primary structure effects on peptide group hydrogen exchange. Proteins 17, 75–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Engen J. R., and Wales T. E. (2015) Analytical aspects of hydrogen exchange mass spectrometry. Annu. Rev. Anal. Chem. (Palo Alto, Calif.) 8, 127–148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Trelle M. B., Dupont D. M., Madsen J. B., Andreasen P. A., and Jørgensen T. J. (2014) Dissecting the effect of RNA aptamer binding on the dynamics of plasminogen activator inhibitor 1 using hydrogen/deuterium exchange mass spectrometry. ACS Chem. Biol. 9, 174–182 [DOI] [PubMed] [Google Scholar]
- 50. Sheff J. G., Rey M., and Schriemer D. C. (2013) Peptide-column interactions and their influence on back exchange rates in hydrogen/deuterium exchange-MS. J. Am. Soc. Mass Spectrom. 24, 1006–1015 [DOI] [PubMed] [Google Scholar]
- 51. Cuff J. A., and Barton G. J. (2000) Application of multiple sequence alignment profiles to improve protein secondary structure prediction. Proteins 40, 502–511 [DOI] [PubMed] [Google Scholar]
- 52. Buchan D. W., Minneci F., Nugent T. C., Bryson K., and Jones D. T. (2013) Scalable Web services for the PSIPRED protein analysis workbench. Nucleic Acids Res. 41, W349–W357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Pauling L., Corey R. B., and Branson H. R. (1951) The structure of proteins: Two hydrogen-bonded helical configurations of the polypeptide chain. Proc. Natl. Acad. Sci. U.S.A. 37, 205–211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Kendrew J. C., Dickerson R. E., Strandberg B. E., Hart R. G., Davies D. R., Phillips D. C., and Shore V. C. (1960) Structure of myoglobin: A three-dimensional Fourier synthesis at 2 Å resolution. Nature 185, 422–427 [DOI] [PubMed] [Google Scholar]
- 55. Varki A., and Lowe J. (2009) Biological roles of glycans, in Essentials of Glycobiology (Varki A., Cummings R., and Esko J., eds) 2nd Ed., Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY [Google Scholar]
- 56. Tsuda E., Kawanishi G., Ueda M., Masuda S., and Sasaki R. (1990) The role of carbohydrate in recombinant human erythropoietin. Eur. J. Biochem. 188, 405–411 [DOI] [PubMed] [Google Scholar]
- 57. Skehel J. J., and Wiley D. C. (2000) Receptor binding and membrane fusion in virus entry: The influenza hemagglutinin. Annu. Rev. Biochem. 69, 531–569 [DOI] [PubMed] [Google Scholar]
- 58. Yang Z., Halim A., Narimatsu Y., Jitendra Joshi H., Steentoft C., Schjoldager K. T., Alder Schulz M., Sealover N. R., Kayser K. J., Paul Bennett E., Levery S. B., Vakhrushev S. Y., and Clausen H. (2014) The GalNAc-type O-glycoproteome of CHO cells characterized by the SimpleCell strategy. Mol. Cell. Proteomics 13, 3224–3235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Croset A., Delafosse L., Gaudry J. P., Arod C., Glez L., Losberger C., Begue D., Krstanovic A., Robert F., Vilbois F., Chevalet L., and Antonsson B. (2012) Differences in the glycosylation of recombinant proteins expressed in HEK and CHO cells. J. Biotechnol. 161, 336–348 [DOI] [PubMed] [Google Scholar]
- 60. Vestrheim A. C., Moen A., Egge-Jacobsen W., Bratlie D. B., and Michaelsen T. E. (2013) Different glycosylation pattern of human IgG1 and IgG3 antibodies isolated from transiently as well as permanently transfected cell lines. Scand. J. Immunol. 77, 419–428 [DOI] [PubMed] [Google Scholar]
- 61. Geoghegan K. F., Song X., Hoth L. R., Feng X., Shanker S., Quazi A., Luxenberg D. P., Wright J. F., and Griffor M. C. (2013) Unexpected mucin-type O-glycosylation and host-specific N-glycosylation of human recombinant interleukin-17A expressed in a human kidney cell line. Protein Expr. Purif. 87, 27–34 [DOI] [PubMed] [Google Scholar]
- 62. Jensen P. H., Kolarich D., and Packer N. H. (2010) Mucin-type O-glycosylation: Putting the pieces together. FEBS J. 277, 81–94 [DOI] [PubMed] [Google Scholar]
- 63. Steentoft C., Vakhrushev S. Y., Joshi H. J., Kong Y., Vester-Christensen M. B., Schjoldager K. T., Lavrsen K., Dabelsteen S., Pedersen N. B., Marcos-Silva L., Gupta R., Bennett E. P., Mandel U., Brunak S., Wandall H. H., et al. (2013) Precision mapping of the human O-GalNAc glycoproteome through SimpleCell technology. EMBO J. 32, 1478–1488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. López-Llano J., Campos L. A., and Sancho J. (2006) α-Helix stabilization by alanine relative to glycine: Roles of polar and apolar solvent exposures and of backbone entropy. Proteins 64, 769–778 [DOI] [PubMed] [Google Scholar]
- 65. Walters B. T., Ricciuti A., Mayne L., and Englander S. W. (2012) Minimizing back exchange in the hydrogen exchange-mass spectrometry experiment. J. Am. Soc. Mass Spectrom. 23, 2132–2139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Ibáñez C. F., Ebendal T., and Persson H. (1991) Chimeric molecules with multiple neurotrophic activities reveal structural elements determining the specificities of NGF and BDNF. EMBO J. 10, 2105–2110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Christensen S., and Egebjerg J. (2005) Cloning, expression and characterization of a sialidase gene from Arthrobacter ureafaciens. Biotechnol. Appl. Biochem. 41, 225–231 [DOI] [PubMed] [Google Scholar]
- 68. van Maarschalkerweerd A., Pedersen M. N., Peterson H., Nilsson M., Nguyen T., Skamris T., Rand K., Vetri V., Langkilde A. E., and Vestergaard B. (2015) Formation of covalent di-tyrosine dimers in recombinant α-synuclein. Intrinsically Disord. Proteins 3, e1071302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Rand K. D., Pringle S. D., Morris M., Engen J. R., and Brown J. M. (2011) ETD in a traveling wave ion guide at tuned Z-spray ion source conditions allows for site-specific hydrogen/deuterium exchange measurements. J. Am. Soc. Mass Spectrom. 22, 1784–1793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Mysling S., Salbo R., Ploug M., and Jørgensen T. J. (2014) Electrochemical reduction of disulfide-containing proteins for hydrogen/deuterium exchange monitored by mass spectrometry. Anal. Chem. 86, 340–345 [DOI] [PubMed] [Google Scholar]
- 71. Kraj A., Brouwer H. J., Reinhoud N., and Chervet J. P. (2013) A novel electrochemical method for efficient reduction of disulfide bonds in peptides and proteins prior to MS detection. Anal. Bioanal. Chem. 405, 9311–9320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Guttman M., Scian M., and Lee K. K. (2011) Tracking hydrogen/deuterium exchange at glycan sites in glycoproteins by mass spectrometry. Anal. Chem. 83, 7492–7499 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.