

Abstract
Aryl-substituted bis(imino)pyridine cobalt dinitrogen compounds, (RPDI)CoN2, are effective precatalysts for the intramolecular [2π + 2π] cycloaddition of α,ω-dienes to yield the corresponding bicyclo[3.2.0]heptane derivatives. The reactions proceed under mild thermal conditions with unactivated alkenes, tolerating both amine and ether functional groups. The overall second order rate law for the reaction, first order with respect to both the cobalt precatalyst and the substrate, in combination with electron paramagnetic resonance (EPR) spectroscopic studies established the catalyst resting state as dependent on the identity of the precatalyst and diene substrate. Planar S =1/2 κ3-bis(imino)pyridine cobalt alkene and tetrahedral κ2-bis(imino)pyridine cobalt diene complexes were observed by EPR spectroscopy and in the latter case structurally characterized. The hemilabile chelate facilitates conversion of a principally ligand-based singly occupied molecular orbital (SOMO) in the cobalt dinitrogen and alkene compounds to a metal-based SOMO in the diene intermediates, promoting C–C bond-forming oxidative cyclization. Structure–activity relationships on bis(imino)pyridine substitution were also established with 2,4,6-tricyclopentyl-substituted aryl groups, resulting in optimized catalytic [2π + 2π] cycloaddition. The cyclopentyl groups provide a sufficiently open metal coordination sphere that encourages substrate coordination while remaining large enough to promote a challenging, turnover-limiting C(sp3)–C(sp3) reductive elimination.
Introduction
Metal-catalyzed cycloaddition reactions have emerged as important and highly versatile strategies to generate molecular complexity in a single synthetic step. The combination of multiple π components provides atom-economical and often selective access to various hetero- and carbocycles that are pervasive in organic synthesis.1−5 Among these, [2π + 2π] alkene cycloaddition reactions, which are thermodynamically favorable but thermally forbidden,6 are attractive methods for the synthesis of cyclobutanes, offering a direct, atom-economical route to these structures. The utility of [2π + 2π] reactions has been recognized because of both the prevalence of cyclobutanes in a diverse family of bioactive natural products and the potential of strained ring systems as molecular building blocks in synthesis.7−15
In comparison with six-membered rings, cycloaddition strategies applied to the synthesis of four-membered rings are far less developed. Approaches include photochemical activation methods16−22 and the use of activated or highly strained π components in conjunction with transition metals to overcome the inherent orbital symmetry constraints.23−39 Enantioselective routes to cyclobutanes are even more rare, highlighted by several recent photocatalytic methods that rely on chromophores such as enones as a component of the substrate40−45 or chiral Lewis acid catalysts.46,47
The established catalytic olefin oligomerization chemistry of nickel48 has inspired numerous studies of the stoichiometric and catalytic dimerization activity of various reduced transition metal compounds with ethylene and other alkenes. Grubbs and Miyashita49 observed substoichiometric amounts of cyclobutane following decomposition of bis(phosphine)-ligated nickel metallacyclopentanes. Among the earliest examples of Ni-catalyzed [2π + 2π] cycloadditions, Ni(COD)2 was used in the dimerization of 1,3-butadiene50 or methylenecyclopropane51 and the heterodimerization of norbornadiene and methylenecyclopropane.52 Smith and co-workers also reported nickel-catalyzed dimerization of isoprene to form cyclobutane products in low yield in the context of the synthesis of grandisol.53 Other transition metals, such as Fe,54 Mn,55 and Ti,56 have also been reported in catalytic [2π + 2π] cycloadditions; however, these examples require norbornadiene or 1,3-butadiene as one of the coupling partners and typically suffer from low selectivity.
The bis(imino)pyridine iron dinitrogen complexes (iPrPDI)Fe(N2)2 ([Fe], shown in Scheme 1, top)57 and [(MePDI)Fe(N2)]2(μ2-N2)58 are unique examples of metal catalysts that promote the [2π + 2π] cycloaddition of unactivated alkenes under mild thermal conditions.59 Examples of the intramolecular cyclization of diallyl amines, ethyl diallyl malonate, and 1,6-heptadiene cleanly yield the corresponding bicyclo[3.2.0]heptane products (Scheme 1). Diallyl ether was identified as a limitation in the substrate scope, as irreversible C–O bond cleavage resulted in deactivation of the iron catalyst.60 Important catalytic intermediates, including S = 1 high-spin iron diene compounds and metallacycles, have since been isolated.61 Combined structural, spectroscopic, magnetic, and computational data were used to establish the electronic structures of these compounds and to determine the role of the redox-active bis(imino)pyridine chelate. The data indicate that the bis(imino)pyridine maintains a radical anion form throughout the catalytic cycle, supporting an Fe(I)–Fe(III) redox couple. An intermolecular example has been discovered in which addition of ethylene and butadiene to 5 mol % [(MePDI)Fe(N2)]2(μ2-N2) results in selective conversion to vinylcyclobutane.62 In contrast to the intramolecular examples, a diamagnetic iron allyl alkyl metallacycle intermediate was isolated and structurally characterized (Scheme 1, bottom).
Scheme 1. Alkene [2π + 2π] Cycloadditions Catalyzed by Bis(imino)pyridine Iron Compounds.

Iron complexes bearing redox-active bis(imino)pyridines have thus far proven to be unique catalysts for promoting alkene [2π + 2π] cycloadditions. These observations raise the question as to whether the redox-active bis(imino)pyridine63 enables cyclobutane formation. Correlating a singular property of a metal complex to catalytic performance is a challenging endeavor. Disparate reactivity may be observed for reasons outside the variables being evaluated. Nevertheless, we have been actively exploring this issue not only to determine the electronic structure requirements for catalytic cycloaddition but also to discover new catalysts with improved scope and function. As part of this effort, Danopolous’s iron dinitrogen complex, (iPrCNC)Fe(N2)2,64 was treated with an excess of N,N-diallyl-tert-butylamine, one of the most reactive substrates for (iPrPDI)Fe(N2)2-catalyzed alkene [2π + 2π] cycloaddition. No turnover was observed, and the corresponding iron diene complex was isolated and crystallographically characterized.65 In contrast to bis(imino)pyridine iron diene complexes that have S = 1 ground states and contain ligand-centered radicals, the CNC variant is diamagnetic with the pincer acting as a classical π acceptor. These observations support an enabling role of bis(imino)pyridines in iron-catalyzed alkene [2π + 2π] cycloaddition.
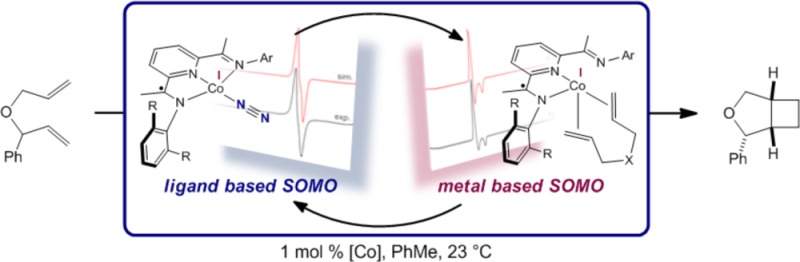
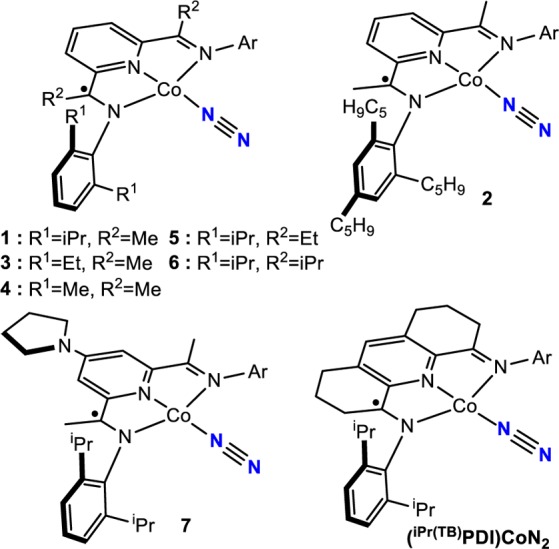
A related objective is to determine whether other transition metals, in combination with redox-active bis(imino)pyridines, would also promote [2π + 2π] cycloaddition. Cobalt was of interest because of its high terrestrial abundance and ample precedent in a diverse range of cycloadditions involving various π components.66 Despite this prior work, the cobalt-catalyzed thermal cycloaddition of two unactivated alkenes to yield cyclobutane products has not been demonstrated.67 Aryl-substituted bis(imino)pyridine cobalt dinitrogen complexes, (RPDI)CoN2, have been prepared by our laboratory68,69 and that of Budzelaar,70 and the role of the bis(imino)pyridine chelate in their electronic structures has been established.71 The overall S = 1/2 compounds are best described as low-spin Co(I) d8 complexes with a bis(imino)pyridine-based singly occupied molecular orbital (SOMO), the same redox state of the chelate found during iron-catalyzed α,ω-diene [2π + 2π] cycloaddition. Catalysis with the cobalt dinitrogen compounds may also offer practical benefits. The cobalt compounds are generally more straightforward to handle and synthesize than the corresponding iron derivatives and may overcome limitations in substrate scope. Here we describe the catalytic alkene [2π + 2π] cycloaddition reactivity of a family of bis(imino)pyridine cobalt dinitrogen compounds. The availability of a range of cobalt dinitrogen compounds (Scheme 2) has enabled the evaluation of structure–reactivity relationships and provided key insights for new ligand design in order to expand the scope of the reaction. A combination of kinetic measurements, deuterium labeling experiments, and in situ observations by electron paramagnetic resonance (EPR) spectroscopy has provided detailed insight into the identity of catalytically competent intermediates and highlighted the role of hemilabile ligands to alter the ligand field strength, ultimately enabling catalytic carbon–carbon bond formation.
Scheme 2. Bis(imino)pyridine Cobalt Dinitrogen Compounds Used in This Study.
Results and Discussion
Evaluation of Catalytic Performance
The catalytic performance of (iPrPDI)CoN2 (1) was evaluated for the [2π + 2π] cycloaddition of a family of α,ω-dienes, including substituted allyl amines and ethers (Table 1). Also included in Table 1 are results of the same cycloaddition chemistry using [Fe] as the precatalyst. These investigations established the efficiency of the iron precatalyst at a loading of 1 mol % in order to provide a comparison with the cobalt catalysts.
Table 1. Intramolecular Alkene [2π + 2π] Cycloaddition Performance of Bis(imino)pyridine Iron and Cobalt Dinitrogen Complexes.


Isolated yields.
The reaction was run at 0.2 M in C6D6, and the yield was determined by 1H NMR analysis using diiodomethane as an internal standard.
2.5 mol % [Fe] was added to the 1 mol % experiment.
Stoichiometric C–O bond cleavage with respect to [Fe].
A 22% yield of 3-methyl-4-methylenetetrahydrofuran was also dectected.
The diastereomeric ratio was determined by 1H NMR analysis; the major diastereomer is shown.
The reaction was run at 50 °C.
The results in Table 1 establish 1 as an active and selective precatalyst for α,ω-diene [2π + 2π] cycloaddition. In toluene solution, efficient cyclization of N,N-diallyltritylamine (entry 1) was observed over the course of 6.5 h with 2.5 mol % 1. Both tert-butyl (entry 2) and aryl amines (entry 3) were effective in the reaction and furnished the desired products in high yields. As with iron, cis diastereomers of the azabicyclo[3.2.0]heptane products were exclusively observed as judged by NMR spectroscopy and gas chromatography (GC).
Allyl ethers also underwent [2π + 2π] cycloaddition in the presence of 10 mol % 1. The observation of catalytic turnover with allyl ether in the presence of 1 is noteworthy given that deactivation by irreversible C–O bond cleavage was observed with [Fe]. The introduction of a phenyl substituent adjacent to the oxygen (entry 6) increased the observed turnover frequency and the selectivity of the reaction, as the [2π + 2π] cycloaddition product was obtained exclusively as the all-cis diastereomer after 23 h at 23 °C. Again the iron precatalyst was ineffective for this reaction, suffering from catalyst deactivation by C–O bond cleavage. Introduction of a second phenyl substituent (entry 7) improved the performance of the cobalt-catalyzed reaction and also enabled turnover with [Fe]. Hydrocarbon substrates (entries 8–10) were unreactive with 1, and no turnover was observed even upon heating to 50 °C for 24 h.
Although 1 exhibited a tolerance for allyl ethers that was absent in iron catalysis, the catalytic activity was poor, and no products were observed in entries 8–10. To overcome these limitations, alteration of the bis(imino)pyridine ligand was explored. Substitution of the isopropyl groups in the 2,6-diisopropylaryl substituents with cyclopentyl groups was initially targeted with the goal of opening the coordination sphere of the cobalt while maintaining a sufficient steric profile to promote the C(sp3)–C(sp3) reductive elimination. The direct analogue of 1, (C5H9PDI)CoN2 (C5H9PDI = 2,6-bis{1-[N-(2,6-dicyclopentylphenyl)imino]ethyl}pyridine), was synthesized and offered promising catalytic [2π + 2π] cycloaddition reactivity. The requisite aniline, 2,6-dicyclopentylaniline, has proven challenging to synthesize or obtain commercially. Because of the more straightforward synthesis, the preparation of 2,4,6-tricyclopentylaniline was pursued. Friedel–Crafts trialkylation of benzene with C5H9Br followed by nitration and reduction yielded the desired aniline. The free bis(imino)pyridine (TricPDI) was conveniently obtained on multigram scales following straightforward condensation with 2,6-diacetylpyridine. Metalation with CoCl2 in tetrahydrofuran (THF) followed by reduction with excess 0.5% sodium amalgam gave (TricPDI)CoN2 (2) as a teal solid in 88% isolated yield.
Both (C5H9PDI)CoN2 and 2 have S = 1/2 ground states and exhibit broad and featureless 1H NMR spectra at room temperature. The solid-state structures of both compounds were determined by X-ray diffraction (Figure 1). As in 1, an essentially planar geometry about cobalt is observed, with distortions of the bond distances of the bis(imino)pyridine chelate diagnostic of one-electron reduction.68,72
Figure 1.

Representations of the molecular structures of (left) (C5H9PDI)CoN2 and (right) 2 with 30% probability ellipsoids. Hydrogen atoms have been omitted for clarity.
The catalytic [2π + 2π] cycloaddition performance of 2 proved to be superior to that of 1 and approached the activity of [Fe] in some cases. The cycloaddition of N,N-diallyl-tert-butylamine (Table 1, entry 2) was complete within 5 min using only 1 mol % 2 or [Fe] but required 3 h to reach full conversion with 2.5 mol % 1. The higher sensitivity of [Fe] compared with the library of reduced bis(imino)pyridine cobalt dinitrogen compounds reported herein is highlighted with N,N-diallylbenzylamine (Table 1, entry 4). Upon mixing of 1 mol % [Fe] with this substrate, visible catalyst decomposition was observed and was attributed to trace amounts of water in the amine. No product was detected by GC even after 24 h. Further addition of 2.5 mol % [Fe] to the reaction mixture resulted in full conversion to the cyclobutane in 30 min. By contrast, 1 mol % 2 cleanly cyclized N,N-diallylbenzylamine in 12 h without observable catalyst deactivation, demonstrating the increased fidelity of the cobalt precursors.
Influence of Bis(imino)pyridine Substitution on Catalytic Performance and Establishment of Chelate Electronic Properties
Additional modifications to the bis(imino)pyridine were made in order to explore possible structure–reactivity relationships and gain insight into the mechanism of turnover. Such experiments are enabled by the synthetic accessibility of a range of bis(imino)pyridine cobalt dinitrogen compounds. For these studies, N,N-diallyl-4-fluoroaniline and N,N-diallyl-tert-butylamine were chosen as representative substrates, and catalytic reactions were conducted with a 5 mol % loading of the cobalt precursor in 0.2 M toluene solution at 23 °C. The results for the former substrate are reported in Table 2, and the data for the latter are reported in the Supporting Information.
Table 2. Bis(imino)pyridine Substituent Effects on the Time to Reach >98% Conversion in the [2π + 2π] Cycloaddition of N,N-Diallyl-4-fluoroaniline.

| entry | [Co] | time (h)a |
|---|---|---|
| 1 | (iPrPDI)CoN2 (1) | 8 |
| 2 | (TricPDI)CoN2 (2) | 1 |
| 3 | (EtPDI)CoN2 (3) | 44 |
| 4 | (MePDI)CoN2 (4) | 60 |
| 5 | (iPrEtPDI)CoN2 (5) | >72b |
| 6 | (iPriPrPDI)CoN2 (6) | >72c |
| 7 | 4-pyrr-(iPrPDI)CoN2 (7) | 36d |
Time to >98% conversion.
41% conversion at 72 h.
7% conversion at 72 h.
48% cycloaddition and 52% alkene isomerization products.
The identity of the groups at the 2- and 6-positions in the N-arylimino substituents has a significant influence on the catalytic performance, with larger alkyl groups enabling higher rates of [2π + 2π] cycloaddition. The cyclopentyl-substituted precatalyst 2 reached >98% conversion to product in 1 h, while the ethyl- (3) and methyl-substituted (4) variants required 44 and 60 h, respectively. Alteration of the imine carbon substituents also has a profound effect on catalyst performance, but a trend opposite to that for the aryl substituents was observed. Replacement of the methyl groups on the imine carbons with ethyl (5) or isopropyl (6) groups significantly decreased the turnover, as only partial conversion was observed after 72 h. Electronic effects were also briefly examined. The introduction of an electron-donating 4-pyrrolidinyl substituent,73 a group known to accelerate cobalt-catalyzed alkene hydroboration,74 decreased the rate of [2π + 2π] cycloaddition, as the time to reach >98% conversion with 7 was 36 h and the azabicyclo[3.2.0]heptane accounted for 48% of the reaction mixture, with the balance of the product arising from isomerization of the alkene.
Electrochemical studies were carried out on the free bis(imino)pyridines as well as the cobalt dinitrogen compounds to assess the electronic differences among the catalyst precursors. A summary of the measured reduction potentials (vs Cp2Fe/Cp2Fe+) and infrared stretching frequencies of the terminal N2 ligand for representative cobalt compounds is reported in Table 3. Each bis(imino)pyridine exhibits a single reversible reduction wave in THF solution with 0.1 M [Bu4N][PF6] as the electrolyte, a silver wire reference electrode, a platinum counter elecrode, and a glassy carbon working electrode. There is little variance in the reduction potentials, with a difference of just 90 mV observed within the series of imine-substituted ligands. The introduction of an amino group at the 4-position of the pyridine has a larger effect, as a 200 mV difference in the potentials of 7 and 2 was measured. With the exception of 2, each of the bis(imino)pyridine cobalt dinitrogen compounds exhibits three separate and reversible one-electron oxidation waves. Only two reversible waves were observed at −1.70 and −2.11 V for 2, with the absence of the expected reversible wave at ∼1 V that was evident for the other compounds. A comparison of the first reduction potentials across the series of bis(imino)pyridine cobalt dinitrogen compounds reinforces the trend observed for the free ligands, although the electronic difference between 1 and 7 is only 90 mV. The infrared stretching frequencies of the cobalt dinitrogen compounds also reflect this trend, as little variance is observed between the values for 1, 2, 4, and 5. The pyrrolidinyl-substituted compound 7 exhibits the most reduced N2 stretching frequency, with a value of 2091 cm–1 in pentane solution. Because there is no clear trend correlating the electrochemical data and the catalytic cycloaddition activity, the observed catalyst effects are not primarily based on chelate electronic effects.
Table 3. Reduction Potentials of Free Bis(imino)pyridine Ligands and Reduction Potentials and Infrared Stretching Frequencies of the N2 Band in (PDI)CoN2 Compounds.
| reduction potentialsa |
IR νNNb | ||
|---|---|---|---|
| compound | free ligand | (PDI)CoN2 | (PDI)CoN2 |
| (iPrPDI)CoN2 (1) | –2.62c | –0.97 | 2101 |
| (TricPDI)CoN2 (2) | –2.67 | –d | 2106 |
| (MePDI)CoN2 (4) | –2.71c | –0.93 | 2105 |
| (iPrEtPDI)CoN2 (5) | –2.65 | –0.87 | 2100 |
| 4-pyrr-(iPrPDI)CoN2 (7) | –2.82c,e | –1.08 | 2091 |
All reductions potentials are reported in V vs Fc/Fc+.
All IR spectra were recorded in pentane and are reported in cm–1.
Value from ref (72).
Not observed.
The value is for 4-NMe2-(iPrPDI).
Deuterium Labeling Experiments
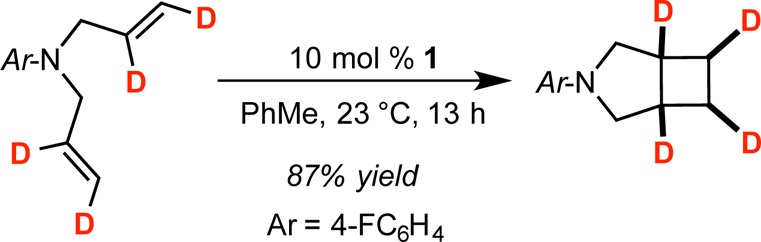
The stereochemistry of the cobalt-catalyzed α,ω-diene [2π + 2π] cycloaddition was investigated using the d4 isotopologue of N,N-diallyl-4-fluoroaniline. The desired substrate, shown in Scheme 3, was prepared by Lindlar reduction of the corresponding N,N-dipropargyl-4-fluoroaniline with 1 atm D2. Stirring a toluene solution of N,N-diallyl-4-fluoroaniline-d4 with 10 mol % 1 afforded the azabicyclo[3.2.0]heptane product with the all-cis configuration of the deuterium atoms preserved. Analysis of the free bis(imino)pyridine by 1H and 2H NMR spectroscopy following hydrolysis of the catalytic mixture established that no deuterium incorporation into liberated ligand occurred, demonstrating that rapid cyclometalation pathways are not competitive with [2π + 2π] cycloaddition, unlike previous observations in iron catalysis.75 The retention of the cis stereochemistry supports an organometallic pathway involving metallacycle formation and C–C reductive elimination. While a radical pathway cannot be definitively excluded, the 4-exo-cyclobutane-forming cyclization would have to be faster than Co–C bond rotation.
Scheme 3. Deuterium Labeling Experiments Establishing Stereospecific Cobalt-Catalyzed [2π + 2π] Cycloaddition of N,N-Diallyl-4-fluoroaniline-d4 with 1.
Kinetic Experiments and Determination of Reaction Order
The kinetics of N,N-diallyl-4-fluororaniline [2π + 2π] cycloaddition catalyzed by 1 was determined by GC analysis of aliquots taken throughout the reaction. Representative plots for the disappearance of the diene as a function of time at three different precatalyst loadings, 2.5 (green), 5 (pink), and 10 (blue) mol % 1, are presented in the left panel of Figure 2. Clean exponential decay was observed in each case, establishing a first order dependence on substrate at 23 °C. A plot of kobs versus the concentration of the precatalyst was also obtained (Figure 2, right) and established a first order dependence on 1 at the same reaction temperature. Similar behavior was observed for the [2π + 2π] cycloaddition of N,N-diallyl-tert-butylamine with 1 (see the Supporting Information). In both cases, the data clearly establish overall second order reactions, first order with respect to both the substrate and the cobalt precatalyst.
Figure 2.

(left) Representative kinetic data for the [2π + 2π] cycloaddition of N,N-diallyl-4-fluororaniline with different catalyst loadings of 1 and (right) establishment of the reaction order with respect to cobalt. All of the data were recorded at 23 °C. Data for the plot on the left were recorded in triplicate, and error bars are shown.
A different kinetic profile was observed using the less sterically protected cobalt dinitrogen compound 4. Plots of the disappearance of N,N-diallyl-tert-butylamine versus time at 23 °C were fit to linear decay, establishing a zeroth order dependence on the substrate (Figure 3, left), while a plot of kobs versus the concentration of 4 (Figure 3, right) established a first order dependence on the cobalt precatalyst at the same reaction temperature. These data are consistent with saturation behavior where the bis(imino)pyridine cobalt complex contains coordinated diene in the turnover-limiting step.
Figure 3.

(left) Representative kinetic data for the [2π + 2π] cycloaddition of N,N-diallyl-tert-butylamine with different catalyst loadings of 4 and (right) establishment of the reaction order with respect to cobalt. All of the data were recorded at 23 °C.
In Situ Monitoring of Catalyst Turnover by EPR Spectroscopy
Because the S = 1/2 cobalt dinitrogen compounds have largely uninformative NMR spectra, ambient-temperature X-band EPR spectroscopy was used to monitor the catalytic [2π + 2π] cycloadditions with the goal of identifying the catalyst resting state. As reported previously,68 the EPR spectrum of 1 exhibits an isotropic signal with giso = 2.003, consistent with a ligand-centered SOMO with relatively small coupling to the I = 7/2 59Co nucleus (Aiso = 24 MHz). Similar features were observed in fluid toluene solution EPR spectra of all of the other cobalt dinitrogen compounds prepared in this work (see the Supporting Information).
To reproduce the catalytic conditions, 40 equiv of N,N-diallyl-tert-butylamine was added to a toluene solution of 1, and the EPR spectrum was recorded at 23 °C (Figure 4, left). Two distinct signals were observed after 10 min. The major species was identified as 1 (red simulation, giso = 2.00), and a second, new species (labeled as A) was observed with giso = 2.17 (blue simulation). The presence of 1 was also confirmed by analysis of the reaction mixture by IR spectroscopy, as a strong N–N stretching frequency was observed at 2093 cm–1 in toluene.68 The relative concentrations of 1 and A were determined by double integration of the initial absorption EPR spectra (Table 4; vide infra).
Figure 4.

X-band EPR spectra upon the addition of (left) 0.2 M N,N-diallyl-tert-butylamine or (right) 0.2 M N,N-diallyl-4-fluoroaniline to a toluene solution of 1.
Table 4. Summary of EPR Parametersa.
|
N,N-diallyl-tert-butylamine |
N,N-diallyl-4-fluoroaniline |
|||||
|---|---|---|---|---|---|---|
| N2 g = 2.00 |
B g = 2.05 |
A g = 2.17 |
[Co] | N2 g = 2.00 |
B g = 2.05 |
A g = 2.17 |
| 57% | – | 43% | (iPrPDI)CoN2 (1) | 63% | 33% | 4% |
| 13% | – | 87% | (TricPDI)CoN2 (2)b | – | 100% | – |
| – | 92% | 8% | (MePDI)CoN2 (4) | – | 100% | – |
| 100% | – | – | (iPrEtPDI)CoN2 (5) | 100% | – | – |
| 60% | – | 40% | 4-pyrr-(iPrPDI)CoN2 (7) | 63% | 31% | 6% |
Mixtures of bis(imino)pyridine cobalt dinitrogen compound under standard catalytic conditions (40 equiv of substrate, 0.2 M in PhMe at 295 K). The relative contributions of species A, B, and N2 were calculated by double integration of the absorption curves. See the Supporting Information for a full table of spectra.
The spectrum was recorded with 100 equiv of substrate.
Repeating the experiment with 40 equiv of N,N-diallyl-4-fluoroaniline also produced an EPR spectrum with 1 as the major component (Figure 4, right). A trace quantity of the signal at giso = 2.17 (A) was observed, with a second product appearing at giso = 2.05 (green simulation). Upon full consumption of the starting material, as determined by GC, the EPR spectrum exhibits a single component, identified as 1.
Efforts were devoted to identifying the intermediates responsible for these signals. Our laboratory previously reported that use of the “tied-back” bis(imino)pyridine (iPr(TB)PDI) in iron-catalyzed α,ω-diene cycloaddition slowed turnover and imparted crystallinity to catalytic intermediates, facilitating characterization and electronic structure determination of iron diene compounds and metallacycles.61 This approach was applied to cobalt catalysis, and treatment of a toluene solution of (iPr(TB)PDI)CoN2 with an excess of N,N-diallyl-4-methoxyaniline furnished purple crystals suitable for X-ray diffraction. The solid-state structure is presented in Figure 5 and establishes the formation of the desired cobalt diene complex, 8. The measured solid-state magnetic moment of 1.8 μB (23 °C) is consistent with an S = 1/2 ground state. Notably, the solid-state structure reveals that one of the imine arms of the bis(imino)pyridine has dissociated from the metal center, with a Co(1)–N(3) distance of 2.567(3) Å, which is much longer than the Co(1)–N(1) distance of 2.152(3) Å. The toluene solution EPR spectrum of the isolated crystals at 23 °C exhibits a strong, broadened signal at giso = 2.17. Cooling the sample to 10 K and recording the spectrum in toluene glass produced an axial signal that was readily simulated using relatively small anisotropic g values (gx = 2.254, gy = 2.245, gz = 2.012) and A values (Axx = 42 MHz, Ayy = 38 MHz, Azz = 2 MHz). The observation of the peak at giso = 2.17 in fluid toluene solution 23 °C suggests that compound A formed from treatment of 1 with excess N,N-diallyl-tert-butylamine or N,N-diallyl-4-fluoroaniline is the analogous cobalt diene complex with a κ2-bis(imino)pyridine. Similar results were obtained with N,N-diallyl-tert-butylamine, as addition of excess diene to a toluene solution of (iPr(TB)PDI)CoN2 also generated the EPR signal at giso = 2.17, supporting the formation of the cobalt diene complex. The significant deviation from ge (ge = 2.003) and the giso value observed for the chelate-centered radicals of bis(imino)pyridine cobalt dinitrogen compounds (giso = 2.00) suggests a change in electronic structure upon the formation of 8.
Figure 5.

(left) Representation of the molecular structure of 8 with 30% probability ellipsoids. Hydrogen atoms and the isopropyl groups on the left aryl ring have been omitted for clarity. (middle) EPR spectra of 8 in toluene solution at 23 °C (right) and in a toluene glass at 10 K.
Full-molecule density functional theory studies were conducted on 8 using the B3LYP functional,76 which has been used successfully to determine the electronic structures of other cobalt compounds bearing redox-active bis(imino)pyridine chelates.68 Unrestricted Kohn–Sham (UKS) and broken-symmetry (BS) possibilities were calculated to account for the experimentally observed S = 1/2 spin state. Both inputs converged to a BS(2,1) solution, and a qualitative molecular orbital diagram and spin density plot are presented in Figure 6. The preferred electronic structure corresponds to an unusual high-spin Co(I) center (d8, SCo = 1) with one electron in a metal dyz orbital engaged in antiferromagnetic coupling (S = 0.35) with the b2 orbital of the bis(imino)pyridine. The SOMO is principally cobalt-based with small contributions from both the bis(imino)pyridine and the diene, which accounts for the larger g anisotropy observed in 8 than in the corresponding cobalt dinitrogen compounds. The computed EPR parameters (g1 =2.263, g2 = 2.252, g3 = 2.066) are in good agreement with the experimental values at 10 K, further validating the computational findings.
Figure 6.
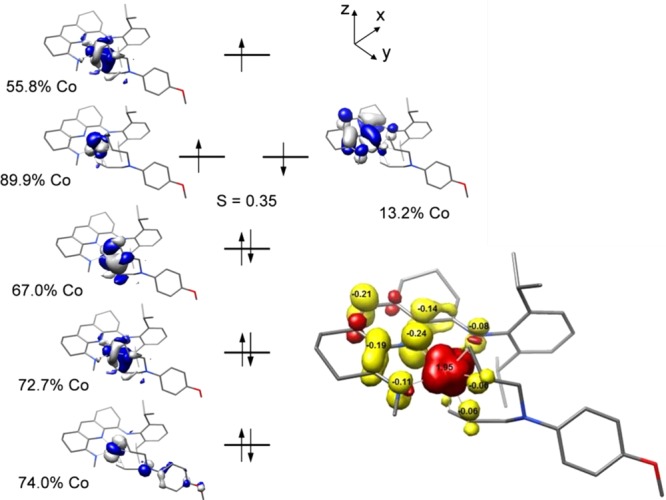
(left) Qualitative molecular orbital diagram for 8 from a BS(2,1) calculation at the B3LYP level. (right) Spin density plot obtained from Mulliken population analysis (red, positive spin density; yellow, negative spin density).
Having established the identity of the signal at giso = 2.17 as κ2-bis(imino)pyridine cobalt diene complexes, attention was devoted to identifying the compound responsible for the signal at giso = 2.05, denoted as B in Figure 4. Addition of N,N-diallyl-4-methoxyaniline to a solution of 4 resulted in the isolation of a green solid, 9, that exhibited the g = 2.05 EPR signal. This cobalt dinitrogen complex was selected because it is one of the most open compounds in the series and also because in situ monitoring of catalytic [2π + 2π] cycloaddition with both N,N-diallyl-4-fluoroaniline and N,N-diallyl-tert-butylamine revealed a catalyst resting state that is predominantly an S = 1/2 intermediate with giso = 2.05. Although 9 was isolated and fully characterized, single crystals were not obtained, precluding elucidation of the substrate–cobalt interaction. One possibility is coordination of the amine nitrogen of the substrate to form a classic Werner-type complex with a σ interaction between the metal and the substrate. To evaluate this possibility, a toluene solution of 4 was treated with a large excess of N,N-dipropyl-4-fluoroaniline, the saturated version of one of the diene substrates. The addition of a large excess of saturated amine produced no change in the EPR spectrum of 4 (Scheme 4, top), suggesting that the signal at giso = 2.05 does not arise from simple amine σ coordination.
Scheme 4. Toluene Solution X-Band EPR Spectra of (top) 4 with 40 equiv of N,N-Dipropyl-4-fluoroaniline Recorded at 23 °C, (middle) 4 with 40 equiv of N-Allyl-N-propyl-4-fluororaniline Recorded at (left) 23 °C or (right) 10 K, and (bottom) 4 with 40 equiv of (1-(Allyloxy)allyl)benzene Recorded at 23 °C (Conditions: Microwave Frequency, 9.37 GHz; Power, 2.0 mW; Modulation, 0.6325 mT/100 kHz).

Another possibility for the identity of B is the formation of an olefin complex with coordination of only one alkene arm of the substrate, where the κ3 coordination of the bis(imino)pyridine is maintained. Mixing experiments wherein an excess of N-allyl-N-(3-butenyl)-4-fluoroaniline, N-allyl-N-crotyl-4-fluoroaniline, or N-allyl-N-propyl-4-fluoroaniline was added to a toluene solution of 4 in each case generated a signal at giso = 2.05 at 23 °C. No catalytic turnover was observed with any of these substrates after extended periods. The observation of the giso = 2.05 signal at 23 °C with N-allyl-N-propyl-4-fluoroaniline (Scheme 4, middle) is particularly informative and strongly supports formation of bis(imino)pyridine cobalt alkene complexes as intermediates during catalytic [2π + 2π] cycloaddition. Another possibility for B is κ1,η2 coordination of the amino substrate, where the nitrogen acts as a σ donor and the olefin forms a π complex with the metal. The addition of 40 equiv of (1-(allyloxy)allyl)benzene to a toluene solution of 4 generated the EPR signal at g = 2.05 at 23 °C (Scheme 4, bottom), arguing against the formation of a κ1,η2 adduct with the substrate. These experiments support the identity of B as a bis(imino)pyridine cobalt η2-alkene complex. However, the analogous mixing experiment of 4 and an excess of 1-octene produced no change as judged by EPR spectroscopy, suggesting that simple hydrocarbons are poor ligands for bis(imino)pyridine cobalt compounds.
Computational studies were conducted to provide further support for this formulation of B. N-Allyl-N-propyl-4-fluoroaniline was selected as the model substrate. Geometry optimizations of this molecule proved to be challenging because of the many degrees of freedom associated with the propyl substituent. In an attempt to circumvent this complication, additional calculations were carried out using N,N-dimethylallylamine as the model substrate. Both unrestricted UKS and BS possibilities were calculated at the B3LYP level, in agreement with the experimentally observed S = 1/2 ground state of B. Both calculations converged to a BS(2,1) solution analogous to that obtained for 8, although maintaining the κ3 coordination mode for the bis(imino)pyridine chelate resulted in a significant distortion from a planar geometry. The computed EPR parameters (g1 =2.264, g2 = 2.277, g3 = 2.189) are significantly different from the experimental values, discounting the validity of this computational output. Beginning the geometry optimizations of N,N-dimethylallylamine with the olefin within the idealized square plane, perpendicular to the chelate, also converged to a BS(2,1) solution with a principally cobalt-centered spin, resulting in similar computed EPR parameters that also contradict the experimental findings. Calculations with these model substrates using other functionals, including BP86, TPSS, and M06,77 also resulted in solutions that are inconsistent with our experimental values.
The absence of large anisotropy in the ambient-temperature EPR spectrum of B as well as in the spectrum at 10 K strongly suggests that while there is likely an increased contribution from electron density at cobalt to the observed signal in comparison with the bis(imino)pyridine cobalt dinitrogen compounds, the SOMO remains principally chelate-based.78 In the absence of experimental metrical parameters, our attempts to determine the electronic structure of B computationally have been unsuccessful, but our experimental evidence suggests the formation of a κ3-bis(imino)pyridine cobalt alkene complex having a similar geometry and electronic structure as the cobalt dinitrogen compounds.
Mechanistic Proposal for Cobalt-Catalyzed [2π + 2π] Cycloaddition
On the basis of the isolated and calculated catalytic intermediates, a proposed catalytic cycle that accounts for the observed reactivity, resting states as determined by in situ EPR spectroscopic monitoring, steric and electronic catalyst effects, and deuterium labeling is presented in Scheme 5.
Scheme 5. Proposed Mechanism of Bis(imino)pyridine Cobalt Dinitrogen-Catalyzed [2π + 2π] Cycloaddition.

The proposed mechanism involves initial, reversible displacement of dinitrogen by an alkene to yield B, an intermediate observed with several cobalt–substrate combinations by room-temperature fluid solution EPR spectroscopy. Coordination of the second olefin displaces an imine arm, forming the cobalt diene complex A with a κ2-bis(imino)pyridine. The catalytic resting states observed by EPR spectroscopy are highly dependent on the cobalt precatalyst as well as the substrate. The observed trend establishes that with increasing steric bulk at the imine carbon position (R group in Scheme 6) and larger substituents on the N-aryl ring of the imine (Ar in Scheme 6), the equilibrium among the cobalt dinitrogen compound, A, and B favors the dinitrogen compound. Oxidative cyclization furnishes the resulting unobserved, proposed cobalt metallacycle C. Key to catalytic turnover is the conversion of the planar cobalt dinitrogen and alkene (B) compounds to tetrahedral diene derivatives (A). These reactions highlight the ability of the ligand hemilability to transform the principally bis(imino)pyridine-based SOMO of the precatalyst and the alkene compound into a primarily metal-based orbital upon dissociation of one of the imine arms and coordination of a second alkene. From this cobalt-based SOMO, C–C bond-forming oxidative cyclization occurs via a metal-based redox event. Subsequent turnover-limiting directional C(sp3)–C(sp3) reductive elimination79 releases the cyclobutane product and regenerates the cobalt dinitrogen compound in the presence of an atmosphere of N2.
Scheme 6. Structure–Reactivity Relationships in Bis(imino)pyridine Cobalt Dinitrogen Precatalysts for the [2π + 2π] Cycloaddition of α,ω-Dienes.
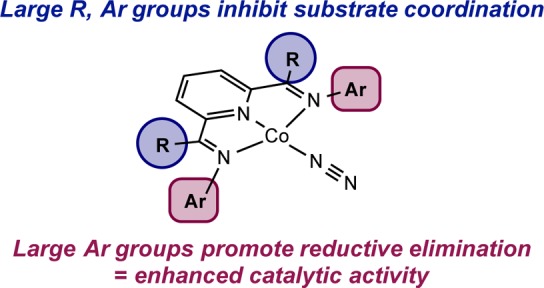
Although cobalt metallacycles (C) have not been directly observed, structural manipulation of the cobalt precatalysts supports turnover-limiting reductive elimination (Scheme 5). The over 4-fold decrease in the [2π + 2π] cycloaddition turnover frequency upon introduction of a 4-pyrrolidonyl substituent (7 vs 1) is consistent with lower rates of C–C bond-forming reductive elimination from more electron-rich metal centers,80 while no change in resting state was observed by EPR spectroscopy. It should be noted that while deuterium labeling experiments support a two-electron reductive elimination process, a Co–C bond homolysis event followed by C–C bond formation on a time scale faster than Co–C bond rotation cannot be definitively excluded. If such a pathway were operative, similar electronic influences would expected.
Other precatalyst substituent effects reported in Table 4 are consistent with the mechanism proposed in Scheme 5. The observed trend in catalytic activity with the size of the imine carbon substituents (Me > Et > iPr) is consistent with the first order dependence on the substrate and observations from EPR spectroscopy. Increasing the size of the substituents above and below the idealized metal–ligand plane decreases the equilibrium constant for substrate coordination. Because this equilibrium occurs prior to the rate-determining step, the overall turnover frequency for catalytic [2π + 2π] cycloaddition is affected by this coordination. As illustrated with 3 and 4, reducing the size of the imine aryl substituents increases the equilibrium constant for substrate coordination, making the overall rate zeroth order in diene, but poor catalytic performance is still observed. This observation further supports reductive elimination as turnover-limiting. Why then is the cyclopentyl-substituted cobalt precatalyst 2 the most active in the series? The steric profile of the ring generates a sufficiently open environment to encourage substrate coordination yet exhibits a large enough profile to encourage reductive elimination (Scheme 6).
Concluding Remarks
A family of cobalt dinitrogen compounds bearing redox-active bis(imino)pyridine ligands are active for the catalytic [2π + 2π] cycloaddition of α,ω-dienes to form the corresponding bicyclic products. Systematic evaluation of the steric and electronic properties of the bis(imino)pyridine chelates established that relatively electron-poor cobalt compounds with substituents that favor both diene coordination and C–C reductive elimination are the most active for cyclobutane formation. Deuterium labeling studies established a stereospecific reaction, while EPR spectroscopic studies were used to identify the catalyst resting state. Isomeric cobalt–substrate complexes were identified, including κ3-bis(imino)pyridine cobalt η2-alkene compounds and κ2-bis(imino)pyridine cobalt η2,η2-diene derivatives. An example of the latter was structurally characterized. The combined spectroscopic, structural, and computational studies support hemilability as a modulator of the ligand field strength as key to the observed catalytic chemistry. Although S = 1/2 ground states are maintained throughout turnover, the parentage of the SOMO changes from principally ligand-based in the planar intermediates to essentially metal-based in the tetrahedral complexes, which ultimately enables C–C bond formation
Experimental Section81
Synthesis of (TricPDI)CoN2 (2)
A mixture of (TricPDI) (0.790 g, 1.09 mmol, 1.05 equiv) and CoCl2 (0.135 g, 1.04 mmol, 1.0 equiv) in THF (10 mL) was stirred at room temperature for 16 h before filtration to collect (TricPDI)CoCl2 (0.726 g, 0.852 mmol, 82% yield) as a mustard-yellow solid. To a round-bottom flask containing mercury (4.71 g, 23.5 mmol, 100 equiv) and toluene (25 mL) was added sodium (27 mg, 1.2 mmol, 5 equiv) in small pieces. The mixture was allowed to amalgamate for 10 min, and then (TricPDI)CoCl2 (0.200 g, 0.235 mmol, 1 equiv) was added. A color change to mauve-purple was initially observed before a slow color change to dark teal over the course of 8 h. The solution was then filtered through Celite and washed with pentane, and a concentrated solution was chilled to −35 °C to afford (TricPDI)CoN2 (2) (0.167 g, 0.206 mmol, 88% yield) as a dark-teal solid. X-ray-quality crystals were obtained by chilling a concentrated toluene solution of 2. Anal. Calcd for C51H67N5Co: C, 75.71; H, 8.35; N, 8.66. Found: C, 75.78; H, 8.24; N, 8.34. IR (pentane): νNN = 2106 cm–1.
Preparation of (iPr(TB)PDI)Co(N,N-diallyl-4-methoxyaniline) (8)
A 20 mL scintillation vial was charged with 0.075 g (0.12 mmol) of (iPr(TB)PDI)CoN2 and approximately 2 mL of pentane. A solution containing 0.074 g (0.36 mmol) of N,N-diallyl-4-methoxyaniline and approximately 1 mL of pentane was added. The resulting slurry was mixed for 5 min, and the solid was collected by filtration on a glass frit. The solid was washed with pentane (2 × 0.5 mL), affording 0.062 g (78% yield) of a purple solid identified as (iPr(TB)PDI)Co(N,N-diallyl-4-methoxyaniline) (8). Anal. Calcd for C50H65N4OCo: C, 75.35; H, 8.22; N, 7.03. Found: C, 75.11; H, 8.14; N, 6.80. Magnetic Susceptibility Balance: μeff = 1.8 μB (23 °C).
General Procedure for Cobalt-Catalyzed Diene [2π + 2π] Cycloaddition
In a nitrogen-filled glovebox, a 20 mL scintillation vial was charged with (iPrPDI)CoN2 (1) (5 mg, 0.00879 mmol, 0.025 equiv) and PhMe (0.2M, 1.7 mL) before the α,ω-diene (0.352 mmol, 1 equiv) was added via microsyringe. The mixture was stirred at room temperature in the glovebox until the substrate was completely consumed as judged by GC. The reaction mixture was then quenched by exposure to air, addition of 500 μL MeOH, and allowing the crude mixture to sit for 1 h. Filtration through a short silica plug and flushing with additional MeOH afforded analytically pure cyclobutane product upon removal of the solvent under reduced pressure.
Acknowledgments
V.A.S. thanks the NIH for a Ruth L. Kirschstein National Research Service Award (F32 GM109594) and the Princeton University Intellectual Property Accelerator Fund for financial support. We thank Dr. Carsten Milsmann for helpful discussions on EPR analyses.
Supporting Information Available
Additional experimental details; electrochemical characterization; spectroscopic, kinetic, and computational details; and crystallographic data for 2, (C5H9PDI)CoN2, 5, 7, and 8 in CIF format. The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.5b04034.
The authors declare no competing financial interest.
Supplementary Material
References
- Lautens M.; Klute W.; Tam W. Chem. Rev. 1996, 96, 49–92. [DOI] [PubMed] [Google Scholar]
- Ojima I.; Tzamarioudaki M.; Li Z.; Donovan R. J. Chem. Rev. 1996, 96, 635–662. [DOI] [PubMed] [Google Scholar]
- Aubert C.; Buisine O.; Malacria M. Chem. Rev. 2002, 102, 813–834. [DOI] [PubMed] [Google Scholar]
- Watson I. D. G.; Toste F. D. Chem. Sci. 2012, 3, 2899–2919. [Google Scholar]
- Yamamoto Y. Chem. Rev. 2012, 112, 4736–4769. [DOI] [PubMed] [Google Scholar]
- Woodward R. B.; Hoffmann R. Angew. Chem., Int. Ed. Engl. 1969, 8, 781–853. [Google Scholar]
- Dembitsky V. M. J. Nat. Med. 2008, 62, 1–33. [DOI] [PubMed] [Google Scholar]
- Apers S.; Vlietinck A.; Pieters L. Phytochem. Rev. 2003, 2, 201–217. [Google Scholar]
- Davis R. A.; Barnes E. C.; Longden J.; Avery V. M.; Healy P. C. Bioorg. Med. Chem. 2009, 17, 1387–1392. [DOI] [PubMed] [Google Scholar]
- Seiser T.; Saget T.; Tran D. N.; Cramer N. Angew. Chem., Int. Ed. 2011, 50, 7740–7752. [DOI] [PubMed] [Google Scholar]
- Namyslo J. C.; Kaufmann D. E. Chem. Rev. 2003, 103, 1485–1538. [DOI] [PubMed] [Google Scholar]
- Lee-Ruff E.; Mladenova G. Chem. Rev. 2003, 103, 1449–1484. [DOI] [PubMed] [Google Scholar]
- Singh J.; Bisacchi G. S.; Ahmad S.; Godfrey J. D. Jr.; Kissick T. P.; Mitt T.; Kocy O.; Vu T.; Papaioannou C. G.; Wong M. K.; Heikes J. E.; Zahler R.; Mueller R. H. Org. Process Res. Dev. 1998, 2, 393–399. [Google Scholar]
- Marrero J.; Rodríguez A. D.; Baran P.; Raptis R. G. Org. Lett. 2003, 5, 2551–2554. [DOI] [PubMed] [Google Scholar]
- Gutekunst W. R.; Baran P. S. J. Org. Chem. 2014, 79, 2430–2452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prier C. K.; Rankic D. A.; MacMillan D. W. C. Chem. Rev. 2013, 113, 5322–5363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crimmins M. T. Chem. Rev. 1988, 88, 1453–1473. [Google Scholar]
- Demuth M.; Mikhail G. Synthesis 1989, 145–162. [Google Scholar]
- Narayanam J. M. R.; Stephenson C. R. J. Chem. Soc. Rev. 2011, 40, 102–113. [DOI] [PubMed] [Google Scholar]
- Hurtley A. E.; Lu Z.; Yoon T. P. Angew. Chem., Int. Ed. 2014, 53, 8991–8994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du J.; Skubi K. L.; Schultz D. M.; Yoon T. P. Science 2014, 344, 392–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ischay M. A.; Ament M. S.; Yoon T. P. Chem. Sci. 2012, 3, 2807–2811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canales E.; Corey E. J. J. Am. Chem. Soc. 2007, 129, 12686–12687. [DOI] [PubMed] [Google Scholar]
- Jiang X.; Cheng X.; Ma S. Angew. Chem., Int. Ed. 2006, 45, 8009–8013. [DOI] [PubMed] [Google Scholar]
- Chao K. C.; Rayabarapu D. K.; Wang C.-C.; Cheng C.-H. J. Org. Chem. 2001, 66, 8804–8810. [DOI] [PubMed] [Google Scholar]
- Huang D.-J.; Rayabarapu D. K.; Li L.-P.; Sambaiah T.; Cheng C.-H. Chem.—Eur. J. 2000, 6, 3706–3713. [DOI] [PubMed] [Google Scholar]
- Rayabarapu D. K.; Cheng C.-H. Acc. Chem. Res. 2007, 40, 971–983. [DOI] [PubMed] [Google Scholar]
- Treutwein J.; Hilt G. Angew. Chem., Int. Ed. 2008, 47, 6811–6813. [DOI] [PubMed] [Google Scholar]
- Burton R. R.; Tam W. J. Org. Chem. 2007, 72, 7333–7336. [DOI] [PubMed] [Google Scholar]
- Shibata T.; Takami K.; Kawachi A. Org. Lett. 2006, 8, 1343–1345. [DOI] [PubMed] [Google Scholar]
- Ohara H.; Itoh T.; Nakamura M.; Nakamura E. Chem. Lett. 2001, 30, 624–625. [Google Scholar]
- Baik T.-G.; Luis A. L.; Wang L.-C.; Krische M. J. J. Am. Chem. Soc. 2001, 123, 6716–6717. [DOI] [PubMed] [Google Scholar]
- Wang L.-C.; Jang H.-Y.; Roh Y.; Lynch V.; Schultz A. J.; Wang X.; Krische M. J. J. Am. Chem. Soc. 2002, 124, 9448–9453. [DOI] [PubMed] [Google Scholar]
- Yang J.; Cauble D. F.; Berro A. J.; Bauld N. L.; Krische M. J. J. Org. Chem. 2004, 69, 7979–7984. [DOI] [PubMed] [Google Scholar]
- Trost B. M.; Yanai M.; Hoogsteen K. J. Am. Chem. Soc. 1993, 115, 5294–5295. [Google Scholar]
- Mitsudo T.; Naruse H.; Kondo T.; Ozaki Y.; Watanabe Y. Angew. Chem., Int. Ed. Engl. 1994, 33, 580–581. [Google Scholar]
- Alcaide B.; Almendros P.; Aragoncillo C. Chem. Soc. Rev. 2010, 39, 783–816. [DOI] [PubMed] [Google Scholar]
- Gulías M.; Collado A.; Trillo B.; López F.; Oñate E.; Esteruelas M. A.; Mascareñas J. L. J. Am. Chem. Soc. 2011, 133, 7660–7663. [DOI] [PubMed] [Google Scholar]
- Noucti N. N.; Alexanian E. J. Angew. Chem., Int. Ed. 2015, 54, 5447–5450. [DOI] [PubMed] [Google Scholar]
- Sarkar N.; Nayek A.; Ghosh S. Org. Lett. 2004, 6, 1903–1905. [DOI] [PubMed] [Google Scholar]
- Bach T.; Bergmann H.; Grosch B.; Harms K. J. Am. Chem. Soc. 2002, 124, 7982–7990. [DOI] [PubMed] [Google Scholar]
- Müller C.; Bauer A.; Maturi M. M.; Cuquerella M. C.; Miranda M. A.; Bach T. J. Am. Chem. Soc. 2011, 133, 16689–16697. [DOI] [PubMed] [Google Scholar]
- Parra A.; Reboredo S.; Alemán J. Angew. Chem., Int. Ed. 2012, 51, 9734–9736. [DOI] [PubMed] [Google Scholar]
- Brimioulle R.; Bach T. Science 2013, 342, 840–843. [DOI] [PubMed] [Google Scholar]
- Du J.; Skubi K. L.; Schultz D. M.; Yoon T. P. Science 2014, 344, 392–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narasaka K.; Hayashi Y.; Shimadzu H.; Niihata S. J. Am. Chem. Soc. 1992, 114, 8869–8885. [Google Scholar]
- Conner M. L.; Xu Y.; Brown M. K. J. Am. Chem. Soc. 2015, 137, 3482–3485. [DOI] [PubMed] [Google Scholar]
- Speiser F.; Braunstein P.; Saussine L. Acc. Chem. Res. 2005, 38, 784–793. [DOI] [PubMed] [Google Scholar]
- Grubbs R. H.; Miyashita A. J. Am. Chem. Soc. 1978, 100, 7416–7418. [Google Scholar]
- Heimbach P.; Brenner W. Angew. Chem., Int. Ed. Engl. 1967, 6, 800. [Google Scholar]
- Binger P. Angew. Chem., Int. Ed. Engl. 1972, 11, 309–310. [Google Scholar]
- Noyori R.; Ishigami T.; Hayashi N.; Takaya H. J. Am. Chem. Soc. 1973, 95, 1674–1676. [Google Scholar]
- Billups W. E.; Cross J. H.; Smith C. V. J. Am. Chem. Soc. 1973, 95, 3438–3439. [Google Scholar]
- Greco A.; Carbonaro A.; Dall’Asta G. J. Org. Chem. 1970, 35, 271–274. [Google Scholar]
- Carbonaro A.; Cambisi F.; Dall’Asta G. J. Org. Chem. 1971, 36, 1443–1445. [Google Scholar]
- Cannell L. G. J. Am. Chem. Soc. 1972, 94, 6867–6869. [Google Scholar]
- Bart S. C.; Lobkovsky E.; Chirik P. J. J. Am. Chem. Soc. 2004, 126, 13794–13807. [DOI] [PubMed] [Google Scholar]
- Russell S. K.; Darmon J. M.; Lobkovsky E.; Chirik P. J. Inorg. Chem. 2010, 49, 2782–2792. [DOI] [PubMed] [Google Scholar]
- Bouwkamp M. W.; Bowman A. C.; Lobkovsky E.; Chirik P. J. J. Am. Chem. Soc. 2006, 128, 13340–13341. [DOI] [PubMed] [Google Scholar]
- Trovitch R. J.; Lobkovsky E.; Bouwkamp M. W.; Chirik P. J. Organometallics 2008, 27, 6264–6278. [Google Scholar]
- Hoyt J. M.; Sylvester K. T.; Semproni S. P.; Chirik P. J. J. Am. Chem. Soc. 2013, 135, 4862–4877. [DOI] [PubMed] [Google Scholar]
- Russell S. K.; Lobkovsky E.; Chirik P. J. J. Am. Chem. Soc. 2011, 133, 8858–8861. [DOI] [PubMed] [Google Scholar]
- Chirik P. J. Inorg. Chem. 2011, 50, 9737–9740. [DOI] [PubMed] [Google Scholar]
- Danopoulos A. A.; Wright J. A.; Motherwell W. B. Chem. Commun. 2005, 784–786. [DOI] [PubMed] [Google Scholar]
- Darmon J. M.; Stieber S. C. E.; Sylvester K. T.; Fernández I.; Lobkovsky E.; Semproni S. P.; Bill E.; Weighardt K.; DeBeer S.; Chirik P. J. J. Am. Chem. Soc. 2012, 134, 17125–17137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gandeepan P.; Cheng C.-H. Acc. Chem. Res. 2015, 48, 1194–1206. [DOI] [PubMed] [Google Scholar]
- For examples with activated alkenes and alkynes, see ref (25).
- Bowman A. C.; Milsmann C.; Atienza C. C. H.; Lobkovsky E.; Chirik P. J. J. Am. Chem. Soc. 2010, 132, 1676–1684. [DOI] [PubMed] [Google Scholar]
- Bowman A. C.; Milsmann C.; Bill E.; Lobkovsky E.; Weyhermuller T.; Weighardt K.; Chirik P. J. Inorg. Chem. 2010, 49, 6110–6123. [DOI] [PubMed] [Google Scholar]
- Zhu D.; Budzelaar P. H. M. Organometallics 2010, 29, 5759–5761. [Google Scholar]
- Knijnenburg Q.; Hetterscheid D.; Kooistra T. M.; Budzelaar P. H. M. Eur. J. Inorg. Chem. 2004, 1204–1211. [Google Scholar]
- Bart S. C.; Chlopek K.; Bill E.; Bouwkamp M. W.; Lobkovsky E.; Neese F.; Wieghardt K.; Chirik P. J. J. Am. Chem. Soc. 2006, 128, 13901–13912. [DOI] [PubMed] [Google Scholar]
- Darmon J. M.; Turner Z. R.; Lobkovsky E.; Chirik P. J. Organometallics 2012, 31, 2275–2285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obligacion J. V.; Chirik P. J. J. Am. Chem. Soc. 2013, 135, 19107–19110. [DOI] [PubMed] [Google Scholar]
- Sylvester K. T.; Chirik P. J. J. Am. Chem. Soc. 2009, 131, 8772–8774. [DOI] [PubMed] [Google Scholar]
- Neese F. Wiley Interdiscip. Rev.: Comput. Mol. Sci. 2012, 2, 73–78. [Google Scholar]
- Brogden D. W.; Berry J. F. Chem. Commun. 2015, 51, 9153–9156. [DOI] [PubMed] [Google Scholar]
- Knijnenburg Q.; Horton A. D.; van der Heijden H.; Kooistra T. M.; Hetterscheid D. G. H.; Smits J. M. M.; de Bruin B.; Budzelaar P. H. M.; Gal A. W. J. Mol. Catal. A: Chem. 2005, 232, 151–159. [Google Scholar]
- For an example of C(sp3)–C(sp3) bond-forming reductive elimination from Co(III) to Co(I), see:Xu H.; Bernskoetter W. H. J. Am. Chem. Soc. 2011, 133, 14956–14959. [DOI] [PubMed] [Google Scholar]
- Hartwig J. F. Inorg. Chem. 2007, 46, 1936–1947. [DOI] [PubMed] [Google Scholar]
- Representative procedures are reported here; complete details, including general considerations, are reported in the Supporting Information.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.