

Abstract
Hepatitis B virus (HBV) infection afflicts millions worldwide, causing cirrhosis and liver cancer. HBV e-antigen (HBeAg), a clinical marker for disease severity, is a soluble variant of the viral capsid protein. HBeAg is not required for viral replication but is implicated in establishing immune tolerance and chronic infection. The structure of recombinant e-antigen (rHBeAg) was recently determined, yet to date, the exact nature and quantitation of HBeAg still remain uncertain. Here, to further characterize HBeAg, we used phage display to produce a panel of chimeric rabbit/human monoclonal antibody fragments (both Fab and scFv) against rHBeAg. Several of the Fab/scFv, expressed in Escherichia coli, had unprecedentedly high binding affinities (Kd ∼10−12 m) and high specificity. We used Fab/scFv in the context of an enzyme-linked immunosorbent assay (ELISA) for HBeAg quantification, which we compared with commercially available kits and verified with seroconversion panels, the WHO HBeAg standard, rHBeAg, and patient plasma samples. We found that the specificity and sensitivity are superior to those of existing commercial assays. To identify potential fine differences between rHBeAg and HBeAg, we used these Fabs in microscale immunoaffinity chromatography to purify HBeAg from individual patient plasmas. Western blotting and MS results indicated that rHBeAg and HBeAg are essentially structurally identical, although HBeAg from different patients exhibits minor carboxyl-terminal heterogeneity. We discuss several potential applications for the humanized Fab/scFv.
Keywords: antibody engineering; hepatitis B virus (HBV, Hep B); phage display; single-domain antibody; surface plasmon resonance (SPR); viral immunology; ELISA; Fab; core antigen (HBcAg); e-antigen (HBeAg)
Introduction
Hepatitis B virus (HBV)3 is a small, partially double-stranded DNA pathogen that poses a health burden on a global scale. Viral hepatitis is the seventh leading cause of death in the world and may be increasing (1). HBV is non-cytopathic but chronic infection, which affects over 350 million people, can ultimately lead to liver cirrhosis and hepatocellular carcinoma (HCC). Liver cirrhosis and HCC do not stem from acute infection but from repeated cycles of hepatocyte destruction and regeneration during the immune clearance phase of chronic infection. The first recombinant vaccine was the HBV surface antigen (HBsAg)4 produced in yeast and has proven to be highly effective. However, in many parts of the world the vaccine is either not available or it is too expensive. In areas with a large population of HBV carriers, more than 90% of perinatal transmissions result in chronic hepatitis B (CHB). The factors that appear to establish CHB are the HBV e-antigen (HBeAg) and subviral particles composed of HBsAg (2, 3). The relationship between these antigens and the progression of CHB is complex (4).
HBV is an enveloped virus with a proteolipid surface glycoprotein (HBsAg) and a core composed of the core antigen (HBcAg), which forms an icosahedral structure containing the viral genome. The structural biology of HBV has been recently reviewed (5), and this study focuses on HBcAg and the closely related HBeAg (Figs. 1 and supplemental Fig. S1). Both proteins are derived from the C gene but initiated from two different start codons. The HBcAg is expressed as a 183-residue protein that polymerizes to form the viral capsid. HBeAg is expressed as a preprotein and, compared with HBcAg, has a 29-residue signal sequence upstream that targets the protein to the endoplasmic reticulum (ER). Following processing of the signal sequence by the ER signal peptidase, oxidation and additional carboxyl-terminal processing occur in the ER lumen (6). The secreted HBeAg retains 10 amino-terminal residues from the preprotein, but the position of the carboxyl-terminal processing is unclear, occurring between Val-149 and Arg-154 (7). Although this processing scenario is true for most of HBeAg, about 15% of the protein in the endoplasmic reticulum returns to the cytoplasm (8). Cytoplasmic forms of HBeAg appear to be able to form DNA-deficient capsids, due to the lack of carboxyl-terminal arginine residues, and can be enveloped and released as decoy particles, thereby playing a role in maintaining viral persistence (9, 10). It has been shown that reduction of the soluble oxidized rHBeAg in vitro can cause a conformational switch leading to the assembly of capsid-like structures (11, 12). The different redox conditions in the ER (oxidizing) and the cytoplasm (reducing) could mediate similar structural changes in vivo.
Figure 1.

Structures of rHBcAg and rHBeAg. A, sequence schematics for HBcAg and HBeAg. In addition to the assembly domain (residues 1–149), HBcAg has an arginine-rich nucleic acid-binding domain (residues 150–183). HBeAg shares the assembly domain with HBcAg but has a 10-residue propeptide (magenta) and a potentially variable carboxyl terminus, as discussed in the text. B, atomic structure of rHBcAg dimer (residues 1–149 only) in surface representation. The two chains are joined at Cys-61 by an intermolecular disulfide bond. C, atomic structure of rHBeAg dimer (residues (−10)–149) in surface representation. Each chain has a Cys-(-7)–Cys-61 intramolecular disulfide bond. D, atomic structure of rHBcAg (capsid, T = 4 symmetry) assembled from 120 rHBcAg dimers. The structure is considered analogous to HBcAg (nucleocapsid, core-antigen). One dimer is highlighted. E, rHBeAg dimer shown at the same scale as rHBcAg in D. The differences in conformation, size, and polymeric state are all not adequately communicated by the terms HBcAg and HBeAg.
The HBeAg is not an essential structural component of the virion nor does it appear to take part in the viral replication cycle (13). Its role appears to be linked to long-term viral persistence in the host, as manifested in chronic HBV infection. Unlike the related HBcAg, which activates type 1 T helper (Th1) cells leading to immune attack, the HBeAg activates Th2 cells that promote immune tolerance (14). HBeAg may cross the placenta and establish immune tolerance in the developing fetus and thereby suppress innate signaling pathways (15). The molecular details of these complex events are not fully elucidated, and better tools are required to assess the specific roles of HBeAg. Identification and structural determinations of the key interactions of HBeAg would provide a clearer path to targeted therapeutics.
In CHB infection, the long-term persistence of HBeAg is associated with the development of HCC (16). In contrast, HBeAg seroconversion (from HBeAg carrier to anti-HBeAg carrier) is a marker for the successful therapy of CHB patients (16, 17). Therefore, the quantitative assay of HBeAg is of clinical importance. All currently available HBeAg assays have two shortcomings as follows: 1) they are non-quantitative in that their readout is a value relative to a defined but arbitrary standard rather than a mass quantity; and 2) neither the antigen nor the antibodies are structurally defined, and the latter often cross-react with the HBcAg. This situation persists despite the advances made in defining in structural detail the antigenic determinants of rHBcAg capsids and rHBeAg dimers (12, 18).
Chimeric rabbit/human Fabs consist of rabbit variable domains VH and VK and human constant domains CH1 and CL (19, 20). It has been shown that libraries of such chimeric hybrid antibodies can be generated from the spleen and bone marrow of immunized rabbits and subsequently selected by phage display (20, 21). These Fabs can be conveniently expressed in Escherichia coli using an expression cassette with two signal sequences, pelB and ompA, which direct secretion of the two chains into the oxidizing periplasmic space. A carboxyl-terminal His tag on the VH–CH1 chain allows purification of the soluble protein. Such chimeric antibodies often have both high affinity and specificity and can be fully humanized, making them both powerful research tools and of therapeutic potential. Here, we describe the preparation of a panel of chimeric Fabs against the rHBeAg. We have studied these antibodies in terms of their binding affinity and stoichiometry and cross-reactivity with the closely related rHBcAg. From these characterizations, we developed a sensitive and quantitative assay for the HBeAg and compared its clinical performance with commercial tests. The protein constituents of the assay are of known sequence and structure and can be produced indefinitely by recombinant expression. These features are unique and constitute a powerful incentive for additional development and application. For further characterization of HBeAg, we immunoaffinity-purified the protein from single-patient plasma samples for mass spectrometry. The results indicate that the carboxyl terminus extends beyond Val-149, as determined previously, to at least Arg-151.
Results
Panel of rHBeAg-specific Fab
The Fab complementary-determining region (CDR) sequences of a panel of 24 clones are shown in Table 1. Included in Table 1, for comparison, are the murine/human Fab me6 derived from the murine Mab e6, which binds rHBeAg with high specificity (22), and chimeric rabbit/human Fab Rev, which binds to the HIV-1 Rev protein with picomolar affinity (23). We selected 17 of the clones from Table 1 for further analysis based on their sequence diversity. The chimeric rabbit/human Fabs were expressed in E. coli using the expression cassette pC3C, which included the following: ompA-Vk-Ck-pelB-VH-CH1-polyHis where the ompA and pelB leader sequences direct secretion of the light and heavy chains into the periplasm. The carboxyl-terminal His tag on the heavy chain enables protein purification by immobilized metal-affinity chromatography on Ni2+-nitrilotriacetic acid-agarose. Usually, this one step of purification was adequate, but for structural studies, an additional gel-filtration step was included. With Western blot analysis, none of the Fab panel detected rHBeAg following SDS-PAGE, indicating that they were directed against conformational epitopes.
Table 1.
Anti-rHBeAg chimeric rabbit/human Fab CDR sequences
Fab refers to the clone number of anti-rHBeAg except rev, which is anti-HIV-1 Rev. All clones are rabbit/human chimeric Fabs except me6, which is a murine/human chimeric. LCDR1, HCDR1, etc. refer to V-Light Chain and V-Heavy Chain CDR regions, respectively. The anti-rHBeAg clones are not listed sequentially as they were re-sorted during multiple sequence alignment.

Characterization of chimeric Fab antibodies
ELISA screening
The characterization of the Fab panel had two initial goals as follows: first, to assess affinity against the rHBeAg standard; and second, to check specificity, primarily against the closely related rHBcAg (Fig. 1) but also against HBsAg, which is often present together with HBeAg in clinical samples (24, 25). For initial screening, we used an indirect ELISA microtiter plate assay where rHBeAg was coated by adsorption onto the plate, and the chimeric rabbit/human Fab binding was detected using HRP-labeled human IgG. The strongest responses were from Fabs e1, e8, e13, e21, and e38 (Fig. 2). The Fab panel was then assayed with plates coated with either rHBcAg (capsids) or HBsAg. As expected, the panel only presented background-level binding to HBsAg. In contrast, many Fabs displayed above-background binding to rHBcAg, especially Fabs e1, e5, e8, e13, e21, e29, and e38 (Fig. 2). Typically, Fab binding to rHBeAg was higher than to rHBcAg, except for Fabs e5, e16, and e30, which all gave low responses to either antigen. From this basic survey, Fabs e13, e21, and e38 gave the highest response to rHBeAg and the lowest cross-reactivity to rHBcAg. To put the specificity of these selected Fabs in context, we can compare with the murine/human chimeric Fab me6, which does not bind to rHBcAg (capsids) (22). For example, with Fabs e13 and e38, which are the strongest rHBeAg binders, the ratios of binding to rHBeAg versus rHBcAg (capsids) were both ∼2.5, compared with 4.5 obtained with the Fab me6. In general, when antigens are adsorbed onto a plate, some protein denaturation can occur, which may change the presentation of epitopes, potentially decreasing or increasing antibody binding.
Figure 2.

Screening the affinity and specificity of Fabs. Microtiter plates were coated with 10 μg/ml dimeric rHBeAg, HBcAg capsid, or rHBsAg, washed, blocked, and then treated with 2 μg/ml of the indicated Fab. Following additional washing the bound Fab was detected with anti-human IgG as described under “Experimental procedures.” The samples indicated (−) were probed with two different HBV-negative human plasmas. This survey was performed twice, with similar results.
Binding kinetics studied by SPR
The binding characteristics of the Fabs that appeared to be the strongest binders to rHBeAg in the ELISA were next examined in more detail using the Biacore system, which detects binding by SPR. Fab was immobilized on the chip (ligand) and titrated with antigen (analyte). The dissociation constants (kd) are given in Table 2. Binding affinities to the rHBeAg ranged from Kd ∼10−7–10−12 m and were ranked (high to low affinity) as follows: Fab e38 > e13 > e21 > e1 > e8, which closely matches the ELISA data (Fig. 2). The tightest binders (Fab e13 and Fab e38) exhibited very low dissociation rates, which is a characteristic of high-affinity binding (supplemental Fig. S2). However, we note that with such low off-rates, and with the technical limitations of the method, there is some uncertainty about the actual kd values, but there is no doubt that they exhibit the high affinities typical of antibodies selected from rabbit immune repertoires by phage display (26).
Table 2.
Binding of rHBeAg and rHBcAg to chimeric Fab as measured by SPR
Fabs (ligands) were immobilized on the Biacore chip. The recombinant dimeric antigens (analytes) were titrated over at least a 100-fold concentration range, and the indicated kinetic constants were determined. Statistics associated with these measurements are provided in supplemental Table S1. To determine their stoichiometry, the immune complexes of Fab and dimeric rHBeAg were resolved by gel filtration, and the masses were determined by analytical ultracentrifugation (ND is not determined).
| Ligand Fab | Analyte | ka | kd | Kd | Stoichiometry Fab-rHBAg |
|---|---|---|---|---|---|
| m−1 s−1 | s−1 | m | |||
| e1 | rHBeAg | 9.01 × 103 | 3.61 × 10−5 | 4.01 × 10−9 | 2:2 |
| rHBcAg | 1.62 × 103 | 1.37 × 10−4 | 8.43 × 10−7 | ND | |
| e8 | rHBeAg | 6.8 × 103 | 8.2 × 10−4 | 1.22 × 10−7 | 1:2 |
| rHBcAg | >10−5 | ND | |||
| e13 | rHBeAg | 8.50 × 103 | 2.61 × 10−7 | 3.07 × 10−11 | 2:2 |
| rHBcAg | 2.31 × 104 | 2.09 × 10−3 | 9.05 × 10−8 | ND | |
| e21 | rHBeAg | 1.60 × 103 | 1.78 × 10−4 | 1.11 × 10−10 | 1:2 |
| rHBcAg | 2.70 × 102 | 5.73 × 10−5 | 2.12 × 10−7 | ND | |
| e38 | rHBeAg | 8.18 × 103 | 7.19 × 10−8 | 8.79 × 10−12 | 2:2 |
| rHBcAg | 3.67 × 103 | 1.10 × 10−4 | 2.99 × 10−8 | ND | |
| me6 | rHBeAg | 1.6 × 104 | 1.6 × 10−4 | 1.08 × 10−8 | 2:2 |
| rHBcAg | 0 | 0 | 0 | No binding |
Binding of Fabs to rHBcAg capsids was generally lower, ranging from Kd ∼10−5–10−8 m, and ranked as follows: Fab e38, e13 > e1, e21 > e8, which again is similar to the ELISA data (Fig. 2). For example, with Fab e38 and Fab e13, the off-rates are approximately 4 orders of magnitude higher compared with their binding to rHBeAg. Therefore, although binding to rHBeAg is not absolutely specific, it is significantly stronger than to rHBcAg capsid. The murine/human chimeric Fab me6 exhibited no binding to rHBcAg capsid (Table 2). This confirms our previous finding with the murine Mab e6 (22). To generate chimeric Fab me6, we sequenced Mab e6 (12) and used this for gene synthesis and E. coli expression. The SPR data clearly show that specificity was retained, and this was also true for the scFv e6 (data not shown).
Binding stoichiometry
As the rHBeAg is a homodimer, in principle it has two copies of most epitopes. This was studied by mixing rHBeAg with a 2-fold molar excess of Fab and resolving complexes from excess reagents by gel filtration and then analyzing by analytical ultracentrifugation. The sedimentation equilibrium profile, for example, from Fab e13 (Fig. 3) was used to determine a mass of 127 kDa, which is close to that predicted for a complex of two Fabs and one rHBeAg dimer (131 kDa). The 2:2 binding stoichiometry was also observed with Fabs e38, e1, and Fab me6. For Fabs e8 and e21, 1:2 binding was observed indicating that only one Fab bound to dimeric rHBeAg (Table 2). The structural implications of these binding modes are discussed later.
Figure 3.
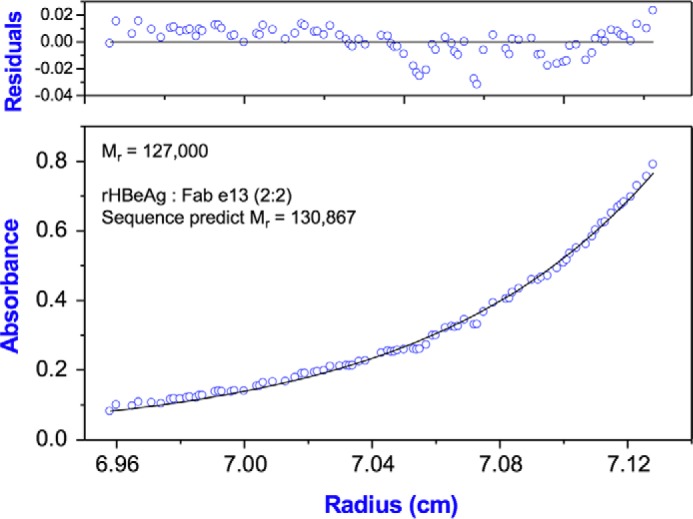
Sedimentation equilibrium of rHBeAg-Fab e13 complex. The panels show absorbance (bottom panel) and residuals (upper panel). Open circles show the UV absorbance gradient in the centrifuge cell. The solid line indicates the calculated fit for an ideal single species. Residuals show the difference between the fitted and experimental values as a function of radial position. The determined molecular weight 127,000 (±1500) is indicated, and this compares with a molecular weight of 130,867 calculated for a complex of 2 Fab e13 and one rHBeAg dimer.
New and specific HBeAg assay
Assay development
One of the aims of this study was to apply the high-affinity Fabs toward the development of a specific and quantitative assay for the HBeAg. We used the traditional ELISA sandwich format for the formulation of this assay. Based on its specificity for the rHBeAg, we used a murine monoclonal antibody Mab e6 (IgG2a) (22, 27) for antigen capture even though it does not have the highest binding affinity (Table 2). We found in initial screening trials that microtiter plates coated with Mab e6 gave more reliable results than the chimeric Fab me6, suggesting that more correctly oriented binding sites are presented with the larger adsorbed molecule. For detection, we screened the highest-affinity binders identified in Table 2, namely Fabs e38, e21, and e13, and used HRP-coupled anti-human IgG for detection. Over the rHBeAg concentration range 0–1 μg/ml, Fab e13 gave the strongest response, and Fab e21, which binds rHBeAg with 1:2 stoichiometry, gave only a weak signal (Fig. 4). Based on these results, the pairing of Mab e6 and Fab e13 for capture and detection, respectively, of HBeAg was further developed.
Figure 4.

Secondary antibody pairing. A sandwich ELISA was used that incorporated Mab e6-coated plates for antigen capture and Fabs e13, e21, or e38 for detection. Anti-human IgG-HRP was used to generate the absorbance signal at 450 nm. The assay was performed over the range of 0–1 μg/ml rHBeAg. The experiment was performed three times with similar results.
Epitope mapping
In a sandwich ELISA, the capture and detection antibodies should ideally have different, non-overlapping epitopes. Although this requirement is not essential for antibodies that can bind to both epitopes of a dimeric protein, we sought to combine specific capture with high-affinity detection, and as rationalized above, the combination of Mab e6 and Fab e13 was the combination of choice. The cross-reactivity of these two antibodies was studied by rHBeAg neutralization as monitored by SPR. First, immobilized Fab e13 was titrated with a fixed amount of rHBeAg together with increasing concentrations of Mab e6. There was a positive non-competitive response proportional to Mab e6 concentration (Fig. 5A), namely rHBeAg with bound Mab e6 could still bind to the Fab e13 ligand. Second, immobilized Mab e6 was titrated with a fixed amount of rHBeAg together with varying concentrations of either Fab e13 or Fab me6, and non-competitive and competitive bindings, respectively, were observed (Fig. 5B). These results indicate that Mab e6 and Mab e13 bind to rHBeAg, and presumably HBeAg, at non-overlapping epitopes.
Figure 5.

Epitope mapping using surface plasmon resonance. A, Biacore sensograms indicating binding kinetics generated from immobilized Fab e13 (ligand) titrated with analyte mixtures containing a fixed amount of rHBeAg (1.5 μm) and a variable amount of Fab me6 (0–133 nm). B, immobilized Fab me6 (ligand) titrated with analyte mixtures containing either a fixed amount (0.4 μm) of rHBeAg and a variable amount of Fab e13, or a fixed amount (1.5 μm) rHBeAg and a variable amount of Fab me6. The steady-state maximum binding is plotted as a function of analyte Fab concentration (abscissa). A and B, ordinate scales indicate SPR response in response units (RU).
Calibration curves
A sandwich ELISA, which incorporated Mab e6 for antigen capture, Fab e13 for detection, and HRP-anti-human IgG for signal generation, was used to titrate rHBeAg over the range of 0–100 μg/ml (Fig. 6). The absorbance response was linear from 0.1 to 1 μg/ml, and at >50 μg/ml the “hook effect” (28) was observed indicating saturation. To simplify the assay, rather than using secondary antibody detection, HRP was conjugated to Fab e13. Over the linear response range (0.1–1 μg/ml), the conjugate gave only a slightly lower response than the non-conjugated antibody; therefore, it was adopted for all subsequent studies (supplemental Fig. S3). Following further optimization, including, for example, antibody concentrations, selection of blocking reagents, and incubation times, the ELISA protocol was established as described under “Experimental procedures” and as shown in the schematic of supplemental Fig. S4.
Figure 6.

ELISA response curve with rHBeAg. With the optimized sandwich ELISA (supplemental Fig. S4), rHBeAg is titrated over the concentration range 0–100 μg/ml (0–5.6 μm). The experiment was performed in triplicate, and a sigmoidal fit to the data is shown with an EC50 ∼12 nm.
For standardization of the calibration curves we determined a cutoff (CO) value for the assay (see “Experimental procedures”) and as illustrated in supplemental Fig. S5A. We also checked intra- and inter-plate variations using samples with negative, low, medium, and high signals (supplemental Fig. S5B). The intra- and inter-assay coefficient of variance (CV) values were <5% and <7.2%, respectively, which indicated high reproducibility (supplemental Fig. S5C). Using rHBeAg, calibration curves of signal (S) to cutoff ratio (S/CO) (supplemental Fig. S6A) indicated a linear response up to 1 μg/ml (56 nm, monomer) with a lower detection limit of ∼4 ng/ml (0.2 nm). A calibration curve was also prepared using the WHO HBeAg international standard from the Paul-Ehrlich-Institut, which has an activity of 100 PE IU/ml. The linear plots for 0–1.0 PE IU/ml and 0–10 PE IU/ml are shown (supplemental Fig. S6, B and C), respectively. The lower detection level is ∼0.02 PE IU/ml. From these plots, 1 PE IU/ml corresponds to ∼0.2 μg/ml (∼10 nm) of the rHBeAg (Fig. 7).
Figure 7.

Calibration curves, rHBeAg and WHO standard. Using the sandwich ELISA (supplemental Fig. S4), calibration curves for the HBeAg WHO reference sample from the Paul-Ehrlich-Institut (PE IU/ml) and rHBeAg were prepared where the S/CO ratio was determined as described under “Experimental procedures.” From the plots, 1 PE IU/ml corresponds to ∼0.05 μg/ml (2.8 nm) rHBeAg. The experiment was performed in triplicate, and the individual plots and linear fittings are shown in supplemental Fig. S6.
Assay clinical performance
To test the utility of the assay on patient samples, we first checked the matrix effect by titrating rHBeAg to 1 μg/ml in the presence of undiluted and 1:1 diluted non-immune human plasma. No differences were observed in the linear plots (supplemental Fig. S5C). Our next check of the assay was to detect HBeAg content in patient plasma using a negative-to-positive seroconversion panel. Two commercial panels from ZeptoMetrix were used, which also included assay data for HBsAg and HBeAg determined using the Abbott Laboratory assay systems. There was a high correlation between our assay and the Abbott assay in detecting (or not) patient plasma HBeAg (supplemental Fig. S7).
The 67 HBV patient plasma samples previously assayed for HBeAg by the Clinical Center at the National Institutes of Health were assayed again with two commercial kits and with the system described above. A comparison of a subset of the data (25 samples) is shown in supplemental Table S2. The data show that the detection of positive samples is similar with all three systems. The Vitros system had the strongest signal as it employs chemiluminescent detection rather than the chromogenic detection used in the other two.
The assay data described above, albeit limited in number, indicate that our assay matches the commercial systems in terms of the simple detection of HBeAg in plasma; however, our system appears superior in the following ways. First, our system is more sensitive based on detection limits using both the WHO PE standard and the rHBeAg (supplemental Fig. S8A). Second, our assay, based on capture with Mab e6, is more selective for rHBeAg than rHBcAg (capsids) and therefore presumably HBeAg and HBcAg. It might be assumed that commercial systems would be highly specific for the target antigen, but two of the systems that we tested showed high cross-reactivity with rHBcAg (capsids) (supplemental Fig. S8B). For the testing of clinical plasma, this is not critical as HBcAg (i.e. free nucleocapsids) is usually not present (discussed further below), but for basic research purposes this may be of importance. Third, our system is quantitative, and we have shown calibration curves for the rHBeAg and the WHO PE standard (Fig. 7 and supplemental Fig. S6). The quantitative aspect of our assay should be useful for monitoring the treatment of CHB (24) and for basic research, as discussed below.
Identification of HBeAg in patient plasma
Another aim of this study was to isolate HBeAg from patient plasma to confirm its sequence, especially at the carboxyl-terminal end. The antigen was isolated from HBeAg-positive plasma samples (S/CO ∼1000–3000; ∼1–3 μg/ml HBeAg (Fig. 8)) by affinity chromatography using Mab e6 immobilized on resin. SDS-PAGE of the eluted protein under reducing and non-reducing conditions followed by Western blot analysis showed an ∼19-kDa species that migrated slightly faster under the non-reducing condition (Fig. 8A), consistent with the presence of an intramolecular disulfide bond, as occurs in rHBeAg between Cys-(-7) and Cys-61 (11, 12). The protein bands were digested with trypsin and analyzed by mass spectrometry. The peptides were compared with a database of peptides derived from rHBeAg and rHBcAg where full coverage had been established. Three peptides were identified, corresponding to residues 28–40, 82–98, and 127–151 (supplemental Fig. S9B). The 127–151 peptide corresponds to the carboxyl-terminal sequence of the rHBeAg (146TTVV149) plus an additional two arginine residues (147TTVVRR151). This result was obtained with HBV genotypes C (two independent determinations) and F. We failed to detect this peptide in the E genotype.
Figure 8.

Immunoaffinity purification of HBeAg from HBV patient plasma. A, SDS-PAGE/Western blot of rHBeAg run under reducing (R) conditions (+DTT) and non-reducing conditions (O). The position of the reduced monomer is indicated with M, and the oxidized monomer with the higher mobility with M*. HBeAg was immunoaffinity-purified from an individual HBV-positive patient (see “Experimental procedures”) and analyzed under reducing (+R) and non-reducing conditions (+O). An HBV-negative plasma sample was analyzed under reducing conditions (−R). B, SDS-PAGE with Coomassie Blue staining: rHBeAg analyzed under reducing (lane 1) and non-reducing conditions (lane 2) and rHBcAg analyzed under reducing (lane 3) and non-reducing conditions (lane 4). Because of the intramolecular disulfide bond (Cys-(−7)–Cys-61) in rHBeAg, the protein in lane 2 has a slightly higher mobility than the reduced form, although due to the intermolecular disulfide bond (Cys-61–Cys-61) in rHBcAg, the protein in lane 4 has a substantially lower mobility than the reduced from. The molecular weights of a standard protein mixture are indicated.
Discussion
Antibodies and the HBeAg protein
We have generated chimeric rabbit/human monoclonal antibody fragments directed against the rHBeAg through phage display. Rabbit monoclonal antibodies often have both high affinity and specificity and can recognize epitopes conserved between human, rat, and mouse antigens (26, 29). In a previous study, we produced a chimeric rabbit/human Fab with an exceptionally high affinity for the HIV-1 protein Rev, which served as a crystallization chaperone mediating the first structural determination of this key protein (30). Here, we have generated a repertoire of chimeric Fab molecules, consisting of rabbit variable domains and human constant domains, against the rHBeAg. The 50-kDa chimeric Fab molecules can be fully humanized (19, 31) and converted to an IgG by fusion with Fc-coding sequences (31). This can be advantageous in applications where greater size and/or bivalency is necessary. Alternatively, the 50-kDa Fab can be downsized to a 25-kDa scFv, which is sometimes more suitable as a crystallization chaperone as it can form a more closely packed crystal (32), as discussed below.
The specificities of commercially available antibodies and assay systems against HBcAg and HBeAg are usually either not clearly defined or not known. For example, detailed binding kinetics using two well-known commercial anti-HBeAg antibodies (904 and 905) indicated that they did not discriminate between the rHBeAg and rHBcAg (22). Although commercial kits have longstanding utility for assaying these antigens in the clinic, they may be unsuited for research purposes due to such cross-reactivity. In part, the problem lies in the close sequence similarity between the two proteins, although at a structural level they are vastly different: a 4-MDa 240-subunit capsid (HBcAg) versus a soluble 35-kDa dimeric protein (Fig. 1). Also, until recently (12), the structure of the rHBeAg was unknown, and selection of the protein preparation for any immunization protocol and screening may not have been correct.
In characterizing the Fab panel, we have paid particular attention to the binding stoichiometry. Using sedimentation equilibrium ultracentrifugation, we determined the mass of Fab-rHBeAg complexes. In previous work, we used the murine Fab e6 (obtained by digestion of Mab e6) as a molecular chaperone to solve the structure of the rHBeAg (12). One Fab binds to the carboxyl-terminal helix-loop region of each of the two subunits; two Fabs are accommodated without any steric hindrance. Although most of the chimeric Fabs also bind with 2:2 stoichiometry (Table 2), they do not necessarily share the same epitope, and Fab e13, for example, shows no cross-reactivity with Fab me6 (Fig. 5). Two of the Fabs (e8 and e22) exhibited 1:2 binding; these are of interest because they mimic the monovalent binding of the traditional anti-HBeAg antibodies described in the literature (33). This mode of binding suggested that HBeAg was a monomeric protein, and this belief persisted until recently. As the rHBeAg is a dimeric molecule, the binding of one antibody either sterically blocks binding of a second antibody, or binding of one antibody is accompanied by an allosteric change sensed in the second binding site thereby reducing its affinity. With the rHBcAg capsid, we have previously described examples of these binding characteristics (18, 34, 35).
The rHBcAg expressed in E. coli appears structurally and antigenically similar to HBcAg and is used as a positive standard in HBcAg assay kits (36). However, attempts to isolate the HBeAg from HBV-positive plasma (37–39) have not to date yielded protein for detailed characterization to guide recombinant expression. The HBeAg is derived from an alternatively translated C-gene with a 29-residue signal sequence that is subsequently processed at the endoplasmic reticulum to a 10-residue propeptide (6, 40). The processing at the carboxyl terminus is not completely understood (as discussed below), but the protein extends to at least residue 149 (37). Hence, we used the consensus (−10)-149 sequence (supplemental Fig. S1), which also corresponds to the published structure (12). Based on our understanding of the rHBeAg structure, we have carefully monitored the oxidation and physical state of the protein to ensure that it is dimeric and that the intramolecular Cys-(-7)–Cys-61 disulfide bond is formed. The 10-residue propeptide hinders formation of rHBcAg-like dimers that can assemble to form capsid-like structures. Although the intramolecular Cys-(-7)–Cys-61 disulfide bond stabilizes the position of the propeptide, this is not essential as the reduced protein can maintain its fold and tertiary structure; however, upon warming, the rHBeAg dimer can rearrange into the assembly-competent rHBcAg dimer-like form (12). Thus, rHBeAg purification requires careful characterization to ensure that preparations are not contaminated with structural isomers, different oxidation states, and aggregated protein, work that is rarely performed, or at least not reported. We have used physically homogeneous, dimeric, and fully-oxidized rHBeAg for immunization of rabbits used for construction of the phage libraries, for screening of antibody panels, for generating calibration curves, and for all other studies described. It should be noted that the WHO HBeAg standard used in clinical assays is not a purified protein but rather patient(s) plasma with a high HBeAg titer.
Assay for HBeAg
By applying the anti-rHBeAg Fab panel, we have devised a sensitive and specific assay for HBeAg. In principle, during both acute and chronic HBV disease, the clinical assay of HBeAg does not require high specificity because the closely related HBcAg, although present in viral particles (Dane particles), is normally not exposed. However, disruption of the fragile viral particles during sample acquisition and handling could expose viral nucleocapsids, thereby leading to an artificially elevated measurement. With commercial kits, potential non-specific binding is unlikely to change the basic assessment of whether a plasma sample is HBeAg-positive or -negative (see for example, supplemental Table S2), whereas in research this selectivity may be more important. The basis of selectivity in our ELISA is derived from the Mab e6 used for HBeAg capture. We have previously shown that this antibody does not bind to assembled rHBcAg (22). In that study, we also observed that Mab 3120 (obtained from the Institute of Immunology, Tokyo, Japan) has the opposite specificity, only binding to assembled rHBcAg and not to the dimeric protein. In combination with specific antigen capture, we used for detection the very high affinity chimeric Fab e13, which does not cross-react with Mab e6 (Fig. 5). Although the sensitivity of our ELISA appears to match, and in some instances even exceed, the commercial systems that we have compared it with, the assay is by no means optimized yet. First, the affinity of the capture Mab e6 (Kd ∼ 10−8 m) is not as high as many of the other chimeric Fabs, including Fab e13 (Table 2), but unlike the others, it has the highest specificity (as defined above). In this respect, the rabbit phage display library is a valuable resource for further screening for unique HBeAg epitopes. As the conversion of murine Mab e6 to chimeric murine/human Fab me6 (and to scFv e6) had no effect on either affinity or specificity, the phage system can also be used for affinity maturation (41–43). Furthermore, as direct adsorption of Fab me6 to microtiter plates reduced efficacy compared with the Mab e6, a generic Fc domain, for example, could be fused to the chimeric molecule (44, 45). Another assay limitation, namely the detection signal, could be enhanced by switching from the current chromogenic system to either a fluorescent or chemiluminescent system.
Assay performance
The assay was validated by assessing the matrix effect, reproducibility, and analytical sensitivity (supplemental Fig. S5). The assay detection limit of 0.02 PE IU/ml, which corresponds to 0.2 nm rHBeAg, is lower than that of any of the commercial systems that we tested. Also, the clinical performance, albeit limited in absolute numbers, closely matches that of the Ortho-Clinical Vitros Eci immunodiagnostic system. Our assay can be improved; it was not our intention to make a commercial grade system but rather a useful research tool with high sensitivity and specificity. We consider the most important attribute of our system to be that it is well-defined and accessible. The expression, purification, and structure of the assay constituents have been detailed for Fab e6 + rHBeAg (12) and will be made available elsewhere for scFv e13 + rHBeAg.5
Assay of anti-HBeAg
The seroconversion of HBeAg status is of clinical importance, and commercial kits are available for detecting this (46). These kits employ a variation of the ELISA format; plasma samples are mixed with (usually) a fixed amount of rHBeAg. Excess or non-neutralized HBeAg is then captured with solid-phase anti-rHBeAg antibody, and the second conjugated anti-rHBeAg antibody is used for detection. This is termed a neutralization anti-e-antigen immunoassay, and a positive sample gives no color reaction. A previous study compared commercial systems and discussed their limitations; the major limitation is that anti-HBeAg can only be detected in the absence of excess HBeAg (46). Circumventing this by increasing rHBeAg used for neutralization led the authors to conclude that the HBeAg in blood is antigenically different from the rHBeAg used in the commercial kits. Although we have not applied our assay for antibody detection, the well-defined antibodies and highly characterized rHBeAg that we have described could be applied to develop an antibody detection system that would give less ambiguous performance than those currently in use.
Immunoaffinity purification of HBeAg
One of our initial aims was to purify HBeAg from patient plasma for structural comparison with the rHBeAg used here and in all our earlier studies. It may seem surprising that this has not been accomplished before and may indeed have been; but if so, this achievement is buried deep in the vast HBV literature. Our first task was to see whether we could directly identify HBeAg in positive plasma using Western blotting (supplemental Fig. S9A). The best commercial antibody we found for this task was a polyclonal rabbit anti-rHBcAg. Parenthetically, this antibody, from Dako, was generated using rHBcAg from E. coli and must detect linear epitopes of rHBeAg and rHBcAg exposed during SDS-PAGE. We used a small-scale immunoaffinity adsorption method to purify and concentrate HBeAg from individual plasma samples. Although the amounts of protein obtained in this way were limited, we identified the HBeAg according to the following criteria. 1) The antibody we used for purification (Mab e6 or Fab me6) is specific for rHBeAg (and likely for HBeAg). Even if HBcAg were present due to, for example, disruption of any Dane particles, this protein would not bind. 2) SDS-PAGE and Western blot of reduced versus oxidized protein (Fig. 8A) showed a shift to higher mobility in the latter. This indicates the presence of the intramolecular (Cys-(-7)–Cys-61) disulfide bond; in comparison, oxidized rHBcAg migrates as a dimer due to an intermolecular (Cys-61–Cys-61) disulfide bond at the dimer interface (Fig. 8B). 3) Mass spectrometric analysis of the SDS-PAGE band detected peptides corresponding to the HBeAg (supplemental Fig. S9B). Although the sequence coverage was incomplete, the carboxyl-terminal region was identified as extending to Arg-151. There is a consensus that the HBeAg amino-terminal region contains the residual 10 residues from processing of the 29-residue signal sequence, but there is some debate regarding the carboxyl-terminal sequence. The earliest determination identified cleavage at Val-149 (37). This site also corresponds to the boundary of the HBcAg assembly domain, and structural studies have consequently focused on residues 1–149 (5). Subsequent work with cell culture models showed that cleavage via furin processing occurs at residues 154 and 159 and perhaps 164 and 169 (7, 47). The difficulty with these studies is that the sequence (150RRRGRSPRRRTPSPRRRRSQ169) is susceptible to variable processing by enzymes with serine protease-like activity. With cell culture expression, even if the initial processing is specific, subsequent processing may occur by endogenous protease activity, and this is also true of the plasma-derived protein. Despite differences between these reports, they do indicate some in vivo heterogeneity in the HBeAg. The microscale purification from individual plasma samples coupled with mass spectrometry, as we have described, indicates that the carboxyl terminus of HBeAg extends to at least Arg-151 (in at least two genotypes) and perhaps even further, as suggested by the cell culture studies mentioned above (7, 47). It was noted that HBcAg expressed in E. coli can be partially processed at Arg-151 by bacterial proteases, and it was speculated that analogous activity in vivo could proceed following uncoating of the capsid (the carboxyl terminus is sequestered inside the capsid) (48).
Therapeutic applications
The therapeutic targeting of HBeAg is attractive given its key role in regulating immune tolerance, establishing viral persistence and establishing CHB (49). Furthermore, given the global magnitude of the problem (1), any new approach should be considered, including high-affinity antibody fragments or the smaller mimetics, administered especially to HBeAg-positive women to block perinatal transmission. In cell culture, delivery of scFv against HBcAg inhibited HBV replication, illustrating the potential of direct antibody treatment (50).
Included in the treatments of CHB, which are under development and recently summarized (51), are drugs that target capsid assembly and multi-antigen vaccines (52). Related to this, it is worth noting that Fab me6, which does not bind to rHBcAg (capsids) and was used in the ELISA for specific capture of HBeAg, binds to residues at the dimer–dimer (assembly) interface of rHBcAg and are also present on rHBeAg (see supplemental Fig. S4 in Ref. 12). The Mab e6 was first derived from the immunization of mice with HBcAg treated with SDS (27). Historically, Murray and co-workers first demonstrated that HBeAg could be detected by treating HBcAg with SDS, thereby exposing cryptic HBeAg-like epitopes (53). Based on our previous structural studies, it would be predicted that Fab me6 can block rHBcAg assembly, and this has been verified experimentally.6 We are currently screening members of our chimeric Fab panel, especially those binding with the same stoichiometry as Fab me6, for their inhibitory effects on rHBcAg assembly. These antibodies could be useful as they may target the HBeAg as well as HBcAg assembly.
Apart from the direct targeting of HBeAg with antibodies and related mimetics, there is of course classical vaccination. This was in fact attempted over 30 years ago by Murray et al. (54), who immunized chimpanzees with HBcAg and SDS-treated HBcAg (55). Despite the limited number of animals tested, the positive results supported further vaccination trials based upon or containing HBcAg, and its HBeAg-like derivative, and perhaps a general role for internal antigens in generating immunity against viral infection. This combinational approach is currently being evaluated against HBV (52), and we propose that new vaccine formulations should include rHBeAg.
Conclusions
We have produced a panel of chimeric rabbit/human Fabs specific for rHBeAg, some with unprecedented high affinities (Kd ∼10−12 m). These antibodies have both diagnostic and therapeutic potential. We have described a quantitative assay for HBeAg in which both the epitopes and paratopes are known and which is superior to existing commercial assays, both in sensitivity and specificity. On a microscale, we have immunoaffinity-purified HBeAg from individual patient plasmas and subjected it to proteomic analysis. Our results suggest that the carboxyl terminus is heterogeneous and can extend to at least Arg-151, consistent with recent cell culture studies and contrary to the earlier determination from plasma that the terminus is Val-149. Future studies will determine whether the additional residues have any structural and functional consequences. We have discussed the untested strategy of targeting HBeAg with antibodies and related mimetics.
Experimental procedures
HBV patient plasmas
A total of 67 HBV patient plasma samples, 35 HBeAg-positive and 32 HBeAg-negative, were obtained from the Clinical Center, National Institutes of Health, in accordance with institutional policies for protection of human subjects. All plasma samples had been screened by the Clinical Center on the Ortho Clinical Vitros Eci immunodiagnostic system, and the HBeAg data corresponding to each of the plasma samples were provided.
rHBeAg
The E. coli expression and purification were performed as described previously (22). We used the construct Cp(-10)–149.C48C/C107A (supplemental Fig. S1). If the protein was not fully oxidized as judged by non-reducing SDS-PAGE (Fig. 8B), it was incubated with 1 μm CuCl2 for 30 min, and 5 mm EDTA was added and re-chromatographed on Superdex 200 to remove any aggregated protein.
rHBeAg with carboxyl-terminal Avi tag
An rHBeAg construct with a 17-residue peptide biotin ligase substrate domain (Avi tag, GGGLNDIFEAQKIEWHE) appended to the carboxyl terminus was expressed in E. coli. Protein purification was as described above. Biotinylation with biotin ligase (Avidity, LLC) was done according to the manufacturer's protocol. Following the reaction, the protein was gel-filtrated on Superdex S200. The protein was characterized by mass spectrometry to confirm protein labeling.
rHBcAg
The construct Cp1–149.C48A/C107A (supplemental Fig. S1), which corresponds to the capsid assembly domain, was produced as described previously and assembled into capsids (22). For many of the assays we used capsids assembled from Cp1–149.C48A/C107A dimers as surrogate HBcAg.
rHBsAg
The Small form (genotype D) was purified from recombinant yeast by immunoaffinity chromatography (56).
Selection of anti-rHBeAg chimeric rabbit/human Fab by phage display
The methods used were as described previously (23, 57). In brief, rabbits homozygous for immunoglobulin allotypes VHal and CKb9 were immunized with purified rHBeAg, and spleen and bone marrow were collected and processed for total RNA preparation. RT-PCR amplification of rabbit VL, CK, and VH encoding sequences was performed using established primer combinations and protocols (20). VL–CK–VH cassette assembly and asymmetric Sfil ligation into the phage display vector pC3C was performed as described previously (20, 31). The library consisting of transformed rabbit/human Fab clones was selected by phage display on rHBeAg, which had been selectively biotinylated on the carboxyl-terminal Avi tag (see above) and immobilized on streptavidin-coated plates. After several rounds of panning, selected clones were screened for rHBeAg binding by ELISA, and hits were sequenced as described previously (20).
Expression and purification of Fab and scFv antibody fragments
A modified ompA-VK-CK-pelB-VH-CH1-polyHis cassette was transferred from pC3C into E. coli expression vector pET11a (Novagen) between the NdeI–BamHI restriction sites. The Fab VK and VH sequences were also cloned into pET11a such that the single-chain variable fragment (scFv) versions of them would be expressed joined by the 18-residue linker GGSSRSSSSGGGGSGGGG, i.e. ompA-VK-linker-VH-polyHis. Similar to Fab, these scFv also had a carboxyl-terminal polyHis tag to facilitate purification. The expression plasmids for Fab or scFv production were transfected into E. coli strain BL21-CodonPlusRIL (Stratagene), and the resulting transfectants were grown in a 1-liter fermenter as described previously (23). Bacterial cultures were clarified by centrifugation at 14,000 × g for 1 h. The secreted antibody in the supernatant had a carboxyl-terminal His tag and was captured using 75 ml of nickel-Sepharose resin (GE Healthcare) at 4 °C for 1–2 h, washed with PBS, pH 7.4, plus 20 mm imidazole. The Fabs were eluted with PBS plus 0.5 m imidazole and then dialyzed against 25 mm HEPES buffer, pH 7.4, 0.15 m NaCl, 0.2 mm tris(2-carboxyethyl)phosphine, 10% glycerol. To maintain solubility during purification, ∼1 m urea was often included in buffers. In the studies described, the carboxyl-terminal His tag was not removed.
Kinetics of antibody binding using surface plasmon resonance
All experiments were performed on a Biacore X100 (GE Healthcare) instrument at 25 °C. HBS-EP (10 mm HEPES, pH 7.4, 150 mm sodium chloride, 3 mm EDTA, 0.05% Polysorbate 20) was used as the running buffer, and data were analyzed using Biacore X100 evaluation software (GE Healthcare). Cell 1 was left untreated to serve as a reference surface, and cell 2 was used as the experimental surface. Fabs were diluted (10–20 μg/ml) in 10 mm sodium acetate buffer, pH 4.5–5.0, and immobilized on CM5 sensor chips by the standard amine coupling method (Amine Coupling kit, GE Healthcare) at a flow rate of 5 μl/min. The immobilization levels of the proteins on the sensor chip surfaces were ∼1500 response units. For kinetic analysis, analytes were prepared by serial dilution with HBS-EP buffer over a range typically 10 nm to 1 μm and injected over both the reference and experimental surfaces at a flow rate of 30 μl/min. Sensor chips were regenerated by a 60-s injection of 50 mm sodium hydroxide. Signals from the reference surface and an ensemble of buffer blank injections were subtracted to correct for non-specific binding and injection artifacts. The corrected results were globally fitted to a 1:1 binding model, and the association rate constant (ka) and dissociation rate constant (kd) were used to determine the equilibrium dissociation constant (Kd) in molar units.
Sandwich ELISA
Non-treated 96-well microplates (Thermo Fisher Scientific) were coated with 100 μl per well of 10 μg/ml murine monoclonal antibody Mab e6 (27) in 0.2 m sodium carbonate buffer, pH 9.4, by incubation either at 37 °C for 1 h or at 4 °C overnight. The plates were then washed with 200 μl/well PBST (8 mm Na2HPO4, 150 mm KH2PO4, 3 mm KCl, 0.05% Tween 20, pH 7.4) for 5 min and repeated three times at room temperature with shaking. Plates were blocked with 10% fetal bovine serum/PBS, 300 μl/well, at 37 °C for 1 h and then washed with PBST as above. Test samples, diluted if necessary with PBS, and positive and negative controls were added, 100 μl/well, and incubated at 37 °C for 1 h. Plates were then washed with PBST as above. Fab e13-HRP, 1 μg/ml in 2% BSA/PBS, was added at 100 μl/well and incubated at 37 °C for 1 h. Plates were then washed with PBST as above. Color was developed with tetramethylbenzidine (TMB) 100 μl/well and incubated at room temperature for 5–15 min. The reaction was quenched by addition of 100 μl of 2 n H2SO4, and then absorbance at 450 nm was measured using a Bio-Tek Synergy HT plate reader. Test results were calculated as the S/CO.
In the process of assay development, we used goat anti-human IgG-HRP (SeraCare-KPL) to detect the chimeric secondary Fab antibody, and this was performed as described below. Also, for blocking we used either 5% BSA in PBS or 10% FBS in PBS, although the latter was preferred as it gave cleaner backgrounds.
Fab e13 HRP conjugation
This was performed using an EZ-link Plus activated peroxidase kit (Thermo Fisher Scientific) according to the manufacturer's instructions.
Assay quantification
The intra-assay and inter-assay coefficients of variation (CV) were determined with several different concentrations of HBV-positive patient plasma samples, in triplicate. The procedure was repeated three times independently and each run was performed on different days. The CV was defined as (%CV = STD/average × 100), where STD is the standard deviation.
Determination of CO value
25 HBV patient samples, which were determined to be HBeAg-negative with the Ortho-Clinical Vitros Eci immunodiagnostic system in the Clinical Center at the National Institutes of Health, were assayed in triplicate using the sandwich ELISA described above. The CO value was defined as (CO = average + (3 × STD)).
Indirect ELISA for screening cross-reactivity
The microtiter plates were coated with 10 μg/ml rHBeAg, rHBcAg (capsids), or HBsAg and then washed and blocked as described above. Negative controls were healthy patient plasmas. Fabs were added to 100 μl (2 μg/ml) and incubated at 37 °C for 1 h followed by washing. Bound antibody was detected using 100 μl of anti-human IgG-HRP (KPL, Inc.) diluted 1:5000 with 2% BSA in PBS. Following incubation for 1 h in the dark at 37 °C and then washing, the color was developed with TMB as described above. The HBeAg reference preparation was obtained from the Paul-Ehrlich-Institut (Langen, Germany, code 129097/12). This is HBV-positive patient plasma and is used as an international standard. The preparation has a defined HBeAg activity of 100 PE IU/ml. Two hepatitis B seroconversion panels (catalogue numbers 6278 and 6282) were obtained from ZeptoMetrix.
Immunoaffinity purification of HBeAg from HBV patient plasma
Mab e6 was linked to AminoLink resin (Thermo Fisher Scientific) by amine coupling according to the manufacturer's instructions. HBV patient plasmas (∼1 ml) were incubated with Mab e6 resin at 4 °C overnight and then washed three times with PBST. Protein was eluted from the resin either using glycine buffer, pH 3.0, or by boiling in SDS-PAGE loading buffer. The samples were separated on a 4–12% SDS-polyacrylamide gel, and gel slices were analyzed for HBeAg by the Taplin Mass Spectrometry Facility at the Harvard Medical School.
Western blotting
Samples were separated on a 4–12% SDS-polyacrylamide gel and electrotransferred to a PVDF membrane (Invitrogen). After blocking with 5% nonfat milk solution for 1 h, the membrane was incubated with anti-rHBcAg polyclonal antibody (Dako, B0586) to recognize HBeAg protein in the samples. After washing, the membrane was further incubated with peroxidase-conjugated anti-rabbit IgG (KPL). Signals were then detected with a chemiluminescence reagent (Thermo Fisher Scientific) and exposure on X-ray film (Eastman Kodak).
Preparation of immune complexes for structural studies
A 2-fold or greater molar excess of Fab or scFv was mixed with rHBeAg, and the mixture was then applied to a Superdex S200 column. The column fractions were monitored by SDS-PAGE, and the immune complex was identified (usually the main peak). This was used for characterization and for crystallization screening. Alternatively, immune complexes were applied to a nickel-Sepharose 6 Fast Flow column and processed as described previously (57).
Analytical ultracentrifugation
A Beckman Optima XL-I analytical ultracentrifuge, absorption optics, an An-60 Ti rotor, and standard double-sector centerpiece cells were used. Equilibrium measurements were made at 20 °C at 11,500 rpm for Fab complexes and 14,500 rpm for scFv complexes. Concentration profiles were recorded every 4 h for 16 h and then baselines were established by overspeeding at 45,000 rpm for 3 h. Data (the average of 8–10 scans collected using a radial step size of 0.001 cm) were analyzed using the standard Optima XL-I data analysis software. Protein partial specific volumes (v̄), calculated from the amino acid compositions, and solvent densities were estimated using the program SEDNTERP.
Author contributions
X. Z. and P. T. W. designed the experiments. X. Z., N. R. W., I. W. P., J. D. K., J. L. T., E. E., and P. T. W carried out the experiments. N. R. W., A. D. D., E. E., A. C. S., C. R., and P. T. W. analyzed the data. N. R. W. and P. T. W. wrote the paper.
Supplementary Material
Acknowledgment
We thank Dr. Stephen J. Stahl for the invaluable contributions to this laboratory.
This work was supported by the Intramural Research Program of the NIAMS, and the Center for Cancer Research of the NCI, National Institutes of Health. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains supplemental Figs. S1–S9 and Tables S1–S2.
In the hepatitis B virus literature, there are different terms for each of the proteins below, and these can also have different meanings, causing considerable ambiguity and potential for confusion: the terms HBcAg, HBc, and core antigen may refer to a protein (either natural or recombinant), an antigen, or the viral nucleocapsid; the terms HBeAg, HBe, and e-antigen may refer to a protein, an antigen, or a disease state. In addition, there are ambiguities of sequence (subtype, laboratory mutation, and both the amino and carboxyl termini), oxidation state, and assembly state (monomer, dimer, and capsid). To avoid such ambiguities, but at the expense of some fluidity, in this communication we will refer to these proteins only by the terms given below. The assembly state of HBcAg (either dimer or capsid) will be stated where necessary. The sequences for rHBcAg and rHBeAg are provided in the supplemental material. The sequence for rHBsAg is as indicated under “Experimental procedures.” The terms used in this paper are as follows: HBcAg, HBV core antigen, the viral nucleocapsid; HBeAg, HBV e-antigen, a soluble protein produced during infection, largely collinear with HBcAg; HBsAg, HBV surface antigen, a component of the virion envelope; rHBcAg, recombinant HBV core antigen, a dimeric protein corresponding to core antigen residues 1–149 and having an intermolecular Cys-61–Cys-61 disulfide bond; rHBeAg, recombinant HBV e-antigen, a dimeric protein corresponding to core antigen residues 1–149, but preceded by a 10-residue propeptide and having an intramolecular Cys-(-7)–Cys-61 disulfide bond; rHBsAg, recombinant HBV surface antigen, corresponding to the Small form of HBsAg, genotype D.
E. Eren, N. R. Watts, A. D. Dearborn, J. Kaufman, I. Palmer, A. C. Steven, and P. T. Wingfield, manuscript in preparation.
N. Watts, unpublished data.
- HBV
- hepatitis B virus
- HCC
- hepatocellular carcinoma
- CHB
- chronic hepatitis B
- SPR
- surface plasmon resonance
- ER
- endoplasmic reticulum
- HBeAg
- HBV e-antigen
- r
- recombinant
- HBsAg
- HBV surface antigen
- HBcAg
- HBV core antigen
- S/CO
- signal to cutoff ratio
- CV
- coefficient of variation
- WHO
- World Health Organization
- CO
- cutoff
- TMB
- tetramethylbenzidine
- CDR
- complementarity-determining region
- PE
- Paul Ehrlich.
References
- 1. Maini M. K., and Bertoletti A. (2017) HBV in 2016: Global and immunotherapeutic insights into hepatitis B. Nat. Rev. Gastroenterol. Hepatol. 14, 71–72 [DOI] [PubMed] [Google Scholar]
- 2. Chen M. T., Billaud J. N., Sällberg M., Guidotti L. G., Chisari F. V., Jones J., Hughes J., and Milich D. R. (2004) A function of the hepatitis B virus precore protein is to regulate the immune response to the core antigen. Proc. Natl. Acad. Sci. U.S.A. 101, 14913–14918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Vanlandschoot P., and Leroux-Roels G. (2003) Viral apoptotic mimicry: an immune evasion strategy developed by the hepatitis B virus? Trends Immunol. 24, 144–147 [DOI] [PubMed] [Google Scholar]
- 4. Brunetto M. R. (2010) A new role for an old marker, HBsAg. J. Hepatol. 52, 475–477 [DOI] [PubMed] [Google Scholar]
- 5. Venkatakrishnan B., and Zlotnick A. (2016) The structural biology of hepatitis B virus: form and function. Annu. Rev. Virol. 3, 429–451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ou J. H., Laub O., and Rutter W. J. (1986) Hepatitis B virus gene function: the precore region targets the core antigen to cellular membranes and causes the secretion of the e antigen. Proc. Natl. Acad. Sci. U.S.A. 83, 1578–1582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Messageot F., Salhi S., Eon P., and Rossignol J. M. (2003) Proteolytic processing of the hepatitis B virus e antigen precursor. Cleavage at two furin consensus sequences. J. Biol. Chem. 278, 891–895 [DOI] [PubMed] [Google Scholar]
- 8. Duriez M., Rossignol J. M., and Sitterlin D. (2008) The hepatitis B virus precore protein is retrotransported from endoplasmic reticulum (ER) to cytosol through the ER-associated degradation pathway. J. Biol. Chem. 283, 32352–32360 [DOI] [PubMed] [Google Scholar]
- 9. Kimura T., Ohno N., Terada N., Rokuhara A., Matsumoto A., Yagi S., Tanaka E., Kiyosawa K., Ohno S., and Maki N. (2005) Hepatitis B virus DNA-negative dane particles lack core protein but contain a 22-kDa precore protein without C-terminal arginine-rich domain. J. Biol. Chem. 280, 21713–21719 [DOI] [PubMed] [Google Scholar]
- 10. Duriez M., Thouard A., Bressanelli S., Rossignol J. M., and Sitterlin D. (2014) Conserved aromatic residues of the hepatitis B virus precore propeptide are involved in a switch between distinct dimeric conformations and essential in the formation of heterocapsids. Virology 462, 273–282 [DOI] [PubMed] [Google Scholar]
- 11. Watts N. R., Conway J. F., Cheng N., Stahl S. J., Steven A. C., and Wingfield P. T. (2011) Role of the propeptide in controlling conformation and assembly state of hepatitis B virus e-antigen. J. Mol. Biol. 409, 202–213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. DiMattia M. A., Watts N. R., Stahl S. J., Grimes J. M., Steven A. C., Stuart D. I., and Wingfield P. T. (2013) Antigenic switching of hepatitis B virus by alternative dimerization of the capsid protein. Structure 21, 133–142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Tong S., and Revill P. (2016) Overview of hepatitis B viral replication and genetic variability. J. Hepatol. 64, S4–S16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Milich D., and Liang T. J. (2003) Exploring the biological basis of hepatitis B e antigen in hepatitis B virus infection. Hepatology 38, 1075–1086 [DOI] [PubMed] [Google Scholar]
- 15. Dandri M., and Locarnini S. (2012) New insight in the pathobiology of hepatitis B virus infection. Gut 61, i6–i17 [DOI] [PubMed] [Google Scholar]
- 16. Liaw Y. F., Kao J. H., Piratvisuth T., Chan H. L., Chien R. N., Liu C. J., Gane E., Locarnini S., Lim S. G., Han K. H., Amarapurkar D., Cooksley G., Jafri W., Mohamed R., Hou J. L., et al. (2012) Asian-Pacific consensus statement on the management of chronic hepatitis B: a 2012 update. Hepatol. Int. 6, 531–561 [DOI] [PubMed] [Google Scholar]
- 17. Liaw Y. F. (2009) HBeAg seroconversion as an important end point in the treatment of chronic hepatitis B. Hepatol. Int. 3, 425–433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Steven A. C., Conway J. F., Cheng N., Watts N. R., Belnap D. M., Harris A., Stahl S. J., and Wingfield P. T. (2005) Structure, assembly, and antigenicity of hepatitis B virus capsid proteins. Adv. Virus Res. 64, 125–164 [DOI] [PubMed] [Google Scholar]
- 19. Rader C., Ritter G., Nathan S., Elia M., Gout I., Jungbluth A. A., Cohen L. S., Welt S., Old L. J., and Barbas C. F. 3rd. (2000) The rabbit antibody repertoire as a novel source for the generation of therapeutic human antibodies. J. Biol. Chem. 275, 13668–13676 [DOI] [PubMed] [Google Scholar]
- 20. Rader C. (2009) Generation and selection of rabbit antibody libraries by phage display. Methods Mol. Biol. 525, 101–128 [DOI] [PubMed] [Google Scholar]
- 21. Popkov M., Jendreyko N., Gonzalez-Sapienza G., Mage R. G., Rader C., and Barbas C. F. 3rd. (2004) Human/mouse cross-reactive anti-VEGF receptor 2 recombinant antibodies selected from an immune b9 allotype rabbit antibody library. J. Immunol. Methods 288, 149–164 [DOI] [PubMed] [Google Scholar]
- 22. Watts N. R., Vethanayagam J. G., Ferns R. B., Tedder R. S., Harris A., Stahl S. J., Steven A. C., and Wingfield P. T. (2010) Molecular basis for the high degree of antigenic cross-reactivity between hepatitis B virus capsids (HBcAg) and dimeric capsid-related protein (HBeAg): insights into the enigmatic nature of the e-antigen. J. Mol. Biol. 398, 530–541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Stahl S. J., Watts N. R., Rader C., DiMattia M. A., Mage R. G., Palmer I., Kaufman J. D., Grimes J. M., Stuart D. I., Steven A. C., and Wingfield P. T. (2010) Generation and characterization of a chimeric rabbit/human Fab for co-crystallization of HIV-1 Rev. J. Mol. Biol. 397, 697–708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Nguyen T., Desmond P., and Locarnini S. (2009) The role of quantitative hepatitis B serology in the natural history and management of chronic hepatitis B. Hepatol. Int. 3, 5–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Chan H. L., Wong V. W., Wong G. L., Tse C. H., Chan H. Y., and Sung J. J. (2010) A longitudinal study on the natural history of serum hepatitis B surface antigen changes in chronic hepatitis B. Hepatology 52, 1232–1241 [DOI] [PubMed] [Google Scholar]
- 26. Weber J., Peng H., and Rader C. (2017) From rabbit antibody repertoires to rabbit monoclonal antibodies. Exp. Mol. Med. 49, e305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ferns R. B., and Tedder R. S. (1984) Monoclonal antibodies to hepatitis Be antigen (HBeAg) derived from hepatitis B core antigen (HBcAg): their use in characterization and detection of HBeAg. J. Gen. Virol. 65, 899–908 [DOI] [PubMed] [Google Scholar]
- 28. Fernando S. A., and Wilson G. S. (1992) Studies of the ‘hook’ effect in the one-step sandwich immunoassay. J. Immunol. Methods 151, 47–66 [DOI] [PubMed] [Google Scholar]
- 29. Mage R. G., Lanning D., and Knight K. L. (2006) B cell and antibody repertoire development in rabbits: the requirement of gut-associated lymphoid tissues. Dev. Comp. Immunol. 30, 137–153 [DOI] [PubMed] [Google Scholar]
- 30. DiMattia M. A., Watts N. R., Stahl S. J., Rader C., Wingfield P. T., Stuart D. I., Steven A. C., and Grimes J. M. (2010) Implications of the HIV-1 Rev dimer structure at 3.2 A resolution for multimeric binding to the Rev response element. Proc. Natl. Acad. Sci. U.S.A. 107, 5810–5814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Hofer T., Tangkeangsirisin W., Kennedy M. G., Mage R. G., Raiker S. J., Venkatesh K., Lee H., Giger R. J., and Rader C. (2007) Chimeric rabbit/human Fab and IgG specific for members of the Nogo-66 receptor family selected for species cross-reactivity with an improved phage display vector. J. Immunol. Methods 318, 75–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. DiMattia M. A., Watts N. R., Cheng N., Huang R., Heymann J. B., Grimes J. M., Wingfield P. T., Stuart D. I., and Steven A. C. (2016) The structure of HIV-1 rev filaments suggests a bilateral model for rev-RRE assembly. Structure 24, 1068–1080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Imai M., Nomura M., Gotanda T., Sano T., Tachibana K., Miyamoto H., Takahashi K., Toyama S., Miyakawa Y., and Mayumi M. (1982) Demonstration of two distinct antigenic determinants on hepatitis B e antigen by monoclonal antibodies. J. Immunol. 128, 69–72 [PubMed] [Google Scholar]
- 34. Wu W., Chen Z., Cheng N., Watts N. R., Stahl S. J., Farci P., Purcell R. H., Wingfield P. T., and Steven A. C. (2013) Specificity of an anti-capsid antibody associated with hepatitis B virus-related acute liver failure. J. Struct. Biol. 181, 53–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Bereszczak J. Z., Rose R. J., van Duijn E., Watts N. R., Wingfield P. T., Steven A. C., and Heck A. J. (2013) Epitope-distal effects accompany the binding of two distinct antibodies to hepatitis B virus capsids. J. Am. Chem. Soc. 135, 6504–6512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Cohen B. J., and Richmond J. E. (1982) Electron microscopy of hepatitis B core antigen synthesized in E. coli. Nature 296, 677–679 [DOI] [PubMed] [Google Scholar]
- 37. Takahashi K., Machida A., Funatsu G., Nomura M., Usuda S., Aoyagi S., Tachibana K., Miyamoto H., Imai M., Nakamura T., Miyakawa Y., and Mayumi M. (1983) Immunochemical structure of hepatitis B e antigen in the serum. J. Immunol. 130, 2903–2907 [PubMed] [Google Scholar]
- 38. Howard C. R., and Zuckerman A. J. (1979) The separation and analysis of hepatitis B e antigen. J. Med. Virol. 4, 303–314 [DOI] [PubMed] [Google Scholar]
- 39. Neurath A. R., Strick N., Szmuness W., Stevens C. E., and Harley E. J. (1979) Radioimmunoassay of hepatitis B e-antigen (HBeAg): identification of HBeAg not associated with immunoglobulins. J. Gen. Virol. 42, 493–504 [DOI] [PubMed] [Google Scholar]
- 40. Standring D. N., Ou J. H., Masiarz F. R., and Rutter W. J. (1988) A signal peptide encoded within the precore region of hepatitis B virus directs the secretion of a heterogeneous population of e antigens in Xenopus oocytes. Proc. Natl. Acad. Sci. U.S.A. 85, 8405–8409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Rader C., and Barbas C. F. 3rd. (1997) Phage display of combinatorial antibody libraries. Curr. Opin. Biotechnol. 8, 503–508 [DOI] [PubMed] [Google Scholar]
- 42. Bradbury A. R., Sidhu S., Dübel S., and McCafferty J. (2011) Beyond natural antibodies: the power of in vitro display technologies. Nat. Biotechnol. 29, 245–254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Hu D., Hu S., Wan W., Xu M., Du R., Zhao W., Gao X., Liu J., Liu H., and Hong J. (2015) Effective optimization of antibody affinity by phage display integrated with high-throughput DNA synthesis and sequencing technologies. PLoS ONE 10, e0129125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Ying T., Chen W., Gong R., Feng Y., and Dimitrov D. S. (2012) Soluble monomeric IgG1 Fc. J. Biol. Chem. 287, 19399–19408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Völkers M., Rohde D., Zelniker T., Weiss C. S., Giannitsis E., Katus H. A., and Meyer F. J. (2013) High-sensitive troponin T increase after exercise in patients with pulmonary arterial hypertension. BMC Pulm. Med. 13, 28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Baumeister M. A., Medina-Selby A., Coit D., Nguyen S., George-Nascimento C., Gyenes A., Valenzuela P., Kuo G., and Chien D. Y. (2000) Hepatitis B virus e antigen specific epitopes and limitations of commercial anti-HBe immunoassays. J. Med. Virol. 60, 256–263 [PubMed] [Google Scholar]
- 47. Ito K., Kim K. H., Lok A. S., and Tong S. (2009) Characterization of genotype-specific carboxyl-terminal cleavage sites of hepatitis B virus e antigen precursor and identification of furin as the candidate enzyme. J. Virol. 83, 3507–3517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Wingfield P. T., Stahl S. J., Williams R. W., and Steven A. C. (1995) Hepatitis core antigen produced in Escherichia coli: subunit composition, conformational analysis, and in vitro capsid assembly. Biochemistry 34, 4919–4932 [DOI] [PubMed] [Google Scholar]
- 49. Walsh R., and Locarnini S. (2012) Hepatitis B precore protein: pathogenic potential and therapeutic promise. Yonsei Med. J. 53, 875–885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Xun Y., Pan Q., Tang Z., Chen X., Yu Y., Xi M., and Zang G. (2013) Intracellular-delivery of a single-chain antibody against hepatitis B core protein via cell-penetrating peptide inhibits hepatitis B virus replication in vitro. Int. J. Mol. Med. 31, 369–376 [DOI] [PubMed] [Google Scholar]
- 51. Chen G. F., Wang C., and Lau G. (2017) Treatment of chronic hepatitis B infection-2017. Liver Int. 37, 59–66 [DOI] [PubMed] [Google Scholar]
- 52. King T. H., Kemmler C. B., Guo Z., Mann D., Lu Y., Coeshott C., Gehring A. J., Bertoletti A., Ho Z. Z., Delaney W., Gaggar A., Subramanian G. M., McHutchison J. G., Shrivastava S., Lee Y. J., et al. (2014) A whole recombinant yeast-based therapeutic vaccine elicits HBV X, S and core specific T cells in mice and activates human T cells recognizing epitopes linked to viral clearance. PLoS ONE 9, e101904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. MacKay P., Lees J., and Murray K. (1981) The conversion of hepatitis B core antigen synthesized in E. coli into e antigen. J. Med. Virol. 8, 237–243 [DOI] [PubMed] [Google Scholar]
- 54. Murray K., Bruce S. A., Hinnen A., Wingfield P., van Erd P. M., de Reus A., and Schellekens H. (1984) Hepatitis B virus antigens made in microbial cells immunise against viral infection. EMBO J. 3, 645–650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Murray K., Bruce S. A., Wingfield P., van Eerd P., de Reus A., and Schellekens H. (1987) Protective immunisation against hepatitis B with an internal antigen of the virus. J. Med. Virol. 23, 101–107 [DOI] [PubMed] [Google Scholar]
- 56. Wingfield P. T., Graber P., and Payton M. A. (1987) Biochemical estimation of hepatitis B surface antigen in recombinant yeast. Yeast 3, 43–49 [DOI] [PubMed] [Google Scholar]
- 57. Stahl S. J., Watts N. R., and Wingfield P. T. (2014) Generation and use of antibody fragments for structural studies of proteins refractory to crystallization. Methods Mol. Biol. 1131, 549–561 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.