

Abstract
Oral squamous cell carcinoma (OSCC) develops through a multistep carcinogenic process involving field cancerization. The DEK gene is a proto‐oncogene with functions in genetic and epigenetic modifications, and has oncogenic functions, including cellular proliferation, differentiation, and senescence. DEK overexpression is associated with malignancies; however, the functional roles of DEK overexpression are unclear. We demonstrated that DEK‐expressing cells were significantly increased in human dysplasia/carcinoma in situ and OSCC. Furthermore, we generated ubiquitous and squamous cell‐specific doxycycline (DOX)‐inducible Dek mice (iDek and iDek‐e mice respectively). Both DOX+ iDek and iDek‐e mice did not show differences in the oral mucosa compared with DOX‐ mice. In the environment exposed to carcinogen, DOX‐treated (DOX+) iDek mice showed field cancerization and OSCC development. Microarray analysis revealed that DEK overexpression was mediated by the upregulation of DNA replication‐ and cell cycle‐related genes, particularly those related to the G 1/S transition. Tongue tumors overexpressing DEK showed increased proliferating cell nuclear antigen and elongator complex protein 3 expression. Our data suggest that DEK overexpression enhanced carcinogenesis, including field cancerization, in OSCC by stimulating the G 1/S phase transition and promoting DNA replication, providing important insights into the potential applications of DEK as a target in the treatment and prevention of OSCC.
Keywords: DEK, field cancerization, oncogene, squamous cell carcinoma
Introduction
Head and neck cancer is the sixth most common human cancer 1 and occurs in the oral cavity in 48% of cases; 90% of these cases are oral squamous cell carcinoma (OSCC) 2. The development of OSCC is a multistep process involving progression from normal mucosa to papillary hyperplasia, dysplasia, carcinoma in situ (CIS), and invasive squamous cell carcinoma (SCC) 3. This progression of OSCC requires the accumulation of multiple genetic, epigenetic, and chromosomal alterations, which are influenced by a patient's genetic/epigenetic predisposition and by environmental influences, including tobacco, alcohol, chronic inflammation, and human papilloma virus (HPV) infection 4, 5, 6. Tobacco use and alcohol consumption are major risk factors for oral cancer 7.
Field cancerization (also known as field defects) is a term first proposed by Slaughter et al. in 1953 8. The principle of field cancerization encompasses carcinogen‐induced early genetic/epigenetic changes in the mucosa of the oral cavity, leading to the development of precancerous lesions and additional multifocal tumors, even though the epithelium exposed to the carcinogen may have an appearance similar to that of the normal mucosa 9. Several studies have shown that tobacco smoking increases the likelihood of malignant changes toward OSCC in the proliferating epithelium 10, 11. Furthermore, increased proliferation of the epithelium is observed, even after cessation of smoking 11, suggesting that smoking‐related high proliferative activity could be a potential event involved in field cancerization. Proliferating cell nuclear antigen (PCNA), minichromosome maintenance protein (MCM) family, and cell division cycle 6 (CDC6), which are associated with regulation of the cell cycle, particularly the G 1/S phase transition, are significantly upregulated in precancerous lesions and OSCC in the human oral cavity 12, 13, 14, 15, 16, 17. A key step in the regulation of cell proliferation is the control of the initiation of DNA synthesis by the G 1/S transition 18, 19.
The human DEK oncogene was first reported to be the target of a recurrent t(6;9) translocation that generates a fusion protein with the nucleoporin CAN in a subset of patients with acute myeloid leukemia (AML) 20, 21. DEK, a highly conserved nuclear factor, is the only member of its protein class 22 and plays an important role in various cell processes and cellular metabolic functions, such as maintenance of global heterochromatin integrity, transcriptional control, mRNA splicing, DNA replication, DNA damage repair, and susceptibility 23. While the regulation of DEK has not been well‐studied, E2 factor (E2F), nuclear transcription factor Y (NF‐Y), Yin Yang 1 (YY‐1), and estrogen receptor α are thought to directly modulate the transcription of the DEK gene 24, 25, 26. Furthermore, DEK has been proposed to be a potential target gene of the p16‐pRB‐E2F pathway 27, 28, a key regulator of the G 1/S transition in mammalian cells 29. The regulation of DEK expression by E2F transcription factors provides an explanation for the finding that DEK expression is induced by the activity of the high‐risk HPV E7 protein. However, the target genes of DEK and the mechanisms through which DEK affects carcinogenesis are still unclear.
Owing to its frequent upregulation in various human malignancies, DEK is thought to have oncogenic activities 30; additionally, targeted suppression of DEK may represent a new strategic approach to the treatment of cancers 31. Interestingly, Dek‐knockout mice do not exhibit any abnormal phenotypes compared with wild‐type mice 31, suggesting that DEK may be an attractive drug target.
Recently, Adams et al. 32 used an HPV16 E7‐induced transgenic mouse model of OSCC and demonstrated that Dek was required for the growth of head and neck SCCs. Moreover, DEK protein was universally upregulated in both HPV‐positive and ‐negative human SCCs relative to adjacent normal tissue 32. Furthermore, DEK has been shown to be upregulated in tobacco chewing‐mediated OSCC 33. Thus, DEK is thought to be closely associated with the carcinogenesis of OSCC through multiple mediators, including HPV and tobacco. However, it is unclear whether DEK is an actual proto‐oncogene or oncogene in OSCC.
In this study, we generated a doxycycline (DOX)‐inducible Dek transgenic mouse model for controlling the timing and localization of DEK overexpression. Using this model, we investigated the role of DEK in OSCC in both humans and mice.
Materials and Methods
Mice
Krt14‐Cre and Rosa26‐LSL‐rtTA‐IRES‐GFP mice were obtained from The Jackson Laboratory (Bar Harbor, ME, USA). Rosa26‐M2rtTA and Tet‐O‐Dek mice were generated as described in Supplementary Methods. All experiments were performed in accordance with the Gifu University International Animal Care and Use Committee guidelines for the use of animals.
Human samples
All human samples were obtained from patients who had undergone surgery for resection at Gifu University Hospital. All patients provided informed consent for the use of their tissues. This study was approved by the Institutional Review Board of Gifu University.
DOX treatment, 4‐nitroquinoline 1‐oxide (4NQO) carcinogen exposure, preparation of tissue samples for counting and histological analysis, immunohistochemistry (IHC), evaluation of immunohistochemical staining, RNA extraction, and real‐time reverse transcription (RT)‐PCR, PCR array, western blot analysis, and microarray were performed using standard methods, as detailed in Data S1. PCR array data and the primers used for real‐time RT‐PCR and R are listed in Tables S1 and S2 respectively.
Results
DEK expression was associated with human OSCC
To investigate the relationship between DEK expression and OSCC in humans, we performed IHC for DEK protein in human normal mucosa adjacent to OSCC (n = 5), papilloma (hyperplasia; n = 12), CIS (n = 16), and OSCC (n = 34). DEK protein predominantly showed a nuclear staining pattern with slight cytoplasmic staining in normal epithelium, papilloma, CIS, and OSCC tissues (Fig. 1A). Except in the epithelium, DEK‐positive cells were observed in inflammatory cells, but not muscle tissues (Fig. 1B). In the epithelium, DEK‐positive cells were localized at the basal layer in normal epithelium and papilloma. The DEK‐positive cells spread to the upper layer in CIS and were broadly extended, reaching the invasive front, in invasive SCC. The DEK‐positive index in OSCC was significantly higher than that of normal epithelium, papilloma, and CIS (Fig. 1C). Moreover, the DEK‐positive index in CIS was significantly higher than that of normal epithelium and papilloma. There were no significant differences between normal epithelium and papilloma, which are benign lesions. These results suggested that DEK was associated with malignant epithelial lesions in OSCC.
Figure 1.
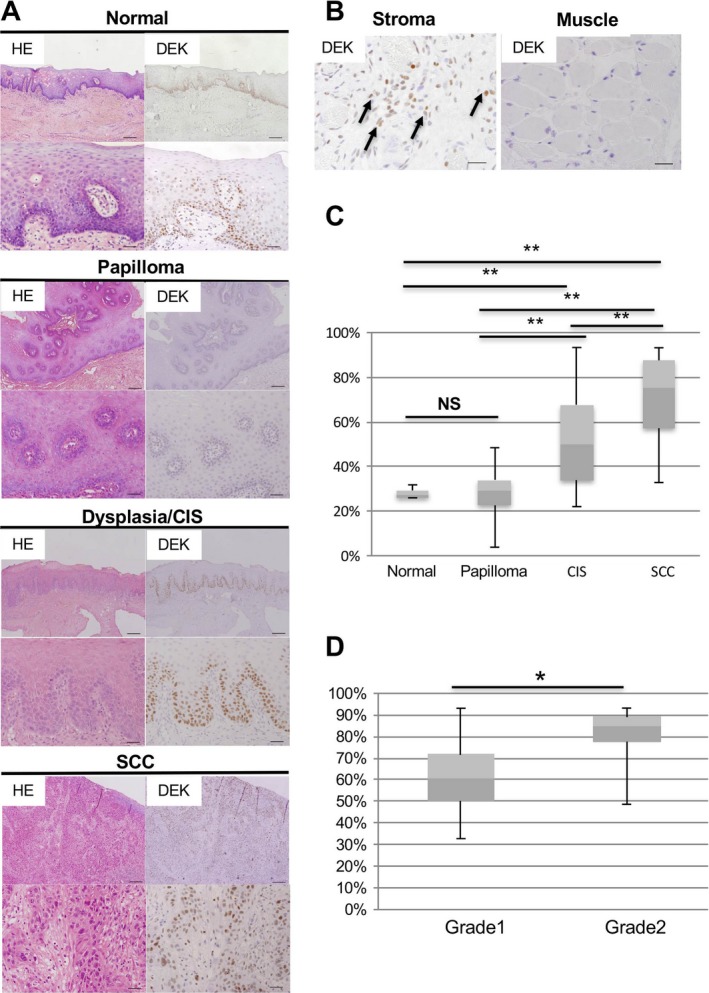
DEK overexpression in human OSCC. (A) Representative images of HE staining and IHC analysis of DEK expression in human oral tissues. Normal, normal mucosa adjacent to OSCC; CIS, carcinoma in situ; SCC, squamous cell carcinoma. Scale bars, 200 and 40 μm in the upper and lower images respectively. (B) DEK expression in the stroma and muscle layer. Arrows indicate DEK‐positive cells. Scale bars, 40 μm. (C) The positive index of DEK in normal mucosa, papilloma, CIS, and SCC in oral tissues. Boxes: 25th–75th percentiles; the median is the central line in each box (**P < 0.01; NS, not significant). (D) The positive index of DEK in grade 1 and grade 2 OSCC. Boxes: 25th–75th percentiles; the median is the central line in each box (*P < 0.05; NS, not significant).
Next, we evaluated the DEK‐positive index based on histological grade (e.g., grade 1 [well‐differentiated type, n = 18], grade 2 [moderately differentiated type, n = 16], and grade 3 (poorly differentiated type, n = 0]), in OSCCs. The DEK‐positive index in grade 2 OSCC was significantly higher than that in grade 1 OSCC (Fig. 1D). These data indicated that DEK overexpression was closely associated with OSCC in humans.
Inducible expression of Dek in ES cells and mice
It is unclear whether DEK overexpression can directly affect OSCC in vivo. Thus, to clarify the role of DEK, we generated a DOX‐inducible DEK overexpression mouse model 34, 35 to control Dek transcription. We first generated DOX‐inducible Dek ES cell clones based on the KH2 ES cell line, which harbors a modified reverse tetracycline transactivator (M2‐rtTA) targeted to the ROSA26 locus (Fig. 2A). Subsequent addition of DOX to these ES cell clones resulted in upregulation of Dek‐FLAG (Fig. S1) and induction of the FLAG‐tagged protein (Fig. 2B). Next, Dek‐inducible ES cells were injected into murine blastocysts, and mice with the TetO‐Dek allele were obtained; heterozygous mice were used in this study.
Figure 2.
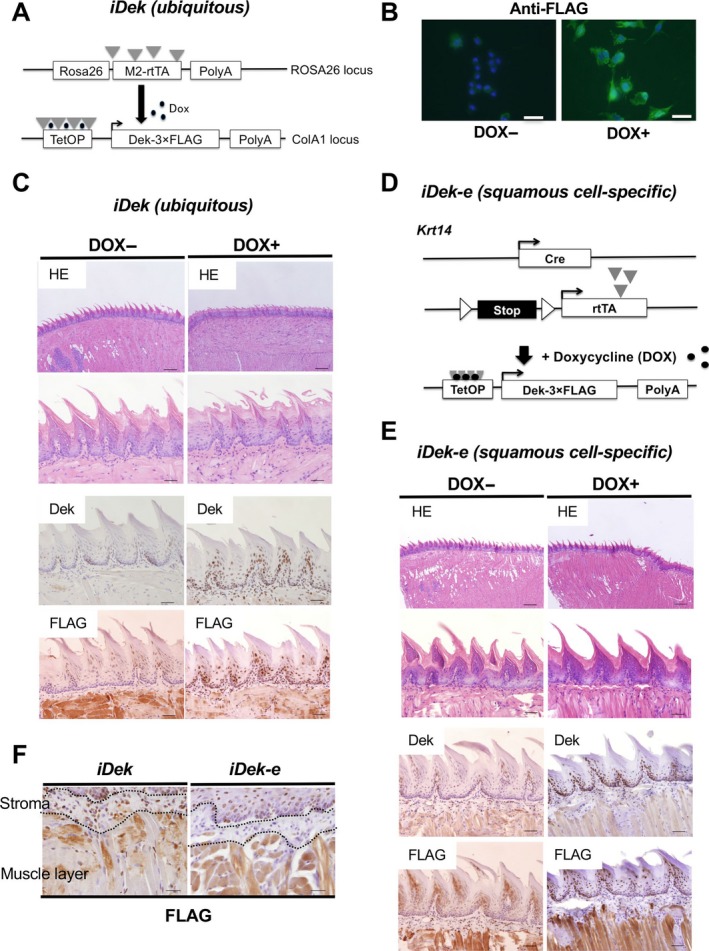
Inducible DEK expression in the tongues of iDek and iDek‐e mice. (A) Schematic representation of transgenes used to produce Dek‐inducible ES cells and iDek mice. PolyA, polyadenylation signal; TetOP, tetracycline/doxycycline‐responsive operator. (B) Immunofluorescent analysis of FLAG expression in DOX‐ and DOX+ inducible Dek ES cells after treatment with DOX (1 mg/L) for 48 h. (C) Representative images of HE and IHC staining for DEK and FLAG in the tongues of DOX‐ and DOX+ iDek mice. The concentration of DOX was 2 mg/L in the water. Scale bars, 200 and 40 μm in the upper and lower images respectively. (D) Schematic representation of transgenes used to produce squamous cell epithelium‐specific iDek (iDek‐e) mice. (E) Representative images of HE staining and IHC analysis for DEK and FLAG expression in the tongues of DOX‐ and DOX+ iDek mice. Scale bars, 200 and 40 μm in the upper lower images respectively. (F) FLAG expression in the stroma of tongue tissues from DOX+ iDek and DOX+ iDek‐e mice. Scale bars, 40 μm.
We crossed TetO‐Dek mice with Rosa26‐M2rtTA mice to generate DOX‐inducible Dek expression (iDek) mice; Dek could be induced with the addition of DOX in a dose‐dependent and reversible manner (data not shown). iDek mice ubiquitously expressed M2‐rtTA driven by the murine Rosa26 promoter and exhibited ubiquitous DEK expression in vivo. To determine DEK‐FLAG expression in the tongue and esophagus of adult mice, we analyzed DOX‐treated (DOX+) and untreated (DOX‐) iDek mice for a month. There were no obvious macroscopic or microscopic changes in the tongue or esophagus between DOX+ and DOX‐ iDek mice (n = 20 each; Fig. 2C and Fig. S2A). Both DEK and FLAG were predominantly expressed in the basal epithelial layer of the tongue and esophagus. There were no phenotypes in the induced skin, thymus, and other tissues of i‐Dek mice (Fig. S3). These results suggested that forced and ubiquitous Dek expression in adult iDek mice may not contribute to visible changes under normal conditions.
Next, we generated squamous cell epithelium‐specific iDek (iDek‐e) mice by crossing TetO‐Dek mice with Krt14‐Cre and Rosa26‐LSL‐rtTA‐IRES‐EGFP mice (Fig. 2D). The iDek‐e mice overexpressed DEK protein only in tissues producing Krt14 protein under DOX treatment. We administered DOX to iDek‐e mice for 1 month and observed no obvious macroscopic or microscopic changes in the tongue and esophagus between DOX‐ and DOX+ iDek‐e mice (Fig. 2E, Fig. S2B). Expression of DEK and FLAG was observed only in the basal epithelial layer of the tongue and esophagus. Even when the observation time was extended to 1 year, there was no obvious differences in the tongue or esophagus between DOX+ iDek and DOX+ iDek‐e mice (n = 10 each, data not shown). These data indicated that both squamous epithelium‐specific and ubiquitous DEK overexpression in adult mice may not contribute to visible changes under normal conditions.
In human tissues, DEK was expressed in both epithelial cells and inflammatory cells (Fig. 1B). iDek mice broadly expressed DEK protein in both epithelial cells and inflammatory cells, whereas iDek‐e mice did not (Fig. 2F), thus indicating that iDek mice phenocopied human DEK expression whereas iDek‐e mice did not. Therefore, we used iDek mice in the carcinogenesis experiment in this study.
DEK overexpression promoted the development of malignant lesions in the tongues of 4NQO‐treated mice
The 4NQO‐induced oral carcinogenesis model is very useful for elucidating the effects of carcinogens, for example, tobacco and alcohol, in human oral carcinogenesis 36 because the development of premalignant lesions and OSCC can be observed for a time even after stopping 4NQO exposure 3. To investigate whether DEK overexpression induced neoplastic changes in carcinogen exposure, we performed 4NQO treatment followed by DOX administration to iDek mice (Fig. 3A). All 4NQO‐treated mice survived during this experiment, and there were no differences in survival rates between DOX‐ and DOX+ iDek mice (n = 13 and 8 respectively). There were no detectable pathological differences in the liver, kidney, lung, or heart between DOX‐ and DOX+ iDek mice (data not shown).
Figure 3.
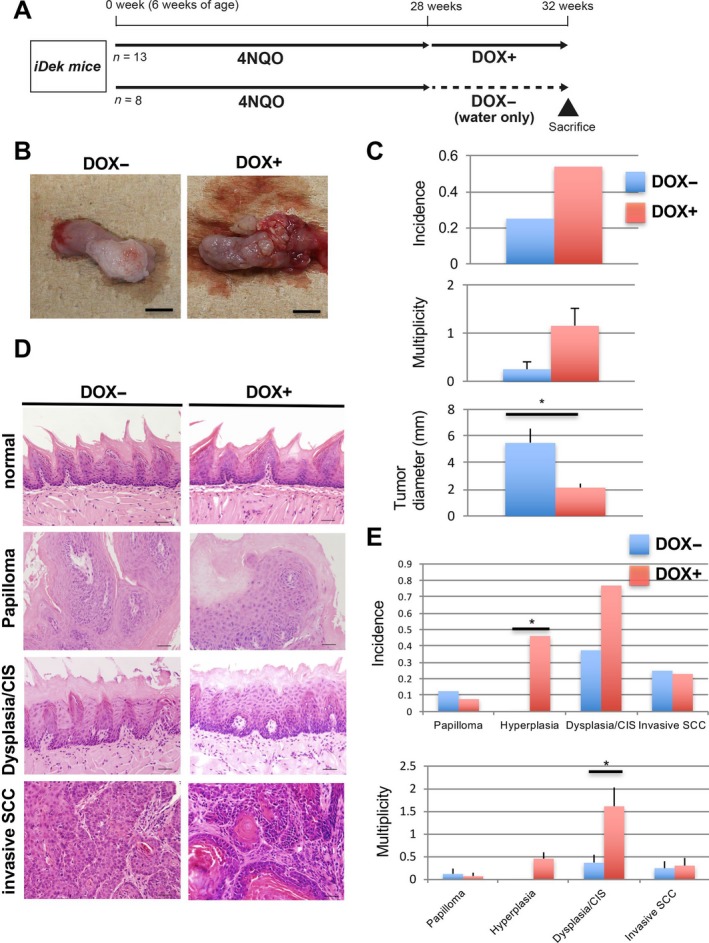
Forced DEK expression promoted early malignant lesions (dysplasia/CIS) in 4NQO‐treated mice. (A) Experimental protocol for this study. The concentration of 4NQO was 20 ppm. SAC, sacrifice. (B) Representative macroscopic images of tongue samples in DOX+ and DOX‐ iDek mice. Scale bars, 5 mm. (C) Incidence, multiplicity, and size (diameter) of macroscopic tumors of the tongue in DOX+ and DOX‐ iDek mice. Data are means (±SDs). * P < 0.05. (D) Representative microscopic images of HE staining in lesions of the tongue in DOX+ and DOX‐ iDek mice. Scale bars, 40 μm. (E) Incidence and multiplicity of microscopic lesions of the tongue in DOX+ and DOX‐ iDek mice. Data are means (±SDs). * P < 0.05.
All mice were sacrificed at 32 weeks, and tumor development was evaluated. Macroscopically nodular and polypoid tumors were found in the dorsum and lateral edge of the tongue in both cohorts (Fig. 3B). The incidences of macroscopic tongue tumors were 53% (7/13) and 25% (2/8) in DOX+ and DOX‐ iDek mice respectively (Fig. 3C). The multiplicities of macroscopic tongue tumors were 1.15 ± 1.29 and 0.25 ± 0.43 in DOX+ and DOX‐ iDek mice respectively.
In the esophagus, the incidence of macroscopic tumors was 15.4% (2/13) in DOX+ iDek mice; in contrast, no tumors were observed in DOX‐ iDek mice (0/8; Fig. S4A). There were no significant differences between DOX+ and DOX‐ mice in both the incidence and multiplicity of macroscopic tumors of the tongue. Microscopically, hyperplasia, dysplasia/CIS, and SCC were developed in the tongue and esophagus of both groups of mice (Fig. 3D and Fig. S4B). In the tongue, the multiplicity of dysplasia/CIS in DOX+ iDek mice was significantly higher than that in DOX‐ iDek mice (Fig. 3E). The incidence of hyperplasia in the tongues of DOX+ iDek mice was significantly higher than that in DOX‐ iDek mice. The incidence of dysplasia/CIS in the tongues of DOX+ iDek mice was obviously higher than that in DOX‐ iDek mice; however, this difference was not significant. DEK expression in dysplasia/CIS and invasive SCC were increased compared with normal and papilloma in 4NQO‐oral carcinogenesis like humans (Fig. S5 and Fig. 1). These data suggested that DEK overexpression was associated with the development of malignant lesions in an OSCC mouse model in the context of environmental carcinogen exposure.
DEK upregulated both PCNA and elongator complex protein 3 (ELP3) during OSCC progression
DEK promotes cancer cell proliferation, thereby enhancing cancer growth 24. Thus, to determine the proliferation status of tumor cells in each group of mice, we performed Ki67 immunostaining in tongue lesions (Fig. S6A). There were no significant differences in Ki67 staining of tongue tissues between DOX+ iDek and DOX‐ iDek mice for tissues representing normal epithelium, dysplasia, and invasive SCC. However, the number of Ki67‐positive cells was significantly increased during tumorigenesis in each group (Fig. S6B). We analyzed the p53 positive cells in oral carcinogenesis by IHC, however, there were not significant differences during the carcinogenesis (Fig. S7A and B). These data indicated that DEK overexpression was unlikely to induce Ki67‐proliferating cancer cells, which are known to be present during all phases of the active cell cycle (G 1, S, G 2, and M), in the progression of OSCC.
DEK is thought to act as an antagonist of senescence, and overexpression of DEK extends the life span of primary human keratinocytes 37. To clarify whether DEK overexpression contributed to the progression of malignant lesions via cellular senescence in our model, we investigated mRNA expression profiles using an RT2 Profiler PCR Array (Mouse Aging set) in tongue tumors from DOX+ iDek and DOX‐ iDek mice (n = 3 each). Seven (C1qb, C1qc, Elp3, Tfam, Tfb2 m, Angle2, and Vwa5a) of 84 aging‐related genes in the set were significantly upregulated in tumors from DOX+ iDek mice compared with those from DOX‐ iDek mice (Fig. 4A and Table S2).
Figure 4.
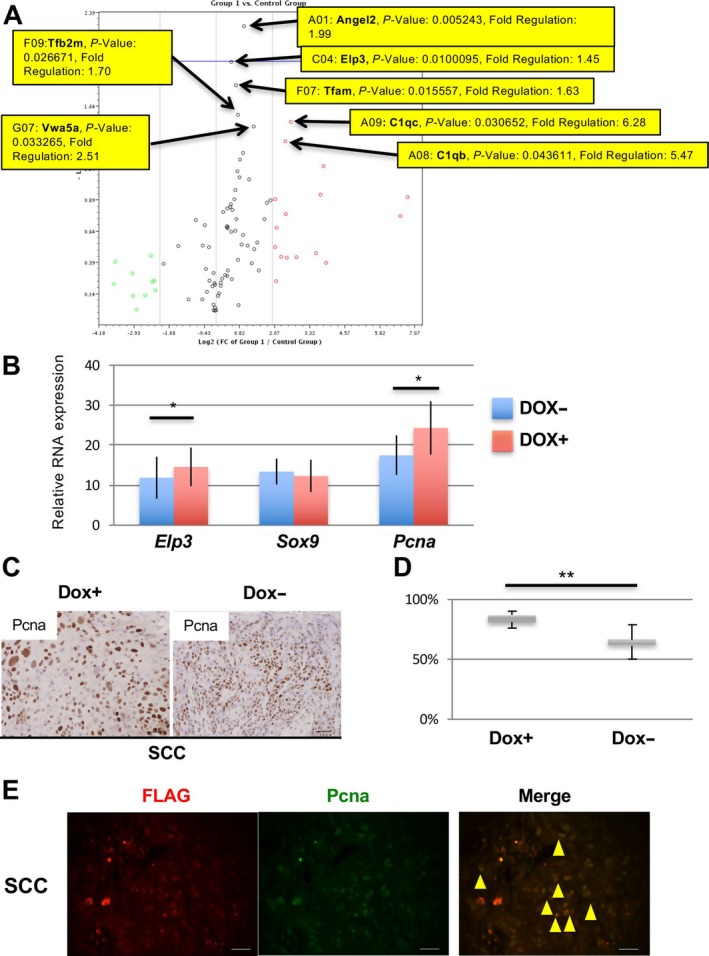
Upregulation of PCNA and ELP3 in tumor cells from DOX+ iDek mice treated with 4NQO. (A) Volcano plot of genes analysed using an RT2 Profiler PCR Array with a Mouse Aging set. Arrows indicate the seven genes (P < 0.05) with P values and fold changes. (B) Relative mRNA expression of Elp3, Sox9, and Pcna in tongue tumors of DOX‐ and DOX+ iDek mice (n = 5 each). Data are presented as the means ± SDs. * P < 0.05. (C) Representative images of IHC analysis of PCNA expression in tongue tumors from DOX‐ and DOX+ iDek mouse. Scale bars, 40 μm. (D) The PCNA‐positive index in tongue tumors. Data are means ± SDs. * P < 0.05, ** P < 0.01. (E) Double immunofluorescent staining with anti‐FLAG (red) and anti‐PCNA (green) antibodies in tongue tumors from DOX+ iDek mice treated with 4NQO. Scale bars, 20 μm. Yellow arrowheads indicate representative double‐positive cells.
ELP3 is induced by Wnt signaling and promotes SOX9 translation, which is necessary for maintenance of intestinal cancer stem cells 38, and SOX9 is upregulated in stem cell population in tongue SCC cells 39. Furthermore, ELP3 maintains genomic stability and regulates DNA replication and DNA repair by directly interacting with PCNA, which may also recruit the elongator complex to modulate chromatin structure during DNA replication or in response to DNA damage 40, 41. To confirm these findings in our model, we analysed the expression of Sox9, Pcna, and Elp3 mRNAs in DOX+ iDek and DOX‐ iDek mice (n = 5 each) by real‐time RT‐RCR. Pcna and Elp3 mRNA levels were significantly upregulated in tongue tumors from DOX+ iDek mice compared with those from DOX‐ iDek mice (Fig. 4B). However, PCNA, Elp3, and Sox9 protein levels were not significant differences (Fig. S8). To determine whether PCNA was functionally upregulated by DEK overexpression, we performed IHC for PCNA (Fig. 4C). The PCNA‐positive cells in tongue tumors of DOX+ iDek mice were significantly higher than those of DOX+ iDek mice (Fig. 4D). Furthermore, to clarify whether exogenous DEK‐FLAG protein induced PCNA overexpression in cancer cells, we performed double immunofluorescent staining for the FLAG tag and PCNA in tongue tumors of DOX+ iDek mice (Fig. 4E). Most FLAG and PCNA expression was detected in cancer cells, with obvious colocalization, in cancer cells in the tongues of DOX+ iDek mice treated with 4NQO, indicating that DEK may interact with PCNA in cancer cells during tumor progression.
DEK‐dependent enhancement of field cancerization was mediated by cell cycle‐related genes, particularly during the G 1/S transition
PCNA‐positive cells increased during carcinogenesis. Thus, to investigate whether the occurrence of oral field cancerization was associated with carcinogen exposure in the context of DEK overexpression, we assessed mRNA expression profiles in the precancerous epithelium, that is, microscopically normal tissues, of the tongue in 4NQO‐treated mice with or without DOX treatment (Fig. 5A). After 10 weeks, we obtained tongue epithelial tissues showing a histologically normal appearance by HE staining (Fig. 5B) from each cohort and confirmed the absence of tumors. mRNA expression profiles were then obtained using microarray analysis. A total of 136 genes were identified as differentially upregulated (Fig. 5C). Gene ontology (GO) analysis revealed upregulation of terms associated with the cell cycle and DNA replication (seven and nine genes respectively). The seven genes belonging to the cell cycle category (i.e., Cdc6, Mcm2–6, and E2f1) also belonged to the cell cycle pathway in KEGG pathway analysis. Notably, for significantly downregulated genes, no significantly enriched pathways were identified. We confirmed the relative RNA expression of the upregulated genes by real‐time RT‐PCR (Fig. 5D). Interestingly, in this pathway, Mcm7, cyclin E, and Pcna showed a fold‐change in greater than 1.5, with a p value of less than 0.01 (Fig. 5E). Consequently, DEK‐induced cell cycle‐associated and upregulated genes were mainly related to the G 1/S phase in the cell cycle pathway. These results indicated that DEK overexpression may be a risk factor promoting the precancerous environment, that is, field cancerization, in the context of carcinogen exposure by accelerating the cell cycle, particularly the G 1/S transition, despite the apparent normal appearance of the microenvironment.
Figure 5.
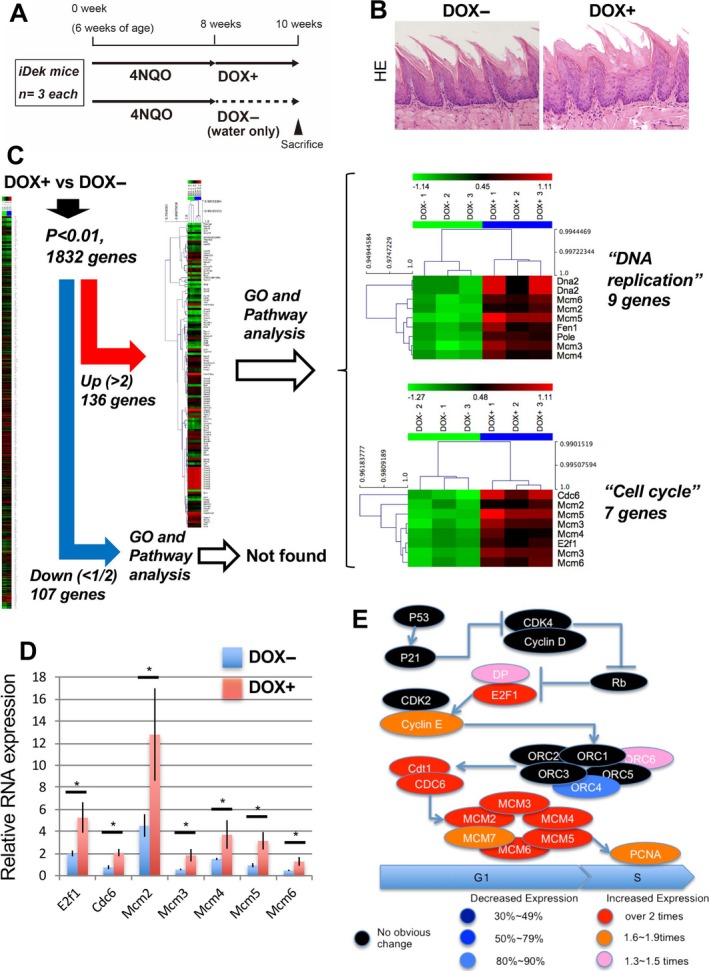
DEK enhanced field cancerization via upregulation of G 1 check point‐related genes. (A) Experimental protocol for the short‐term experiment. (B) Representative images of HE staining of tongue tissues from 4NQO‐treated iDek mice followed treatment with or without DOX (DOX+ and DOX‐ respectively). Scale bars, 40 μm. (C) Heat map representation of microarray analysis with GO and pathway analysis for DOX+ and DOX‐ iDek mice. Hierarchical clustering was performed on log2 signal intensity data. The values were resized to distance from the median for single genes (green, low expression to red, high expression). (D) RNA expression was measured by real‐time RT‐PCR for cell cycle‐associated genes. Data are means ± SDs. * P < 0.05. (E) The modified cell cycle pathway based on the KEGG cell cycle pathway. G 1 and S indicate phase of the cell cycle.
Next, to determine whether MCM2 and PCNA protein levels increased in the normal‐appearing epithelium under carcinogen exposure, we performed IHC for MCM2 and PCNA in the tongue epithelium of DOX+ and DOX‐ mice following 4NQO treatment (using the same epithelial tissues used for the microarray analysis in the short‐term experiment; Fig. 5A). MCM2‐positive cells were localized to the basal layer of the epithelium in each cohort (Fig. 6A); however, the MCM2‐positive cells in DOX+ iDek mice were spread to the upper layer of the epithelium (Fig. 6A), and the MCM2‐positive index in DOX+ iDek mice was significantly higher than that in DOX‐ iDek mice (Fig. 6B). In PCNA staining, similar results were obtained (Fig. 6C and D). Furthermore, to clarify whether the exogenous DEK‐FLAG protein induced PCNA overexpression in squamous cells exposed to carcinogen, we performed double immunofluorescent staining for the FLAG tag and PCNA in the normal‐appearing epithelium from DOX+ iDek mice (Fig. 6E). Most FLAG and PCNA expression was detected in the basal layer in the tongue epithelium, with obvious colocalization, in DOX+ iDek mice treated with 4NQO, indicating that DEK may interact with PCNA in the normal‐appearing epithelium following exposure to a carcinogen.
Figure 6.
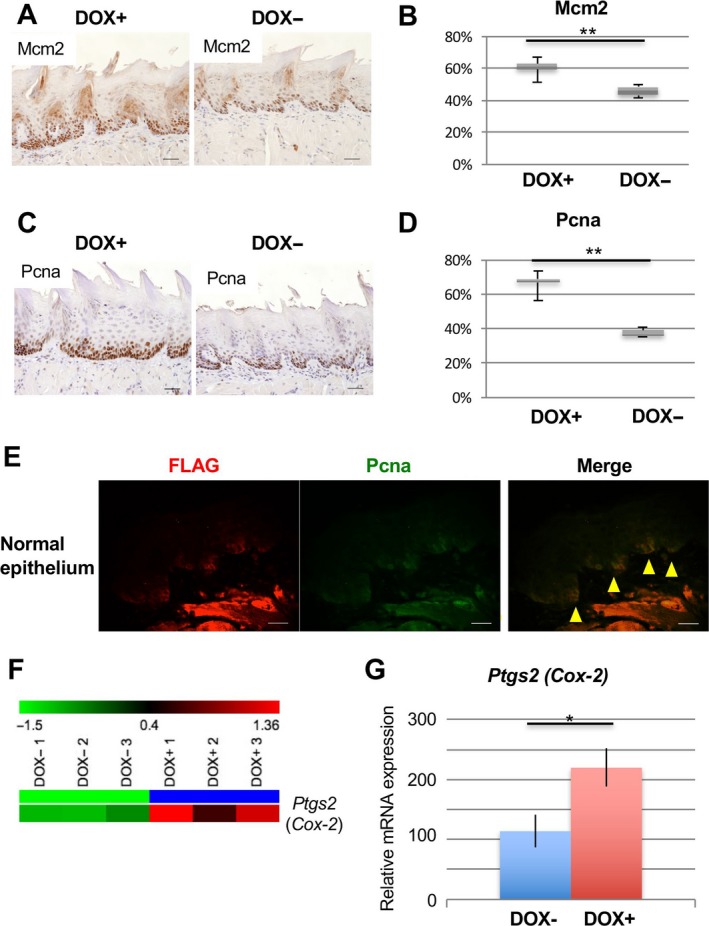
Induced DEK expression enhanced MCM2 and PCNA protein expression in normal‐appearing tongue epithelium exposed to 4NQO. (A) Representative images of IHC analysis for detection of MCM2 in tongue tissues from 4NQO‐treated mice with or without DOX treatment (DOX+ and DOX‐ respectively). Scale bars, 40 μm. (B) The positive index of MCM2 in normal tongue epithelium from mice. Data are means ± SDs. ** P < 0.01. (C) Representative images of IHC analysis for detection of PCNA in the tongues of 4NQO‐treated mice with or without DOX (DOX+ and DOX‐ respectively). Scale bars, 40 μm. (D) The positive index of PCNA in normal tongue epithelium from mice. Data are means ± SDs. ** P < 0.01. (E) Double immunofluorescent staining for FLAG (red) and PCNA (green) in normal tongue epithelium from DOX+ iDek mice. Yellow arrows indicate double‐positive epithelial cells. Scale bars, 20 μm. (F) Heat map representations of microarray analysis of Ptgs2 (Cox‐2) in DOX+ and DOX‐ iDek mice treated with 4NQO. These values were resized to the distance from the median for single genes (green, low expression to red, high expression). (G) RNA expression was measured by real‐time RT‐PCR for Ptgs2 (Cox‐2). Data are means ± SDs. * P < 0.05.
Among genes found to be significantly upregulated in the microarray analysis, we found that Ptgs2 (Cox‐2) was upregulated in the tongues of DOX+ iDek mice treated with 4NQO compared with that in DOX‐ iDek mice treated with 4NQO in the short‐term experiment (Figs. 5A and 6F). We further confirmed Cox‐2 upregulation by real‐time RT‐PCR (Fig. 6G). These data suggested that Cox‐2 upregulation was also associated with the promotion of field cancerization mediated by DEK overexpression. Cox‐2, encoding cyclooxygenase 2, is known to be strongly associated with carcinogenesis in many types of cancers, including OSCC. A previous report showed that COX‐2 protein is upregulated in the normal appearing oral mucosa of tobacco smokers 42, consistent with our findings. Thus, Cox‐2 may be related to promotion of DEK‐dependent field cancerization in OSCC (Fig. 7).
Figure 7.
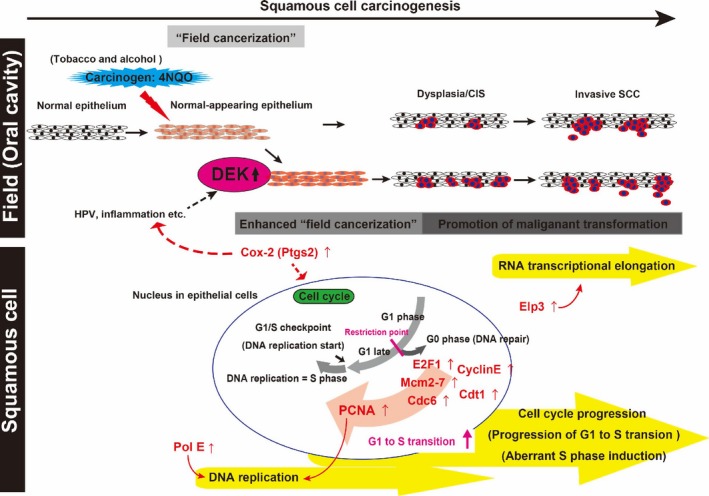
Schematic illustration of the proposed role of DEK in carcinogenesis of OSCC. OSCC progresses through a multistep process from normal mucosa to normal‐appearing mucosa (exposed to carcinogens, tobacco, or alcohol), hyperplasia, dysplasia, CIS, and invasive SCC. When DEK is upregulated and overexpressed by HPV, inflammation, or other insults, field cancerization and cancer development are promoted. Aberrant activation of the cell cycle, DNA replication, and RNA transcriptional elongation is involved in this oncogenic mechanism caused by DEK overexpression in OSCC. Upregulation of cell cycle progression‐, DNA replication‐, and RNA transcriptional elongation‐related genes (red) may contribute to the genomic, epigenetic, and chromosomal instabilities involved in carcinogenesis.
Discussion
In this study, we demonstrated that DEK was increased in human OSCC. Moreover, in iDek (DOX‐inducible Dek) mice exposed to a carcinogen, DEK overexpression stimulated crucial oncogenic processes, such as DNA replication and cell cycle progression, in precancerous lesions, thereby promoting malignant transformation of tongue tumors.
DEK overexpression has been reported to be associated with human SCC, including the uterine cervix, head and neck, and lung 43, 44, 45, 46, 47. These studies have shown that expansion of DEK‐expressing cells occurs during the early phases of SCC development, e.g., in dysplasia and CIS. Our human data indicated that DEK‐expressing cells expanded during the early phases of SCC in the oral cavity.
We generated ubiquitous and squamous cell‐specific DOX‐inducible Dek mice. Ubiquitous DEK overexpression led to aberrant cell proliferation owing to increased DNA replication, cell cycle progression, and expression of transcriptional elongation‐related genes in epithelial tissues exposed to 4NQO, although no changes were observed in normal epithelium in either strain following DOX alone, even after 1 year. These results may be explained by the localization of cellular DEK. Indeed, DEK predominately localizes in the nucleus under steady‐state conditions; however, induction of DNA damage, apoptosis, and pro‐inflammatory factors can cause translocation of DEK to the extracellular space following release from inflammatory cells as a chemotactic agent 48, 49. In our study, enhancement of field cancerization by DEK overexpression may be related to the intracellular localization of DEK because there were no visible differences, e.g., inflammation, in mucosal appearance between DOX+ and DOX‐ mice following 4NQO. However, after 4NQO exposure, the number of tumors in DOX+ iDek mice increased compared with that in DOX‐ iDek mice. These findings could be related to the function of extracellular DEK released by inflammatory cells because many inflammatory cells are located in and around tumor tissues. Unfortunately, we were not able to distinguish intracellular and extracellular DEK in the tumor environment. It is necessary to consider the potential role of extracellular DEK in the tumor microenvironment, immunity, and/or autoimmunity and the function of both intracellular and extracellular DEK will be needed to be studied further in order to develop targeted therapies.
We have realized that the function of DEK is highly complex. Several studies have shown that DEK has multiple functions in DNA replication, transcriptional regulation, mRNA processing, and DNA architecture modulation 50, 51. Similarly, we found that DEK overexpression upregulated nine cell cycle‐related genes, particularly G 1/S checkpoint‐related genes, and eight DNA replication‐related genes in the context of carcinogen exposure. The majority of these genes are involved in the replication‐ and transcription‐related functions of DEK. Furthermore, we demonstrated that ELP3, the catalytic subunit of the histone acetyltransferase elongator complex, was upregulated in tongue tumors of DOX+ iDek mice. ELP3, an elongator histone acetyltransferase, is required for transcriptional silencing and maintenance of genome stability and has been shown to bind directly to PCNA 41. Furthermore, the elongator complex is transported with PCNA during DNA replication to modulate chromatin structure and/or proteins involved in DNA replication to ensure that DNA synthesis is coupled to nucleosome assembly during the S phase of the cell cycle. Consequently, our data suggested that DEK promoted OSCC progression by mediating DNA replication and transcriptional elongation. Moreover, we showed that DEK could accelerate DNA replication and transcription (including RNA transcriptional elongation) in OSCC.
There were no significant differences in Ki67 staining between DOX‐ and DOX+ iDek mice in OSCC tissues induced by 4NQO (Fig. S5). However, PCNA‐ and MCM2‐positive cells were more abundant in the tongues of DOX+ iDek mice than in those of DOX‐ iDek mice. These findings indicated early entry of squamous cells into the G 1 phase of the cell cycle. MCMs and PCNA are expressed throughout G 1 phase, while Ki67 may not be expressed until late G 1 phase 52. Furthermore, PCNA is a less useful marker for cell cycle entry than MCMs 53. PCNA shows maximum expression during the S phase, with weak staining observed in G 1, G 2, and M phases 54. PCNA is involved in the excision and replacement of abnormal nucleotides and is thus also expressed in nonproliferating cells undergoing DNA repair. This result was different from Ki67‐proliferative cells, which are all proliferating cells. Thus, PCNA‐positive proliferating cells, at least and in part, may not induce hyperplastic change in the epithelium related to Dek overexpression. Overall, our data clearly demonstrated that disruption and acceleration of the G 1/S phase transition, resulting in promotion of carcinogenesis 54, occurred in OSCC. Although the functions of DEK have not been fully elucidated, DEK may have potential applications in therapeutic approaches for the prevention and treatment of OSCC.
Conflict of Interest
The authors have no conflict of interest to disclose.
Supporting information
Figure S1. Dek‐FLAG (exogenous) expression induced by DOX increased in three DOX‐inducible Dek‐FLAG KH2 ES clones.
Figure S2. No difference between DOX+ and DOX‐ iDek mice in esophagus.
Figure S3. No difference between DOX+ and DOX‐ iDek mice in skin and thymus after treatment with DOX(1 mg/L) for 2 weeks.
Figure S4. The high frequency of esophagus tumors in DOX+ and DOX‐ iDek mice.
Figure S5. DEK expression increased in the 4NQO‐ tongue carcinogenesis in both DOX+ and DOX‐iDek mice.
Figure S6. Ki67 proliferation marker increased in the tongue carcinogenesis in both DOX+ and DOX‐ iDek mice, but showed no significant difference between DOX+ and DOX‐ iDek mice.
Figure S7. No difference between p53 positive cells in tongue lesions of DOX+ and DOX‐ iDek mice.
Figure S8. Western blot analyses for Sox9, PCNA, and Elp3 of tumor between overexpression of Dek or not.
Table S1. PCR array data in the tongue tumors between DOX+ and DOX‐ iDek mice.
Table S2. Primers used for quantitative real‐time RT‐PCR.
Data S1. Details of experimental methods in this study.
Acknowledgments
We thank K. Takahashi, A. Suga, M. Shimizu, and R. Kitazumi for assistance with the experiments. We also thank NPO Biotechnology Research and Development for technical assistance. This work was supported by grants from the Ministry of Education, Culture, Sports, Science, and Technology of Japan: grant nos. 15K11289, 26430111, and 26305035 (A. H., H. T., and T. S. respectively).
Cancer Medicine 2017; 6(10):2424–2439
This work was supported by grants from the Ministry of Education, Culture, Sports, Science, and Technology of Japan: grant nos. 15K11289, 26430111, and 26305035 (A. H., H. T., and T. S., respectively).
References
- 1. Pai, S. I. , and Westra W. H.. 2009. Molecular pathology of head and neck cancer: implications for diagnosis, prognosis, and treatment. Annu. Rev. Pathol. 4:49–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Jemal, A. , Siegel R., Ward E., Hao Y., Xu J., and Thun M. J.. 2009. Cancer statistics, 2009. CA Cancer J. Clin. 59:225–249. [DOI] [PubMed] [Google Scholar]
- 3. Tanaka, T. , and Ishigamori R.. 2011. Understanding carcinogenesis for fighting oral cancer. J. Oncol. 2011:603740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Choi, S. , and Myers J. N.. 2008. Molecular pathogenesis of oral squamous cell carcinoma: implications for therapy. J. Dent. Res. 87:14–32. [DOI] [PubMed] [Google Scholar]
- 5. Gasche, J. A. , and Goel A.. 2012. Epigenetic mechanisms in oral carcinogenesis. Future Oncol. 8:1407–1425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Gillison, M. L. , Koch W. M., Capone R. B., Spafford M., Westra W. H., Wu L, et al. 2000. Evidence for a causal association between human papillomavirus and a subset of head and neck cancers. J. Natl Cancer Inst. 92:709–720. [DOI] [PubMed] [Google Scholar]
- 7. Blot, W. J. , McLaughlin J. K., Winn D. M., Austin D. F., Greenberg R. S., Preston‐Martin S., et al. 1988. Smoking and drinking in relation to oral and pharyngeal cancer. Cancer Res. 48:3282–3287. [PubMed] [Google Scholar]
- 8. Slaughter, D. P. , Southwick H. W., and Smejkal W.. 1953. Field cancerization in oral stratified squamous epithelium; clinical implications of multicentric origin. Cancer 6:963–968. [DOI] [PubMed] [Google Scholar]
- 9. Braakhuis, B. J. , Tabor M. P., Kummer J. A., Leemans C. R., and Brakenhoff R. H.. 2003. A genetic explanation of Slaughter's concept of field cancerization: evidence and clinical implications. Cancer Res. 63:1727–1730. [PubMed] [Google Scholar]
- 10. Shin, D. M. , Voravud N., Ro J. Y., Lee J. S., Hong W. K., and Hittelman W. N.. 1993. Sequential increases in proliferating cell nuclear antigen expression in head and neck tumorigenesis: a potential biomarker. J. Natl Cancer Inst. 85:971–978. [DOI] [PubMed] [Google Scholar]
- 11. van Oijen, M. G. , Gilsing M. M., Rijksen G., Hordijk G. J., and Slootweg P. J.. 1998. Increased number of proliferating cells in oral epithelium from smokers and ex‐smokers. Oral Oncol. 34:297–303. [DOI] [PubMed] [Google Scholar]
- 12. Zakaria, S. H. , Farag H. A., and Khater D. S.. 2016. Immunohistochemical expression of MCM‐2 in oral epithelial dysplasias. Appl. Immunohistochem. Mol. Morphol. [DOI] [PubMed] [Google Scholar]
- 13. Valverde, L. F. , de Freitas R. D., Pereira T. A., de Resende M. F., Agra I. M., Dos Santos J. N., et al. 2016. MCM3: a novel proliferation marker in oral squamous cell carcinoma. Appl. Immunohistochem. Mol. Morphol. [DOI] [PubMed] [Google Scholar]
- 14. Ohnishi, Y. , Fujii T., Ugaki Y., et al. 2016. Usefulness of a fluorescence visualization system for the detection of oral precancerous and early cancerous lesions. Oncol. Rep. 36:514–520. [DOI] [PubMed] [Google Scholar]
- 15. Tamura, T. , Shomori K., Haruki T., Nosaka K., Hamamoto Y., Shiomi T., et al. 2010. Minichromosome maintenance‐7 and geminin are reliable prognostic markers in patients with oral squamous cell carcinoma: immunohistochemical study. J. Oral Pathol. Med. 39:328–334. [DOI] [PubMed] [Google Scholar]
- 16. Li, J. N. , Feng C. J., Lu Y. J., Li H. J., Tu Z., Liao G. Q., et al. 2008. mRNA expression of the DNA replication‐initiation proteins in epithelial dysplasia and squamous cell carcinoma of the tongue. BMC Cancer 8:395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Feng, C. J. , Li H. J., Li J. N., Lu Y. J., and Liao G. Q.. 2008. Expression of Mcm7 and Cdc6 in oral squamous cell carcinoma and precancerous lesions. Anticancer Res. 28:3763–3769. [PubMed] [Google Scholar]
- 18. Brand, N. , Faul T., and Grummt F.. 2007. Interactions and subcellular distribution of DNA replication initiation proteins in eukaryotic cells. Mol. Genet. Genomics 278:623–632. [DOI] [PubMed] [Google Scholar]
- 19. Bell, S. P. , and Dutta A.. 2002. DNA replication in eukaryotic cells. Annu. Rev. Biochem. 71:333–374. [DOI] [PubMed] [Google Scholar]
- 20. von Lindern, M. , Breems D., van Baal S., Adriaansen H., and Grosveld G., 1992. The translocation (6;9), associated with a specific subtype of acute myeloid leukemia, results in the fusion of two genes, dek and can, and the expression of a chimeric, leukemia‐specific dek‐can mRNA. Mol. Cell. Biol. 12:1687–1697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. von Lindern, M. , Breems D., van Baal S., Adriaansen H., and Grosveld G.. 1992. Characterization of the translocation breakpoint sequences of two DEK‐CAN fusion genes present in t(6;9) acute myeloid leukemia and a SET‐CAN fusion gene found in a case of acute undifferentiated leukemia. Genes Chromosom. Cancer 5:227–234. [DOI] [PubMed] [Google Scholar]
- 22. Lin, L. , Piao J., Gao W., Piao Y., Jin G., Ma Y., et al. 2013. DEK over expression as an independent biomarker for poor prognosis in colorectal cancer. BMC Cancer 13:366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Piao, J. , Shang Y., Liu S., Piao Y., Cui X., Li Y., et al. 2014. High expression of DEK predicts poor prognosis of gastric adenocarcinoma. Diagn. Pathol. 9:67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Privette Vinnedge, L. M. , Ho S. M., Wikenheiser‐Brokamp K. A., and Wells S. I.. 2012. The DEK oncogene is a target of steroid hormone receptor signaling in breast cancer. PLoS ONE 7:e46985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Carro, M. S. , Spiga F. M., Quarto M., Di Ninni V., Volorio S., Alcalay M., et al. 2006. DEK expression is controlled by E2F and deregulated in diverse tumor types. Cell Cycle 5:1202–1207. [DOI] [PubMed] [Google Scholar]
- 26. Sitwala, K. V. , Adams K., and Markovitz D. M.. 2002. YY1 and NF‐Y binding sites regulate the transcriptional activity of the Dek and Dek‐can promoter. Oncogene 21:8862–8870. [DOI] [PubMed] [Google Scholar]
- 27. Vernell, R. , Helin K., and Muller H.. 2003. Identification of target genes of the p16INK4A‐pRB‐E2F pathway. J. Biol. Chem. 278:46124–46137. [DOI] [PubMed] [Google Scholar]
- 28. Muller, H. , Bracken A. P., Vernell R., Moroni M. C., Christians F., Grassilli E., et al. 2001. E2Fs regulate the expression of genes involved in differentiation, development, proliferation, and apoptosis. Genes Dev. 15:267–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Sherr, C. J. 1996. Cancer cell cycles. Science 274:1672–1677. [DOI] [PubMed] [Google Scholar]
- 30. Privette Vinnedge, L. M. , Kappes F., Nassar N., and Wells S. I.. 2013. Stacking the DEK: from chromatin topology to cancer stem cells. Cell Cycle 12:51–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Wise‐Draper, T. M. , Mintz‐Cole R. A., Morris T. A., Simpson D. S., Wikenheiser‐Brokamp K. A., Currier M. A., et al. 2009. Overexpression of the cellular DEK protein promotes epithelial transformation in vitro and in vivo. Cancer Res. 69:1792–1799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Privette Vinnedge, L. M. , Benight N. M., Wagh P. K., Pease N. A., Nashu M. A., Serrano‐Lopez J., et al. 2015. The DEK oncogene promotes cellular proliferation through paracrine Wnt signaling in Ron receptor‐positive breast cancers. Oncogene 34:2325–2336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Nagpal, J. K. , and Das B. R.. 2007. Identification of differentially expressed genes in tobacco chewing‐mediated oral cancer by differential display‐polymerase chain reaction. Eur. J. Clin. Invest. 37:658–664. [DOI] [PubMed] [Google Scholar]
- 34. Beard, C. , Hochedlinger K., Plath K., Wutz A., and Jaenisch R.. 2006. Efficient method to generate single‐copy transgenic mice by site‐specific integration in embryonic stem cells. Genesis 44:23–28. [DOI] [PubMed] [Google Scholar]
- 35. Hochedlinger, K. , Yamada Y., Beard C., and Jaenisch R.. 2005. Ectopic expression of Oct‐4 blocks progenitor‐cell differentiation and causes dysplasia in epithelial tissues. Cell 121:465–477. [DOI] [PubMed] [Google Scholar]
- 36. Tang, X. H. , Knudsen B., Bemis D., Tickoo S., and Gudas L. J.. 2004. Oral cavity and esophageal carcinogenesis modeled in carcinogen‐treated mice. Clin. Cancer Res. 10:301–313. [DOI] [PubMed] [Google Scholar]
- 37. Wise‐Draper, T. M. , Allen H. V., Thobe M. N., Jones E. E., Habash K. B., Munger K., et al. 2005. The human DEK proto‐oncogene is a senescence inhibitor and an upregulated target of high‐risk human papillomavirus E7. J. Virol. 79:14309–14317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Ladang, A. , Rapino F., Heukamp L. C., Tharun L., Shostak K., Hermand D., et al. 2015. Elp3 drives Wnt‐dependent tumor initiation and regeneration in the intestine. J. Exp. Med. 212:2057–2075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Misuno, K. , Liu X., Feng S., and Hu S.. 2013. Quantitative proteomic analysis of sphere‐forming stem‐like oral cancer cells. Stem Cell Res. Ther. 4:156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Chen, C. , Huang B., Eliasson M., Ryden P., and Bystrom A. S.. 2011. Elongator complex influences telomeric gene silencing and DNA damage response by its role in wobble uridine tRNA modification. PLoS Genet. 7:e1002258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Li, Q. , Fazly A. M., Zhou H., Huang S., Zhang Z., and Stillman B.. 2009. The elongator complex interacts with PCNA and modulates transcriptional silencing and sensitivity to DNA damage agents. PLoS Genet. 5:e1000684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Moraitis, D. , Du B., De Lorenzo M. S., Boyle J. O., Weksler B. B., Cohen E. G., et al. 2005. Levels of cyclooxygenase‐2 are increased in the oral mucosa of smokers: evidence for the role of epidermal growth factor receptor and its ligands. Cancer Res. 65:664–670. [PubMed] [Google Scholar]
- 43. Adams, A. K. , Hallenbeck G. E., Casper K. A., Patil Y. J., Wilson K. M., Kimple R. J., et al. 2015. DEK promotes HPV‐positive and ‐negative head and neck cancer cell proliferation. Oncogene 34:868–877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Wise‐Draper, T. M. , Morreale R. J., Morris T. A., Mintz‐Cole R. A., Hoskins E. E., Balsitis S. J., et al. 2012. Future directions and treatment strategies for head and neck squamous cell carcinomas. Transl. Res. 160:167–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Wise‐Draper, T. M. , Morreale R. J., Morris T. A., et al. 2009. DEK proto‐oncogene expression interferes with the normal epithelial differentiation program. Am. J. Pathol. 174:71–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Rampias, T. , Sasaki C., Weinberger P., and Psyrri A.. 2009. E6 and e7 gene silencing and transformed phenotype of human papillomavirus 16‐positive oropharyngeal cancer cells. J. Natl Cancer Inst. 101:412–423. [DOI] [PubMed] [Google Scholar]
- 47. Wu, Q. , Li Z., Lin H., Han L., Liu S., and Lin Z.. 2008. DEK overexpression in uterine cervical cancers. Pathol. Int. 58:378–382. [DOI] [PubMed] [Google Scholar]
- 48. Tabbert, A. , Kappes F., Knippers R., Kellermann J., Lottspeich F., and Ferrando‐May E.. 2006. Hypophosphorylation of the architectural chromatin protein DEK in death‐receptor‐induced apoptosis revealed by the isotope coded protein label proteomic platform. Proteomics 6:5758–5772. [DOI] [PubMed] [Google Scholar]
- 49. Mor‐Vaknin, N. , Punturieri A., Sitwala K., Faulkner N., Legendre M., Khodadoust M. S., et al. 2006. The DEK nuclear autoantigen is a secreted chemotactic factor. Mol. Cell. Biol. 26:9484–9496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Ko, S. I. , Lee I. S., Kim J. Y., Kim S. M., Kim D. W., Lee K. S., et al. 2006. Regulation of histone acetyltransferase activity of p300 and PCAF by proto‐oncogene protein DEK. FEBS Lett. 580:3217–3222. [DOI] [PubMed] [Google Scholar]
- 51. Alexiadis, V. , Waldmann T., Andersen J., Mann M., Knippers R., and Gruss C.. 2000. The protein encoded by the proto‐oncogene DEK changes the topology of chromatin and reduces the efficiency of DNA replication in a chromatin‐specific manner. Genes Dev. 14:1308–1312. [PMC free article] [PubMed] [Google Scholar]
- 52. Scholzen, T. , and Gerdes J.. 2000. The Ki‐67 protein: from the known and the unknown. J. Cell. Physiol. 182:311–322. [DOI] [PubMed] [Google Scholar]
- 53. Gonzalez, M. A. , Tachibana K. E., Laskey R. A., and Coleman N.. 2005. Control of DNA replication and its potential clinical exploitation. Nat. Rev. Cancer 5:135–141. [DOI] [PubMed] [Google Scholar]
- 54. Celis, J. E. , and Celis A.. 1985. Cell cycle‐dependent variations in the distribution of the nuclear protein cyclin proliferating cell nuclear antigen in cultured cells: subdivision of S phase. Proc. Natl Acad. Sci. USA 82:3262–3266. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Dek‐FLAG (exogenous) expression induced by DOX increased in three DOX‐inducible Dek‐FLAG KH2 ES clones.
Figure S2. No difference between DOX+ and DOX‐ iDek mice in esophagus.
Figure S3. No difference between DOX+ and DOX‐ iDek mice in skin and thymus after treatment with DOX(1 mg/L) for 2 weeks.
Figure S4. The high frequency of esophagus tumors in DOX+ and DOX‐ iDek mice.
Figure S5. DEK expression increased in the 4NQO‐ tongue carcinogenesis in both DOX+ and DOX‐iDek mice.
Figure S6. Ki67 proliferation marker increased in the tongue carcinogenesis in both DOX+ and DOX‐ iDek mice, but showed no significant difference between DOX+ and DOX‐ iDek mice.
Figure S7. No difference between p53 positive cells in tongue lesions of DOX+ and DOX‐ iDek mice.
Figure S8. Western blot analyses for Sox9, PCNA, and Elp3 of tumor between overexpression of Dek or not.
Table S1. PCR array data in the tongue tumors between DOX+ and DOX‐ iDek mice.
Table S2. Primers used for quantitative real‐time RT‐PCR.
Data S1. Details of experimental methods in this study.