

Abstract
The aminopeptidase PfA-M1 is a key contributor to peptide catabolism in the human malaria parasite Plasmodium falciparum. PfA-M1 substrate specificity is shaped by the cylindrical S1 subsite, which accommodates the sidechain of the substrate P1 residue. At the top of the S1 subsite are two “cap” residues, E572 and M1034, that are positioned to influence S1 subsite specificity. In this study, we have mutated the cap residues, individually and together, and have evaluated the effects on PfA-M1 specificity and catalytic efficiency. When the P1 residue was too small to engage the cap residues, the mutations had no effect on catalysis. Hydrolysis of dipeptide substrates with a basic P1 residue was significantly impaired in the E572A mutant, most likely due to the loss of a stabilizing salt bridge between E572 and the P1 sidechain. With M1034A, a substantial reduction in catalytic efficiency was observed when the P1 sidechain was large and non-polar. The double E572A/M1034A exhibited significant decreases in catalytic efficiency for most substrates. This effect was not reversed with the polar substitutions E572N/M1034Q, which replaced the PfA-M1 cap residues with those of Escherichia coli aminopeptidase N. Both E572 and M1034 contributed to the binding of the competitive aminopeptidase inhibitor bestatin.
Keywords: aminopeptidase, enzyme, malaria, specificity
Graphical abstract
Mutation of the S1-subsite cap residues E572 and M1034 reveals their contributions to the efficient catalysis of substrates with basic and large non-polar P1 sidechains.
1. Introduction
M1-aminopeptidases constitute a diverse and widely distributed family of zinc-dependent hydrolytic enzymes [1]. They are strict exopeptidases that catalyze the hydrolysis of the amino-terminal residue from a peptide substrate. We are interested in understanding the biological roles of the M1-family aminopeptidase termed “PfA-M1” [2] during the pathogenic asexual intraerythrocytic stage of the malaria parasite Plasmodium falciparum. PfA-M1 exhibits a complex subcellular distribution in the parasite, residing in the food vacuole, the nucleus and the parasitophorous vacuole [3–5]. The best-characterized function of PfA-M1 is the catabolism of hemoglobin-derived peptides in the lumen of the acidic food vacuole. During its residence in the erythrocyte, P. falciparum ingests and degrades to amino acids up to 75% of soluble host cell cytosol, which consists mostly of hemoglobin [6]. A series of cleavages by vacuolar endo- and exo-peptidases results in the generation of large quantities of sequence-diverse short peptides [7]. By virtue of its vacuolar localization and its catalytic activity at the acidic pH of the vacuole [5], PfA-M1 is positioned to be a key catalyst for degrading these short globin peptides to individual amino acids. Treatment of parasites with a PfA-M1-selective inhibitor resulted in swelling of the food vacuole, accumulation of globin peptides, and, importantly, parasite death [8], which together with the biochemical evidence cited above support an essential role for PfA-M1 in vacuolar peptide catabolism. The inability to disrupt the gene encoding PfA-M1 in the haploid blood stage parasite is further evidence of its importance [4].
The essential role of PfA-M1 in disease-causing, asexual parasites has stimulated efforts to develop PfA-M1 inhibitors into novel anti-malarials [9, 10]. The natural product bestatin, a Phe-Leu analog with an N-terminal α-hydroxy-β-amino acid [11], is a sub-micromolar inhibitor of PfA-M1 [5, 12, 13] that has provided a convenient scaffold for optimization of inhibitor potency and selectivity [8, 14]. Phosphonate analogs of amino acids have also been explored as transition state mimics [15, 16]. The hydroxamate moiety, which chelates the catalytic zinc atom, has been successfully exploited to generate potent PfA-M1 inhibitors [17–20].
To better understand the biological functions of PfA-M1 and to facilitate the design of potent inhibitors, we are interested in elucidating principles of substrate selection and catalysis. The S1 subsite is a well-defined, cylindrical pocket that receives the substrate’s P1 residue sidechain1 [13]. The S1 subsite plays a major role in substrate selection: basic and non-β-branched, non-polar P1 sidechains such as those of Leu, Met, Phe, Arg and Lys are accommodated, whereas substrates with acidic, β-branched or cyclic P1 residues (Asp, Glu, Val, Ile, Pro) are not efficiently hydrolyzed [2, 13, 21, 22]. The structure of the PfA-M1 S1 subsite occupied by the sidechain of the Phe-Leu analog bestatin, a competitive inhibitor of PfA-M1, is shown in Fig. 1 [13]. At the bottom is the catalytic zinc atom liganded by three enzyme sidechains. The largely non-polar “walls” of the cylinder are formed by four amino acid sidechains: E319 (β- and γ-methylene groups), V459, M462 and Y575. As the cylinder wall residues define the shape and polarity of the base of the S1 subsite, they likely contribute to the selection against substrates with acidic or β-branched sidechains; however, they cannot explain how the subsite can tolerate both positively charged and large non-polar P1 sidechains.
Figure 1. Architecture of the PfA-M1 S1 subsite.
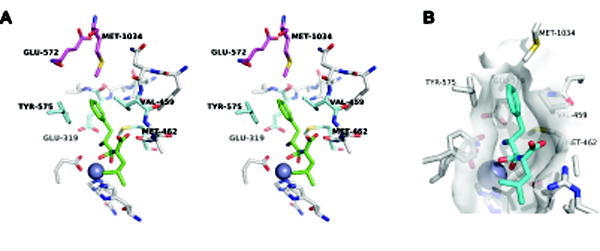
(A) The S1 subsite from the structure of the PfA-M1:bestatin complex [13] is shown in a stereo pair. The catalytic Zn(II) atom is represented as a dark blue sphere. The residues forming the walls of the S1 subsite cylinder (E319, V459, M462 and Y575) are colored light blue and the cap residues (E572 and M1034) are colored magenta. Bestatin is colored green; the phenylmethylene sidechain occupies the S1 subsite. The sidechain of M1034 forms van der Waals contacts with the P1 phenyl ring, while that of E572 is oriented away from the S1 subsite. (B) Surface topology of the S1 subsite derived from residues within 5 Å of bestatin (cyan). The E572 sidechain is not visible because it is oriented away from the bestatin phenyl ring (i.e., not within 5 Å). Figure adapted from Dalal et al [23].
At the top of the S1 subsite cylinder are two “cap” residues: E572 and M1034 [13]. Atomic structures of PfA-M1 have revealed important clues into potential roles for the cap residues in substrate selection. In the PfA-M1:bestatin co-crystal structure ([13]; Fig. 1), the P1 phenyl ring occupies the S1 subsite. M1034 is positioned to make stabilizing van der Waals contacts with the phenyl ring. Notably, the E572 sidechain swings away from the S1 subsite, presumably to avoid situating a polar group in a non-polar environment. Strikingly different conformations of the two cap residues are observed when a positively-charged group occupies the S1 subsite. In the co-crystal structure of the hydrolysis product arginine in the active site of the PfA-M1 mutant V459P, the E572 sidechain carboxylate points into the S1 subsite and forms a salt bridge with the Arg guanidinyl group, while the M1034 sidechain adopts two conformations, both of which orient it away from the S1 subsite [23]. These observations suggest a “complementarity” model for the contributions of the two cap residues to substrate selection: large, non-polar P1 sidechains engage M1034, whereas the positively-charged P1 sidechains of Arg and Lys are stabilized by electrostatic interaction with E572. Conformational flexibility of the cap residue sidechains is a key attribute of this model as it allows for the mitigation of unfavorable interactions.
Here, we have undertaken a mutagenic study to define the contributions of the S1 subsite cap residues E572 and M1034 to substrate selection and catalytic efficiency. The two residues were mutated to alanine, both independently and together. In addition, both cap residues were mutated to those in Escherichia coli PepN (EcPepN), a distantly-related M1-family aminopeptidase, yielding the double mutant E572N/M1034Q. Analysis of the E572N/M1034Q mutant was undertaken to assess the context dependence of cap residue function. We predicted that the E572N/M1034Q mutant would retain high catalytic efficiency with substrates having an Arg or Lys P1 residue, as the cap residues of EcPepN (N373, Q821) have been shown to directly interact with P1-sidechains of Lys (N373 only) and Arg (N373 and Q821) [24]. Effects of the mutations on S1 subsite preferences were assessed by determining the steady-state kinetic parameters for catalysis of hydrolysis of a series of model dipeptide substrates. To inform future inhibitor development efforts, we also determined the effects of cap residue mutations on the binding affinity of the aminopeptidase inhibitor bestatin.
2. Materials and Methods
2.1. Expression and purification of PfA-M1 cap mutants
The PfA-M1 expression construct consisted of the four conserved M1-family aminopeptidase domains (residues 192 – 1085) and an N-terminal hexahistidine tag; the signal peptide and Plasmodium-specific N-terminal extension were omitted as previously described [5]. Mutations were introduced at the codons for the cap residues E572 and M1034 by QuikChange mutagenesis (Stratagene). PfA-M1 cap mutants were expressed and purified as previously described [21], a procedure that included immobilized metal affinity chromatography, tobacco etch virus protease removal of the hexahistidine tag, reverse metal affinity chromatography, dialysis against 100 μM ZnCl2 and gel filtration chromatography. Concentrations of purified PfA-M1 mutants were determined by absorbance using a calculated extinction coefficient of 1.15 × 105 M−1•cm−1. Aliquots of enzymes were snap frozen in liquid nitrogen and stored at −80 °C.
2.2. Steady-state kinetic analysis
Steady-state kinetic parameters for the hydrolysis of the dipeptides X-Ala, where X is one of Arg, Lys, Leu, Met or Trp (Bachem), and for Ala-Leu (Sigma), were determined as previously described for wild-type PfA-M1 [21]. Ala-Leu was used as a substrate with a small P1 sidechain instead of Ala-Ala because the latter has a high Km value for PfA-M1 (9 mM; [21]) which required very high peptide concentrations and yielded high variability in independent determinations of steady-state kinetic parameters. In contrast, the Km value of Ala-Leu is 1.4 mM and use of this substrate provided high quality kinetic data (Supplemental Table S1). Briefly, enzyme assays were conducted in 100 mM sodium 4-(2-hydroxyethyl)-1-piperazineethanesulfonate, 110 mM NaCl, pH 7.5 at 30 °C for 15 minutes. Nine substrate concentrations (including no substrate) were used to construct Michaelis-Menten plots, with the substrate concentration range adjusted for each enzyme-substrate combination such that data points above and below the Km value were obtained. Enzyme concentration was adjusted such that substrate consumption was not greater than 10% by the end of the assay. The reaction product Ala was quantified by derivatization with 6-aminoquinolyl-N-hydroxysuccinimidly carbamate (Waters) followed by ultra-high pressure liquid chromatography as previously described [5]. An alanine standard was included in each experiment to convert Ala peak areas to concentrations. Km and kcat were determined by non-linear regression fitting to the Michaelis-Menten equation ν = Vs/(Km + s) using Kaleidagraph 4.5 (Synergy Software), where V is the limiting velocity and s is the substrate concentration. kcat was calculated from V = kcat[E]. kcat/Km was calculated with propagation of error. All kinetic parameters were determined in at least three independent experiments and are reported as the mean and standard deviation. Statistical comparisons were made using Welch’s unequal variances two-tailed t-test [25]. Inhibition constants for bestatin were determined by the Dixon method [26].
2.3. Calculation of ΔΔGb values
The change in binding free energy during the transition state elicited by cap residue mutations was calculated as described by Wilkinson et al [27]: ΔΔGb = RT ln[(kcat/Km)mut/(kcat/Km)wt]. The condition that the mutation not affect the chemical steps of catalysis is valid as demonstrated by the analysis of Ala-Leu hydrolysis described below.
3. Results
3.1. Cap residue mutations do not affect catalysis when the P1 residue is Ala
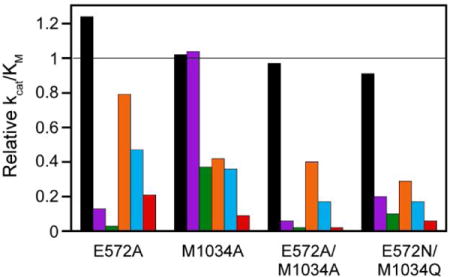
As the cap residues E572 and M1034 are situated at the top of the elongated S1 subsite cylinder (Fig. 1), they are not expected to interact with small P1 sidechains such as the methyl group of Ala. To assess the effects of the cap mutations on the catalytic properties of PfA-M1 independent of interactions with the substrate P1 sidechain, we determined the steady state kinetic constants for hydrolysis of the dipeptide Ala-Leu. Unlike the other substrates in this study, which have a P1′-Ala residue, Ala-Leu possesses a P1′ Leu residue. This choice was driven by practical considerations: use of a P1′-Leu residue lowered the Km value ~9-fold [21], which brought it into a more experimentally tractable range. For all mutants, the steady-state kinetic parameters for Ala-Leu hydrolysis did not change significantly from wild-type values of 1.4 mM for Km (p > 0.19), 26 s−1 for kcat (p > 0.12) and 1.9 × 104 M−1•s−1 for kcat/Km (p > 0.55). All parameters determined in this study are represented graphically in Fig. 2 and 3A and are listed in Supplemental Table S1. Fig. 3B depicts the relative catalytic efficiencies for each mutant ((kcat/Km)(mut) divided by kcat/Km(wt)) to permit facile assessment of the changes in S1 subsite specificity. These results indicate that the cap residues are not directly involved in the chemical steps of catalysis.
Figure 2. Steady-state kinetic parameters for wild-type PfA-M1 and four cap mutants organized by substrate.
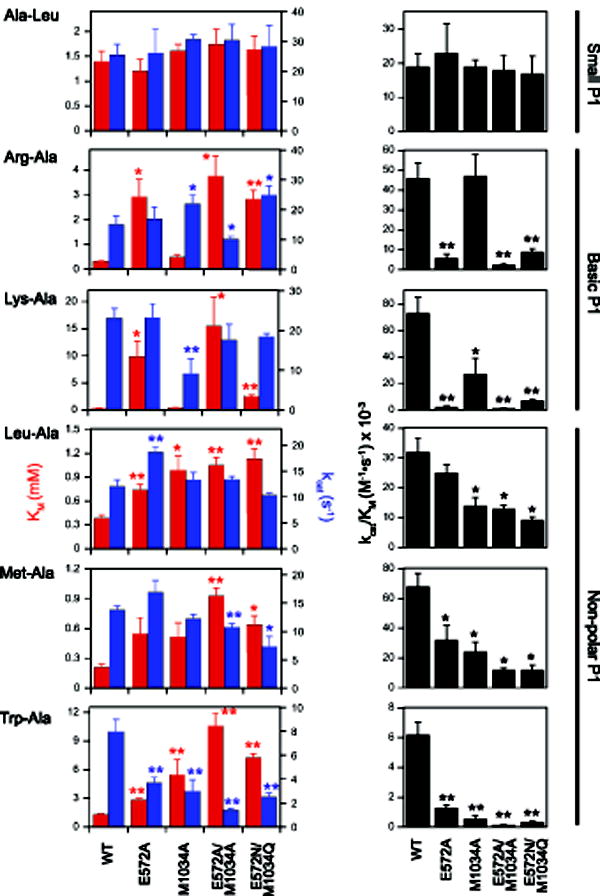
Km (red) and kcat (blue) values are shown on the left side and kcat/Km is shown on the right side. Note that the ordinate scales vary with substrate. All values are reported as means with standard deviations indicated by error bars and are provided in Supplemental Table S1. Statistically significant differences relative to wild-type (WT) are indicated with one (0.05>p>0.01) or two (p<0.01) asterisks.
Figure 3. Catalytic efficiencies of PfA-M1 and cap mutants organized by enzyme.
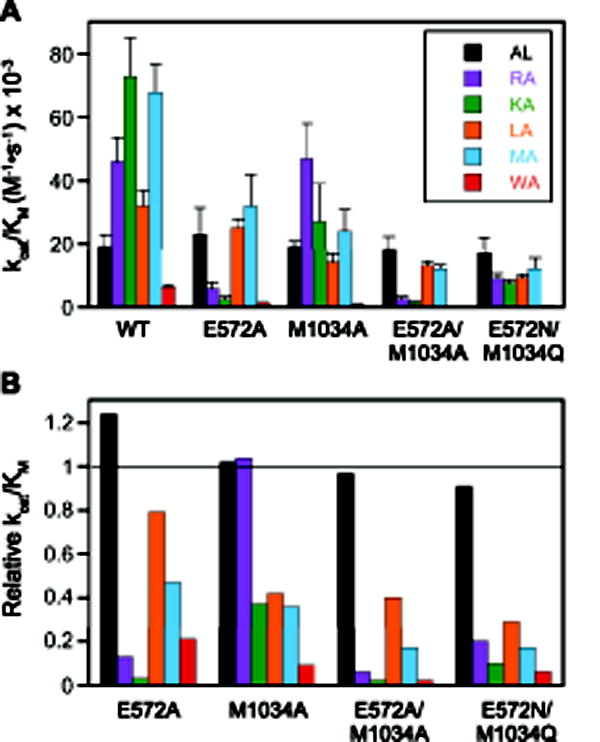
(A) Comparison of specificity profiles for wild-type PfA-M1 and four cap mutants. Error bars represent standard deviation. (B) Relative kcat/Km values (defined as [kcat/Km]mut/[kcat/Km]wt). The horizontal line indicates a value of 1, i.e., identity with wild-type. Dipeptide substrates are indicated in single-letter code in the legend.
3.2. Effects of cap mutations on hydrolysis of substrates with basic P1 sidechains
The dipeptides Arg-Ala and Lys-Ala were selected to probe the effects of cap mutations on substrates with extended, positively-charged P1 sidechains (Figs. 2 and 3 and Supplemental Table S1). The E572A mutation had a dramatic effect on Km, increasing this parameter 9-fold for Arg-Ala and 31-fold for Lys-Ala. In contrast, the kcat values for these substrates were not significantly different from that of wild-type PfA-M1. The net effect of these changes was a reduction in catalytic efficiency (kcat/Km) of 8-fold or Arg-Ala and 30-fold for Lys-Ala, which corresponds to reductions in binding energy (ΔΔGb) [27] of 5.2 kJ/mol and 8.6 kJ/mol, respectively. The effect of the M1034A mutation was distinct from that of E572A. There were no significant differences in Km values for the two substrates, but there were modest yet statistically significant changes in kcat. The double mutation E572A/M1034A, like E572A, exhibited large increases in Km, whereas the effects on kcat were slight.
Interestingly, introduction of the cap residues of EcPepN (E572N/M1034Q) did not significantly alter the Km value for Arg-Ala when compared to the value for the E572A/M1034A mutant (p = 0.18). This result was somewhat surprising, given that the EcPepN:Arg co-crystal structure indicates that the Arg guanidinium group hydrogen bonds with EcPepN cap residues N373 and Q821 [24], and suggests that the cap residues in the E572N/M1034Q PfA-M1 mutant are not able to interact with the Arg sidechain in the same fashion that they do in EcPepN. In contrast, a six-fold drop in Km for Lys-Ala hydrolysis by the E572N/M1034Q mutant vs. E572A/M1034A implies that the P1-Lys sidechain is able to interact with at least one of the cap residues; however, the Km value remained 6-fold higher than that of wild-type PfA-M1. The observation that the Lys-Ala kcat values for wild-type PfA-M1 and the E572A/M1034A and E572N/M1034Q mutants were not significantly different (Fig. 2) indicates that interactions of the P1 sidechain with the cap residues were not critical for aligning the scissile amide bond of this substrate with the catalytic machinery.
3.3. Effects of cap mutations on hydrolysis of substrates with non-polar P1 sidechains
To interrogate the effects of cap mutations on the hydrolysis of substrates with non-polar P1 residues, we used two substrates with large aliphatic P1 sidechains (Leu-Ala, Met-Ala) and one with a bulky aromatic P1 sidechain (Trp-Ala). With Leu-Ala and Met-Ala, all of the cap mutants exhibited modestly reduced catalytic efficiency, the primary driver for which was an increase in Km values. More dramatic was the effect of the mutations on Trp-Ala hydrolysis. Individually, E572A and M1034A mutations caused significant increases in Km and decreases in kcat. Combination of the two mutations exacerbated these trends and decreased catalytic efficiency to a value 50-fold lower than that of wild-type PfA-M1. The introduction of EcPepN cap residues (E572N/M1034Q mutant) either did not increase catalytic efficiency over that observed with the E572A/M1034A mutant (Leu-Ala, Met-Ala) or afforded only a marginal increase (Trp-Ala).
3.4. Effects of cap mutations on the bestatin inhibition constant
To assess the contribution of the cap residues to ligand binding independently of catalysis, we determined the inhibition constant (Ki) values for the competitive inhibitor bestatin with the mutants E572A, M1034A and the double mutant E572A/M1034A (Table 1). The Ki for the M1034A mutant increased about four-fold, an observation that accords with the known interaction of the phenyl ring of the P1 sidechain of bestatin with the sidechain of M1034 [13]. More surprising was the effect of the E572A mutation, which resulted in an increase in Ki of a similar magnitude. In the PfA-M1:bestatin crystal structure, the sidechain of E572 rotates away from the S1 subsite and is not thought to make contact with the bestatin ligand [13]. The effect of the two mutations was approximately additive, as the increase in Ki in the E572A/M1034A mutant was 14-fold.
Table 1. Bestatin inhibition constants.
Means and standard deviations (SD) are reported from three independent experiments. WT, wild-type.
| Enzyme | Mean Ki ± SD (μM) |
|---|---|
| WT | 0.16 ± 0.02 |
| E572A | 0.69 ± 0.08 |
| M1034A | 0.75 ± 0.05 |
| E572A/M1034A | 2.3 ± 0.3 |
4. Discussion
The view emerging from these studies is that the two PfA-M1 cap residues E572 and M1034 contribute to efficient catalysis of peptide hydrolysis by stabilizing the binding of chemically diverse P1 sidechains in the S1 subsite. This effect was most pronounced for substrates with basic and large aromatic sidechains. The stabilizing effect of E572 on the binding of substrates with positively-charged P1 sidechain guanidinyl and amino groups (Arg and Lys, respectively) is likely due to the formation of a salt bridge, such as that previously observed in the PfA-M1(V459P):Arg co-crystal structure [23]. In contrast, the stabilization of non-polar P1 sidechains likely occurs through two possible mechanisms: direct van der Waals interactions with the sidechain of M1034A and/or exclusion of water from the top of the S1 subsite (i.e., the hydrophobic effect). The effect of the M1034A mutation was greatest for the substrate with the largest non-polar P1 sidechain (Trp-Ala); this may reflect the ability of the M1034 sidechain to form more extensive interactions with the bulky Trp sidechain as compared to the smaller Leu and Met sidechains.
At the same time, the results did not accord with the simple “complementarity” model described in the Introduction. For example, in the case of Trp-Ala and the E572A mutant, a statistically significant increase in Km and decreases in kcat and kcat/Km were observed. One possible explanation is that the indole NH forms a hydrogen bond with the E572 sidechain. While we can’t rule this out, we note that in the EcPepN:Trp co-crystal structure, the indole NH forms a hydrogen bond to a water molecule and does not directly interact with the cap residues [24]. Another example is the increase in bestatin Ki in the E572A mutant. Available structural evidence [13] indicates that the E572 sidechain does not interact directly with bestatin. One possible explanation for these findings is that water molecules fill the space vacated by the E572A mutation, altering the electrostatic properties of the S1 subsite. More generally, the bestatin data indicate that productive interactions with the cap residues can substantially increase the binding affinity of inhibitors. Strategies to develop potent PfA-M1 inhibitors should take into consideration the benefits of occupying the S1 subsite such that E572 and/or M1034 are engaged in inhibitor binding.
The functional significance of the cap residues with regard to the established role of PfA-M1 in hemoglobin catabolism in the food vacuole is a key question that is difficult to address with in vitro studies. We note that three of the dipeptide substrates used in our study are found in the sequences of human α- and β-globin: Leu-Ala (5 occurrences), Lys-Ala (two occurrences) and Ala-Leu (7 occurrences). However, we do not know whether these dipeptides are generated in the food vacuole. Perhaps the best way to evaluate the biological significance of the cap residues will be to attempt to introduce mutations in the parasite’s genomic copy of PfA-M1.
Previously, we have shown that mutation of a weakly conserved residue that contributes to the formation of the S1 cylinder (V459 in PfA-M1) can dramatically alter the S1 subsite preferences of the enzyme [23]. Here, we have identified a second, independent determinant of S1 subsite specificity. To explore the level of conservation in cap residues, we aligned ten homologous M1-family aminopeptidases from eight genera of apicomplexan parasites. The residue corresponding to E572 was conserved either as an acidic residue (Glu or Asp) or a polar residue (Asn). In contrast, the residue corresponding to M1034 was highly variable, with Val, Ile, Leu, Gln, His and Arg found in this position. Although the functional implications of this natural variation are unknown, it is conceivable that variation of the two cap residues is a means by which the S1 subsite preferences can be modulated in response to selective pressures, allowing the rapid evolution of novel specificities.
Finally, we note that there appears to be a context dependence to the specificity imparted through the cap residues. In previous work, we found that the catalytic efficiencies of EcPepN with Arg-Ala and Lys-Ala matched or exceeded those of PfA-M1, even when all four cylinder “wall” residues were identical [23]. However, when we substituted PfA-M1 cap residues with those of EcPepN (i.e., in the E572N/M1034Q mutant), the catalytic efficiencies of the E572N/M1034Q mutant were 5- to 10-fold lower than those of wild-type PfA-M1 (Fig. 3). Clearly, there are factors beyond the identities of the six S1 subsite residues contributing to overall catalytic efficiency.
Supplementary Material
Highlights.
PfA-M1 S1-subsite cap residues E572 and M1034 were mutated independently and together.
Steady-state kinetic parameters were determined for model dipeptide substrates.
E572 and M1034 contributed to efficient catalysis of substrates with positively charged and large non-polar P1 sidechains.
Mutation of both cap residues substantially reduced the binding affinity of the aminopeptidase inhibitor bestatin.
Acknowledgments
This work was funded by the National Institutes of Health (grant AI077638) and USDA National Institute of Food and Agriculture (project VA-139761). We appreciate the support of M.R. through a Summer Undergraduate Research Fellowship from Fralin Life Science Institute, Virginia Tech. We are grateful to Feng Li for contributing to the initiation of this project.
Abbreviations
- EcPepN
Escherichia coli aminopeptidase N
- PfA-M1
Plasmodium falciparum aminopeptidase-M1 family
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
The nomenclature of Schechter and Berger [28] is used here. P1 and P1′ refer to substrate residues while S1 and S1′ refer to the corresponding enzyme subsites. For aminopeptidases, the scissile peptide bond is between the P1 and P1′ residues.
References
- 1.Page MJ, Di Cera E. Evolution of peptidase diversity. J Biol Chem. 2008;283:30010–4. doi: 10.1074/jbc.M804650200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Allary M, Schrevel J, Florent I. Properties, stage-dependent expression and localization of Plasmodium falciparum M1 family zinc-aminopeptidase. Parasitology. 2002;125:1–10. doi: 10.1017/s0031182002001828. [DOI] [PubMed] [Google Scholar]
- 3.Azimzadeh O, Sow C, Geze M, Nyalwidhe J, Florent I. Plasmodium falciparum PfA-M1 aminopeptidase is trafficked via the parasitophorous vacuole and marginally delivered to the food vacuole. Malar J. 2010;9:189–205. doi: 10.1186/1475-2875-9-189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dalal S, Klemba M. Roles for two aminopeptidases in vacuolar hemoglobin catabolism in Plasmodium falciparum. J Biol Chem. 2007;282:35978–87. doi: 10.1074/jbc.M703643200. [DOI] [PubMed] [Google Scholar]
- 5.Ragheb D, Dalal S, Bompiani KM, Ray WK, Klemba M. Distribution and biochemical properties of an M1-family aminopeptidase in Plasmodium falciparum indicate a role in vacuolar hemoglobin catabolism. J Biol Chem. 2011;286:27255–65. doi: 10.1074/jbc.M111.225318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hanssen E, Knoechel C, Dearnley M, Dixon MW, Le Gros M, Larabell C, Tilley L. Soft X-ray microscopy analysis of cell volume and hemoglobin content in erythrocytes infected with asexual and sexual stages of Plasmodium falciparum. J Struct Biol. 2012;177:224–32. doi: 10.1016/j.jsb.2011.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Goldberg DE. Hemoglobin degradation. Curr Top Microbiol Immunol. 2005;295:275–91. doi: 10.1007/3-540-29088-5_11. [DOI] [PubMed] [Google Scholar]
- 8.Harbut MB, Velmourougane G, Dalal S, Reiss G, Whisstock JC, Onder O, Brisson D, McGowan S, Klemba M, Greenbaum DC. Bestatin-based chemical biology strategy reveals distinct roles for malaria M1- and M17-family aminopeptidases. Proc Natl Acad Sci USA. 2011;108:E526–34. doi: 10.1073/pnas.1105601108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gonzalez-Bacerio J, Fando R, del Monte-Martinez A, Charli JL, de Chavez ML. Plasmodium falciparum M1-aminopeptidase: a promising target for the development of antimalarials. Curr Drug Targets. 2014;15:1144–65. doi: 10.2174/1389450115666141024115641. [DOI] [PubMed] [Google Scholar]
- 10.Skinner-Adams TS, Stack CM, Trenholme KR, Brown CL, Grembecka J, Lowther J, Mucha A, Drag M, Kafarski P, McGowan S, Whisstock JC, Gardiner DL, Dalton JP. Plasmodium falciparum neutral aminopeptidases: new targets for anti-malarials. Trends Biochem Sci. 2010;35:53–61. doi: 10.1016/j.tibs.2009.08.004. [DOI] [PubMed] [Google Scholar]
- 11.Suda H, Takita T, Aoyagi T, Umezawa H. The structure of bestatin. J Antibiot. 1976;29:100–1. doi: 10.7164/antibiotics.29.100. [DOI] [PubMed] [Google Scholar]
- 12.Flipo M, Florent I, Grellier P, Sergheraert C, Deprez-Poulain R. Design, synthesis and antimalarial activity of novel, quinoline-based, zinc metallo-aminopeptidase inhibitors. Bioorg Med Chem Lett. 2003;13:2659–62. doi: 10.1016/s0960-894x(03)00550-x. [DOI] [PubMed] [Google Scholar]
- 13.McGowan S, Porter CJ, Lowther J, Stack CM, Golding SJ, Skinner-Adams TS, Trenholme KR, Teuscher F, Donnelly SM, Grembecka J, Mucha A, Kafarski P, Degori R, Buckle AM, Gardiner DL, Whisstock JC, Dalton JP. Structural basis for the inhibition of the essential Plasmodium falciparum M1 neutral aminopeptidase. Proc Natl Acad Sci USA. 2009;106:2537–42. doi: 10.1073/pnas.0807398106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Velmourougane G, Harbut MB, Dalal S, McGowan S, Oellig CA, Meinhardt N, Whisstock JC, Klemba M, Greenbaum DC. Synthesis of new (−)-bestatin-based inhibitor libraries reveals a novel binding mode in the S1 pocket of the essential malaria M1 metalloaminopeptidase. J Med Chem. 2011;54:1655–66. doi: 10.1021/jm101227t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kannan Sivaraman K, Paiardini A, Sienczyk M, Ruggeri C, Oellig CA, Dalton JP, Scammells PJ, Drag M, McGowan S. Synthesis and structure-activity relationships of phosphonic arginine mimetics as inhibitors of the M1 and M17 aminopeptidases from Plasmodium falciparum. J Med Chem. 2013;56:5213–7. doi: 10.1021/jm4005972. [DOI] [PubMed] [Google Scholar]
- 16.Cunningham E, Drag M, Kafarski P, Bell A. Chemical target validation studies of aminopeptidase in malaria parasites using alpha-aminoalkylphosphonate and phosphonopeptide inhibitors, Antimicrob. Agents Chemother. 2008;52:3221–8. doi: 10.1128/AAC.01327-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Drinkwater N, Vinh NB, Mistry SN, Bamert RS, Ruggeri C, Holleran JP, Loganathan S, Paiardini A, Charman SA, Powell AK, Avery VM, McGowan S, Scammells PJ. Potent dual inhibitors of Plasmodium falciparum M1 and M17 aminopeptidases through optimization of S1 pocket interactions. Eur J Med Chem. 2016;110:43–64. doi: 10.1016/j.ejmech.2016.01.015. [DOI] [PubMed] [Google Scholar]
- 18.Mistry SN, Drinkwater N, Ruggeri C, Sivaraman KK, Loganathan S, Fletcher S, Drag M, Paiardini A, Avery VM, Scammells PJ, McGowan S. Two-pronged attack: dual inhibition of Plasmodium falciparum M1 and M17 metalloaminopeptidases by a novel series of hydroxamic acid-based inhibitors. J Med Chem. 2014;57:9168–83. doi: 10.1021/jm501323a. [DOI] [PubMed] [Google Scholar]
- 19.Flipo M, Beghyn T, Leroux V, Florent I, Deprez BP, Deprez-Poulain RF. Novel selective inhibitors of the zinc plasmodial aminopeptidase PfA-M1 as potential antimalarial agents. J Med Chem. 2007;50:1322–34. doi: 10.1021/jm061169b. [DOI] [PubMed] [Google Scholar]
- 20.Deprez-Poulain R, Flipo M, Piveteau C, Leroux F, Dassonneville S, Florent I, Maes L, Cos P, Deprez B. Structure-activity relationships and blood distribution of antiplasmodial aminopeptidase-1 inhibitors. J Med Chem. 2012;55:10909–17. doi: 10.1021/jm301506h. [DOI] [PubMed] [Google Scholar]
- 21.Dalal S, Ragheb DR, Klemba M. Engagement of the S1, S1′ and S2′ subsites drives efficient catalysis of peptide bond hydrolysis by the M1-family aminopeptidase from Plasmodium falciparum. Mol Biochem Parasitol. 2012;183:70–7. doi: 10.1016/j.molbiopara.2012.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Poreba M, McGowan S, Skinner-Adams TS, Trenholme KR, Gardiner DL, Whisstock JC, To J, Salvesen GS, Dalton JP, Drag M. Fingerprinting the substrate specificity of M1 and M17 aminopeptidases of human malaria, Plasmodium falciparum. PLoS ONE. 2012;7:e31938. doi: 10.1371/journal.pone.0031938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dalal S, Ragheb DR, Schubot FD, Klemba M. A naturally variable residue in the S1 subsite of M1 family aminopeptidases modulates catalytic properties and promotes functional specialization. J Biol Chem. 2013;288:26004–12. doi: 10.1074/jbc.M113.465625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Addlagatta A, Gay L, Matthews BW. Structural basis for the unusual specificity of Escherichia coli aminopeptidase N. Biochemistry. 2008;47:5303–11. doi: 10.1021/bi7022333. [DOI] [PubMed] [Google Scholar]
- 25.Ruxton GD. The unequal variance t-test is an underused alternative to Student’s t-test and the Mann-Whitney U test. Behav Ecol. 2006;17:688–690. [Google Scholar]
- 26.Dixon M. The determination of enzyme inhibitor constants. Biochem J. 1953;55:170–171. doi: 10.1042/bj0550170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wilkinson AJ, Fersht AR, Blow DM, Winter G. Site-directed mutagenesis as a probe of enzyme structure and catalysis: tyrosyl-tRNA synthetase cysteine-35 to glycine-35 mutation. Biochemistry. 1983;22:3581–6. doi: 10.1021/bi00284a007. [DOI] [PubMed] [Google Scholar]
- 28.Schechter I, Berger A. On the size of the active site in proteases. I. Papain. Biochem Biophys Res Commun. 1967;27:157–62. doi: 10.1016/s0006-291x(67)80055-x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.