

Summary
Regulatory T cells (Tregs) play a pivotal role in the inhibition of anti-tumor immune responses. Understanding the mechanisms governing Treg homeostasis may therefore be important for development of effective tumor immunotherapy. We have recently demonstrated a key role for the canonical nuclear factor κB (NF-κB) subunits, p65 and c-Rel, in Treg identity and function. In this report, we show that NF-κB c-Rel ablation specifically impairs the generation and maintenance of the activated Treg (aTreg) subset, which is known to be enriched at sites of tumors. Using mouse models, we demonstrate that melanoma growth is drastically reduced in mice lacking c-Rel, but not p65, in Tregs. Moreover, chemical inhibition of c-Rel function delayed melanoma growth by impairing aTreg-mediated immunosuppression and potentiated the effects of anti-PD-1 immunotherapy. Our studies therefore establish inhibition of NF-κB c-Rel as a viable therapeutic approach for enhancing checkpoint-targeting immunotherapy protocols.
Graphical abstract
NF-κB c-Rel is critical for the function of activated Tregs and serves as a target to reduce Treg function in the tumor microenvironment without compromising systemic tolerance or causing autoimmunity.

Introduction
Tumors employ a wide variety of strategies to grow, metastasize, and avoid recognition and elimination by the immune system. One of the major mechanisms of immune evasion by tumors is through the engagement of immunosuppressive receptors, such as PD-1 and CTLA-4, on effector T cells by their ligands expressed in the tumor microenvironment. Emerging cancer immunotherapy approaches are aimed toward overcoming such tolerance mechanisms and promoting a productive anti-tumor immune response. Although these immune checkpoint blockade therapies are revolutionizing cancer care (Sharma and Allison, 2015), there are a significant number of tumors that do not benefit from such approaches alone. One possible explanation for such resistance is that other immunosuppressive mechanisms are engaged in such tumors. For example, CD4+Foxp3+ regulatory T cells (Tregs) suppress anti-tumor responses (Nishikawa and Sakaguchi, 2010; Tang and Bluestone, 2008). Tregs are highly enriched in patient melanomas (Jandus et al., 2008; Ouyang et al., 2016) and their presence in tumors correlates with poor prognosis (Baumgartner et al., 2009).
Tregs, that either develop in the thymus (nTreg) or are induced in the periphery (iTreg), normally represent 5%–15% of the CD4+ T cell population. They can inhibit immune responses through diverse mechanisms that include secretion of inhibitory cytokines, direct cytotoxicity, disruption of metabolic pathways in target cells, or inhibition of antigen-presenting cells (Tang and Bluestone, 2008). The expression of the forkhead-box transcription factor Foxp3 is crucial for the acquisition of the Treg suppressive program and maintenance of Treg identity (Samstein et al., 2012). Tregs may exist as specialized subsets that have distinct functions. In mice, the resting Treg population (rTreg) that resides in lymphoid tissues acts to prevent lymphoproliferation and autoimmunity and is maintained by the transcription factor Foxo1 (Huehn et al., 2004; Luo et al., 2016). In contrast, the “effector-memory like” activated Treg subset (aTreg) migrates to inflamed tissues and tumors and has been shown to act as a potent inhibitor of anti-tumor responses (Darrasse-Jèze et al., 2009; Levine et al., 2014; Luo et al., 2016). It was recently proposed that IRF-4 and Myb could help maintain aTreg homeostasis (Levine et al., 2014) (Dias et al., 2017). However, the regulators of aTreg differentiation and function are poorly characterized. Hence, understanding the molecular signals that drive Treg development and homeostasis remains of great interest.
Several studies have shown that the transcription factor NF-κB, in particular the c-Rel subunit, is crucial for the expression of FoxP3 and the thymic development of Tregs (Isomura et al., 2009; Long et al., 2009; Ruan et al., 2009). Activation of nuclear factor κB (NF-κB) occurs through two pathways, the canonical and the non-canonical pathway. The canonical pathway leads to activation of NF-κB heterodimers consisting of p50 and p65, or p50 and c-Rel, and is primarily involved in immune activation and cell survival. The non-canonical pathway leads to nuclear translocation of p52-RelB heterodimers and is mainly involved in lymphoid organogenesis. NF-κB complexes containing either p65 or c-Rel can perform distinct biological roles. While p65 containing NF-κB complexes are mainly responsible for cellular activation responses, c-Rel containing NF-κB complexes play more specialized rolesinthe immune response and lymphoid development. Germline deletion of p65 leads to embryonic lethality, and cells lacking p65 show broad defects in survival, proliferation, and function (Beg et al., 1995). In contrast, mice lacking c-Rel are viable and despite strong c-Rel expression in lymphocytes, show limited immunological defects (Köntgen et al., 1995). Hence, availability of inhibitors that could target only NF-κB c-Rel would likely avoid the undesirable side effects that have halted advancement of NF-κB-inhibitors for therapeutic applications in inflammatory diseases and cancer (DiDonato et al., 2012).
With our discovery that NF-κB c-Rel and p65 have divergent roles in the maintenance of Treg function (Oh et al., 2017), we tested the hypothesis that inhibiting c-Rel might be a viable method of selectively modulating Treg activity in cancer. We first observed that c-Rel, but not p65, was an important regulator of aTreg maintenance. Using genetic models with specific deletion of either p65 or c-Rel in Tregs, we found that c-Rel was selectively required for Treg-mediated melanoma tolerance. We have confirmed that Pentoxfylline (PTXF), an FDA-approved drug, can cause selective degradation of c-Rel, without affecting p65. Inhibition of c-Rel using PTXF delays tumor growth by altering Treg identity and function. Finally, we show that PTXF administration, along with PD-1-checkpoint blockade, has an additive effect in treating established melanoma in mice. These results confirm recent work suggesting that prevention of tumor tolerance and suppression of lymphoproliferation and autoimmunity are the result of distinct Treg functions (Luo et al., 2016; Sugiyama et al., 2013) and provide preclinical evidence for the use of c-Rel inhibitors in conjunction with other immune checkpoint inhibitors.
Results
NF-κB c-Rel Regulates Differentiation and Gene Expression of the Activated Treg Population
In the accompanying study (Oh et al., 2017), we show that mice lacking p65 in Tregs develop a lethal autoimmune syndrome by 6–12 weeks, whereas deletion of c-Rel leads to mild inflammation, but only after 20 weeks of age. These phenotypes were associated with distinct transcriptional programs governed by p65 and c-Rel and suggested a more prominent role for p65 in Tregs for the maintenance of peripheral tolerance and prevention of autoimmunity. We wondered whether the NF-κB subunits played distinct roles in the different Treg subsets and therefore analyzed gene expression profiles of WT aTreg (CD44hiCD62Llow) and rTreg (CD44lowCD62Lhigh) by RNA sequencing (RNA-seq) and compared them to the gene expression profiles from wild-type (WT), as well as p65 and c-Rel-deficient total Tregs (Oh et al., 2017). We first identified gene sets enriched in aTreg (841 genes) and rTreg (320 genes) populations. Ablation of p65 only marginally affected aTreg and rTreg gene signatures (Figure 1A). However, in the absence of c-Rel, the numbers of affected genes increased dramatically (Figure 1B). Hallmarks of aTregs, such as Itgae, Tigit, Klrg1, Il1r2, or Tnfrsf8, were downregulated in c-Rel-deficient Tregs, while their expression was unaffected in p65-deficient Tregs (Figure 1C). The majority of the affected genes have been suggested to regulate Treg homeostasis and/or function, especially during cancer. We wanted to know whether this effect on aTreg genes was due to a reduction in aTreg numbers or rather due to an intrinsic defect in gene expression. Fluorescence-activated cell sorting (FACS) analysis of splenic T cells from Foxp3CREp65F/F animals showed a slight increase in the proportion of aTreg among total Tregs compared to controls; in contrast, we observed a profound decrease of aTregs in Foxp3CREc-RelF/F mice (Figures 1D and 1E), whereas rTreg numbers were unchanged (Figure 1F). Similarly, the proportion of Tregs expressing CD103 was reduced, whereas the Ki-67+ and Ly6C+ population were expanded in these mice (Figures 1G, S1A, and S1B). Therefore, c-Rel, but not p65, appears to be important for the maintenance of aTreg numbers. We next assessed whether NF-κB was required for establishment of the aTreg transcriptional landscape in already differentiated aTregs, by performing RNA-seq on sorted aTreg and rTreg from p65- and c-Rel-deficient mice. Interestingly, deletion of either subunit led to impairment of the aTreg gene signature, as 103 or 104 genes, respectively, out of the 841 genes that characterize aTregs, were significantly downregulated (Figures 1H, S1C, and S1D). However, careful examination of these genes revealed a more important role for c-Rel in the expression of aTreg hallmark genes such as Tnfrsf8, Klrg1, Il1r2, Tigit, or Ccr8 that were all downregulated in c-Rel knockout (KO) aTregs, but affected to a lesser extent in p65-deficient aTregs (Figure 1H). A significant decrease in Klrg1 and PD-1 protein expression in c-Rel-deficient, but not p65-deficient aTreg was also detected by FACS (Figures S1G and S1H). Finally, analysis of the rTreg transcriptome did not reveal a critical involvement of either p65 or c-Rel (Figures S1E and S1F), suggesting that other transcription factors, such as Foxo 1, might be more important for the maintenance of the rTreg population. Taken together, these results demonstrate that c-Rel plays a central role in aTreg biology and suggests that c-Rel deletion might have effects on anti-tumor responses.
Figure 1. NF-κB c-Rel Regulates the Activated-Treg Differentiation and Gene Expression.
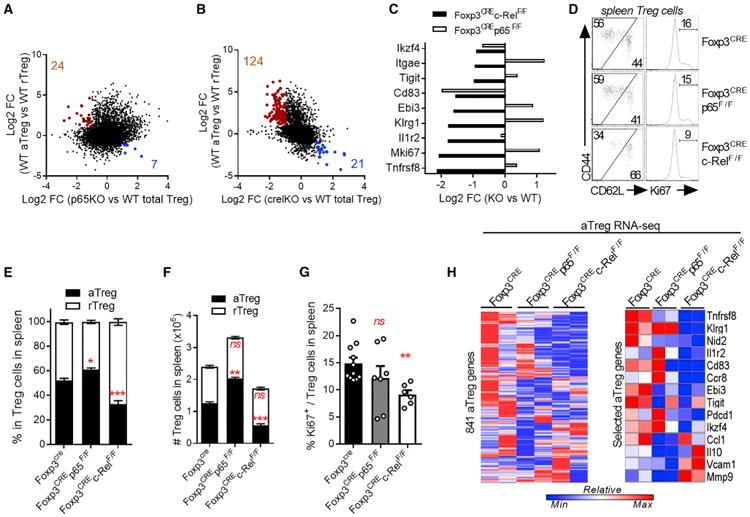
(A–C) CD62Llow CD44high aTreg and CD62Lhigh CD44low rTreg were sorted from Foxp3CRE-YFP (WT) mice and submitted to RNA-seq analysis. Gene expression was compared to that in stimulated total Foxp3crep65F/F (p65KO) and Foxp3crec-RelF/F (c-RelKO) Treg (Oh et al., 2017). (A and B) Gene expression changes in WT aTreg versus WT rTreg were plotted against those in p65KO versus WT total Tregs (A) and c-RelKO versus WT total Tregs (B). Numbers and colored dots indicate genes upregulated in aTreg while downregulated in KO Treg (red) and downregulated in aTreg while upregulated in KO Treg (blue) (fold change >2, p < 0.01).
(C) Expression of selected aTreg genes.
(D) Representative FACS profiles in spleen Tregs from 5- to 7-week-old Foxp3cre, Foxp3crep65F/F, and Foxp3crec-RelF/F mice.
(E and F) Cumulative % (E) and absolute numbers (F) of rTreg and aTreg in spleen Tregs.
(G) Cumulative % of Ki67+ in spleen Tregs.
(H) RNA-seq analysis of aTregs sorted from Foxp3cre, Foxp3crep65F/F, and Foxp3crec-RelF/F. Left heatmap shows expression of all 841 aTreg genes (fold change >2, p < 0.01 in WT aTreg versus WT rTreg) in each genotype. Right heatmap shows expression of selected aTreg genes. All RNA-seq data come from 2 independent experiments. FACS data is shown as mean ± SEM of 3 experiments with >6 mice/group. *p < 0.05, **p < 0.01, **p < 0.001; ns, non-significant. See also Figure S1.
Tregs Specifically Require c-Rel to Inhibit Anti-tumor Effector Responses
To test the role of p65 and c-Rel in Tregs during anti-tumor responses, we compared the growth of B16F1 melanoma cells in WT mice versus mice lacking either NF-κB subunit. We observed exponential melanoma growth in control Foxp3CRE transgenic animals (Figure 2A), and despite the functional defects in Tregs lacking p65 (Oh et al., 2017), tumor growth was unaltered in Foxp3CREp65F/F mice (Figure 2A). In contrast, only 50% of the Foxp3CREc-RelF/F animals exhibited detectable tumors after 2 weeks, and these tumors were significantly smaller than those seen in control littermates (Figure 2A). We next measured T cell infiltration 2 weeks after tumor inoculation and observed an equal increase of CD8+ T cells in p65- and c-Rel-deficient mice in tumor infiltrating lymphocytes (TILs). CD4+ T cells, however, were only increased in Foxp3CREc-RelF/F mice (Figures 2B and 2C), but the intrinsic expression of IFN-γ by T cells remained unchanged between strains (Figure 2D). We observed an equivalent reduction in the proportion of TIL Tregs in both c-Rel- and p65-deficient animals when compared to WT littermates (Figure 2B). Therefore, the difference in tumor growth between the p65 and c-Rel KO strains reflects divergence in the functional capability of Tregs. This fits our initial observation on the important role of c-Rel in the homeostasis of the aTreg population, which was suggested to specifically control tumor immunity (Figure 1).
Figure 2. c-Rel Expression in Tregs Restricts Anti-tumor Immune Responses.

Five- to 7-week-old Foxp3cre, Foxp3crep65F/F, and Foxp3crec-RelF/F were transplanted subcutaneously with B16F1 cells.
(A) Tumor growth over time. Numbers indicate the number of mice with detectable tumors at the end of the experiment.
(B–H) Flow cytometry analysis 16 days after tumor challenge, with (B–D, G, and H) or without (E–G) PMA-ionomycin re-stimulation. (B) Numbers of T cells among 106 live cells. (C) CD4/CD8 T cells ratio. (D) Percent of IFN-γ+ in gated CD4+Foxp3− (CD4) and CD8+ (CD8) live T cells. (E) Representative FACS profiles in TILs. Numbers indicate % in gate. (F) Percent of Foxp3+ cells among CD4+ T cells. (G and H) Percent of Helios+ and IFN-γ+ among Tregs in TILs.
(I–K) Splenocytes were stained immediately (I and J) or after PMA-ionomycin re-stimulation, 4 days after tumor challenge. Proportion of Ki67+ (I), CD44high (J), and TNFhighIFN-γ+ (K) cells is shown.
(L) Mice were transplanted as in (A) and were injected at D0 and D3 with anti-CD8 or IgG isotype control. Tumor growth over time is shown. All data are represented as mean ± SEM of at least 3 experiments. *p < 0.05, **p < 0.01; n.s., non-significant.
See also Figure S2.
Among the 688 genes whose expression was modified by the absence of c-Rel but not p65, we observed a striking loss of Treg signature genes. For example, the expression of Ikzf2 (Helios) or Ppar-γ, which are important regulators of Treg homeostasis, were significantly decreased; while expression of Pde3b, which is deleterious for Foxp3 stability, was increased (Figure S2A). On the other hand, the expression of effector transcription factors and cytokines, such as Eomes, Tbx21, Il2, or Ifng, were strongly increased in Tregs lacking c-Rel, but not p65 (Figure S2A). The modulation of Helios and IFN-g expression in Tregs was also specifically observed by flow cytometry in the TILs of Foxp3CREc-RelF/F mice, upon melanoma transplantation (Figures 2E, 2G, and 2H). This strongly suggests that the molecular program driven by c-Rel, in contrast to p65, allows Tregs to inhibit anti-tumor responses at early stages following tumor implantation and raises the possibility that Tregs lacking c-Rel could actually promote tumor immunity through effector cytokine production.
To better understand how c-Rel deficiency in Tregs affects early anti-tumor responses, we assessed T cell phenotype 4 days after melanoma inoculation. We observed enhanced expression of Ki67 and CD44 in splenic CD8 T cells of animals with conditional deletion of c-Rel in Tregs, compared to WT littermates (Figures 2I and 2J). This was associated with increased levels of polyfunctional (IFN-γTNFhigh) T cells upon polyclonal restimulation ex vivo (Figure 2K). These parameters remained mostly unchanged in p65-deficient mice. This may explain the different outcomes in tumor growth between c-Rel- and p65-deficient animals. Enhanced CD8 T cell activation in Foxp3CREc-RelF/F mice was observed following transplantation of the tumor, but not in unmanipulated mice (Figure S2B). Moreover, we observed increased secretion of IFN-γ by CD8 T cells upon gp100/p-mel restimulation (Figure S2C). In line with these results, CD8 depletion restored normal tumor growth in all the Foxp3CREc-RelF/F animals (Figure 2L). We confirmed these findings with 2 additional melanoma models. In mice injected intravenously with B16F10 melanoma cells, examination of the lungs showed significantly reduced number of metastatic foci in c-Rel-deficient animals compared to controls (Figure S2D). We also observed decreased growth when we transplanted subcutaneous BRAFCAPten−/− melanomas into c-Rel-deficient animals (Figure S2E). Taken together, our results demonstrate that c-Rel controls a specific genetic program in Tregs that is required for inhibition of the anti-melanoma protective immune response mediated by CD8 T cells.
c-Rel Inhibition Impairs Treg Identity
Although canonical NF-κB signaling is important for conventional T cell responses, the role of c-Rel in cellular immunity in vivo is not well established (Kontgen et al., 1995). Therefore, we hypothesized that inhibition of c-Rel would selectively block Treg-mediated tumor tolerance with minimal inhibition of antitumor responses. Pentoxifylline (PTXF) is a xanthine derivative that has been approved by FDA for clinical use for a wide variety of clinical conditions over the past 30 years. PTXF has been proposed to act through multiple targets in vivo, including as a potent inhibitor of c-Rel expression and activation in T cells (Neo et al., 2014; Wang et al., 1997). In our hands, treatment of Tregs with PTXF led to a significant reduction of c-Rel protein, whereas levels of p65 were unaffected (Figure 3A). We observed significantly decreased expression of Treg markers such as Foxp3, CD25, and Helios (Figure 3B), genes that were also reduced in Tregs upon conditional deletion of c-Rel (Figure S2A). To assess whether the decrease of these Treg markers was due to the effect of PTXF on c-Rel, we overexpressed c-Rel in Tregs using retroviral transduction. Interestingly, c-Rel overexpression alone increased steady-state expression of Foxp3 and Helios (Figure 3C). Whereas PTXF treatment led to a significant decrease in Foxp3, Helios and CD25 expression in GFP-transduced cells, it did not in c-Rel-transduced Tregs. We next used the converse approach of treating WT and c-Rel-deficient Tregs with PTXF. As expected, the levels of Foxp3, Helios and CD25 were reduced in c-Rel-deficient Tregs compared to WT; also, PTXF failed to further decrease the expression of these proteins (Figure 3D). This suggested that PTXF induced a perturbation of the Treg-associated signature through its effect on c-Rel. To determine the extent to which PTXF inhibited the c-Rel-mediated transcriptional program in Tregs, we next compared the transcriptomes of control, PTXF-treated, and c-Rel-deficient Tregs. PTXF treatment led to a more profound alteration of the Treg transcriptome than genetic ablation of c-Rel (Figures 3E, S3A, and S3B), which was not surprising, given that PTXF likely affects additional signaling pathways (Deree et al., 2007; Kamran and Gude, 2013; Pinzani et al., 1996). Of note, the expression of the c-Rel mRNA itself was unaffected by PTXF, suggesting that PTXF was affecting c-Rel through a post-translational mechanism (Figure 3F). A number of genes described to maintain Treg function and homeostasis were altered upon both chemical and genetic inhibition of c-Rel (Figures 3F, S3C, and S3D), such as Gzmb and Tgfb1, both involved in Treg function during cancer. Moreover, known NF-κB target genes, such as Tnfrsf8, Tnaip2, or Pde3b, also displayed altered expression in both PTXF-treated and c-Rel-deficient Tregs. In contrast, Cbl-b and Satb1, which destabilize Tregs, were upregulated in both conditions. Confirmation of gene expression by qPCR showed that these genes were, in fact, affected to a comparable extent by both chemical and genetic inhibition of NF-κB (Figure S3D). Interestingly, we observed only a minor overlap between the WT Treg+PTXF and Foxp3CREp65F/F transcriptomes, reinforcing the idea that PTXF affects c-Rel but not p65 (Figure S3B). Gene set enrichment analysis (GSEA) indicated that, in addition to unique signatures found in the PTXF and c-Rel KO samples, 107 gene signatures overlapped between the 2 samples (Figure 3G). Among them, we detected a loss of Treg-associated genes and an impairment of the T cell activation processes and memory phenotype (Figure S3E). Ingenuity Pathway Analysis revealed that inhibition of the NF-κB pathway was a key feature of the c-Rel-deficiency and PTXF-induced phenotype (Figure S3F). We next assessed whether PTXF treatment also affected the aTreg/rTreg signature. A total of 159 aTreg genes were significantly downregulated in PTXF-Tregs (Figure 3H), including Tnfrsf8, Il1r2, or Nid2. In contrast, 55 rTreg genes were enhanced, such as Klf2 or S1pr1. This indicates an enrichment of rTreg genes at the expense of aTreg genes. Thus, PTXF induced a substantial shift in Treg transcriptional program that is consistent with the change seen upon loss of c-Rel activity.
Figure 3. Chemical c-Rel Inhibition Impairs Treg Identity.

(A and B) CD4+GFP+ Tregs were sorted form Foxp3eGFP spleens and stimulated for 16 hr with anti-CD3/CD28 and mIL-2 in the presence of 500 μg/mL PTXF or H20. (A) Western Blot on total cell lysates and cumulative expression of c-Rel and RelA across 3 experiments. (B) Representative FACS profiles in gated live CD4+ cells.
(C) Tregs sorted from Foxp3RFP mice were infected with GFP or c-Rel-GFP-encoding retroviruses and treated with PTXF or H2O as in (A). Data is shown as MFI relative to the GFP+H2O sample.
(D) Tregs sorted from Foxp3cre (WT) and Foxp3crec-RelF/F (c-RelKO) were treated as in (A) and analyzed by FACS. Data is shown as MFI relative to the WT+H2O sample.
(E–H) CD4+YFP+Tregs sorted from Foxp3cre (WT) and Foxp3crec-RelF/F were activated as in (B) and submitted to RNA-seq analysis. (E) Heatmap of differentially expressed genes (changed in at least one condition, using a fold-change cut-off >2 and a p value < 0.05 when compared to the WT+H2O samples). Gene expression in normalized for each row. (F) Expression of selected genes in each condition. (G) RNA-seq datasets were analyzed for signature enrichment using the C7 GSEA Collection (ImmunoSigDB). The proportion of unique and overlapping signatures (p value < 0.05) is shown. (H) Gene expression in WT aTreg versus WT rTreg (see Figure 1) was plotted against that in PTXF-treated Tregs versus H20-treated Tregs. Numbers and colored dots indicate genes upregulated in aTreg while downregulated in PTXF Treg (red) and downregulated in aTreg while upregulated in PTXF-Treg (blue) (fold change >2, p < 0.01).
(I–K) CD4+GFP+ Tregs were stimulated as in (B) and subsequently tested for in vitro suppression (I) and in vivo colitis assays (J and K). (I) Suppression of responder T cells proliferation. (J) Weight changes upon cell transfer, shown as % of the initial weight at D0. (K) Total colon length at D35 after transfer. In (A)–(D) and (I)–(K), data are shown as mean ± SEM and are cumulative of 3–4 experiments. In (E)–(H), RNA-seq data is from 2 independent experiments. *p < 0.05, **p < 0.01, ***p < 0.001; n.s., non-significant.
See also Figure S3.
Tregs lacking NF-κB c-Rel exhibit little change in suppressive ability in in vitro T cell suppression assays, despite a complete loss of function in in vivo assays (Oh et al., 2017). In an in vitro suppression assay, PTXF-treated Tregs exhibited modest, but significant, reduction in suppression of effector T cell proliferation when compared to control Tregs (Figure 3E) suggesting that PTXF likely affected some additional targets besides those regulated by c-Rel. However, in an in vivo suppression assay, PTXF-treated Treg completely failed to prevent colitis (Figures 3F, 3G, and S3H) similar to c-Rel-deleted Tregs (Oh et al., 2017). In both experiments, PTXF did not affect Treg survival but rather impaired their intrinsic function (Figures S3G and S3I). Taken together, these results show that PTXF treatment impacts the homeostasis and function of Tregs similar to deletion of c-Rel.
c-Rel Inhibition Suppresses Tumor Growth
We predicted that administration of a c-Rel-inhibiting compound like PTXF would break Treg-mediated tumor immune tolerance and augment anti-tumor responses, similar to c-Rel deletion in Tregs. WT mice were transplanted with B16F1 melanoma cells and received daily injections of 50 mg/kg PTXF or PBS starting 1 day before tumor cell inoculation. Consistent with previous reports (Duaet al., 2007),we observed a significant reduction in the rate of tumor growth in PTXF-treated animals as compared to control animals (Figure 4A). No observable differences in the size or composition of lymphoid tissues were detected, and no adverse events were observed (data not shown). As it has been reported that PTXF can induce tumor cell apoptosis in vitro (Edward and MacKie, 1991; Ratheesh et al., 2007), we wanted to determine whether the effects of PTXF on tumor growth were tumor intrinsic or related to improved immune responses. Analysis of tumor infiltrates at D16 revealed an increased infiltration of total T cells, as well as IFN-γ producing CD4 and CD8 T cells (Figures 4B–4D). Additionally, there was a trend toward a decreased proportion of Tregs in the tumors, which also displayed an altered phenotype (Figures 4E, 4F, and S4A). As observed in the Foxp3CREc-RelF/F model, tumor-infiltrating Tregs exhibited increased IFN-γ production (Figure 4F). Moreover, qPCR analysis of total tumors revealed an enhanced inflammatory response, including increased expression of activated T cell markers and cytokines (Figure 4G). This suggests that immune cells, especially T cells, may mediate the protective effect of PTXF on melanoma growth. As previously reported (Ratheesh et al., 2007), we also detected a decrease in the expression of certain integrins, such as Itga2b and Itgb3 (Figure S4B), suggesting that PTXF might act through the inhibition of tumor adhesion. To test whether there were direct effects on tumor growth independent of the changes in T cell responses, we transplanted immunodeficient RAG1−/− mice with B16F1 cells and treated them with PBS or PTXF. In this background, PTXF had no effect on tumor growth (Figure 4G). Therefore, lymphocytes are required for the anti-tumor effects of PTXF. Moreover, CD8-depletion in PTXF-treated mice entirely restored tumor growth (Figure 4I). We further confirmed these results in the settings of B16F10 metastatic melanoma. Again, in immunocompetent mice, early PTXF treatment significantly decreased the total number of metastatic foci in lungs (Figure S4C).
Figure 4. c-Rel Inhibition by PTXF Suppresses Melanoma Growth.

(A–G) WT C57BL/6J mice were transplanted subcutaneously with B16F1 cells and treated from D– 1 to D7 with PTXF or PBS. (A) Tumor growth over time. Arrows indicate the days of PTXF or PBS injection. (B–F) TILs were restimulated with PMA and ionomycin 16 days after tumor inoculation. (B) Cumulative numbers of T cells among 106 live cells. (C) CD4/CD8 T cells ratio. (D) Representative expression in CD8+ T cells (left) and cumulative % (right) of IFN-γ in T cells. (E) Percent of Foxp3+Tregs in CD4+T cells. (F) Percent of IFN-γ+ in Tregs.
(G) qPCR analysis of total tumor RNA at D16.
(H) RAG1−/− mice were transplanted and treated as in (A). Tumor growth overtime is shown.
(I) WT C57BL/6J mice were transplanted and treated as in (A) with or without anti-CD8 administration (blue arrows).
(J) Foxp3cre and Foxp3crec-RelF/F were transplanted subcutaneously with 3 × 105 B16F1 cells and treated as in (A).
(K) WT C57BL/6J mice were transplanted subcutaneously with B16F1 cells and treated from D– 1 to D7 with IT-603 or vehicle (arrows). Data are represented as mean ± SEM of 3 experiments. *p < 0.05, ***p < 0.001.
See also Figure S4.
To assess whether the beneficial effect of PTXF relied on its inhibition of c-Rel in Tregs, we next measured the influence of PTXF administration in Foxp3CREc-RelF/F mice. To ensure B16F1 melanoma growth in KO animals, we transplanted 3 × 105 cells instead of 5 × 104 cells. Melanoma growth was accelerated in these settings, but PTXF was still able to significantly reduce tumor growth in WT mice (Figure 4J). Moreover, all Foxp3CREc-RelF/F mice harbored a visible tumor by D10, even though the tumors were smaller than those in control mice. Interestingly, PTXF was unable to further decrease melanoma growth. This suggested that PTXF acted, at least partially, by inhibiting Treg function through the suppression of c-Rel. Finally, to confirm that the observed effects on melanoma growth can be attributed to c-Rel inhibition, we tested whether an unrelated chemical c-Rel inhibitor could recapitulate the effects of PTXF. We used IT-603, a commercially available compound that inhibits c-Rel-mediated transcription and reduces lymphoid leukemia severity in mouse models (Shono et al., 2014). Administration of 200 μg IT-603 every other day, starting at D-1, reduced B16F1 growth modestly but significantly (Figure 4K). Increased T cell infiltration, IFN-γ production, and reduced Tregs were observed in the tumors of IT-603-treated mice (Figure S4D). These results demonstrate that chemical c-Rel inhibition can inhibit Treg function and prevent Treg-mediated tumor tolerance.
PTXF Treatment Potentiates the Effects of Checkpoint Blockade Therapies to Inhibit Melanoma Growth
The emergence of checkpoint blockade therapies has been a major step forward in the field of oncology. Antibody-mediated blockade of the PD-1/PD-L1 is believed to work by relieving exhaustion of CD8 T cells, thereby allowing the re-activation of anti-tumor effector cells (Francisco et al., 2010). However, anti-PD-1 monotherapy is ineffective for the treatment of many tumors, including animal models (e.g., in B16-induced melanoma in mice) (Chen et al., 2015). We therefore wondered whether targeting of Tregs with PTXF could potentiate the beneficial effect of PD-1-blockade. Mice were first transplanted with B16F1 melanoma cells and treated with vehicle, PTXF, and/or anti-PD-1 mAb when the tumors reached a volume >2 mm3 (D5 to D6). PTXF or anti-PD-1 monotherapies did not change tumor growth compared to PBS controls. However, the combination of PTXF and anti-PD-1 significantly flattened the growth curves (Figure 5A). Co-administration of PTXF and anti-PD-1 increased T cell infiltration and expression of IFN-γ by CD4 and CD8 T cells, while the CD4/CD8 ratio was decreased (Figures 5B–5D). The proportion of Tregs was reduced while Treg IFN-γ expression increased (Figures 5E and 5F). Of note, curative PTXF monotherapy did not induce dramatic changes in the tumor infiltrate, underscoring the importance of early therapeutic intervention. RNA expression analysis demonstrated a cumulative effect of PTXF and anti-PD-1 treatments, with inflammatory cytokines and activation markers being strongly upregulated in the tumors of mice receiving the combination therapy (Figure 5G). Finally, CD8 depletion with a monoclonal antibody (mAb) fully restored tumor growth in PTXF+anti-PD-1-treated animals (Figure 5H) demonstrating a requirement for CD8 T cells for improved tumor clearance. We confirmed that these findings were not specific to the anti-PD-1 mAb by blocking the ligand of PD-1, PD-L1. Again, the combination of both PTXF and anti-PDL1 had an additive effect in decreasing and delaying melanoma growth (Figure 5I). We also assessed the effect of PTXF/anti-PD-1 therapy in a different genetic background. BALB/c mice were transplanted with CT-26 colon carcinoma and subsequently treated with PTXF and suboptimal doses of anti-PD-1 (because high doses of anti-PD-1 induced a significant decrease in CT-26 growth, data not shown). Again, only the combination of both drugs led to a significant reduction of tumor growth (Figure 5G). To test whether the inhibition of Treg c-Rel was sufficient to potentiate the curative effect of anti-PD-1 therapy, we next treated melanoma-bearing Foxp3CREc-RelF/F mice with anti-PD-1. Strikingly, unlike WT animals, tumor size was significantly reduced upon checkpoint-blockade therapy in mice whose Tregs lacked c-Rel (Figure S5A). Finally, curative combinatorial therapy with the c-Rel inhibitor IT-603 and anti-PD-1 led to reduced melanoma growth and increased T cell infiltration and activation (Figures 5K and S5B). Taken together, our data highlight the therapeutic potential of c-Rel inhibition in addition to checkpoint-blockade antibodies.
Figure 5. PTXF and PD-1-Blockade Have Additive Inhibitory Effects on Growth of Established Melanoma.

(A–G) WT C57BL/6J mice were transplanted subcutaneously with B16F1 cells and treated from D6 with PTXF or anti-PD-1 mAb or PBS.
(A) Tumor growth over time. Red arrows, PTXF injections; black arrows, anti-PD-1 injections.
(B–F) TILS were restimulated with PMA and ionomycin 16 days after tumor inoculation. (B) Cumulative numbers of T cells among 106 live cells. (C) CD4/CD8 T cells ratio. (D) Representative expression in CD8+ T cells (left) and cumulative % (right) of IFN-γ in T cells. (E) Percent of Foxp3+ Tregs in CD4+ T cells. (F) Percent of IFN-γ+ in Tregs.
(G) qPCR analysis of total tumor RNA at D16.
(H) Mice were treated as in (A) and were injected with anti-CD8 mAb or IgG at D7 and D9 (blue arrows).
(I) Mice were treated as in (A) but with anti-PD-L1 mAb.
(J) WT Balb/C mice were transplanted subcutaneously with CT-26 cells and treated from D6 with PTXF or anti-PD-1 mAb (100 μg) or PBS.
(K) WT C57BL/6J mice were transplanted subcutaneously with B16F1 cells and treated from with IT-603, and/or anti-PD-1, or vehicle. Data are represented as mean ± SEM of 3 to 4 experiments. *p < 0.05, ***p < 0.001.
See also Figure S5.
Discussion
NF-κB c-Rel Controls the Homeostasis of the aTreg Subset that Suppresses Anti-tumor Responses
Multiple lines of evidence demonstrate that the Treg pool can be separated into subsets with differing phenotypes and function. In both mice and humans, Tregs can be split into resting and activated populations (Huehn et al., 2004; Miyara et al., 2009). In mice, it was suggested that transcription factor Foxo1 maintains the CD62LhighCD44low rTreg pool (Luo et al., 2016). These rTregs are retained in lymphoid tissues through expression of high levels of CD62L, CCR7, and S1PR1; and it is believed that they are crucial for the prevention of lymphoproliferative disease, as illustrated by the early splenomegaly and lymphadenopathy observed in Treg-restricted Foxo1 conditional mutants (Ouyang et al., 2012). In contrast, CD62LlowCD44high aTregs, which differentiate upon TCR-stimulation, acquire the ability to migrate to inflamed tissues through expression of homing receptors (Huehn et al., 2004; Rosenblum et al., 2011). Therefore, they are more specialized in the suppression of local tissue inflammation. Recently, Tregs have emerged as crucial regulators of cancer progression (Darrasse-Jèze et al., 2009; Luo et al., 2016). It has been suggested that the expression of aTreg-specific molecules, such as CD103, CCR8, Klrg1, or Tigit, are critical for tumor homing of Tregs and suppression of effector T cells, thereby providing a novel specialized function to the aTreg subset (De Simone et al., 2016). However, the precise molecular mechanisms that regulate aTreg differentiation and function are poorly described. It was proposed that IRF4 and Egr2 may be important for aTreg differentiation, and signaling through the TCR was absolutely required for their maintenance (Levine et al., 2014; Vahl et al., 2014). In a recent report (Messina et al., 2016), it was suggested that p65 may be involved in effector Treg development in the competitive environment of fetal liver recipients. However, the authors only observed a mild decrease in aTreg in the lymph nodes, but not the spleen, of Foxp3CREp65F/F mice. Although we observed some effects of p65 deletion on the expression of genes enriched in aTregs, we did not observe a decrease in the aTreg population in vivo. Instead, we show that c-Rel was required to maintain a normal pool of aTregs and a normal aTreg transcriptome. As NF-κB activation is a key downstream event of TCR/CD28 signaling, this observation is in agreement with previous reports. As p65 was mostly dispensable for aTreg, this reinforces the idea that c-Rel and p65 control separate gene targets and exert different functions in Tregs (Oh et al., 2017), possibly because the activation kinetics are different. A number of genes that are crucial for aTreg homing and homeostasis in inflamed tissues, such as CD83, PD-1, and Klrg1 were impaired in c-Rel-deficient Tregs and also in TCR−/− aTreg and in Foxo1CA, but not in p65-deficient Tregs (Levine et al., 2014; Luo et al., 2016) (Figure 1). Thus, our data suggests that c-Rel is specialized in the maintenance of the aTreg population, while p65 controls crucial suppressive properties of Tregs in both the resting and the activated subsets. Hence, this explains the early lethal lymphoproliferation observed in Foxp3CREp65F/F mice and the relative absence of autoimmunity in Foxp3CREc-RelF/F animals.
These data suggested that c-Rel deletion might have a specific effect on tumor tolerance. Indeed, the ablation of c-Rel, but not p65, in mature Tregs strongly impaired melanoma growth in a CD8 T cell-dependent manner and destabilized Treg-associated gene expression. Expression of Helios, which was recently implicated in the maintenance of Treg homeostasis and the suppression of tumor immunity, was reduced in the absence of c-Rel (Nakagawa et al., 2016). Although the immunological abnormalities present in the Foxp3Crep65f/f mice (Oh et al., 2017) complicate the analysis of tumor growth, it is notable that we did not observe any increase in anti-tumor immune response in Treg p65-deficient mice. In WT mice, Tregs efficiently inhibited antitumor effector CD8 T cell responses, thereby allowing uncontrolled cancer cell proliferation. In Foxp3CREc-RelF/F animals, however, the impaired Treg function allowed increased activation of effector cells and enhanced tumor cell killing. This, combined with the lack of evidence of a role for c-Rel in prevention of autoimmunity (Oh et al., 2017), and in effector CD8 responses, suggested that the therapeutic targeting of c-Rel could selectively ablate Treg-mediated tumor tolerance.
c-Rel Inhibition as a Novel Cancer Immunotherapy
An extensive body of literature describes the roles of NF-κB in the initiation, proliferation, and propagation of tumors. This has led multiple groups to test whether global inhibition of NF-κB could have an effect on tumor growth (DiDonato et al., 2012). For instance, inhibition of IKK activity by thalidomide, by the chemical BAY11-7082, or by the small inhibitory Nemo-Binding Domain (NBD) peptide, decrease severity of lymphomas (reviewed in Kim et al., 2006). Bortezomib (Velcade), which prevents degradation of the NF-κB inhibitor IkBα, is now a Food and Drug Administration (FDA)-approved drug and is used for the treatment of multiple myeloma (Mulligan et al., 2007). Despite these promising results, the use of such non-specific NF-κB inhibitors has been complicated by the observation of multiple adverse effects, such as systemic inflammation through inter-leukin (IL)-1β overexpression or non-immune-related complications (Greten et al., 2007). This could be a consequence of (1) the inhibition of non-NF-κB-related pathways upon inhibition of IKK (Oeckinghaus et al., 2011), or (2) the inhibition of the p65 subunit of NF-κB that is well known to exert central roles in organo-genesis and inflammation. Our results using conditional mutant mice suggested that c-Rel, whose biological function is mainly restricted to the adaptive immune system, could be a specific target for the treatment of cancer through its role in Tregs. In vitro, we observed that PTXF reduced the level of c-Rel protein in Tregs, while impairing the molecular identity of Tregs and Treg suppressive function. In agreement with previously published results, PTXF affected c-Rel, but not other NF-κB subunits (Wang et al., 1997). RNA-seq revealed a profound effect of PTXF on Treg-associated gene expression that was partially overlapping with changes observed in c-Rel-deficient Tregs. Genes required for optimal Treg function and immunosuppression in the tumor microenvironment, such as Tgfb1 or Gzmb (Boissonnas et al., 2010; Donkor et al., 2011), as well as numerous aTreg genes, were directly repressed by PTXF treatment, similar to that seen upon genetic deletion of c-Rel. Thus, PTXF affects Treg identity both directly by modifying c-Rel-dependent transcription and indirectly by forcing the expression of genes that promote anti-tumor responses.
PTXF has previously been described as an anti-cancer drug. Early PTXF administration reduced growth of subcutaneous B16F10 melanomas and decreased their metastatic potential (Dua et al., 2007). When injected together with the alkylating agent thiotepa, PTXF diminished growth of human bladder and breast cancer xenografts (Fingert et al., 1988). This has led to several ongoing clinical trials testing the effect of PTXF, in combination with existing treatments, in leukemia, glioblastoma, and non-small cell lung cancer (https://clinicaltrials.gov). It has been proposed that the anti-cancer properties of PTXF were attributable to reduced expression of adhesion molecules or induction of apoptosis in cancer cells (Bravo-Cuellar et al., 2013; Edward and MacKie, 1991). The effects of PTXF on T cells are still debated, as the drug seems to downregulate T cell effector functions in vitro but has an opposite effect in vivo (Jimenez et al., 2001; Suresh et al., 2002). In this study, we show that PTXF improves effector cells anti-tumor responses likely through inhibition of Treg function.
However, PTXF alone failed to reduce the growth of established tumors. This is because B16F1 melanoma has long been described to be highly aggressive and resistant to most monotherapies, including anti-PD-1/PD-L1 (Kleffel et al., 2015). It has been proposed that such treatments could be improved by the co-administration of two drugs. For instance, anti-CTLA-4 mAbs that are believed to directly target Tregs, and anti-PD-1 mAbs that relieve the exhaustion of effector T cells, could have a synergistic effect in monotherapy-resistant cancers (Curran et al., 2010). Although promising preliminary results have been obtained from clinical trials, one major pitfall has been the frequent (>70%) and multiple adverse effects induced by anti-CTLA-4 mAbs, such as cutaneous inflammation or colitis (Bertrand et al., 2015). In contrast, PTXF is well-tolerated in patients and mice (https://www.fda.gov/Safety/MedWatch/SafetyInformation/ucm314605.htm). Here, we show that PTXF potentiates the effect of PD-1-blockade in our melanoma model without any adverse effects. Thus, we propose that this multi-therapy could be used as a safe novel treatment for solid cancers.
Understanding how Treg homeostasis is controlled during tumor growth may reveal new targets for the development of novel anti-cancer therapies. In this study, we have demonstrated that activation of the c-Rel subunit of NF-κB in Tregs is central to the prevention of protective anti-tumor responses. Hence our findings provide a new rationale for the use of selective NF-κB inhibitors, especially those targeting c-Rel, for the treatment of cancer.
Contact for Reagents and Resources Sharing
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Sankar Ghosh (sg2715@cumc.columbia.edu).
Experimental Model and subject Details
Animals
p65-floxed mice were obtained from R. Schmid (Munich, Germany) (Algul et al., 2007) and c-Rel-floxed mice from U. Klein (Columbia University, New York) (Heise et al., 2014). CD4cre (Tg(CD4-cre)1Cw1), Foxp3CREYFP (Foxp3tm4(YFP/cre)Ayr), Foxp3EGFP (Foxp3tm2Tch), Foxp3RFP (Foxp3tm1Flv), CD45.1 (Ptprca Pepcb/BoyJ) and RAG1−/− were originally purchased from the Jackson Laboratory and maintained in our animal facility. C57BL/6J mice were purchased from the Jackson Laboratory. All mice were kept in specific pathogen-free conditions in the animal care facility at Columbia University (New York, NY), and were used between 5 and 8 weeks of age. Both males and females were used indifferently in the study. All mouse experiments were approved by Institutional Animal Care and Use Committee of Columbia University.
Cells
B16F1, B16F10 and CT-26 cells were obtained from the ATCC and grown in DMEM+10% FBS medium. BRAFCAPten−/− cells were originally derived from an in vivo melanoma induced by topical tamoxifen treatment of TyrcreERT2BRAFCAPtenF/F, as described. All cell lines were periodically tested for murine pathogens.
Method Details
Tumor transfer and treatments
5×104 B16F1 (or 3×105 when indicated) or 5×105 CT-26 or BRAFCAPten−/− cells diluted in sterile PBS1X were injected subcutaneously into the shaved flank of each mouse. For metastasis experiments, 2.5×104 B16F10 cells were injected intravenously in the tail vein. PTXF (Sigma) was diluted in PBS1X and sterile filtered prior to each injection. IT-603 (Calbiochem) was first diluted in EtOH for storage; prior to injections it was further diluted in Cremophor EL and then in warm PBS1X (for a final ratio of 1 EtOH: 1 Cremophor:4 PBS1X). Anti-PD-1 (RMP1-14), anti-PD-L1 (10F.9G2) and anti-CD8 (YTS.169-4) mAbs were obtained from BioXCell. Rat IgG isotype control was from R&D Systems. Mice received intraperitoneal injections of 50mg/kg PTXF, 200 μg IT-603 or 200 μg of the given mAb. In Figure 5, BALB/c mice harboring CT-26 tumors received a suboptimal dose of 100 μg of anti-PD-1.
Flow cytometry
Cells were isolated from thymus, spleen and lymph nodes by mechanical desegregation in PBS+FBS 3%. For tumor-infiltrating cell suspensions, total tumors were digested in DMEM (GIBCO) supplemented with 1 mg/ml collagenase type IV (Sigma) and 1 mg/ml DNase I (Sigma) for 40 min at 37°C, followed by centrifugation in a 36% Percoll solution. For intracellular cytokines analyses, cell suspensions were incubated 3 hr with PMA (Sigma, 50 ng/mL), ionoymycin (Sigma, 1 μg/mL) in the presence of Golgi Plug (BD), or incubated 4 hr with mitomycin C-treated WT splenocytes loaded with 10mg/mL gp100/pmel 17 peptide (Neo Biolab) in the presence of Golgi Plug. Cells were then stained with mAbs in PBS+3% FBS. Foxp3 and cytokine staining were performed using the eBioscience kit and protocol. Cells were acquired on a LSR II (BD Biosciences) and analyzed with FlowJo (Tree Star) software.
In vitro PTXF treatment
CD4+YFP+ Treg cells were FACS-sorted and pre-incubated with 500 μg/mL PTXF or H2O for 15 min at 37C. Total cells suspensions were then activated with 5 μg/mL plate-coated anti-mCD3,1 μg/ml soluble anti-mCD28 (BioLegend) and 10 ng/mL mIL-2, overnight at 37C. Treg cells were then washed for further use.
Suppression assays
For in vitro assays, CD45.1+ naive conventional CD4+ T cells were magnetically isolated (Miltenyi) and labeled with CellTrace Violet Proliferation Tracker (Life Technologies). They were cultured with T cell depleted, mitomycin C-treated WT splenocytes and 2.5 μg/mLanti-mCD3, in the presence or not of treated Treg cells as described above. Proliferation of CD45.1+ T cells was assessed by FACS at D4. The % of suppression was calculated as described. For in vivo assays, 4.105 naive Tconv cells were isolated as above and transferred with or without 1.105 Treg cells, to the retro-orbital sinus of 6-9 week-old RAG1−/− mice. Recipients were then weighed every week and euthanized when weight loss was > 30%.
Retroviral transduction
For overexpression of GFP or c-Rel in Treg cells, Foxp3RFP+ Treg cells were sorted and activated with 5 μg/mL plate-coated anti-mCD3,1 μg/ml soluble anti-mCD28 (BioLegend) and 10 ng/mL mIL-2. At D1 and D2, fresh retrovirus supernatant containing pMIGR or pMIGR-CREL (Addgene) was added and the cells were spun at 2500 rpm for 1.5 hr at 30C. After spin infection, the cells were cultured in the T cell culture medium and harvested on day 5 for sorting of RFP+GFP+ transduced cells.
Western Blotting
Total lysates were extracted using RIPA buffer and protease inhibitors with SDS. 20 μg protein extracts were ran in polyacrylamide gels and transferred onto PVDF membranes. Membranes were incubated with anti-p65, c-Rel (Santa-Cruz) and GAPDH (Fitzgerald) Abs, followed HRP-coupled secondary Abs.
RT-qPCR and RNA-sequencing
Total RNA was extracted using a QIAGEN Rneasy Mini Kit with DNase treatment. For qPCR, RNA was reverse transcribed by Superscript III (Invitrogen). cDNAs were used for PCR with SYBR Green reagents (Quanta Biosciences, Gaithersburg, MD) on a C1000 Touch thermal cycler (Bio Rad, Hercules, CA). The data was normalized to GAPDH expression. Primers sequences can be sent under request. For RNA-sequencing, libraries were prepared using an Illumina TruSeq Library Kit and sequenced by an Illumina 2500 instrument. Upon sequencing, raw FASTQ files were aligned on the mm10 genome using STAR aligner with default parameters. Aligned fragments were then counted and annotated using Rsamtools v3.2 and the TxDb.Mmusculus.UCSC.mm10.knownGene' version 3.1.2 transcript database respectively. Normalized FPKM (fragments per kilobase per million mapped reads) were obtained using the robust FPKM estimate function of DeSeq2 v1.10.1 after removing the batch effect using the ComBat function of the sva package v3.18.0. Differentially expressed genes were obtained using the DESeqResults function of the same package. All p values were adjusted for multiple testing using the Benjamini & Hochberg FDR algorithm. For gene set enrichment analysis, we acknowledge our use of the GSEA software, and Molecular Signature Database (MSigDB) including the ImmuneSigDB (C7 collection) (Godec et al., 2016; Subramanian et al., 2005).
Quantitation and Statistical Analysis
For tumor growth analysis, we used 2-way ANOVA followed by Bonferroni post-test (when more than 2 groups), and the non-parametric Mann-Whitney U test (when only 2 groups). In the 2nd case we did not take into account the measures before D7 as tumors have similar sizes. For other comparisons, we used 1-way ANOVA (when more than 2 groups of samples), and the unpaired Student t test (when only 2 groups).
Data and Software Availability
RNA-sequencing data have been deposited to NCBI (GEO: GSE82008).
Supplementary Material
Figure S1. NF-κB c-Rel Regulates the Activated Treg Cell Differentiation and Gene Expression, Related to Figure 1: (A and B) Flow cytometry analysis on spleen total Tregs; the cumulative percentage of CD103+ and Ly6C+ Tregs is shown.
(C–F) CD62Llow CD44high aTreg and CD62Lhigh CD44low rTreg were sorted from Foxp3CRE-YFP (WT) Foxp3crep65F/F (p65KO), and Foxp3crec-RelF/F (c-RelKO) mice and submitted to RNA-seq analysis. Gene expression changes in WT aTreg versus WT rTreg were plotted against those in KO aTreg versus WT aTreg (A, B) and KO rTreg versus WT rTreg (C, D). Numbers and colored dots indicate genes upregulated in WT aTreg while downregulated in KO Treg (red) and downregulated in aTreg while upregulated in KO Treg (blue) (fold change > 2, p < 0.01).
(G and H) Flow cytometry analysis on spleen CD62Llow CD44high aTregs; the cumulative percentage of CD103+ and Ly6C+ cells is shown. Data are form 2 experiments. *p < 0.05, n.s. non-significant.
Figure S2. c-Rel Expression in Treg Cells Restricts Anti-tumor Immune Responses, Related to Figure 2: (A) Splenic CD4+Foxp3YFP+ Tregs of each genotype were isolated, stimulated or not for 3 hr, and submitted to RNA-seq analysis. Fold Change in the expression of selected genes is shown.
(B) Splenocytes of the indicated genotypes were re-stimulated with PMA-ionomycin 4 days after tumor challenge and analyzed by flow cytometry. Mean ± SEM of Ki67+, CD44high and TNFhighIFN-γ+ cells is shown.
(C) 5-7 weeks-old Foxp3cre and Foxp3crerelF/F were transplanted sub-cutaneously with B16F1 cells. Splenocytes were restimulated ex-vivo with PBS or gp100/pmel peptide, 4 days after tumor inoculation. Representative dot plot of IFN-expression in gated CD8+ T cells cells. Numbers indicate the percentage in the gate; MFI: Mean Fluorescence Intensity of IFN- γ in IFN-γ+ cells.
(D) Foxp3creand Foxp3crerelF/F were injected intravenously with B16F10 cells. Left: representative picture of lungs at D14; right: Number of detectable tumor foci/lung at D14. Each dot represents an individual mouse form 2 independent experiments.
(E) Foxp3cre and Foxp3crerelF/F were transplanted sub-cutaneously with BRAFCAPten−/− cells. The mean ± SEM of tumor growth over time is shown. Numbers indicate the number of mice with detectable tumors at the end of the experiment. All data are from 2 to 3 experiments***p < 0.001.
Figure S3. Chemical c-Rel Inhibition Impairs Treg Identity, Related to Figure 3: (A–E) CD4+YFP+Tregs sorted from Foxp3cre (WT) and Foxp3crec-RelF/F were activated with H2O or PTXF and submitted to RNA-Seq analysis. (A) Principal component analysis. (B) Summary of gene expression changes in WT Treg treated with PTXF and stimulated Foxp3crec-RelF/F (top) or stimulated Foxp3crep65F/F (bottom, from Oh et al., 2017), compared to WT+H2O Tregs. Numbers indicate the genes with a fold-change > 2 and a p value < 0.05 when compared to the WT+H2O samples. Up: upregulated expression when compared to the WT control cells; down: downregulated expression. (C) Heatmap showing expression of genes up-or downregulated in both c-RelKO and WT+PTXF samples. (D) Gene expression of selected genes measured by RNA-seq (top) and qRT-PCR (bottom). Fold changes (mean ± SEM) and statistics in WT+PTXF and c-Rel−/− Treg versus WT+H2O Treg are shown. (E) Gene Set Enrichment Analysis plots showing immune signatures selected from the overlapping ones in Figure 3G.
(F) Selected Top Regulator Effect Networks using Ingenuity Pathway Analysis and showing c-Rel as a central regulator of the altered transcriptome in both PTXF and c-RelKO samples.
(G–I) CD4+GFP+ Tregs were stimulated as with H2O or PTXF and subsequently tested for in vitro suppression (G) and in vivo colitis assays (H, I). (G) Percentage of donor CD45.2+ Tregs in each well. (H) Colon histology (H/E) 40 days after cell transfer; bars: 100mm, original magnification 100×. (I) Tissues were analyzed by flow cytometry 40 days after cell transfer. The proportion of Foxp3+ Tregs in live CD4+T cells is shown. In G and I, data represents the mean of 2 experiments; values were non-significantly changed between groups.
Figure S4. c-Rel Inhibition by PTXF Suppresses Melanoma Growth, Related to Figure 4: (A and B) WT C57BL/6J mice were transplanted sub-cutaneously with B16F1 cells and treated from D-1 to D7 with PTXF or PBS as a control. (A) FACS staining in the TILs at D16; Foxp3 expression and % of Helios+ in gated Tregs is shown. (B) qPCR analysis on total tumor RNA at D16.
(C) WT C57BL/6J mice were injected intravenously with B16F10 cells and with PTXF or PBS from D-1 to D7. The number of macroscopic tumor foci is showed.
(D) WT C57BL/6J mice were transplanted sub-cutaneously with B16F1 cells and treated from D-1 to D7 with IT-603 or EtOH+cremophor as a control. FACS analysis of the TILS at D16 is shown. Data are represented as mean ± SEM from 2-3 experiments. *p < 0.05.
Figure S5. PTXF and PD-1-Blockade Have Additive Effects on Growth of Established Melanoma, Related to Figure 5: (A) 5-7 weeks-old Foxp3crerelF/F mice were transplanted sub-cutaneously with 3×105 B16F1 cells and treated from D6 with anti-PD-1 mAb or PBS (black arrows). Mean ± SEM tumor growth over time is shown.
(B) WT C57BL/6J mice were transplanted sub-cutaneously with B16F1 cells and treated from with IT-603, and/or anti-PD-1, or EtOH+cremophor as a control. FACS analysis of TILs at D16 is shown. *p < 0.05, **p < 0.005, ***p < 0.001.
Key Resources Table.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-mouse TCR-b, Percp-Cy5.5 conjugated, clone H57-507 | Tonbo Biosciences | 65-5961 |
| Anti-mouse CD16/CD32, unconjugated, clone 2.4G2 | Tonbo Biosciences | 70-0161 |
| Anti-mouse CD4, APC-eFluor 780 conjugated, clone RM4-5 | EBioscience | 47-0042-82 |
| Anti-mouse CD8a, PE-CF594 conjugatedm clone 53.6.7 | BD Biosciences | 562315 |
| Anti-mouse CD45, Alexa Fluor 700 conjugated, clone 30F11 | Tonbo Biosciences | 80-0451 |
| Anti-mouse IFN-gamma, PE-Cy7 conjugated, clone XMG1.2 | Tonbo Biosciences | 60-7311 |
| Anti-mouse TNF, FITC conjugated, clone MP6-XT22 | EBioscience | 53-7321-82 |
| Anti-mouse IL-2, PE conjugated, clone JES6 | BD Biosciences | 554428 |
| Anti-mouse/rat Foxp3, eFluor 450 conjugated, clone FJK16S | EBioscience | 48-5773-82 |
| Anti-mouse Helios, PE conjugated, clone 22F6 | BioLegend | 137206 |
| Anti-mouse GITR, PE-Cy7 conjugated, clone DTA-1 | EBioscience | 25-5874-80 |
| Anti-mouse CD25, APC conjugated, clone PC61.5 | Tonbo Biosciences | 20-0251 |
| Anti-mouse CD62L, PE conjugated, clone MEL14 | Tonbo Biosciences | 50-0621 |
| Anti-mouse CD44, APC conjugated, clone IM7 | Tonbo Biosciences | 20-0441 |
| Anti-mouse/human Ki-67, PE-Cy7 conjugated, clone B56 | BD Biosciences | 561283 |
| Anti-mouse CD103, PE conjugated, clone 2E7 | EBioscience | 12-1031082 |
| Anti-mouse Ly6C APC conjugated, clone HK1.4 | EBioscience | 17-593280 |
| Anti-mouse PD-1, PE conjugated, clone J43.1 | Tonbo Biosciences | 50-9985 |
| Anti-mouse Klrg1, APC conjugated, clone 2F1 | Tonbo Biosciences | 20-5893 |
| Polyclonal anti-C-Rel, unconjugated, clone sc71 | Santa Cruz | c-Rel SC-71 |
| Polyclonal anti-RelA p65, unconjugated, clone sc70 | Santa Cruz | RelA SC-70 |
| Monoclonal anti-mouse GAPDH, unconjugated | Fitzgerald | 10R-G109a |
| In vivo mAb anti-mouse PD-1, clone RMP1-14 | BioXCell | BE0146 |
| In vivo mAb anti-mouse CD8, clone YTS.169-4 | BioXCell | BE0117 |
| In vivo mAb anti-mouse CD3, clone 145-2C11 | BioXCell | BE0001-1 |
| Anti-mouse CD28, Na/Le | BioLegend | 102102 |
| In vivo mAb anti-mouse PD-L1, clone 10F.9G2 | BioXCell | BE0101 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Lipofectamine 2000 | Invitrogen | 11668-019 |
| Pentoxifylline, powder | Sigma | P1784 |
| Mouse Interleukin-2 | Peprotech | 212-12A |
| PMA salt | Sigma | P8139 |
| IT-603 | EMD Millipore | 530654 |
| Cremophor EL | EMD Millipore | 238470 |
| Golgi Plug protein transport inhibitor | BD Biosciences | 555029 |
| Collagenase type IV form C.Histolyticum | Sigma | C5138 |
| DNase I from bovine pancreas | Sigma | DN25 |
| gp100/pmel 17 peptide | NeoBiolabs | 58686 |
| Superscript IV reverse transcriptase | Invitrogen | 18090050 |
| Veriquet Fast SYBR Fluor | Affymetrix | 75675 |
| Ionomycin salt | Sigma | 10634 |
| Critical Commercial Assays | ||
| Magnisort mouse CD4 T cells enrichment kit | Invitrogen | 8804-6821 |
| Cell Trace Violet Cell proliferation kit | Life technologies | C34557 |
| QIAGEN RNEasy MiniKit | QIAGEN | 74106 |
| Deposited Data | ||
| RNA-seq data: WT, p65KO, cRelKO rTregs, aTregs | https://www.ncbi.nlm.nih.gov/geo/ | N/A |
| RNA-seq data: stimulated WT+H2O/PTXF and crelKO Tregs | https://www.ncbi.nlm.nih.gov/geo/ | N/A |
| Experimental Models: Cell Lines | ||
| Human Phenix-ECO | ATCC | CRL-3214 |
| Mouse B16-F1 | ATCC | CRL-6323 |
| Mouse B16-F10 | ATCC | CRL-6475 |
| Mouse CT-26.WT | ATCC | CRL-2638 |
| Mouse Braf.V600E.PtenKO | This paper | N/A |
| Experimental Models: Organisms/Strains | ||
| Mouse: C57BL/6J | Jackson Laboratories | 000664 |
| Mouse: RAG1-deficient | Jackson Laboratories | 002216 |
| Mouse: Foxp3-YFP-CRE | Jackson Laboratories | 016959 |
| Mouse: Balb/cJ | Jackson Laboratories | 000651 |
| Mouse: BRafCA, PtenloxP, Tyr∷CreERT2 | Jackson Laboratories | 013590 |
| Mouse: c-Rel-Flox (Ulf Klein) | Heise et al., 2014 | N/A |
| Mouse: RelA-Flox (Roland Schmid) | Algul et al., 2007 | N/A |
| Mouse: Foxp3-eGFP | Jackson Laboratories | 016959 |
| Mouse: Foxp3-RFP | Jackson Laboratories | 016959 |
| Mouse: CD45.1 Ptprca Pepcb/BoyJ | Jackson Laboratories | 016959 |
| Oligonucleotides | ||
| Cblb F | CCATGCTTGACTTGGACGATGAC | N/A |
| Cblb R | TGGCGATGTGACTGGTGAGTTC | N/A |
| Ccl22 F | GTGGAAGACAGTATCTGCTGCC | N/A |
| Ccl22 R | AGGCTTGCGGCAGGATTTTGAG | N/A |
| Ccl4 F | ACCCTCCCACTTCCTGCTGTTT | N/A |
| Ccl4 R | CTGTCTGCCTCTTTTGGTCAGG | N/A |
| Ccr7 F | GCCCAGATGGTTTTTGGGTTC | N/A |
| Ccr7 R | GCAAGGTACGGATGATAATGAGG | N/A |
| Cd4 F | GTTCAGGACAGCGACTTCTGGA | N/A |
| Cd4 R | GAAGGAGAACTCCGCTGACTCT | N/A |
| Cd8a F | ACTACCAAGCCAGTGCTGCGAA | N/A |
| Cd8a R | ATCACAGGCGAAGTCCAATCCG | N/A |
| Gapdh F | TTCACCACCATGGAGAAGGC | N/A |
| Gapdh R | GGCATGGACTGTGGTCATGA | N/A |
| Gzmb F | CAGGAGAAGACCCAGCAAGTCA | N/A |
| Gzmb R | CTCACAGCTCTAGTCCTCTTGG | N/A |
| H2-Aa F | TGGGCACCATCTTCATCATTC | N/A |
| H2-Aa R | GGTCACCCAGCACACCACTT | N/A |
| Ifng F | ATCAACGCTACACTGCATCTTGGCTT | N/A |
| Ifng R | CCTCAAACTTGGCTACTCATGAATGC | N/A |
| Il1r2 F | CCCCTGGAGACAATACCAGC | N/A |
| Il1r2 R | TTAGCCAACCACCACACAATG | N/A |
| Il2 F | TGAGCAGGATGGAGAATTACAGG | N/A |
| Il2 R | GTCCAAGTTCATCTTCTAGGCAC | N/A |
| Itga2b F | TGGACTCAGCCCTTCACTCT | N/A |
| Itga2b R | ACCTCAACCGAGACGGCTAT | N/A |
| Itga5 F | GTCCTATCCAGTGCACCACC | N/A |
| Itga5 R | TACTCCACAGGCTCCTCTCC | N/A |
| Itgav F | TTGCCCTCCTTCTACAATCC | N/A |
| Itgav R | ATTCGCCGTGGACTTCTTC | N/A |
| Itgb1 F | CAGGAAACCAGTTGCAAATTC | N/A |
| Itgb1 R | ACACCGACCCGAGACCCT | N/A |
| Itgb3 F | CGCCTCGTGTGGTACAGAT | N/A |
| Itgb3 R | AGTGGCCGGGACAACTCT | N/A |
| Itih5 F | GAATTGTGACGAGAGCCTCC | N/A |
| Itih5 R | AAACCTCCCTCCTCTACCCA | N/A |
| Nkg7 F | CCACAGGTCCTCACTTCTCTGC | N/A |
| Nkg7 R | CAGCCAGGATACAGAAGCTCTG | N/A |
| Pdcd1 F | CGGTTTCAAGGCATGGTCATTGG | N/A |
| Pdcd1 R | TCAGAGTGTCGTCCTTGCTTCC | N/A |
| Pde3b F | GAGGTCATCGTCTGTGTCACTG | N/A |
| Pde3b R | GTTAGAGAGCCAGCAGACACTG | N/A |
| Prf1 F | ACACAGTAGAGTGTCGCATGTAC | N/A |
| Prf1 R | GTGGAGCTGTTAAAGTTGCGGG | N/A |
| Ptprc F | CTTCAGTGGTCCCATTGTGGTG | N/A |
| Ptprc R | TCAGACACCTCTGTCGCCTTAG | N/A |
| Satb1 F | TCACAGGCAGTATTTGCACGCG | N/A |
| Satb1 R | CGAAGGTTTACCAGCAGAGACTG | N/A |
| Tbx21 F | CCTCTTCTATCCAACCAGTAT | N/A |
| Tbx21 R | CTCCGCTTCATAACTGTGT | N/A |
| Tgfb1 F | CCCTATATTTGGAGCCTGGA | N/A |
| Tgfb1 R | CTTGCGACCCACGTAGTAGA | N/A |
| Tnf F | CTGGGACAGTGACCTGGACTGT | N/A |
| Tnf R | ACTCTCCCTTTGCAGAACTCAGG | N/A |
| Tnfrsf8 F | ACTACGTCAATGAAGACGGGA | N/A |
| Tnfrsf8 R | TCACAGATTCGAGGAGAGTTCC | N/A |
| Recombinant DNA | ||
| MIG(F)-CRel | N/A | Addgene 26984 |
| MIGR1 (ctrl GFP vector) | N/A | Addgene 27490 |
| Software and Algorithms | ||
| FlowJo | https://www.flowjo.com | N/A |
| Graphpad Prism | http://www.graphpad.com/scientific-software/prism | N/A |
| DESEq2 (1.14.1) | http://bioconductor.org/packages/release/bioc/html/DESeq2.html | N/A |
| GenomicAlignments (1.10.1) | https://bioconductor.org/packages/release/bioc/html/GenomicAlignments.html | N/A |
| GenomicFeatures (1.26.4) | http://bioconductor.org/packages/release/bioc/html/GenomicFeatures.html | N/A |
| sva (3.18.0) | https://bioconductor.org/packages/release/bioc/html/sva.html | RRID:SCR_012836 |
| TxDb.Mmusculus.UCSC.mm9.knownGene (3.4.0) | http://bioconductor.org/packages/release/data/annotation/html/TxDb.Mmusculus.UCSC.mm9.knownGene.html | N/A |
| R (3.3.3) | http://www.r-project.org/ | RRID:SCR 001905 |
| Gene Set Enrichment Analysis Software | http://software.broadinstitute.org/gsea/index.jsp | N/A |
| Ingenuity Pathway Analysis | https://www.qiagenbioinformatics.com/products/ingenuity-pathway-analysis/ | N/A |
| Morpheus | https://software.broadinstitute.org/morpheus/ | N/A |
| Other | ||
| GEO accession number (RNaseq data) | GEO: GSE82008 | N/A |
Highlights.
NF-κB c-Rel regulates the transcriptional landscape of activated Tregs
c-Rel activity in Tregs restricts anti-tumor responses
Chemical c-Rel inhibition reduces melanoma growth and potentiates anti-PD-1 therapy
Acknowledgments
We thank Lekha Nair, Alice Lepelley, Thomas S. Postler, John J. Seeley, Crystal Bussey, Gaelle H. Martin, Christian Schindler, Thomas M. Enzler, and Gary K. Schwartz for technical help and constructive discussions on the project. Y.G.B. was supported by a postdoctoral fellowship from the Cancer Research Institute. This work was supported by grants from the NIH (R01-AI068977) and the Herbert Irving Cancer Center at Columbia University and institutional support from Columbia University to S.G.
Footnotes
Author Contributions: Y.G.-B. and S.G. conceived the experiments. Y.G.-B., M.S.H., and S.G. wrote the paper. Y.G.-B., H.O., and R.C., performed experiments. Y.G.-B., A.D., D.B., T.C., M.S.H., and S.G. analyzed the data. U.K. and R.S. provided mice and reagents. S.G. secured funding.
Supplemental Information: Supplemental Information includes five figures and can be found with this article online at http://dx.doi.org/10.1016/j.cell.2017.08.004.
References
- Algul H, Treiber M, Lesina M, Nakhai H, Saur D, Geisler F, Pfeifer A, Paxian S, Schmid RM. Pancreas-specific RelA/p65 truncation increases susceptibility of acini to inflammation-associated cell death following cerulein pancreatitis. J Clin Invest. 2007;117:1490–1501. doi: 10.1172/JCI29882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baumgartner JM, Gonzalez R, Lewis KD, Robinson WA, Richter DA, Palmer BE, Wilson CC, McCarter MD. Increased survival from stage IV melanoma associated with fewer regulatory T Cells. J Surg Res. 2009;154:13–20. doi: 10.1016/j.jss.2008.04.043. [DOI] [PubMed] [Google Scholar]
- Beg AA, Sha WC, Bronson RT, Ghosh S, Baltimore D. Embryonic lethality and liver degeneration in mice lacking the RelA component of NF-kappa B. Nature. 1995;376:167–170. doi: 10.1038/376167a0. [DOI] [PubMed] [Google Scholar]
- Bertrand A, Kostine M, Barnetche T, Truchetet ME, Schaeverbeke T. Immune related adverse events associated with anti-CTLA-4 antibodies: systematic review and meta-analysis. BMC Med. 2015;13:211. doi: 10.1186/s12916-015-0455-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boissonnas A, Scholer-Dahirel A, Simon-Blancal V, Pace L, Valet F, Kissenpfennig A, Sparwasser T, Malissen B, Fetler L, Amigorena S. Foxp3+ T cells induce perforin-dependent dendritic cell death in tumor-draining lymph nodes. Immunity. 2010;32:266–278. doi: 10.1016/j.immuni.2009.11.015. [DOI] [PubMed] [Google Scholar]
- Bravo-Cuellar A, Hernández-Flores G, Lerma-Díaz JM, Domínguez-Rodríguez JR, Jave-Suárez LF, De Célis-Carrillo R, Aguilar-Lemarroy A, Gómez-Lomeli P, Ortiz-Lazareno PC. Pentoxifylline and the proteasome inhibitor MG132 induce apoptosis in human leukemia U937 cells through a decrease in the expression of Bcl-2 and Bcl-XL and phosphorylation of p65. J Biomed Sci. 2013;20:13. doi: 10.1186/1423-0127-20-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S, Lee LF, Fisher TS, Jessen B, Elliott M, Evering W, Logronio K, Tu GH, Tsaparikos K, Li X, et al. Combination of 4-1BB agonist and PD-1 antagonist promotes antitumor effector/memory CD8 T cells in a poorly immunogenic tumor model. Cancer Immunol Res. 2015;3:149–160. doi: 10.1158/2326-6066.CIR-14-0118. [DOI] [PubMed] [Google Scholar]
- Curran MA, Montalvo W, Yagita H, Allison JP. PD-1 and CTLA-4 combination blockade expands infiltrating T cells and reduces regulatory T and myeloid cells within B16 melanoma tumors. Proc Natl Acad Sci USA. 2010;107:4275–4280. doi: 10.1073/pnas.0915174107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darrasse-Jèze G, Bergot AS, Durgeau A, Billiard F, Salomon BL, Cohen JL, Bellier B, Podsypanina K, Klatzmann D. Tumor emergence is sensed by self-specific CD44hi memory Tregs that create a dominant tolerogenic environment for tumors in mice. J Clin Invest. 2009;119:2648–2662. doi: 10.1172/JCI36628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Simone M, Arrigoni A, Rossetti G, Gruarin P, Ranzani V, Politano C, Bonnal RJ, Provasi E, Sarnicola ML, Panzeri I, et al. Transcriptional landscape of human tissue lymphocytes unveils uniqueness of tumor-infiltrating T regulatory cells. Immunity. 2016;45:1135–1147. doi: 10.1016/j.immuni.2016.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deree J, Melbostad H, Loomis WH, Putnam JG, Coimbra R. The effects of a novel resuscitation strategy combining pentoxifylline and hypertonic saline on neutrophil MAPK signaling. Surgery. 2007;142:276–283. doi: 10.1016/j.surg.2007.04.008. [DOI] [PubMed] [Google Scholar]
- Dias S, D'Amico A, Cretney E, Liao Y, Tellier J, Bruggeman C, Almeida FF, Leahy J, Belz GT, Smyth GK, et al. Effector regulatory T cell differentiation and immune homeostasis depend on the transcription factor Myb. Immunity. 2017;46:78–91. doi: 10.1016/j.immuni.2016.12.017. [DOI] [PubMed] [Google Scholar]
- DiDonato JA, Mercurio F, Karin M. NF-κB and the link between inflammation and cancer. Immunol Rev. 2012;246:379–400. doi: 10.1111/j.1600-065X.2012.01099.x. [DOI] [PubMed] [Google Scholar]
- Donkor MK, Sarkar A, Savage PA, Franklin RA, Johnson LK, Jungbluth AA, Allison JP, Li MO. T cell surveillance of oncogene-induced prostate cancer is impeded by T cell-derived TGF-β1 cytokine. Immunity. 2011;35:123–134. doi: 10.1016/j.immuni.2011.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dua P, Ingle A, Gude RP. Suramin augments the antitumor and antimetastatic activity of pentoxifylline in B16F10 melanoma. Int J Cancer. 2007;121:1600–1608. doi: 10.1002/ijc.22843. [DOI] [PubMed] [Google Scholar]
- Edward M, MacKie RM. Pentoxifylline enhances lung colonization and alters cell adhesion and glycosaminoglycan synthesis by metastatic B16 melanoma cells. Int J Cancer. 1991;49:711–716. doi: 10.1002/ijc.2910490514. [DOI] [PubMed] [Google Scholar]
- Fingert HJ, Pu AT, Chen ZY, Googe PB, Alley MC, Pardee AB. In vivo and in vitro enhanced antitumor effects by pentoxifylline in human cancer cells treated with thiotepa. Cancer Res. 1988;48:4375–4381. [PubMed] [Google Scholar]
- Francisco LM, Sage PT, Sharpe AH. The PD-1 pathway in tolerance and autoimmunity. Immunol Rev. 2010;236:219–242. doi: 10.1111/j.1600-065X.2010.00923.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greten FR, Arkan MC, Bollrath J, Hsu LC, Goode J, Miething C, Göktuna SI, Neuenhahn M, Fierer J, Paxian S, et al. NF-kappaB is a negative regulator of IL-1beta secretion as revealed by genetic and pharmacological inhibition of IKKbeta. Cell. 2007;130:918–931. doi: 10.1016/j.cell.2007.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Godec J, Tan Y, Liberzon A, Tamayo P, Bhattacharya S, Butte AJ, Mesirov JP, Haining WN. Compendium of immune signatures identifies conserved and species-specific biology in response toinflammation. Immunity. 2016;44:194–206. doi: 10.1016/j.immuni.2015.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heise N, De Silva NS, Silva K, Carette A, Simonetti G, Pasparakis M, Klein U. Germinal center B-cell maintenance and differentiation are controlled by distinct NF-kappaB transcription factor subunits. J Exp Med. 2014;211:2103–2118. doi: 10.1084/jem.20132613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huehn J, Siegmund K, Lehmann JC, Siewert C, Haubold U, Feuerer M, Debes GF, Lauber J, Frey O, Przybylski GK, et al. Developmental stage, phenotype, and migration distinguish naive- and effector/memory-like CD4+ regulatory T cells. J Exp Med. 2004;199:303–313. doi: 10.1084/jem.20031562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isomura I, Palmer S, Grumont RJ, Bunting K, Hoyne G, Wilkinson N, Banerjee A, Proietto A, Gugasyan R, Wu L, et al. c-Rel is required for the development of thymic Foxp3+ CD4 regulatory T cells. J Exp Med. 2009;206:3001–3014. doi: 10.1084/jem.20091411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jandus C, Bioley G, Speiser DE, Romero P. Selective accumulation of differentiated FOXP3(+) CD4 (+) T cells in metastatic tumor lesions from melanoma patients compared to peripheral blood. Cancer Immunol Immunother. 2008;57:1795–1805. doi: 10.1007/s00262-008-0507-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jimenez JL, Punzón C, Navarro J, Muñoz-Fernández MA, Fresno M. Phosphodiesterase 4 inhibitors prevent cytokine secretionby T lymphocytes by inhibiting nuclear factor-kappaB and nuclear factor of activated T cells activation. J Pharmacol Exp Ther. 2001;299:753–759. [PubMed] [Google Scholar]
- Kamran MZ, Gude RP. Pentoxifylline inhibits melanoma tumor growth and angiogenesis by targeting STAT3 signaling pathway. Biomed Pharmacother. 2013;67:399–405. doi: 10.1016/j.biopha.2013.03.020. [DOI] [PubMed] [Google Scholar]
- Kim HJ, Hawke N, Baldwin AS. NF-kappaB and IKK as therapeutic targets in cancer. Cell Death Differ. 2006;13:738–747. doi: 10.1038/sj.cdd.4401877. [DOI] [PubMed] [Google Scholar]
- Kleffel S, Posch C, Barthel SR, Mueller H, Schlapbach C, Guenova E, Elco CP, Lee N, Juneja VR, Zhan Q, et al. Melanoma cell-intrinsic PD-1 receptor functions promote tumor growth. Cell. 2015;162:1242–1256. doi: 10.1016/j.cell.2015.08.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Köntgen F, Grumont RJ, Strasser A, Metcalf D, Li R, Tarlinton D, Gerondakis S. Mice lacking the c-rel proto-oncogene exhibit defects in lymphocyte proliferation, humoral immunity, and interleukin-2 expression. Genes Dev. 1995;9:1965–1977. doi: 10.1101/gad.9.16.1965. [DOI] [PubMed] [Google Scholar]
- Levine AG, Arvey A, Jin W, Rudensky AY. Continuous requirement for the TCR in regulatory T cell function. Nat Immunol. 2014;15:1070–1078. doi: 10.1038/ni.3004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long M, Park SG, Strickland I, Hayden MS, Ghosh S. Nuclear factor-kappaB modulates regulatory T cell development by directly regulating expression of Foxp3 transcription factor. Immunity. 2009;31:921–931. doi: 10.1016/j.immuni.2009.09.022. [DOI] [PubMed] [Google Scholar]
- Luo CT, Liao W, Dadi S, Toure A, Li MO. Graded Foxo1 activity in Treg cells differentiates tumour immunity from spontaneous autoimmunity. Nature. 2016;529:532–536. doi: 10.1038/nature16486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Messina N, Fulford T, O'Reilly L, Loh WX, Motyer JM, Ellis D, McLean C, Naeem H, Lin A, Gugasyan R, et al. The NF-κB transcription factor RelA is required for the tolerogenic function of Foxp3(+) regulatory T cells. J Autoimmun. 2016;70:52–62. doi: 10.1016/j.jaut.2016.03.017. [DOI] [PubMed] [Google Scholar]
- Miyara M, Yoshioka Y, Kitoh A, Shima T, Wing K, Niwa A, Parizot C, Taflin C, Heike T, Valeyre D, et al. Functional delineation and differentiation dynamics of human CD4+ T cells expressing the FoxP3 transcription factor. Immunity. 2009;30:899–911. doi: 10.1016/j.immuni.2009.03.019. [DOI] [PubMed] [Google Scholar]
- Mulligan G, Mitsiades C, Bryant B, Zhan F, Chng WJ, Roels S, Koenig E, Fergus A, Huang Y, Richardson P, et al. Gene expression profiling and correlation with outcome in clinical trials of the proteasome inhibitor bortezomib. Blood. 2007;109:3177–3188. doi: 10.1182/blood-2006-09-044974. [DOI] [PubMed] [Google Scholar]
- Nakagawa H, Sido JM, Reyes EE, Kiers V, Cantor H, Kim HJ. Instability of Helios-deficient Tregs is associated with conversion to a T-effector phenotype and enhanced antitumor immunity. Proc Natl Acad Sci USA. 2016;113:6248–6253. doi: 10.1073/pnas.1604765113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neo WH, Lim JF, Grumont R, Gerondakis S, Su IH. c-Rel regulates Ezh2 expression in activated lymphocytes and malignant lymphoid cells. J Biol Chem. 2014;289:31693–31707. doi: 10.1074/jbc.M114.574517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishikawa H, Sakaguchi S. Regulatory T cells in tumor immunity. Int J Cancer. 2010;127:759–767. doi: 10.1002/ijc.25429. [DOI] [PubMed] [Google Scholar]
- Oeckinghaus A, Hayden MS, Ghosh S. Crosstalk in NF-κB signaling pathways. Nat Immunol. 2011;12:695–708. doi: 10.1038/ni.2065. [DOI] [PubMed] [Google Scholar]
- Oh H, Grinberg-Bleyer Y, Liao W, Maloney D, Wang P, Wu Z, Wang J, Bhatt DM, Heise N, Schmid RM, et al. An NF-κB transcription factor-dependent, lineage specific transcriptional program promotes regulatory T cell identity and function. Immunity. 2017;47 doi: 10.1016/j.immuni.2017.08.010. http://dx.doi.org/10.1016/j.immuni.2017.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ouyang W, Liao W, Luo CT, Yin N, Huse M, Kim MV, Peng M, Chan P, Ma Q, Mo Y, et al. Novel Foxo1-dependent transcriptional programs control T(reg) cell function. Nature. 2012;491:554–559. doi: 10.1038/nature11581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ouyang Z, Wu H, Li L, Luo Y, Li X, Huang G. Regulatory T cells in the immunotherapy of melanoma. Tumour Biol. 2016;37:77–85. doi: 10.1007/s13277-015-4315-0. [DOI] [PubMed] [Google Scholar]
- Pinzani M, Marra F, Caligiuri A, DeFranco R, Gentilini A, Failli P, Gentilini P. Inhibition by pentoxifylline of extracellular signal-regulated kinase activation by platelet-derived growth factor in hepatic stellate cells. Br J Pharmacol. 1996;119:1117–1124. doi: 10.1111/j.1476-5381.1996.tb16012.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ratheesh A, Ingle A, Gude RP. Pentoxifylline modulates cell surface integrin expression and integrin mediated adhesion of B16F10 cells to extracellular matrix components. Cancer Biol Ther. 2007;6:1743–1752. doi: 10.4161/cbt.6.11.4833. [DOI] [PubMed] [Google Scholar]
- Rosenblum MD, Gratz IK, Paw JS, Lee K, Marshak-Rothstein A, Abbas AK. Response to self antigen imprints regulatory memory in tissues. Nature. 2011;480:538–542. doi: 10.1038/nature10664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruan Q, Kameswaran V, Tone Y, Li L, Liou HC, Greene MI, Tone M, Chen YH. Development of Foxp3(+) regulatory t cells is driven by the c-Rel enhanceosome. Immunity. 2009;31:932–940. doi: 10.1016/j.immuni.2009.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samstein RM, Arvey A, Josefowicz SZ, Peng X, Reynolds A, Sandstrom R, Neph S, Sabo P, Kim JM, Liao W, et al. Foxp3 exploits a pre-existent enhancer landscape for regulatory T cell lineage specification. Cell. 2012;151:153–166. doi: 10.1016/j.cell.2012.06.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma P, Allison JP. Immune checkpoint targeting in cancer therapy: toward combination strategies with curative potential. Cell. 2015;161:205–214. doi: 10.1016/j.cell.2015.03.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shono Y, Tuckett AZ, Ouk S, Liou HC, Altan-Bonnet G, Tsai JJ, Oyler JE, Smith OM, West ML, Singer NV, et al. A small-molecule c-Rel inhibitor reduces alloactivation of T cells without compromising anti-tumor activity. Cancer Discov. 2014;4:578–591. doi: 10.1158/2159-8290.CD-13-0585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci USA. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugiyama D, Nishikawa H, Maeda Y, Nishioka M, Tanemura A, Katayama I, Ezoe S, Kanakura Y, Sato E, Fukumori Y, et al. Anti-CCR4 mAb selectively depletes effector-type FoxP3+CD4+ regulatory T cells, evoking antitumor immune responses in humans. Proc Natl Acad Sci USA. 2013;110:17945–17950. doi: 10.1073/pnas.1316796110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suresh R, Vig M, Bhatia S, Goodspeed EP, John B, Kandpal U, Srivastava S, George A, Sen R, Bal V, et al. Pentoxifylline functions as an adjuvant in vivo to enhance T cell immune responses by inhibiting activation-induced death. J Immunol. 2002;169:4262–4272. doi: 10.4049/jimmunol.169.8.4262. [DOI] [PubMed] [Google Scholar]
- Tang Q, Bluestone JA. The Foxp3+ regulatory T cell: a jack of all trades, master of regulation. Nat Immunol. 2008;9:239–244. doi: 10.1038/ni1572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vahl JC, Drees C, Heger K, Heink S, Fischer JC, Nedjic J, Ohkura N, Morikawa H, Poeck H, Schallenberg S, et al. Continuous T cell receptor signals maintain a functional regulatory T cell pool. Immunity. 2014;41:722–736. doi: 10.1016/j.immuni.2014.10.012. [DOI] [PubMed] [Google Scholar]
- Wang W, Tam WF, Hughes CC, Rath S, Sen R. c-Rel is a target of pentoxifylline-mediated inhibition of T lymphocyte activation. Immunity. 1997;6:165–174. doi: 10.1016/s1074-7613(00)80423-9. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. NF-κB c-Rel Regulates the Activated Treg Cell Differentiation and Gene Expression, Related to Figure 1: (A and B) Flow cytometry analysis on spleen total Tregs; the cumulative percentage of CD103+ and Ly6C+ Tregs is shown.
(C–F) CD62Llow CD44high aTreg and CD62Lhigh CD44low rTreg were sorted from Foxp3CRE-YFP (WT) Foxp3crep65F/F (p65KO), and Foxp3crec-RelF/F (c-RelKO) mice and submitted to RNA-seq analysis. Gene expression changes in WT aTreg versus WT rTreg were plotted against those in KO aTreg versus WT aTreg (A, B) and KO rTreg versus WT rTreg (C, D). Numbers and colored dots indicate genes upregulated in WT aTreg while downregulated in KO Treg (red) and downregulated in aTreg while upregulated in KO Treg (blue) (fold change > 2, p < 0.01).
(G and H) Flow cytometry analysis on spleen CD62Llow CD44high aTregs; the cumulative percentage of CD103+ and Ly6C+ cells is shown. Data are form 2 experiments. *p < 0.05, n.s. non-significant.
Figure S2. c-Rel Expression in Treg Cells Restricts Anti-tumor Immune Responses, Related to Figure 2: (A) Splenic CD4+Foxp3YFP+ Tregs of each genotype were isolated, stimulated or not for 3 hr, and submitted to RNA-seq analysis. Fold Change in the expression of selected genes is shown.
(B) Splenocytes of the indicated genotypes were re-stimulated with PMA-ionomycin 4 days after tumor challenge and analyzed by flow cytometry. Mean ± SEM of Ki67+, CD44high and TNFhighIFN-γ+ cells is shown.
(C) 5-7 weeks-old Foxp3cre and Foxp3crerelF/F were transplanted sub-cutaneously with B16F1 cells. Splenocytes were restimulated ex-vivo with PBS or gp100/pmel peptide, 4 days after tumor inoculation. Representative dot plot of IFN-expression in gated CD8+ T cells cells. Numbers indicate the percentage in the gate; MFI: Mean Fluorescence Intensity of IFN- γ in IFN-γ+ cells.
(D) Foxp3creand Foxp3crerelF/F were injected intravenously with B16F10 cells. Left: representative picture of lungs at D14; right: Number of detectable tumor foci/lung at D14. Each dot represents an individual mouse form 2 independent experiments.
(E) Foxp3cre and Foxp3crerelF/F were transplanted sub-cutaneously with BRAFCAPten−/− cells. The mean ± SEM of tumor growth over time is shown. Numbers indicate the number of mice with detectable tumors at the end of the experiment. All data are from 2 to 3 experiments***p < 0.001.
Figure S3. Chemical c-Rel Inhibition Impairs Treg Identity, Related to Figure 3: (A–E) CD4+YFP+Tregs sorted from Foxp3cre (WT) and Foxp3crec-RelF/F were activated with H2O or PTXF and submitted to RNA-Seq analysis. (A) Principal component analysis. (B) Summary of gene expression changes in WT Treg treated with PTXF and stimulated Foxp3crec-RelF/F (top) or stimulated Foxp3crep65F/F (bottom, from Oh et al., 2017), compared to WT+H2O Tregs. Numbers indicate the genes with a fold-change > 2 and a p value < 0.05 when compared to the WT+H2O samples. Up: upregulated expression when compared to the WT control cells; down: downregulated expression. (C) Heatmap showing expression of genes up-or downregulated in both c-RelKO and WT+PTXF samples. (D) Gene expression of selected genes measured by RNA-seq (top) and qRT-PCR (bottom). Fold changes (mean ± SEM) and statistics in WT+PTXF and c-Rel−/− Treg versus WT+H2O Treg are shown. (E) Gene Set Enrichment Analysis plots showing immune signatures selected from the overlapping ones in Figure 3G.
(F) Selected Top Regulator Effect Networks using Ingenuity Pathway Analysis and showing c-Rel as a central regulator of the altered transcriptome in both PTXF and c-RelKO samples.
(G–I) CD4+GFP+ Tregs were stimulated as with H2O or PTXF and subsequently tested for in vitro suppression (G) and in vivo colitis assays (H, I). (G) Percentage of donor CD45.2+ Tregs in each well. (H) Colon histology (H/E) 40 days after cell transfer; bars: 100mm, original magnification 100×. (I) Tissues were analyzed by flow cytometry 40 days after cell transfer. The proportion of Foxp3+ Tregs in live CD4+T cells is shown. In G and I, data represents the mean of 2 experiments; values were non-significantly changed between groups.
Figure S4. c-Rel Inhibition by PTXF Suppresses Melanoma Growth, Related to Figure 4: (A and B) WT C57BL/6J mice were transplanted sub-cutaneously with B16F1 cells and treated from D-1 to D7 with PTXF or PBS as a control. (A) FACS staining in the TILs at D16; Foxp3 expression and % of Helios+ in gated Tregs is shown. (B) qPCR analysis on total tumor RNA at D16.
(C) WT C57BL/6J mice were injected intravenously with B16F10 cells and with PTXF or PBS from D-1 to D7. The number of macroscopic tumor foci is showed.
(D) WT C57BL/6J mice were transplanted sub-cutaneously with B16F1 cells and treated from D-1 to D7 with IT-603 or EtOH+cremophor as a control. FACS analysis of the TILS at D16 is shown. Data are represented as mean ± SEM from 2-3 experiments. *p < 0.05.
Figure S5. PTXF and PD-1-Blockade Have Additive Effects on Growth of Established Melanoma, Related to Figure 5: (A) 5-7 weeks-old Foxp3crerelF/F mice were transplanted sub-cutaneously with 3×105 B16F1 cells and treated from D6 with anti-PD-1 mAb or PBS (black arrows). Mean ± SEM tumor growth over time is shown.
(B) WT C57BL/6J mice were transplanted sub-cutaneously with B16F1 cells and treated from with IT-603, and/or anti-PD-1, or EtOH+cremophor as a control. FACS analysis of TILs at D16 is shown. *p < 0.05, **p < 0.005, ***p < 0.001.