

Abstract
MicroRNAs (miRNAs) and Transcription Factors (TFs) both influence messenger RNA (mRNA) expression, disrupting biological pathways involved in carcinogenesis and prognosis. As many miRNAs target multiple mRNAs, thus influencing a multitude of biological pathways, deciphering which miRNAs are important for cancer development and survival is difficult. In this study, we (i) determine associations between TF and survival (N = 168 colon cancer cases); (ii) identify miRNAs associated with TFs related to survival; and (iii) determine if factors derived from TF‐specific miRNA principal component analysis (PCA) influence survival. Cox Proportional hazard models were run for each PCA factor to determine Hazard Ratios (HR) and 95% Confidence Intervals (CI) adjusting for age, center, and AJCC stage. Thirty TFs improved survival when differential expression increased; 27 of these were associated significantly with normal colonic mucosa expression of 65 unique miRNAs when an FDR q‐value of <0.05 was applied. Five factors, comprising 21 miRNAs, altered survival in rectal cancer subjects; four of these five factors improved survival and one factor reduced survival. One factor comprising four miRNAs reduced survival in colon cancer subjects. In summary, our data suggest that expression of TFs and their related miRNAs influence survival after diagnosis with colorectal cancer.
Keywords: cancer, colorectal, microRNA, survival, transcription factor
Abbreviations
- CRC
colorectal cancer
- cDNA
complementary DNA
- CI
confidence intervals
- GC
fold change
- HR
hazard ratios
- IPA
ingenuity pathway analysis
- KPMCP
Kaiser Permanente Medical Care Program of Northern California
- mRNA
messenger RNA
- miRNA
microRNA
- NF‐κB
nuclear factor κB
- PCR
polymerase chain reaction
- PCA
principal component analysis
- SEER
surveillance, epidemiology, and end results
- TF
transcription factor
1. INTRODUCTION
Transcription factors (TFs) are proteins that bind to promoter regions of genes, via their DNA‐binding domain, to activate, or repress transcription of genes.1 MicroRNAs (miRNAs) are small, non‐protein coding RNA molecules, able to bind to mRNAs, and repress mRNA translation or cause mRNA degradation.2 MiRNAs may regulate multiple targets, and many genes are targeted by a multitude of miRNAs, creating complex regulatory networks.3, 4 MiRNAs and TFs both have the ability to act as oncogenes or tumor suppressors depending on different conditions, simultaneously regulating many biological pathways, including those important to cancer development, and progression.4, 5 TFs are able to alter miRNA expression as much or more so than somatic changes and epigenetic factors, and while miRNAs may not drastically reduce cellular protein levels, they are known to have a marked influence on cell‐fate determination and contribute to many diseases, including cancer.4 Additionally, miRNAs are known to interact with TFs, forming feed‐forward, and feedback loops that regulate various processes and diseases.5, 6
There are various TFs that are known to be important in human cancers, many of which have been extensively investigated, including nuclear factor κB (NF‐κB), BRCA1, MYC,7 and TP53.4 We previously investigated the role the JAK/STAT pathway plays in cancer development and survival, and found polymorphisms within the genes of this pathway were associated significantly with reduced survival in colon and rectal cancer subjects.8 In protein‐protein interactions within metabolic and biological networks whose degree distribution follows a power law, it has been purported that those networks of a larger diameter and more connectivity have an advantage for survival.9 Martinez et al investigated TF‐miRNA relationships in Caenorhabditis elegans and determined that regulation of miRNA expression by TFs is similar to that of protein‐coding genes.10 In our previous investigations, we have found numerous miRNAs that are associated with altered cancer survival in colon or rectal cases, or in both.11 It is possible that the associations previously detected between miRNAs and survival or between TFs and survival are the results of more complex relationships between these two sets of regulators, the impact they have on one another, and subsequently the biological pathways in which they are involved.
In this study, we investigate the impact differentially expressed TFs have on survival in colon cancer subjects, and identify associations between these TFs and expression of miRNA in normal mucosa from colon and rectal cancer cases. We assess differential expression of TF‐specific miRNAs to determine if they work collaboratively to influence survival. We hypothesize that TFs that regulate a larger number of miRNAs will have a greater influence on survival.
2. METHODS
2.1. Study population
The data come from participants in the population‐based Diet, Activity, and Lifestyle study that were recruited from Utah or the Kaiser Permanente Medical Care Program of Northern California (KPMCP). Colon cancer cases were identified as having a primary adenocarcinoma diagnosed between October 1991 and September 1994, while rectal cancer cases were diagnosed between May 1997 and May 2001. Eligible cases were between 30 and 79 years of age at diagnosis, currently living in the study area, spoke English, were able to complete an interview, and had no prior history of CRC, Crohn's disease, ulcerative colitis, or known familial adenomatous polyposis. This study was approved by the Institutional Review Board at the University of Utah; participants signed an informed consent form.
2.2. miRNA processing
RNA was extracted from formalin‐fixed paraffin embedded tissues and processed as previously described.11 A total of 100 ng total RNA was labeled with Cy3 and hybridized to Agilent Human miRNA Microarrays V19.0 and were scanned on an Agilent SureScan microarray scanner model G2600D using Agilent Feature Extract software v.11.5.1.1. Data were required to pass stringent QC parameters established by Agilent that included tests for excessive background fluorescence, excessive variation among probe sequence replicates on the array, and measures of the total gene signal on the array to assess low signal. Samples that failed to meet QC standards were repeated, and if a sample failed QC assessment a second time the sample was deemed to be of poor quality and was excluded from down‐stream analysis. The Agilent platform was found to be highly reliable (r = 0.98), and to have reasonable agreement with NanoString12 as well as excellent agreement with qRT‐PCR.13 For unpaired samples due to missing normal scans, we imputed values whenever possible for normal mucosa as previously described.14 In order to minimize differences that could be attributed to the array, amount of RNA, location on array, or other factors that could erroneously influence expression, total gene signal was normalized by multiplying each sample by a scaling factor, which was the median of the 75th percentiles of all the samples divided by the 75th percentile of each individual sample.15 This scaling factor was implemented using SAS 9.4.
2.3. RNA‐Seq
MRNA expression data came from 216 study participants with both carcinoma and paired normal mucosa after applying rigid quality control. Of these 216 participants, 168 were diagnosed with colon cancer and 48 were diagnosed with rectal cancer. Total RNA was extracted from formalin‐fixed paraffin embedded tissues and processed as previously described.11 Sequencing library construction was done with the Illumina TruSeq Stranded Total RNA Sample Preparation Kit with Ribo‐Zero. The samples were then fragmented and primed for complementary DNA (cDNA) synthesis, adapters were then ligated onto the cDNA, and the resulting samples were then amplified using polymerase chain reaction (PCR); the amplified library was then purified using Agencount AMPure XP beads. A more detailed description of the methods can be found in our previous work.16
Sequencing was done using an Illumina TruSeq v3 single read flow cell, and a 50 cycle single‐read sequence run was performed on an Illumina HiSeq instrument. Reads were then aligned to a sequence database containing the human genome (build GRCh37/hg19, February 2009 from http://genome.ucsc.edu) and alignment was performed using novoalign v2.08.01. Python and a pysam library were used to calculate counts for each exon and UTR of the genes using a list of gene coordinates obtained from http://genome.ucsc.edu. We dropped features that were not expressed in our data or for which the expression was missing for the majority of samples. A more detailed description of the methods can be found in our previous work.16
2.4. Survival information
Survival information was obtained from Surveillance, Epidemiology, and End Results (SEER) tumor registries in Utah and California. Survival months were calculated from the date of diagnosis to the date of last contact or death. AJCC stage and cause of death were also obtained from the SEER registries. We assessed CRC‐specific mortality. Individuals who died from other causes were censored at the time of death. Individuals alive at the end of follow‐up were also censored alive when calculating survival months.
2.5. Statistical analysis
Figure 1 depicts the study flow. TFs and binding sites were downloaded from UCSC Table Browser,17 using the February 2009 (GRCh37/hg19) alignment and the “Txn Factor ChIP” track and the “wgEncodeTegTfbsClusteredV3” table.18 In total there were 161 unique TFs.
Figure 1.
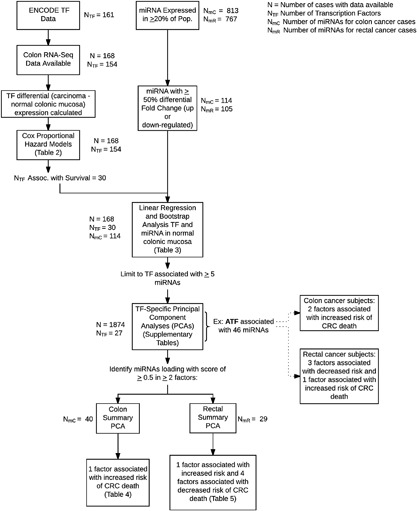
This diagram depicts the study flow
We identified 154 individual TFs with one mapped transcript each that were expressed in colon tissue as determined by RNA‐Seq (N = 168); we did not have sufficient power to discover associations in rectal tissue alone. Differential (carcinoma minus normal mucosa) expression for these TFs was calculated based on paired carcinoma and normal colon mucosa. Cox proportional hazard models were run on those 154 TFs adjusting for age, center, sex, and AJCC stage to determine Hazard Ratios (HR) and 95% Confidence Intervals (CI) with differential expression of TFs using interquartile range as the unit of change for calculation of the HR and CI. A permutation method was implemented using 10 000 permutations to estimate the P‐values. Adjustment for multiple comparisons was conducted using Storey's q‐value.19 The distribution of the raw P‐values from the survival analysis had a clear peak near (but slightly shifted from) zero. In such cases, we have found the Benjamini‐Hochberg FDR adjustment overly conservative, and chose to apply instead the Storey q‐value, which also controls the false discovery rate. We report associations where the q‐value was <0.1.
The 30 TFs that were associated with CRC survival were further evaluated with miRNA expression in normal colonic mucosa. We tested these associations using normal miRNA under the assumption that if a TF altered miRNA expression it would be seen in normal mucosa; by examining differential expression, these associations could be missed if the associations were seen in normal as well as tumor. For this analysis, we ran a series of linear regressions generating raw P‐values from a distribution of 10 000 F statistics derived by resampling the residuals with replacement from the null hypothesis model of no association between miRNA and TF normal mucosa expression using the “boot” package in R. Linear models were adjusted for age, center, and sex. We calculated standardized regression slopes by transforming the TF and miRNA normal mucosa expression to standard normal in order to compare the results across the TF/miRNA pairs. We restricted these analyses to miRNAs that had at least a 50% fold change in expression between carcinoma and normal mucosa, leaving 114 (of 813 total) miRNAs available for analysis for colon cancer and 105 (of 767 total) miRNAs assessed for rectal cancer. A small set of RNA‐Seq data were available for rectal cancer cases (N = 48) to validate the TF/miRNA associations identified in colon cancer; this sample size was too small to validate the TF/survival associations identified in colon cancer. Multiple testing adjustments for TF/miRNA associations were made at the TF level using the FDR by Benjamini and Hochberg,20 using an FDR q‐value <0.05. If the FDR is controlled at 0.05 in each of two (or more) separate sets of comparisons, it will also be controlled at 0.05 in the combined set of all considered comparisons. Since the FDR was controlled at 0.05 within each TF then, by this scalability, the FDR is controlled at 0.05 across all TFs.
For each TF associated with survival that had at least five miRNAs significantly linked with it, we ran a TF‐specific principal component analysis (PCA) on the miRNA differential expression using a varimax rotation. For each individual, a factor summary score was generated. We considered PCA factors as being potentially important if they had an eigenvalue of 1.0 or greater or the total proportion of the variance explained was at least 0.8. We report factor loadings for each miRNA, the eigenvalue, and the proportion of variability in the TF/miRNAs accounted for by that PCA factor, along with the corresponding HR and 95% confidence intervals for the highest quartile compared to the lowest quartile of factor scores. Cox Proportional hazard models were run for each PCA factor with adjustments for age, center, and sex. This analysis was conducted for colon and rectal cancer separately given previously identified differences in survival and miRNA expression levels for colon and rectal cancer, using SAS 9.4.
Given the degree of overlap of important miRNA loadings to the TF‐specific factor scores, we identified miRNAs that loaded with a 0.5 or greater in two or more factors. For these miRNAs, we ran a PCA that summarized the most important miRNAs associated with multiple factors to determine their impact on survival (N = 1874). For instance miR‐210 loaded heavily with 7 TF‐specific factors in the 21 analyzed TFs for rectal cancer. In each of these, it uniformly contributed to any factor that increased the likelihood of dying in rectal cancer. Since many of these important miRNAs were associated with several of the TFs linked to CRC survival in colon cancer cases in our data, a summary PCA allowed us to further consolidate the data. We report the survival associations with each TF in online supplements and report the summary PCA of overlapping and highly‐associated‐with‐survival miRNAs for colon and rectal cancer in the main body of the text.
2.6. Bioinformatics
QIAGEN Ingenuity Pathway Analysis (IPA http://www.qiagen.com/ingenuity) was utilized to perform pathway analysis on the 27 TFs associated significantly with survival as well as normal colonic miRNA expression. We performed a core analysis, including direct and indirect relationships, using the ingenuity knowledge base. We restricted to experimentally verified findings and mammalian species, and considered all data sources, tissues, and mutations.
We determined overlap between Transcription Factor Binding Sites (TFBSs) and primary‐miRNA (pri‐miRNA) transcripts as a means of validation for the associations found. The gene assembly used was GRCh37/hg19 for all coordinates. The UCSC Table browser was utilized to obtain TFBSs as well at match ensembl IDs to known gene names. TFBS coordinates were obtained using the “Regulation” group, the “Txn Factor ChIP” table, and the “wgEncoderRegTfbsClusteredV3.” This was then compared to coordinates of primary‐microRNAs (pri‐miRNAs), downloaded from miRBase v19 archived files. Pairs were made when a TFBS occurred ±300 base pairs (bps) from the start or end of the pri‐miRNA transcript. Pri‐miRNAs were then matched to mature miRNAs, using the same coordinate file from miRBase. All coordinate matching was done using R scripting, and the final table merges and triplet determination was done using SQL commands.
In order to determine whether miRNAs targeted TFs, we searched for pairings between the 5′ seed region of the miRNA and the 3′ UTR of the associated TF. Due to availability, the GRCh38 alignment was used to download 3′ UTR FASTA sequences for TFs that had a CCDS sequence available. Four different mature miRNA seeds, defined as nucleotides 2‐7, 1‐7, 2‐8, and 1‐8, were generated for each miRNA; 3′ UTRs of TFs associated with the respective miRNA in normal colon mucosa were then searched for these seeds. A more detailed description of our methods are described in our previous work.21
Cytoscape (http://www.cytoscape.org)22 was used to visualize interactions between miRNA and TFs as well as TF involvement in canonical pathways discovered with IPA.
3. RESULTS
Characteristics of the study population are described in Table 1. Approximately two‐thirds of the colon and rectal cancer subjects were over the age of 60 and a little over half of each population was male. Colon cases comprised 61% of the data; rectal comprised 39%. We analyzed RNA‐Seq data for 11.5% of the total population; 77.8% of which came from colon cases and 22.2% came from rectal cases. Of the RNA‐Seq data available, approximately 90% were from cases diagnosed at stages 1, 2, and 3, with each stage representing approximately 30%, and a little over 10% was from stage 4 cases.
Table 1.
Description of study participants
| Number of subjects | |||||
|---|---|---|---|---|---|
| With RNAseq | Without RNAseq | ||||
| N | % | N | % | Total N | |
| Total | 216 | 11.5 | 1658 | 88.5 | 1874 |
| Age (>60) | 146 | 67.6 | 1131 | 68.2 | 1277 |
| Sex (% Male) | 118 | 54.6 | 897 | 54.1 | 1015 |
| Site | |||||
| Colon | 168 | 77.8 | 981 | 59.2 | 1149 |
| Rectal | 48 | 22.2 | 677 | 40.8 | 725 |
| Vital status | |||||
| CRC death | 45 | 20.9 | 529 | 31.9 | 574 |
| Other death | 46 | 21.4 | 280 | 16.9 | 326 |
| Alive | 124 | 57.7 | 847 | 51.1 | 971 |
| AJCC stage | |||||
| 1 | 58 | 27.2 | 504 | 30.4 | 562 |
| 2 | 61 | 28.6 | 431 | 26.0 | 492 |
| 3 | 71 | 33.3 | 479 | 28.9 | 550 |
| 4 | 23 | 10.8 | 244 | 14.7 | 267 |
Differential expression of thirty TFs, of the 154 TFs analyzed (∼19.5%), was associated significantly with CRC survival prior to adjustment for multiple comparisons in colon tissue (Table 2). These genes uniformly improved survival when differential expression (carcinoma minus normal mucosa) increased. Most TFs were associated with a two‐thirds reduction in the likelihood of dying when there were greater levels of expression in the tumor than in the normal. The adjusted q‐values for the TFs was 0.08, implying that among these 30 TFs, no more than 8% (or about 3) of them could be expected to be false positives.
Table 2.
Transcription factors significantly associated with altered CRC survival prior to adjustment for multiple comparisons
| Number of miRNAs associated with TF | Differential expression (carcinoma—normal mucosa) | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Gene | P raw < 0.05 | P adj < 0.05 | miRNA with FC > 50% | Q1 | Q3 | HR a | (95% CI) | P‐value | Q‐value |
| ATF1 | 359 | 238 | 46 | −0.01 | 0.75 | 0.54 | (0.33, 0.87) | 0.011 | 0.082 |
| BACH1 | 311 | 160 | 35 | −0.03 | 0.70 | 0.63 | (0.41, 0.96) | 0.039 | 0.082 |
| BCL11A | 232 | 72 | 23 | −0.09 | 0.57 | 0.64 | (0.43, 0.95) | 0.028 | 0.082 |
| BCLAF1 | 200 | 24 | 13 | −0.13 | 1.00 | 0.65 | (0.42, 0.99) | 0.049 | 0.082 |
| CTCF | 306 | 143 | 32 | 0.01 | 0.87 | 0.64 | (0.44, 0.93) | 0.022 | 0.082 |
| EGR1 | 102 | 0 | 0 | −0.87 | 0.92 | 0.58 | (0.39, 0.88) | 0.015 | 0.082 |
| ELF1 | 340 | 226 | 45 | −0.07 | 1.16 | 0.61 | (0.40, 0.94) | 0.030 | 0.082 |
| ELK1 | 239 | 86 | 16 | 0.09 | 0.97 | 0.67 | (0.46, 0.98) | 0.046 | 0.082 |
| ETS1 | 104 | 5 | 3 | −0.43 | 0.57 | 0.56 | (0.36, 0.86) | 0.010 | 0.082 |
| FOS | 97 | 0 | 0 | −1.39 | 0.43 | 0.61 | (0.41, 0.92) | 0.022 | 0.082 |
| GABPA | 340 | 195 | 46 | −0.03 | 0.48 | 0.60 | (0.36, 0.99) | 0.048 | 0.082 |
| HDAC6 | 374 | 283 | 40 | −0.15 | 0.38 | 0.70 | (0.51, 0.97) | 0.042 | 0.082 |
| HDAC8 | 203 | 10 | 3 | −0.06 | 0.55 | 0.53 | (0.32, 0.86) | 0.011 | 0.082 |
| HMGN3 | 314 | 164 | 41 | −0.25 | 0.93 | 0.56 | (0.33, 0.95) | 0.030 | 0.082 |
| IRF1 | 122 | 0 | 0 | −0.38 | 0.71 | 0.43 | (0.25, 0.74) | 0.003 | 0.082 |
| JUN | 204 | 20 | 1 | −0.22 | 1.19 | 0.55 | (0.35, 0.86) | 0.012 | 0.082 |
| JUNB | 222 | 22 | 4 | −0.48 | 1.07 | 0.63 | (0.41, 0.97) | 0.039 | 0.082 |
| JUND | 220 | 60 | 1 | −0.65 | 0.67 | 0.61 | (0.41, 0.90) | 0.017 | 0.082 |
| MEF2A | 278 | 142 | 34 | −0.23 | 0.52 | 0.62 | (0.43, 0.90) | 0.016 | 0.082 |
| MXI1 | 208 | 23 | 2 | −0.32 | 0.35 | 0.63 | (0.44, 0.90) | 0.017 | 0.082 |
| NFYA | 351 | 228 | 37 | −0.06 | 0.77 | 0.65 | (0.44, 0.95) | 0.035 | 0.082 |
| PHF8 | 266 | 120 | 21 | −0.15 | 0.62 | 0.65 | (0.44, 0.98) | 0.046 | 0.082 |
| RBBP5 | 344 | 211 | 43 | −0.04 | 0.64 | 0.61 | (0.38, 0.98) | 0.041 | 0.082 |
| SMARCC1 | 352 | 235 | 46 | 0.32 | 1.51 | 0.65 | (0.44, 0.96) | 0.034 | 0.082 |
| SMARCC2 | 338 | 207 | 38 | −0.22 | 0.98 | 0.63 | (0.41, 0.95) | 0.036 | 0.082 |
| SP1 | 306 | 150 | 34 | −0.18 | 0.97 | 0.62 | (0.41, 0.96) | 0.036 | 0.082 |
| STAT5A | 252 | 68 | 14 | −0.14 | 0.51 | 0.57 | (0.37, 0.87) | 0.010 | 0.082 |
| TCF12 | 379 | 251 | 46 | −0.10 | 0.98 | 0.62 | (0.38, 0.98) | 0.048 | 0.082 |
| ZBTB33 | 320 | 105 | 29 | 0.10 | 1.03 | 0.59 | (0.38, 0.92) | 0.021 | 0.082 |
| ZNF263 | 317 | 174 | 26 | 0.01 | 0.90 | 0.66 | (0.45, 0.97) | 0.040 | 0.082 |
Adjusted for age, center, sex, and AJCC.
Twenty‐seven of the TFs that were associated with survival were associated significantly with the normal mucosa expression of 65 unique miRNAs, creating 719 unique associations, in colon tissue when an FDR q‐value of <0.05 was applied (Table 3); three of the TFs had no significant association with miRNA expression. Twenty‐one TFs were associated with at least five miRNAs that had at least a 50% change in expression between carcinoma and normal tissue. Forty‐four of these miRNAs had increased normal colon mucosa expression with increased normal colon mucosa expression of the respective associated TF (as indicated by a positive beta coefficient), and 21 of the miRNAs had decreased normal colon mucosa expression with increased TF normal colon mucosa expression (as is indicated by a negative beta coefficient). For miRNAs associated with multiple TFs, they were consistently either directly or inversely associated with the respective associated TFs. As a means of data validation in rectal cancer cases, we replicated miRNA/TF associations that were significant in colon normal mucosa after adjustment for multiple comparisons using rectal normal mucosa; these results, along with the specific colon tissue findings can be found in Supplementary Table S1.
Table 3.
Transcription factors associated with normal colonic mucosa miRNA expression in colon cancer subjects
| Number of associated miRNAs | |||
|---|---|---|---|
| Transcription factor | Total | Down reg. | Up reg. |
| ATF1 | 46 | 15 | 31 |
| BACH1 | 35 | 8 | 27 |
| BCL11A | 23 | 9 | 14 |
| BCLAF1 | 13 | 0 | 13 |
| CTCF | 32 | 5 | 27 |
| ELF1 | 45 | 11 | 34 |
| ELK1 | 16 | 5 | 11 |
| ETS1 | 3 | 0 | 3 |
| GABPA | 46 | 10 | 36 |
| HDAC6 | 40 | 16 | 24 |
| HDAC8 | 3 | 0 | 3 |
| HMGN3 | 41 | 10 | 31 |
| JUN | 1 | 0 | 1 |
| JUNB | 4 | 2 | 2 |
| JUND | 1 | 1 | 0 |
| MEF2A | 34 | 7 | 27 |
| MXI1 | 2 | 0 | 2 |
| NFYA | 37 | 7 | 30 |
| PHF8 | 21 | 6 | 15 |
| RBBP5 | 43 | 13 | 30 |
| SMARCC1 | 46 | 15 | 31 |
| SMARCC2 | 38 | 12 | 26 |
| SP1 | 34 | 8 | 26 |
| STAT5A | 14 | 8 | 6 |
| TCF12 | 46 | 14 | 32 |
| ZBTB33 | 29 | 2 | 27 |
| ZNF263 | 26 | 12 | 14 |
Assessment of specific TF‐associated miRNAs using a PCA showed that several factors within these TF‐specific miRNAs were associated with survival (See online Supplements). Over the 27 TFs that showed significant associations with miRNAs, 40 miRNAs expressed in colon tissue loaded highly (factor loading of >0.5) to multiple factors associated significantly with survival (See online Supplemental Tables). These 40 miRNAs were further evaluated using PCA analysis and these factors were subsequently tested for association with survival (Table 4). Of the six factors generated, one factor (Factor 5) was associated significantly with survival after a diagnosis with colon cancer (HR = 1.86, 95% CI 1.36, 2.54). Four miRNAs, hsa‐miR‐133b, hsa‐miR‐145‐5p, hsa‐miR‐99a‐5p and hsa‐miR‐99b‐5p, loaded highly to this factor. This factor, while being statistically significant, accounted for only a small (3%) amount of the variability in TF‐associated miRNAs.
Table 4.
Summary of important TF‐associated miRNAs with survival after diagnosis with colon cancer
| miRNA | Transcription Factors a | Factor 1 | Factor 2 | Factor 3 | Factor 4 | Factor 5 | Factor 6 |
|---|---|---|---|---|---|---|---|
| hsa‐let‐7i‐5p | ATF1, BACH1, CTCF, GABPA, RBBP5, SMARCC1, TCF12, ZBTB33 | 0.32 | −0.10 | 0.56 | 0.58 | 0.15 | −0.11 |
| hsa‐miR‐10a‐5p | BACH1, RBBP5, SMARCC1, TCF12, ZBTB33 | 0.32 | −0.07 | 0.23 | 0.53 | 0.24 | −0.11 |
| hsa‐miR‐124‐3p | ATF1, ELK1, PHF8, SP1 | 0.03 | 0.57 | −0.08 | −0.11 | 0.12 | −0.24 |
| hsa‐miR‐1258 | ATF1, ELK1, SMARCC1 | −0.04 | 0.84 | 0.00 | −0.04 | 0.04 | −0.06 |
| hsa‐miR‐133b | GABPA, HMGN3 | 0.11 | 0.11 | 0.02 | 0.00 | 0.79 | −0.11 |
| hsa‐miR‐145‐5p | CTCF, ELF1, GABPA, HDAC6, HMGN3, MEF2A, NFYA, RBBP5, SMARCC2, SP1, TCF12 | −0.08 | −0.15 | 0.23 | 0.30 | 0.77 | −0.02 |
| hsa‐miR‐17‐5p | CTCF, RBBP5, SMARCC1, TCF12, ZBTB33 | 0.85 | −0.03 | 0.21 | 0.31 | 0.00 | 0.00 |
| hsa‐miR‐1915‐5p | BACH1, MEF2A, NFYA, PHF8, SMARCC1, SP1 | −0.11 | 0.63 | −0.03 | −0.15 | −0.05 | 0.55 |
| hsa‐miR‐193b‐3p | ATF1, BACH1, CTCF, ELF1, GABPA, HMGN3, MEF2A, NFYA, RBBP5, SMARCC1, SMARCC2, TCF12, ZBTB33 | 0.19 | −0.05 | 0.61 | 0.09 | 0.45 | 0.11 |
| hsa‐miR‐199a‐3p | ATF1, BACH1, ELF1, GABPA, MEF2A, RBBP5, SMARCC1, TCF12, ZBTB33 | 0.29 | −0.02 | 0.73 | 0.32 | 0.30 | −0.01 |
| hsa‐miR‐199a‐5p | ATF1, BACH1, CTCF, ELF1, GABPA, HMGN3, MEF2A, RBBP5, SMARCC1, TCF12, ZBTB33 | 0.26 | −0.01 | 0.71 | 0.22 | 0.41 | 0.04 |
| hsa‐miR‐20a‐5p | CTCF, RBBP5, SMARCC1, TCF12, ZBTB33 | 0.84 | −0.07 | 0.21 | 0.33 | 0.02 | 0.00 |
| hsa‐miR‐20b‐5p | CTCF, RBBP5, SMARCC1, TCF12, ZBTB33 | 0.87 | 0.02 | 0.10 | 0.12 | 0.12 | 0.09 |
| hsa‐miR‐214‐3p | ATF1, GABPA | 0.14 | 0.09 | 0.67 | 0.12 | 0.47 | 0.08 |
| hsa‐miR‐21‐3p | RBBP5, SMARCC1, TCF12 | 0.41 | 0.00 | 0.65 | 0.03 | −0.03 | −0.07 |
| hsa‐miR‐21‐5p | BACH1, CTCF, GABPA, RBBP5, SMARCC1, TCF12, ZBTB33 | 0.39 | −0.14 | 0.47 | 0.65 | 0.03 | −0.07 |
| hsa‐miR‐221‐3p | RBBP5, SMARCC1, TCF12, ZBTB33 | 0.67 | −0.03 | 0.34 | 0.05 | 0.21 | 0.04 |
| hsa‐miR‐222‐3p | BACH1, GABPA, RBBP5, SMARCC1, TCF12 | 0.41 | −0.05 | 0.55 | 0.16 | 0.04 | −0.12 |
| hsa‐miR‐23a‐3p | BACH1, CTCF, GABPA, MEF2A, RBBP5, SMARCC1, TCF12, ZBTB33 | 0.44 | −0.16 | 0.38 | 0.68 | 0.27 | −0.08 |
| hsa‐miR‐24‐3p | BACH1, CTCF, GABPA, MEF2A, RBBP5, SMARCC1, TCF12, ZBTB33 | 0.40 | −0.19 | 0.34 | 0.65 | 0.28 | −0.10 |
| hsa‐miR‐25‐3p | CTCF, RBBP5, SMARCC1, TCF12, ZBTB33 | 0.76 | 0.02 | 0.40 | 0.12 | 0.09 | −0.10 |
| hsa‐miR‐27a‐3p | ATF1, BACH1, CTCF, GABPA, RBBP5, SMARCC1, TCF12, ZBTB33 | 0.56 | −0.05 | 0.51 | 0.50 | 0.15 | −0.06 |
| hsa‐miR‐29a‐3p | BACH1, CTCF, RBBP5, SMARCC1, TCF12, ZBTB33 | 0.61 | −0.12 | 0.34 | 0.54 | 0.06 | −0.12 |
| hsa‐miR‐29b‐3p | CTCF, RBBP5, SMARCC1, TCF12, ZBTB33 | 0.65 | −0.04 | 0.39 | 0.32 | 0.08 | −0.07 |
| hsa‐miR‐3181 | BACH1, MEF2A, NFYA, PHF8, SMARCC1, SP1 | −0.09 | 0.66 | −0.03 | −0.23 | −0.07 | 0.27 |
| hsa‐miR‐331‐3p | ATF1, BACH1, CTCF, GABPA, RBBP5, SMARCC1, TCF12, ZBTB33 | 0.37 | −0.10 | 0.51 | 0.23 | 0.25 | −0.13 |
| hsa‐miR‐34a‐5p | ATF1, BACH1, CTCF, GABPA, RBBP5, SMARCC1, TCF12, ZBTB33 | 0.43 | 0.02 | 0.64 | 0.29 | 0.10 | −0.07 |
| hsa‐miR‐3651 | CTCF, RBBP5, SMARCC1, TCF12, ZBTB33 | 0.60 | −0.23 | 0.09 | 0.56 | −0.04 | 0.00 |
| hsa‐miR‐425‐5p | RBBP5, SMARCC1, TCF12 | 0.75 | 0.08 | 0.22 | −0.10 | 0.20 | −0.10 |
| hsa‐miR‐4469 | ATF1, BACH1, ELK1, MEF2A, NFYA, PHF8, SMARCC1, SP1 | −0.02 | 0.77 | −0.01 | −0.08 | 0.06 | 0.13 |
| hsa‐miR‐4492 | ATF1, NFYA, SMARCC1, SP1 | −0.05 | 0.64 | −0.01 | −0.05 | −0.05 | 0.41 |
| hsa‐miR‐4520b‐3p | ATF1, ELK1, SMARCC1 | 0.06 | 0.82 | 0.03 | 0.01 | 0.04 | −0.02 |
| hsa‐miR‐5008‐3p | BACH1, MEF2A, NFYA, PHF8, SMARCC1, SP1 | −0.01 | 0.40 | −0.10 | −0.11 | 0.02 | 0.59 |
| hsa‐miR‐513c‐3p | ATF1, SMARCC1 | 0.00 | 0.80 | 0.00 | 0.04 | 0.02 | −0.10 |
| hsa‐miR‐5685 | ATF1, BACH1, MEF2A, SMARCC1, SP1 | −0.11 | 0.72 | −0.01 | 0.01 | −0.03 | 0.12 |
| hsa‐miR‐6071 | ATF1, BACH1, ELK1, MEF2A, NFYA, PHF8, SMARCC1, SP1 | −0.06 | 0.65 | −0.05 | −0.17 | 0.00 | 0.38 |
| hsa‐miR‐92a‐3p | CTCF, RBBP5, SMARCC1, TCF12, ZBTB33 | 0.65 | −0.35 | −0.03 | 0.46 | 0.09 | 0.15 |
| hsa‐miR‐93‐5p | CTCF, RBBP5, SMARCC1, TCF12, ZBTB33 | 0.80 | −0.03 | 0.32 | 0.23 | 0.01 | −0.13 |
| hsa‐miR‐99a‐5p | GABPA, HMGN3 | 0.15 | 0.05 | 0.26 | 0.08 | 0.76 | 0.05 |
| hsa‐miR‐99b‐5p | ELF1, GABPA, HMGN3 | 0.12 | 0.09 | 0.20 | 0.05 | 0.76 | 0.02 |
| Eigenvalue | 15.36 | 5.81 | 3.10 | 1.38 | 1.18 | 1.01 | |
| Proportion | 0.38 | 0.15 | 0.08 | 0.03 | 0.03 | 0.03 | |
| HR b | 0.78 | 1.24 | 1.22 | 0.93 | 1.86 | 0.83 | |
| (95% CI) | (0.58, 1.05) | (0.92, 1.67) | (0.91, 1.64) | (0.69, 1.25) | (1.36, 2.54) | (0.62, 1.13) |
Bolded values are considered significant.
These TF are displayed because the associated miRNA loaded highly to a factor that was associated with altered CRC survival in the TF‐specific PCA.
Adjusted for age, center, and sex.
Thirty‐one miRNAs loaded highly to multiple TFs associated with survival in a PCA analysis that focused on survival after diagnosis with rectal cancer (Table 5). Of the six factors in this analysis, factors one, three, four, and five were associated with improved survival and factor six was associated with reduced survival. Twenty‐one miRNAs loaded highly to the factors associated significantly with survival. Twelve miRNAs loaded highly to factor 1 (hsa‐miR‐17‐5p, hsa‐miR‐196a‐5p, hsa‐miR‐203a, hsa‐miR‐20a‐5p, hsa‐miR‐20b‐5p, hsa‐miR‐221‐3p, hsa‐miR‐25‐3p, hsa‐miR‐27a‐3p, hsa‐miR‐29a‐3p, hsa‐miR‐29b‐3p, hsa‐miR‐34a‐5p and hsa‐miR‐425‐5p), three miRNAs loaded highly to factor 3 (hsa‐miR‐3651, hsa‐miR‐424‐3p and hsa‐miR‐92a‐3p), three miRNAs loaded highly to factor 4 (hsa‐miR‐146b‐5p, hsa‐miR‐151a‐3p and hsa‐miR‐199b‐5p), two miRNAs loaded highly to factor 5 (hsa‐miR‐146a‐5p and hsa‐miR‐150‐5p), and hsa‐miR‐210 alone loaded highly to factor 6. These factors accounted for 41%, 7%, 4%, 4%, and 3% of the variability in the TF/miRNA summary factors 1, 3‐6, respectively.
Table 5.
Summary of important TF‐associated miRNAs with survival after diagnosis with rectal cancer
| miRNA | Transcription Factor a | Factor 1 | Factor 2 | Factor 3 | Factor 4 | Factor 5 | Factor 6 |
|---|---|---|---|---|---|---|---|
| hsa‐let‐7i‐5p | BCL11A, BCLAF1, ELK1, GABPA, HDAC6, STAT5A, ZNF263 | 0.23 | 0.75 | 0.14 | 0.20 | 0.25 | 0.08 |
| hsa‐miR‐10a‐5p | ELF1, GABPA, MEF2A, NFYA, SP1 | 0.39 | 0.43 | 0.15 | −0.02 | 0.31 | 0.38 |
| hsa‐miR‐145‐5p | GABPA, HDAC6 | −0.32 | 0.65 | 0.03 | 0.17 | 0.10 | −0.05 |
| hsa‐miR‐146a‐5p | ATF1, BCL11A, BCLAF1, CTCF, ELF1, HDAC6, MEF2A, RBBP5, SMARCC2, STAT5A | 0.49 | 0.08 | −0.12 | 0.23 | 0.52 | −0.07 |
| hsa‐miR‐146b‐5p | BACH1, BCLAF1, GABPA, HDAC6, MEF2A, NFYA, RBBP5, SMARCC1, SMARCC2, SP1, STAT5A, TCF12, ZBTB33 | 0.17 | 0.17 | −0.14 | 0.77 | 0.16 | 0.02 |
| hsa‐miR‐150‐5p | ATF1, BCL11A, BCLAF1, CTCF, ELF1, HDAC6, MEF2A, NFYA, RBBP5, SMARCC2 | 0.01 | 0.23 | −0.01 | 0.01 | 0.79 | −0.15 |
| hsa‐miR‐151a‐3p | ATF1, BACH1, HDAC6, MEF2A, NFYA, RBBP5, SMARCC2, SP1, TCF12, ZBTB33 | 0.38 | 0.08 | 0.17 | 0.62 | −0.14 | 0.10 |
| hsa‐miR‐17‐5p | ATF1, BACH1, BCL11A, BCLAF1, CTCF, ELF1, GABPA, HMGN3, MEF2A, NFYA, RBBP5, SMARCC1, SMARCC2, SP1, TCF12, ZBTB33 | 0.76 | 0.28 | 0.39 | 0.00 | 0.01 | 0.07 |
| hsa‐miR‐193b‐3p | GABPA, HDAC6, ZNF263 | 0.13 | 0.71 | −0.01 | 0.14 | 0.02 | −0.07 |
| hsa‐miR‐196a‐5p | ATF1, NFYA | 0.65 | −0.12 | 0.11 | 0.24 | 0.10 | 0.17 |
| hsa‐miR‐199b‐5p | BACH1, GABPA, SMARCC2, TCF12 | 0.02 | 0.31 | 0.01 | 0.77 | 0.06 | −0.09 |
| hsa‐miR‐203a | ELF1, SP1 | 0.62 | 0.06 | −0.02 | 0.26 | −0.01 | 0.25 |
| hsa‐miR‐20a‐5p | ATF1, BACH1, CTCF, ELF1, GABPA, HMGN3, MEF2A, NFYA, RBBP5, SMARCC1, SMARCC2, SP1, TCF12, ZBTB33 | 0.76 | 0.28 | 0.42 | −0.02 | 0.07 | 0.04 |
| hsa‐miR‐20b‐5p | ATF1, BACH1, CTCF, ELF1, ELK1, GABPA, HDAC6, HMGN3, NFYA, PHF8, RBBP5, SMARCC1, SMARCC2, SP1, TCF12, ZBTB33 | 0.72 | 0.12 | 0.38 | 0.31 | 0.01 | −0.08 |
| hsa‐miR‐210 | ATF1, ELK1, HDAC6, SMARCC1, TCF12, ZBTB33, ZNF263 | 0.16 | 0.07 | −0.13 | −0.01 | −0.25 | 0.74 |
| hsa‐miR‐21‐3p | BACH1, HDAC6, SMARCC2, SP1, TCF12, ZNF263 | 0.48 | 0.46 | −0.05 | −0.02 | −0.11 | −0.33 |
| hsa‐miR‐21‐5p | GABPA, HDAC6, MEF2A, SMARCC2, TCF12 | 0.33 | 0.63 | 0.28 | 0.27 | 0.04 | 0.19 |
| hsa‐miR‐221‐3p | ATF1, BACH1, CTCF, ELF1, GABPA, NFYA, PHF8, RBBP5, SMARCC1, SP1, TCF12, ZBTB33 | 0.64 | 0.38 | 0.04 | 0.27 | −0.13 | −0.06 |
| hsa‐miR‐222‐3p | BACH1, BCL11A, BCLAF1, HDAC6, RBBP5, SMARCC2, SP1, STAT5A, TCF12, ZNF263 | 0.45 | 0.61 | −0.01 | 0.04 | −0.18 | −0.13 |
| hsa‐miR‐23a‐3p | CTCF, ELF1, GABPA, HDAC6, SMARCC2, TCF12 | 0.31 | 0.74 | 0.36 | 0.19 | 0.11 | 0.21 |
| hsa‐miR‐24‐3p | BCL11A, BCLAF1, ELK1, GABPA, HDAC6, ZNF263 | 0.25 | 0.77 | 0.34 | 0.16 | 0.09 | 0.20 |
| hsa‐miR‐25‐3p | ATF1, BACH1, BCL11A, BCLAF1, CTCF, ELF1, GABPA, HDAC6, HMGN3, MEF2A, NFYA, PHF8, RBBP5, SMARCC1, SMARCC2, SP1, TCF12, ZBTB33, ZNF263 | 0.78 | 0.33 | 0.21 | −0.02 | 0.04 | 0.00 |
| hsa‐miR‐27a‐3p | ATF1, BACH1, BCLAF1, CTCF, ELF1, GABPA, HDAC6, HMGN3, MEF2A, NFYA, RBBP5, SMARCC1, SMARCC2, SP1, TCF12, ZBTB33 | 0.55 | 0.64 | 0.23 | 0.09 | 0.10 | 0.12 |
| hsa‐miR‐29a‐3p | ATF1, BACH1, BCL11A, BCLAF1, CTCF, ELF1, ELK1, GABPA, HDAC6, HMGN3, MEF2A, NFYA, PHF8, RBBP5, SMARCC1, SMARCC2, SP1, TCF12, ZBTB33, ZNF263 | 0.58 | 0.42 | 0.44 | 0.14 | 0.21 | 0.08 |
| hsa‐miR‐29b‐3p | ATF1, BACH1, CTCF, ELF1, GABPA, HMGN3, MEF2A, NFYA, RBBP5, SMARCC1, TCF12, ZBTB33 | 0.64 | 0.33 | 0.26 | 0.14 | 0.23 | 0.06 |
| hsa‐miR‐331‐3p | GABPA, HDAC6, ZNF263 | 0.28 | 0.68 | 0.10 | −0.02 | 0.13 | 0.04 |
| hsa‐miR‐34a‐5p | BCL11A, BCLAF1, ELF1, ELK1, GABPA, HDAC6, MEF2A, NFYA, PHF8, RBBP5, SMARCC2, SP1, STAT5A, TCF12, ZNF263 | 0.56 | 0.55 | −0.09 | 0.10 | 0.09 | −0.08 |
| hsa‐miR‐3651 | ATF1, BACH1, BCL11A, CTCF, ELF1, ELK1, GABPA, HDAC6, MEF2A, NFYA, SMARCC1, SMARCC2, SP1, ZBTB33, ZNF263 | 0.30 | 0.29 | 0.64 | −0.12 | 0.15 | 0.29 |
| hsa‐miR‐424‐3p | BCL11A, ELK1, HDAC6, NFYA, SMARCC2, SP1, ZNF263 | 0.08 | 0.00 | 0.79 | 0.05 | −0.03 | −0.14 |
| hsa‐miR‐425‐5p | ATF1, CTCF, ELF1, GABPA, HDAC6, NFYA, RBBP5, SMARCC1, SMARCC2, SP1, TCF12 | 0.76 | 0.19 | 0.03 | 0.12 | 0.10 | 0.13 |
| hsa‐miR‐92a‐3p | ATF1, BACH1, BCL11A, BCLAF1, CTCF, ELF1, ELK1, GABPA, HDAC6, HMGN3, NFYA, RBBP5, SMARCC1, SMARCC2, SP1, TCF12, ZBTB33, ZNF263 | 0.31 | 0.22 | 0.74 | −0.05 | −0.14 | −0.10 |
| Eigenvalue | 13.27 | 2.74 | 2.09 | 1.40 | 1.29 | 1.11 | |
| Proportion | 0.41 | 0.09 | 0.07 | 0.04 | 0.04 | 0.03 | |
| HR b | 0.51 | 1.03 | 0.65 | 0.63 | 0.64 | 2.01 | |
| (95% CI) | (0.34, 0.74) | (0.72, 1.48) | (0.45, 0.94) | (0.42, 0.92) | (0.44, 0.91) | (1.33, 3.04) |
Bolded values are considered significant.
These TF are displayed because the associated miRNA loaded highly to a factor that was associated with altered CRC survival in the TF‐specific PCA.
Adjusted for age, center, and sex.
Of the 27 mRNAs used as input to IPA, 16 remained significantly enriched for at least one of 75 different canonical biological pathways after Benjamini‐Hochberg correction for multiple comparisons (Table 6). JUN contributed to the most pathways, 54, and ELK1 contributed to 45. Three mRNAs, TCF12, GABPA, and BACH1, only contributed to one pathway each, and the other 11 mRNAs contributed to between 2 and 18 pathways. JUN, ELK1, and STAT5A have the greatest involvement in canonical pathways, whereas ATF1, BACH1, ELF1, ETS1, GABPA, HDAC6/8, JUNB/D, MEF2A, SP1, SMARCC1/2, and TCF12 were associated with fewer canonical pathways and, in the case of some, larger numbers of miRNAs.
Table 6.
Pathways associated with TFs that are associated with survival and miRNA expression
| TF | Number of pathways | Pathways a |
|---|---|---|
| ATF1 | 7 | ERK/MAPK Signaling, Role of BRCA1 in DNA Damage Response, LPS‐stimulated MAPK Signaling, p38 MAPK Signaling, PI3K Signaling in B Lymphocytes, CD40 Signaling, ATM Signaling |
| BACH1 | 1 | NRF2‐mediated Oxidative Stress Response |
| ELF1 | 3 | Telomerase Signaling, HGF Signaling, ERK/MAPK Signaling |
| ELK1 | 45 | ErbB2‐ErbB3 Signaling, Estrogen‐Dependent Breast Cancer Signaling, Corticotropin Releasing Hormone Signaling, HGF Signaling, Glucocorticoid Receptor Signaling, IL‐2 Signaling, IL‐10 Signaling, ERK/MAPK Signaling, Erythropoietin Signaling, IL‐3 Signaling, LPS‐stimulated MAPK Signaling, Cholecystokinin/Gastrin‐mediated Signaling, p38 MAPK Signaling, PI3K Signaling in B Lymphocytes, Gα12/13 Signaling, HMGB1 Signaling, Oncostatin M Signaling, April Mediated Signaling, B Cell Activating Factor Signaling, B Cell Receptor Signaling, Cardiac Hypertrophy Signaling, EGF Signaling, GM‐CSF Signaling, Toll‐like Receptor Signaling, Regulation of IL‐2 Expression in Activated and Anergic T Lymphocytes, Growth Hormone Signaling, PEDF Signaling, IL‐17 Signaling, FLT3 Signaling in Hematopoietic Progenitor Cells, Neuregulin Signaling, PDGF Signaling, ErbB Signaling, RANK Signaling in Osteoclasts, SAPK/JNK Signaling, IGF‐1 Signaling, T Cell Receptor Signaling, Rac Signaling, Renin‐Angiotensin Signaling, IL‐6 Signaling, GNRH Signaling, 14‐3‐3‐mediated Signaling, Relaxin Signaling, CXCR4 Signaling, Acute Phase Response Signaling, Sertoli Cell‐Sertoli Cell Junction Signaling |
| ETS1 | 7 | Telomerase Signaling, HGF Signaling, ERK/MAPK Signaling, Sumoylation Pathway, B Cell Receptor Signaling, GM‐CSF Signaling, Renal Cell Carcinoma Signaling |
| GABPA | 1 | Agrin Interactions at Neuromuscular Junction |
| HDAC6 | 10 | Telomerase Signaling, Hereditary Breast Cancer Signaling, Huntington's Disease Signaling, Chronic Myeloid Leukemia Signaling, Calcium Signaling, Role of NFAT in Cardiac Hypertrophy, Cell Cycle: G1/S Checkpoint Regulation, Phospholipase C Signaling, Cyclins and Cell Cycle Regulation, Adipogenesis pathway |
| HDAC8 | 10 | Telomerase Signaling, Hereditary Breast Cancer Signaling, Huntington's Disease Signaling, Chronic Myeloid Leukemia Signaling, Calcium Signaling, Role of NFAT in Cardiac Hypertrophy, Cell Cycle: G1/S Checkpoint Regulation, Phospholipase C Signaling, Cyclins and Cell Cycle Regulation, Adipogenesis pathway |
| JUN | 54 | ErbB2‐ErbB3 Signaling, Estrogen‐Dependent Breast Cancer Signaling, Corticotropin Releasing Hormone Signaling, HGF Signaling, Glucocorticoid Receptor Signaling, PCP pathway, IL‐2 Signaling, IL‐10 Signaling, RAR Activation, NRF2‐mediated Oxidative Stress Response, Erythropoietin Signaling, IL‐3 Signaling, Prolactin Signaling, LPS‐stimulated MAPK Signaling, Huntington's Disease Signaling, Sumoylation Pathway, Cholecystokinin/Gastrin‐mediated Signaling, PI3K Signaling in B Lymphocytes, Gα12/13 Signaling, HMGB1 Signaling, April Mediated Signaling, B Cell Activating Factor Signaling, B Cell Receptor Signaling, Thrombopoietin Signaling, Cardiac Hypertrophy Signaling, EGF Signaling, Agrin Interactions at Neuromuscular Junction, Toll‐like Receptor Signaling, CD40 Signaling, Regulation of IL‐2 Expression in Activated and Anergic T Lymphocytes, ATM Signaling, Renal Cell Carcinoma Signaling, JAK/Stat Signaling, IL‐17 Signaling, PDGF Signaling, PPAR Signaling, ErbB Signaling, RANK Signaling in Osteoclasts, SAPK/JNK Signaling, IGF‐1 Signaling, T Cell Receptor Signaling, Rac Signaling, Renin‐Angiotensin Signaling, IL‐6 Signaling, GNRH Signaling, 14‐3‐3‐mediated Signaling, Aryl Hydrocarbon Receptor Signaling, Th2 Pathway, Relaxin Signaling, CXCR4 Signaling, Acute Phase Response Signaling, Sertoli Cell‐Sertoli Cell Junction Signaling, Role of NFAT in Regulation of the Immune Response, Th1 and Th2 Activation Pathway |
| JUNB | 2 | PCP pathway, NRF2‐mediated Oxidative Stress Response |
| JUND | 3 | Corticotropin Releasing Hormone Signaling, PCP pathway, NRF2‐mediated Oxidative Stress Response |
| MEF2A | 9 | Corticotropin Releasing Hormone Signaling, Cholecystokinin/Gastrin‐mediated Signaling, p38 MAPK Signaling, Gα12/13 Signaling, Calcium Signaling, Role of NFAT in Cardiac Hypertrophy, Cardiac Hypertrophy Signaling, Phospholipase C Signaling, Role of NFAT in Regulation of the Immune Response |
| SMARCC1 | 5 | Glucocorticoid Receptor Signaling, Hereditary Breast Cancer Signaling, RAR Activation, Role of BRCA1 in DNA Damage Response, AMPK Signaling |
| SMARCC2 | 5 | Glucocorticoid Receptor Signaling, Hereditary Breast Cancer Signaling, RAR Activation, Role of BRCA1 in DNA Damage Response, AMPK Signaling |
| SP1 | 9 | Telomerase Signaling, ErbB2‐ErbB3 Signaling, Estrogen‐Dependent Breast Cancer Signaling, IL‐10 Signaling, Prolactin Signaling, Huntington's Disease Signaling, Sumoylation Pathway, HMGB1 Signaling, Aryl Hydrocarbon Receptor Signaling |
| STAT5A | 18 | ErbB2‐ErbB3 Signaling, Estrogen‐Dependent Breast Cancer Signaling, Glucocorticoid Receptor Signaling, IL‐2 Signaling, RAR Activation, Erythropoietin Signaling, IL‐3 Signaling, Prolactin Signaling, Chronic Myeloid Leukemia Signaling, Oncostatin M Signaling, Thrombopoietin Signaling, Growth Hormone Signaling, JAK/Stat Signaling, FLT3 Signaling in Hematopoietic Progenitor Cells, Neuregulin Signaling, PPAR Signaling, Th2 Pathway, Th1 and Th2 Activation Pathway |
| TCF12 | 1 | PEDF signaling |
Pathways determined to be significantly enriched in TF dataset, with a −log P‐value of >1.3.
We compared the 27 TFs that were associated significantly with both altered risk of CRC survival and as miRNA expression with the 65 miRNAs that were associated with these TFs in normal colonic mucosa for overlapping coordinates between known TFBSs and pri‐miR coordinates. Fifty‐seven unique matches, between 15 TFs and 22 miRNAs, were identified. Thirty of these findings were for associations with a positive beta coefficient, indicating miRNA transcription enhancement; the other 27 were for associations displaying a negative beta coefficient, indicating miRNA transcription repression. These results can be seen in Supplementary Table S2.
We also identified seed matches between these 27 TFs and 65 miRNAs, which resulted in 294 seed matches identified, between 24 TFs and 52 miRNAs. Nine miRNAs (hsa‐miR‐4469, hsa‐miR‐93‐5p, hsa‐miR‐17‐5p, hsa‐miR‐20b‐5p, hsa‐miR‐27a‐3p, hsa‐miR‐124‐3p, hsa‐miR‐146b‐5p, hsa‐miR‐20a‐5p and hsa‐miR‐24‐3p) had seed pairings with ten or more TFs, including 22 of the 24 TFs with any seed matches. Thirteen TFs (SMARCC1, SP1, GABPA, TCF12, NFYA, RBBP5, BACH1, MEF2A, ZBTB33, PHF8, CTCF, BCL11A and ELF1) were associated with 10 or more miRNAs. These results can be seen in Supplementary Table S3. Cytoscape was used to visualize these findings. In Supplementary Figure S1, all TFs and miRNAs that had a TFBS overlap (N TF = 15; N miRNA = 22) or seed match (N TF = 24; N miRNA = 52) identified are displayed. In Figure 2, all TFs and miRNAs with a TFBS overlap (N TF = 15; N miRNA = 22) and miRNAs with 10 seed matches or more and their corresponding TFs (N TF = 22; N miRNA = 9) are shown (for a total of N TF = 22; N miRNA = 26).
Figure 2.
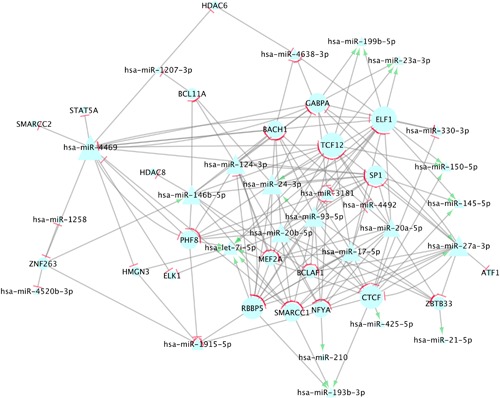
This figure depicts miRNA‐TF TFBS and seed interactions for all TFs that had a TFBS overlap with a pri‐miRNA and miRNAs that had 10 seed matches or more. MiRNAs are shown in triangles; TFs are shown in circles. The size of the molecule corresponds to the number of connections it has, therefore molecules with a greater number of associations are larger. Green arrows (→) depict gene expression enhancement, as indicated by a positive beta coefficient, and red stops (⊣) depict an inhibitory effect, as indicated by a negative beta coefficient. All seed matches are visualized as inhibitory, as this is the typical response
4. DISCUSSION
Thirty TFs were associated significantly with decreased risk of death from CRC, and in all instances the altered risk of dying was associated with increased carcinoma expression compared to normal colonic mucosa expression; of these, 27 TFs were associated with normal colonic miRNA expression. Further evaluation of TF‐specific miRNAs using PCA analysis, showed associations with survival for colon cancer (1 factor) and rectal cancer (4 factors).
Both miRNAs and TFs are known to regulate multiple genes2 and have the ability to be both oncogenes and tumor suppressors,4, 5 depending on cellular conditions. Four miRNAs loaded highly to a factor that was associated significantly with altered CRC survival in colon cancer subjects: hsa‐miR‐133b, hsa‐miR‐145‐5p, hsa‐miR‐99a‐5p and hsa‐miR‐99b‐5p with loading of 0.79, 0.77, 0.76 and 0.76, respectively, were associated significantly with increased risk of death from CRC in colon cancer subjects (Factor 5, HR = 1.86, 95% CI 1.36, 2.54). However, this factor contributed only three percent of the total variance in TF‐associated miRNAs in colon cancer cases. Previously, we found that both hsa‐miR‐99a‐5p and hsa‐miR‐145‐5p were significantly associated with reduced survival in colon cancer subjects when their expression increased in carcinoma tissue.11 As previously stated, interconnectedness of networks and network size have been associated with survival in other studies; as such, we focused on those genes that were associated with differential expression of miRNAs, survival, and with multiple miRNAs in the PCA. Of the genes that were associated in the primary PCA with these miRNAs, GABPA and HMGN3 are associated with all four miRNAs. These TFs were significantly associated with reduced risk of death from CRC in colon cancer subjects when expression in the carcinoma increased (GABPA: HR = 0.60, 95% CI 0.36, 0.99; HMGN3: HR = 0.56, 95% CI 0.33, 0.95). The positive linear association between some TFs and the miRNAs suggest that the miRNAs’ transcription may be activated either directly or indirectly by TFs. There were no TFBS overlaps or seed matches between these TFs and miRNAs.
It has been proposed that TFs that regulate a larger number of genes may be more important for survival, as the likelihood that removal of a given protein in Saccharomyces cerevisiae protein‐protein interaction networks will be fatal is correlated to the number of interactions that protein has.23 The reduction in risk of CRC death associated with each of the 27 TFs was very similar, however, the average number of miRNAs associated with TFs that significantly altered risk of death from CRC was 23.9, and the median was 27.5, compared to the average number of miRNAs associated with TFs that were not associated with survival, 18.42 miRNAs, and the median, 13.5 miRNAs. While this finding is not a statistically significant one, given that the group of TFs associated with survival is a small subset of those analyzed (less than 20%) and this group had a median of 14 more associated miRNAs than those TFs not associated with reduced risk of CRC death, we believe this lends support to the theory that TFs that regulate a larger number of genes are more important for survival.
There is also support for the hypothesis that larger hubs are more important for CRC survival. SMARCC1 was associated with 46 miRNAs as well as with decreased risk of death from CRC in colon cancer subjects (HR = 0.65, 95% CI 0.44, 0.96). Another study on CRC found that patients with higher levels of SMARCC1 protein had significantly better survival overall.24 ATF1 was associated with normal colonic mucosa expression of 46 miRNAs in colon cancer subjects and was associated with at least one miRNA that loaded highly to all of the factors that were associated significantly with altered CRC survival in rectal cancer subjects. A study by Huang et al found that CRC patients with higher levels of ATF1 had improved survival.25 GABPA and HMGN3 were associated with a large number of miRNAs (46 for GABPA and 41 for HMGN3) and decreased risk of death from CRC in colon cancer subjects. The average number of miRNAs associated with TFs that significantly altered risk of death from CRC was 23.9, and the median was 27.5, compared to the average number of miRNA associated with TFs that were not associated with survival, 18.42 miRNAs, and the median, 13.5 miRNAs.
One miRNA that was associated with many of the genes that were associated significantly with altered CRC survival as well as a large number of miRNAs was hsa‐miR‐210. This miRNA loaded highly to summary factor 8 of ATF1, HDAC6, and TCF12, factor 7 of SMARCC1 and to factor 5 of ZBTB33 and ZNF263, which were all associated significantly with reduced CRC survival in rectal cancer patients. Hsa‐miR‐210 was largely the sole contributor to these factors, loading 0.7 or more. Additionally, hsa‐miR‐210 was highly inversely associated with factor 6 of NFYA, loading at −0.70, which was associated significantly with decreased risk of death from CRC in rectal cancer patients (HR = 0.47, 95% CI 0.32, 0.69). This factor only contributed about 3% of the variability for these factors. Hsa‐miR‐210 is known to be upregulated in CRC carcinoma tissues,26 and in this study it was upregulated in carcinoma tissue compared to normal colonic mucosa and its normal mucosa expression was directly associated with ATF1, ELK1, HDAC6, NFYA, SMARCC1, TCF12, ZBTB33 and ZNF263 in colon cancer subjects. These associations were not replicated in rectal tissue; power was considerably less for rectal cancer analysis than for colon cancer analysis with RNA‐Seq data, however, it is also possible that these findings reflect actual differences in pathway participating in colon and rectal cancer. Hsa‐miR‐210 was not independently associated significantly with CRC survival in either colon or rectal cancer cases (HRC = 1.07 95% CI 0.95, 1.20, P = 0.998, q = 0.998; HRR = 1.15 95% CI 0.99, 1.34, P = 0.07, q = 0.08). This suggests that hsa‐miR‐210 works in combination with other factors, such as miRNAs, to alter CRC survival.
It is possible that the 30 TFs that were associated significantly with CRC survival alter pathways important to tumorigenesis. We performed a functional analysis on 27 of these TFs that were associated significantly with CRC survival as well as normal colonic miRNA expression. Of the 27 TFs used as input to IPA, 16 were enriched for canonical pathways. There were 75 canonical pathways significantly enriched after corrections for multiple comparisons, including many pathways known to be involved in cancer development such as MAPK signaling, role of BRCA1 in DNA damage response, various cell cycle pathways, JAK‐Stat signaling, and telomerase activity. As can be seen in Table 6 JUN, ELK1, and STAT5A were associated with the majority of these pathways. These TFs were associated with normal expression of one, 16 and 14 miRNAs respectively. ATF1 and TCF12, which were associated with normal expression of 46 miRNAs each, were conversely only associated with seven and one canonical pathways respectively. This could reflect the larger involvement of JUN, ELK1, and STAT5A in CRC; however, given the significant impact on CRC survival the other TFs have, as well as their numerous associations with miRNA expression, it is more likely that the functional analysis reflects the bias in annotated datasets and previous findings.
This study has many strengths as well as some limitations. One limitation of this study is that we are not able to test direct effects of TF presence on miRNA transcription; as such, it is not possible to assert that these TF directly impact miRNA expression. As miRNAs and TFs are both regulators, it may be that we are not able to detect direction of regulation, only the presence or absence of an association between a given TF and miRNA. Similarly, because RNA‐Seq measures gene expression, we may miss interactions between miRNAs and mRNAs in which the mRNA is blocked from translation but not degraded. However, by determining seed matches and TFBS overlap between TFs and miRNAs, we were able to gain insight into the direction of regulation, and whether the TF regulates the miRNA or the miRNA regulates the TF. Twenty miRNAs that were included in the TFBS analysis had seed matches. In most cases, seed matches accounted for a much larger percentage of associations than TFBS overlap, indicating miRNA regulation of TFs was more prevalent than TF regulation of miRNAs. However, regardless of directionality, TFs that were associated significantly with altered risk of CRC death were at the center of more biological activity than those that were not associated with altered risk. Any disruption in regulation of these TFs would affect more molecules, and the corresponding biological responses, than disruption of a gene associated with less miRNAs, which lends support to our hypothesis. While the TFBSs and seed pairing enables us to identify more direct associations and theorize on the directionality of these observed associations, as illustrated by has‐miR‐210, our PCA results suggest that these associations may not be solely responsible for influences on CRC survival.
A strength of this study is our use of the Agilent miRNA platform. There is a potential bias in the amount of information available for many miRNAs due to lack of investigation, and use of global detection methods contributes more broadly to the field. Additionally, we have shown that our platform has high reliability and good concordance with other methods.12, 13 Similarly, our RNA‐Seq data allow us to identify expression changes in a large amount of mRNAs, and pairing this data with our miRNA expression data allow us to identify colorectal tissue‐specific associations. Another strength of our study is the use of the PCA, which allowed us to consolidate data and analyze the combined effects of the TF‐specific miRNAs and their subsequent effect on survival, rather than investigating each miRNA individually. Given the interconnected nature of both miRNAs and TFs in biological pathways and diseases, this analytical approach more closely represents actuality of coordinated expression. Hsa‐miR‐210 was associated significantly with multiple factors that significantly altered CRC survival in rectal cancer patients in the PCA, but was not independently associated significantly with CRC survival in colon or rectal cancer cases; the PCA was able to detect hsa‐miR‐210's influence on survival, which would not have been possible using traditional methods of analysis.
In summary, our reported associations of TF and TF‐specific miRNAs with survival add to our knowledge of the genomics of colorectal cancer. An important contribution is the combined effect that TF‐defined miRNAs have on altering survival goes beyond that observed for individual miRNAs. We encourage others to replicate and expand upon our findings.
Supporting information
Additional Supporting Information may be found online in the supporting information tab for this article.
Supporting Table S1
Supporting Table S2
Figure S1. This figure depicts all miRNA‐TF TFBS and seed interactions.
Table S1. TF‐miRNA associations in normal colon and rectal tissues for TFs significantly associated with altered risk of CRC death in colon cancer subjects.
Table S2. Transcription Factor Binding Site (TFBS) overlap with miRNA primary‐miRNA (pri‐miRNA) transcript for transcription factors (TFs) and miRNAs associated in normal colonic mucosa.
Table S3. Seed matches found between miRNAs and transcription factors (TFs) associated in normal colonic mucosa.
ACKNOWLEDGMENTS
The contents of this manuscript are solely the responsibility of the authors and do not necessarily represent the official view of the National Cancer Institute. We would like to acknowledge Dr. Bette Caan and the Kaiser Permanente Medical Research Program for sample contributions, Erika Wolff and Michael Hoffman at the University of Utah for miRNA processing, Brett Milash and the Bioinformatics Shared Resource of the Huntsman Cancer Institute and University of Utah for miRNA and mRNA bioinformatics data processing, Sandie Edwards at the University of Utah for her efforts in overall study monitoring and tumor tissue collection, and Daniel Pellatt at the University of Utah for his assistance with statistical analysis. This study was supported by NCI grants CA163683 and CA48998.
Mullany LE, Herrick JS, Wolff RK, Stevens JR, Samowitz W, Slattery ML. Transcription factor‐microRNA associations and their impact on colorectal cancer survival. Molecular Carcinogenesis. 2017; 56: 2512–2526. 10.1002/mc.22698
REFERENCES
- 1. Lee TI, Young RA. Transcription of eukaryotic protein‐coding genes. Annu Rev Genet. 2000; 34:77–137. [DOI] [PubMed] [Google Scholar]
- 2. Macfarlane LA, Murphy PR. MicroRNA: biogenesis, function and role in cancer. Curr Genomics. 2010; 11:537–561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004; 116:281–297. [DOI] [PubMed] [Google Scholar]
- 4. Wu Q, Qin H, Zhao Q, He XX. Emerging role of transcription factor‐microRNA‐target gene feed‐forward loops in cancer. Biomed Rep. 2015; 3:611–616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Delfino KR, Rodriguez‐Zas SL. Transcription factor‐microRNA‐target gene networks associated with ovarian cancer survival and recurrence. PLoS ONE. 2013; 8:e58608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Zhang HM, Kuang S, Xiong X, Gao T, Liu C, Guo AY. Transcription factor and microRNA co‐regulatory loops: important regulatory motifs in biological processes and diseases. Brief Bioinform. 2015; 16:45–58. [DOI] [PubMed] [Google Scholar]
- 7. Darnell JE, Jr . Transcription factors as targets for cancer therapy. Nat Rev Cancer. 2002; 2:740–749. [DOI] [PubMed] [Google Scholar]
- 8. Slattery ML, Lundgreen A, Kadlubar SA, Bondurant KL, Wolff RK. JAK/STAT/SOCS‐signaling pathway and colon and rectal cancer. Mol Carcinog. 2013; 52:155–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Jeong H, Tombor B, Albert R, Oltvai ZN, Barabasi AL. The large‐scale organization of metabolic networks. Nature. 2000; 407:651–654. [DOI] [PubMed] [Google Scholar]
- 10. Martinez NJ, Ow MC, Barrasa MI, et al. A C. elegans genome‐scale microRNA network contains composite feedback motifs with high flux capacity. Genes Dev. 2008; 22:2535–2549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Slattery ML, Herrick JS, Mullany LE, et al. An evaluation and replication of miRNAs with disease stage and colorectal cancer‐specific mortality. Int J Cancer. 2015; 137:428–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Slattery ML, Herrick JS, Pellatt DF, et al. MicroRNA profiles in colorectal carcinomas, adenomas, and normal colonic mucosa: variations in miRNA expression and disease progression. Carcinogenesis. 2016; 37:245–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Pellatt DF, Stevens JR, Wolff RK, et al. Expression profiles of miRNA subsets distinguish human colorectal carcinoma and normal colonic mucosa. Clin Transl Gastroenterol. 2016; 7:e152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Suyundikov A, Stevens JR, Corcoran C, Herrick J, Wolff RK, Slattery ML. Accounting for dependence induced by weighted KNN imputation in paired samples, motivated by a colorectal cancer study. PLoS ONE. 2015; 10:e0119876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Agilent Technologies I. Agilent GeneSpring User Manual. Volume 2015. Santa Clara, CA: Aglient Technologies Inc; 2013. [Google Scholar]
- 16. Slattery ML, Pellatt DF, Mullany LE, Wolff RK, Herrick JS. Gene expression in colon cancer: a focus on tumor site and molecular phenotype. Genes Chromosomes Cancer. 2015; 54:527–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Karolchik D, Hinrichs AS, Furey TS, et al. The UCSC table browser data retrieval tool. Nucleic Acids Res. 2004; 32:D493–D496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Rosenbloom KR, Sloan CA, Malladi VS, et al. ENCODE data in the UCSC genome browser: year 5 update. Nucleic Acids Res. 2013; 41:D56–D63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Storey JD. A direct approach to false discovery rates. J Royal Stat Soc. 2002; 64:479–498. [Google Scholar]
- 20. Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J Royal Stat Soc. 1995; 57:289–300. [Google Scholar]
- 21. Mullany LE, Herrick JS, Wolff RK, Slattery ML. MicroRNA seed region length impact on target messenger RNA expression and survival in colorectal cancer. PLoS ONE. 2016; 11:e0154177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Shannon P, Markiel A, Ozier O, et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003; 13:2498–2504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Jeong H, Mason SP, Barabasi AL, Oltvai ZN. Lethality and centrality in protein networks. Nature. 2001; 411:41–42. [DOI] [PubMed] [Google Scholar]
- 24. Andersen CL, Christensen LL, Thorsen K, et al. Dysregulation of the transcription factors SOX4, CBFB and SMARCC1 correlates with outcome of colorectal cancer. Br J Cancer. 2009; 100:511–523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Huang GL, Guo HQ, Yang F, et al. Activating transcription factor 1 is a prognostic marker of colorectal cancer. Asian Pac J Cancer Prev. 2012; 13:1053–1057. [DOI] [PubMed] [Google Scholar]
- 26. Qu A, Du L, Yang Y, et al. Hypoxia‐inducible MiR‐210 is an independent prognostic factor and contributes to metastasis in colorectal cancer. PLoS ONE. 2014; 9:e90952. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional Supporting Information may be found online in the supporting information tab for this article.
Supporting Table S1
Supporting Table S2
Figure S1. This figure depicts all miRNA‐TF TFBS and seed interactions.
Table S1. TF‐miRNA associations in normal colon and rectal tissues for TFs significantly associated with altered risk of CRC death in colon cancer subjects.
Table S2. Transcription Factor Binding Site (TFBS) overlap with miRNA primary‐miRNA (pri‐miRNA) transcript for transcription factors (TFs) and miRNAs associated in normal colonic mucosa.
Table S3. Seed matches found between miRNAs and transcription factors (TFs) associated in normal colonic mucosa.