

Abstract
Activation of the Ras/MAPK pathway is prevalently involved in the occurrence and development of hepatocellular carcinoma (HCC). However, its effects on the deregulated cellular metabolic processes involved in HCC in vivo remain unknown. In this study, a mouse model of HCC induced by hepatocyte‐specific expression of the Hras12V oncogene was investigated using an integrative analysis of metabolomics and transcriptomics data. Consistent with the phenotype of abundant lipid droplets in HCC, the lipid biosynthesis in HCC was significantly enhanced by (1) a sufficient supply of acetyl‐CoA from enhanced glycolysis and citrate shuttle activity; (2) a sufficient supply of NADPH from enhanced pentose phosphate pathway (PPP) activity; (3) upregulation of key enzymes associated with lipid biosynthesis; and (4) downregulation of key enzymes associated with bile acid biosynthesis. In addition, glutathione (GSH) was significantly elevated, which may result from a sufficient supply of 5‐oxoproline and L‐glutamate as well as an enhanced reduction in the process of GSSG being turned into GSH by NADPH. The high level of GSH along with elevated Bcl2 and Ucp2 expression may contribute to a normal level of reactive oxygen species (ROS) in HCC. In conclusion, our results suggest that the lipid metabolism, glycolysis, PPP, tricarboxylic acid (TCA) cycle, citrate shuttle activity, bile acid synthesis, and redox homeostasis in the HCC induced by ras oncogene are significantly perturbed, and these altered metabolic processes may play crucial roles in the carcinogenesis, development, and pathological characteristics of HCC.
Keywords: Hepatocellular carcinoma, metabolomics, Ras oncogene, transcriptomics
Introduction
Hepatocellular carcinoma (HCC) is a heterogeneous cancer with no promising treatments. It remains one of the most prevalent and lethal malignancies in the world. Approximately 600,000 new cases are diagnosed each year, among which 55% of cases have been identified in China, and an increasing incidence has been observed in Western countries 1. The late diagnosis and consequently poor survival associated with the disease are often attributed to a lack of pathogenomic symptoms and the limitations of diagnostic modalities. Improving both the understanding of HCC etiology and biological processes and the early detection of the disease is an important first step toward the design of effective prevention strategies aimed at early diagnosis and the reduction in HCC.
Ras is a small signal‐transducing guanosine triphosphatase that plays a central role in the control of cell growth and differentiation 2. Activated mutations in ras have been found in all human tumors, and the frequency of ras mutations is the highest among the genes associated with human cancers 3. In the occurrence and development of human HCC, although the mutational activation of Ras protein occurs with an incidence of 5%, hyperactivation of the Ras/MAPK pathway is a frequent event 4. Consistently, murine HCCs also express H‐ras, which is activated in 70% of cases. This evidence supports the view that the Ras pathway plays crucial roles in hepatocarcinogenesis 5. Because animal models are useful tools for dissecting etiological factors and understanding the process of hepatocarcinogenesis, we established Hras12V transgenic (Ras‐Tg) mice lineages in which a hepatocyte‐specific expressed ras oncogene under the control of a mouse albumin enhancer/promoter could induce multicentric spontaneous hepatic tumorigenesis at the appropriate time and with a high level of reproducibility and male prevalence 5. The histopathological characteristics of this disease model are that a nontumor liver shows rare inflammation, no signs of fibrosis or cirrhosis, and significantly higher levels of apoptosis and cell cycle arrest. Altered foci (typically < 2 mm) include the aggregation of atypical hepatocytes with basophilic cytoplasm and hyperchromatic nuclei. Phenotypes of hepatic adenoma are similar to altered foci but with a larger size (typically 2–5 mm) and are relatively well circumscribed; HCCs are large in size (typically >5 mm), have a trabecular arrangement of tumor cells, and have highly anaplastic cells with evidence of necrosis 5. Specifically, these hepatic alterations are in dominant PCNA‐positive cells with abundant droplets but have rare apoptotic signs 5. Moreover, the hyperactivation of the MEK/ERK and PI3K/AKT/mTOR signaling pathways was a marked molecular event in HCC 6. Reports based on the Ras‐Tg mouse demonstrate that it is a valuable model to investigate molecular mechanisms, diagnosis and therapeutic strategies, and biomarkers of hepatocarcinogenesis 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18. For example, by exploring Ras‐Tg mice, we found that differences in the molecular responses to the deregulated Ras oncoprotein between females and males determine the onset of HCC, which may contribute to the striking male prevalence of HCC 5, 6. In addition, we and other researchers found that B lymphocytes, FoxM1, and miR‐221 play crucial roles in HCC by using Ras‐Tg mice 7, 9, 12.
Molecular profiling approaches, such as metabolomics and transcriptomics to monitor pathological processes of disease, have received a great deal of attention. Metabolomic strategies play an increasingly important role in clinical and observational studies, and they will offer new perspectives not only in understanding the processes of disease development but also in the identification of diagnostic/prognostic markers and targeted healthcare 19. Global gene expression studies using RNA‐Seq can provide insights into regulatory genes and critical pathways that might lead to HCC 20. The integrative analysis of metabolomics and transcriptomics data has the potential to greatly increase our understanding of metabolic networks and biological systems, leading to various potential clinical applications 21. Recently, several studies integrating metabolomics and transcriptomics to uncover the biological processes of tumors have been reported and have proven the valuable and powerful applications of this approach 22. However, an analysis of HCC using this approach has rarely been reported.
In this study, we characterized the metabolite profiles of HCC in Ras‐Tg mice by GC‐TOF‐MS analysis to identify the significantly changed metabolic pathways in HCC induced by the ras oncogene. In addition, metabolomic analysis was integrated with transcriptomic analysis of HCC by next‐generation sequencing (NGS) to obtain a better understanding of the metabolic profiles in HCC.
Materials and Methods
Experimental animals, sampling, and histopathological examination
The aim of this study is to investigate the metabolomic and transcriptomic differences between HCC and wild‐type liver tissues. Because Ras‐Tg males develop HCC at a relatively early stage (8–9 months of age) with a high incidence (almost 100%) compared to females (over 15 months of age and 30% incidence) 5, 6, males were chosen for this study. Experimental animals and histopathological examination procedures for animal handling and tissue sampling were conducted in compliance with protocols approved by the Animal Care and Use Committee of Dalian Medical University. Ras‐Tg mice (C57BL/6J genetic background), which are an animal model of hepatic tumors induced by an Hras12V oncogene specifically expressed in hepatocytes 5, and wild‐type mice (C57BL/6J) were bred and housed in the Laboratory Animal Center of Dalian Medical University. Nine‐month‐old male mice were selected for this study. Parts of the hepatic tumor (over 8 mm in diameter) or wild‐type liver tissues of sacrificed mice were removed, rinsed in cold saline, cut into ~ 1 cm3 tissue blocks, and immediately flash frozen in liquid nitrogen. The remaining tissue parts were fixed in 10% neutral buffered formalin, embedded in paraffin, sectioned, and stained with hematoxylin and eosin using standard methods. The histopathology was assessed, and the tissues with a confirmed morphological diagnosis were used for subsequent experimental procedures.
Sample preparation for GC‐MS analysis
The histopathologically identified HCC from 9‐month‐old Ras‐Tg males and liver tissues from 9‐month‐old wild‐type C57BL/6J males (eight individual samples for each group, for a total of over 32 slices with at least 2 per individual, identified by a licensed pathologist) were subjected to two derivatization steps for GC‐MS analysis. Approximately 0.05 mg of each sample was collected in 2‐mL EP tubes and extracted with 0.4 mL methanol–chloroform (V methanol:V chloroform = 3:1). Then, 20 μL of L‐2‐chlorophenylalanine (1 mg/mL stock in dH2O) was added as an internal standard, and the sample was homogenized in a ball mill for 3 min at 65 Hz. After the samples were centrifuged, the supernatant was transferred to a 2‐mL GC‐MS glass vial. An equal volume of 14 μL was taken from each sample and placed into the 2‐mL GC‐MS glass vial as a mixed sample for quality control. The extracts were dried in a vacuum concentrator without heating at 37°C for approximately 2 h. Then, 80 μL of methoxylamine hydrochloride (20 mg/mL) was added to the dried metabolites and incubated at 80°C for 20 min in an oven after mixing and sealing. The lid was opened, and 100 μL of Bis(trimethylsilyl) trifluoroacetamide (BSTFA) (containing 1% trimethylchlorosilane (TMCS), v/v) was added to each sample, which was sealed again and incubated at 70°C for an hour. Then, 5 μL of FAMEs (a standard mixture of fatty acid methyl esters, C8‐C16: 1 mg/mL; C18‐C24: 0.5 mg/mL in chloroform) was added to the mixed sample, cooled to room temperature, and mixed well for GC‐MS analysis.
GC‐TOF‐MS analysis
GC‐TOF‐MS analysis was performed using an Agilent 7890 gas chromatograph system coupled with a Pegasus HT time‐of‐flight mass spectrometer. The system utilized an Rxi‐5Sil MS column (30 m × 250 μm inner diameter, 0.25 μm film thickness; Restek, USA). A 1‐μL aliquot of the analyte was injected in splitless mode. Helium was used as the carrier gas, the front inlet purge flow was 3 mL/min, and the gas flow rate through the column was 20 mL/min. The initial temperature was 50°C for 1 min, then it was increased to 330°C at a rate of 10°C/min, and then maintained for 5 min at 330°C. The injection, transfer line, and ion source temperatures were 280, 280, and 250°C, respectively. The energy was −70 eV in electron impact mode. The mass spectrometry data were acquired in full‐scan mode with an m/z range 30–600 at a rate of 20 spectra per second after a solvent delay of 366 sec.
Analysis of GC‐MS data
First, the chroma TOF4.3X software of the LECO Corporation and LECO‐Fiehn Rtx5 database were used for the extraction of raw peaks, the filtering of data baselines, and the calibration of the baseline, peak alignment, deconvolution analysis, peak identification, and integration of the peak area. The RI (retention time index) method was used in peak identification, and the RI tolerance was 5000. Then, identified GC‐MS data were imported into LineUp (Infometrix, Bothell, WA) and PiroTrans (GL Science) to align the chromatograms based on the peak intensity and the retention time of the internal standard, 2‐hydroxyundecanoic acid. The generated peak lists were imported into Pirouette software (Infometrix, Woodinville, WA) for multivariate statistical analysis. The following composition analysis of the samples was performed using MetaboAnalyst version 3.0 (http://www.metaboanalyst.ca/). The analysis methods included Principal Component Analysis (PCA), Partial Least Squares‐Discriminant Analysis (PLS‐DA), Orthogonal Projections to Latent Structures‐Discriminant Analysis (OPLS‐DA), and model validation.
Metabolite set enrichment
Metabolite set enrichment was investigated using metabolite set enrichment analysis (MSEA) and the overrepresentation analysis (ORA) tools offered by MetaboAnalyst version 3.0. Briefly, all metabolites from the GS‐MS array dataset were entered into MSEA as a list of metabolites. The mouse pathway library was chosen, and the ORA algorithm was selected using a hypergeometric test. The pathway ORA tool then used the proportion of differentially expressed metabolites in the whole array to determine whether a particular pathway was significant. A list of pathways associated with the uploaded metabolites was produced. P < 0.05 was considered to represent significant differences.
Different gene expression analysis by NGS
Different gene expression was detected using NGS analysis. HCC tissues from 9‐month‐old Ras‐Tg mice and wild‐type liver tissues from 9‐month‐old wild‐type C57BL/6J mice (5 for each group) were chosen for total RNA extraction using TRIzol reagent (Invitrogen, Grand Island, NY). Due to the consistent inbred genetic background, the definite etiology (ras oncogene) for HCC development, and the consistent pathological features of the samples in each group, the total RNA samples from the same group were mixed equivalently to generate two composite samples, that is, HCC and wild‐type liver tissues. Preparation of the mRNA sample for RNA‐Seq analysis was performed using the TruSeq RNA LT Sample Prep Kit v2 (Illumina, San Diego, CA). The template molecules were used for cluster generation and sequencing on an Illumina HiSeq 2000 (Illumina, San Diego, CA) instrument. One sample per lane was used to generate 100‐bp paired‐end reads. Read alignment to the UCSC Mouse Reference Genome (mm10, http://genome.ucsc.edu/) was performed using Tophat v1.3.2. Transcript assembly and quantification were performed by Cufflinks v2.0.2 to process the Tophat alignments. The assembled Cufflinks transcripts were processed with Cuffcompare analytical strategies to detect differentially expressed genes. The database for annotation, visualization, and integrated discovery (DAVID) v6.7 was used to interpret the differentially expressed gene pools and KEGG pathway enrichment assay.
Metabolite detection
NADPH: The level of NADPH was detected using a NADP/NADPH Quantification Colorimetric Kit (Cat No. k347‐100; BioVision, Milpitas, USA) according to the instructions of the manufacturer. Briefly, the frozen sample (~50 mg) was washed with ice‐cold 1 × PBS and homogenized in 0.5 mL of NADPH extraction buffer. After keeping the samples on ice for 10 min, the homogenate supernatant was obtained by centrifugation. Two‐hundred microliters of supernatant was transferred into an Eppendorf tube and heated at 60°C for 30 min in a water bath. Then, a 50‐μL aliquot was added to the labeled 96‐well plate in duplicate, and the NADPH level was detected by following the instructions for the kit.
Triglyceride and cholesterol
The frozen samples (~50 mg) were used for lipid extraction. Each frozen tissue was added to a 10‐mL heptane–isopropanol–tween mixture (3:2:0.01 by volume) and homogenized. This homogenate was centrifuged, and the supernatants were collected and evaporated with nitrogen flow. The triglyceride and cholesterol contents were determined using commercial kits (Cat No. A0‐10017 and A0‐10027; Dongou Biological Engineering Co Ltd, Wenzhou, China). All samples were measured in duplicate.
Pyruvate, lactate, and bile acid
The frozen samples (~50 mg) were added to 0.5 mL of 1 × PBS (0°C, pH 7.2–7.4) and homogenized on ice. The homogenate supernatant was obtained by centrifugation, and the levels of pyruvate, lactate, and bile acid were detected using commercial kits according to the instructions of the manufacturer (Cat No. A081, A019‐2, and E003‐2; Nanjing Jiancheng Bioengineering Institute, Nanjing, China). All samples were measured in duplicate.
GSH
Fresh samples (~50 mg) were added to 1 mL of ice‐cold 1 × PBS and homogenized with a glass homogenizer on ice. The resulting suspension was sonicated with an ultrasonic cell disrupter until the solution was clarified. Then, the homogenates were centrifuged for 5 min at 10,000×g. The supernatant was collected and detected immediately using an enzyme‐linked immunosorbent assay kit according to the instructions of the manufacturer (Cat No. CEA294Ge; Cloud‐Clone Corp, Wuhan, China). All samples were measured in duplicate.
ROS
The mouse reactive oxygen species (ROS) ELISA Kit (BH8443, BOYAO Biotechnology, Shanghai, China) was used to detect the ROS levels, and the procedures were performed according to the instructions of the manufacturer. Briefly, the fresh tissue samples were cut, weighed, and placed into a grinding tube containing ceramic beads and 1 mL of pH 7.4 PBS. Then, the tissue was homogenized and centrifuged, and the supernatant was carefully collected and assayed with an ELISA Kit. The final ROS level was calculated by dividing the total ROS content by the original tissue weight.
Statistical analysis
The peak intensities of metabolites and the signal values of gene expression were statistically analyzed by a Student's t‐test between HCC and wild‐type liver tissues. All the data were presented as the mean (), with a level of probability of 0.05 as the criterion for significance. Fold change is a measure describing how much a quantity changes going from the initial to the final value, and we recorded it as a log value.
Results
Gross and histological identification
To investigate the common metabolomics and transcriptomics related to HCC induced by the ras oncogene, Ras‐Tg and wild‐type males were sacrificed at 9 months of age, and the hepatic tumors and wild‐type liver tissues were sampled. The gross and histological findings were the same as our previous reports 5. The hepatic tumors were well developed with visible yellowish‐whitish lesions that were round or oval with enriched blood vessels, had soft and fragile qualities, and were of various sizes. The lesions were protruding onto or embedded in the livers of Ras‐Tg males (Fig. 1B). In contrast, the liver tissues from wild‐type males were bright red in color and flexible in texture with no lesions (Fig. 1A). Histopathological diagnosis showed that the HCC (typically over 5 mm in diameter) had a trabecular arrangement of tumor cells, which were highly anaplastic cells with evidence of necrosis and signs of lymphocytic infiltration (Fig. 1D). In contrast, the wild‐type liver cells were arranged neatly with no inflammatory cell infiltration or hepatic necrosis (Fig. 1C). In all of the sampled mice, lesions were not found in any other organs. The histopathologically identified HCC from eight individual Ras‐Tg males, and liver tissues from eight individual wild‐type males were chosen for further analysis.
Figure 1.
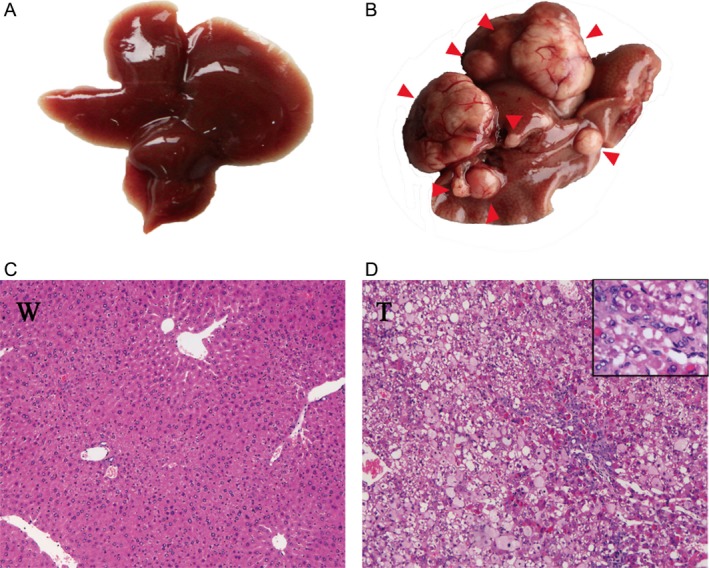
Gross anatomic and histopathological analysis of hepatic alterations. (A) Representative liver stereogram image of a liver from a 9‐month‐old wild‐type male. (B) Representative liver stereogram image of a liver carrying multiple tumors from a 9‐month‐old Ras‐Tg male. The red arrows indicate the hepatic alterations. (C) Relative histopathological H&E staining images (100 × ) showing liver tissue from a wild‐type mouse. (D) Relative histopathological H&E staining images (100 × ; upright corner: 400 × ) showing HCC from a Ras‐Tg mouse. W: wild‐type liver tissue; T: hepatocellular carcinoma (HCC).
GC‐MS chromatograms of HCC and wild‐type liver tissues
Typical GC‐MS total ion current (TIC) chromatograms of HCC and wild‐type liver tissue extracts are shown in Figure S1. The retention time of the internal standard was consistent for each run. The data showed marked differences in the chromatogram patterns between HCC and wild‐type liver tissues.
Clustering of HCC and wild‐type liver tissue samples
First, the missing values of raw data were calculated by using half of the minimum value; then, 631 peaks were detected, and 76 metabolites were excluded through the interquartile range denoising method. In addition, the internal standard normalization method was employed in this data analysis. The resulting two‐dimensional data involving the peak number, sample name, and normalized peak area were entered into the SIMCA‐P 14.0 software package (Umetrics, Umea, Sweden) for principal component analysis (PCA), partial least squares‐discriminant analysis (PLS‐DA), and orthogonal projections to latent structure‐discriminant analysis (OPLS).
PCA is an unsupervised analysis method reflecting the original state of the data. The PCA algorithm generates a single point that represents the metabolites in a sample and their concentrations (each dot in the score plots shown in Fig. 2A represents a single mouse). The close clustering of dots indicates that the samples have similar compositions (classification parameters: R2X (cum) = 0.587; Q2 (cum) = 0.221). The score plots of samples were clustered into two distinct groups (Fig. 2A), which indicates that the profiles of the metabolites in HCC tissues derived from Ras‐Tg males differed from those in liver tissues derived from wild‐type males.
Figure 2.
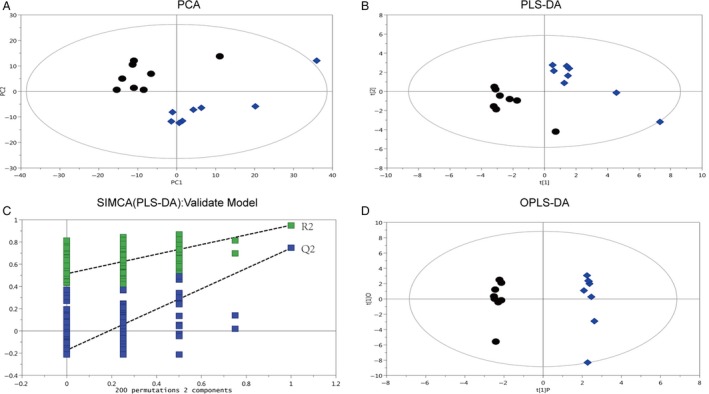
Score plots of PCA, PLS‐DA, and OPLS‐DA based on the metabolite profile data of HCC and wild‐type liver tissues. (A) Score plots of PCA based on the metabolite profile data for wild‐type liver tissues (black circles) and HCC (blue rhombi). The principal components PC1 (t[1]) and PC2 (t[2]) described 33.6% and 15.6% of the variation, respectively (n = 8). (B) The plot of PLS‐DA scores showing almost complete separation of wild‐type liver tissues (black circles) and HCC (blue rhombi). The classification parameters were R2X (cum) = 0.539, R2Y (cum) = 0.951, and Q2 (cum) = 0.746. (C) Validation model of PLS‐DA for wild‐type liver tissues (blue squares) and HCC (green squares) for 200 permutations of the data showing the degradation of R2 to below 0.515 and Q2 to below −0.173. (D) OPLS‐DA showing the contribution of variables to the difference between wild‐type liver tissues (black circles) and HCC (blue rhombi). The classification parameters were R2X (cum) = 0.816, R2Y (cum) = 0.996, and Q2 (cum) = 0.874.
PLS‐DA was applied to obtain a higher level of group separation and produce a better understanding of the variables responsible for classification (Fig. 2B). The classification parameters were R2X (cum) = 0.539; R2Y (cum) = 0.951; and Q2 (cum) = 0.746. Seven‐fold cross‐validation was used to estimate the robustness and the predictive ability of our model, and this permutation test was used to further validate the model. The R2 and Q2 intercept values were 0.515 and −0.173, respectively, after 200 permutations (Fig. 2C). The low values of the Q2 intercept indicate the robustness of the models and therefore show a low risk of overfitting and reliability.
OPLS‐DA was applied to show the contribution of variables to the difference between the two groups (Fig. 2D). The classification parameters were R2X (cum) = 0.816; R2Y (cum) = 0.996; and Q2 (cum) = 0.874. The results also showed important variables that were situated far from the origin, but the loading plot was complex because of many variables. To refine this analysis, the first‐principal component of variable importance projection (VIP) was obtained. The VIP values exceeding 1.0 were first selected as altered metabolites. In step 2, the remaining variables were then assessed by a Student's t‐test (t‐test), P > 0.05, and variables were discarded between the two comparison groups. In addition, commercial databases, including KEGG (http://www.genome.jp/kegg/) and NIST (http://www.nist.gov/index.html), were utilized to search for the metabolite pathways.
Metabolite differences between HCC and wild‐type liver tissues screened by PCA
In addition to the score plot, which is based on the composition of the samples, the PCA algorithm also creates a loading plot, which is based on metabolite values. Loading plots identify metabolites that contribute to the differential clustering of HCC and wild‐type liver tissues in the score plots. The electron impact spectra of these discriminatory metabolites were then compared with the NIST 05 Mass Spectral Library (NIST). Putatively identified discriminatory metabolites with matching NIST spectra are shown in Table 1. Fold changes and P‐values of these metabolites between HCC and wild‐type liver tissues were calculated by the TICs of GC‐MS chromatograms. Overall, the metabolic profiles in HCC and wild‐type liver tissues were different. Among the detected 555 metabolites, 36 common discriminatory metabolites contributed to this difference. However, the array of changes in the individual discriminatory metabolites in HCC and wild‐type liver tissues was more diverse.
Table 1.
Fold change and relative P‐value of the discriminatory metabolites characterizing hepatic carcinoma and wild‐type liver tissues were calculated by the TICs of GC‐MS chromatograms
| Metabolites | Retention time (min) | W mean | T mean | Fold change | P‐value |
|---|---|---|---|---|---|
| Glycolysis; TCA cycle; Pentose phosphate pathway; Lipid biosynthesis | |||||
| Glucose 1‐phosphate (glucose‐1p) | 16.7179 | 1.09E‐01 | 2.14E‐01 | 9.73E‐01 | 2.81E‐03 |
| Adenosine | 24.0355 | 6.81E‐03 | 4.06E‐03 | −7.46E‐01 | 1.17E‐02 |
| Cytidine diphosphate (CDP) | 22.5710 | 3.47E‐03 | 1.08E‐03 | −1.68E+00 | 2.81E‐03 |
| Citrate | 17.2200 | 1.00E‐02 | 4.55E‐02 | 2.19E+00 | 3.18E‐02 |
| Cytidine monophosphate (CMP) | 19.0875 | 3.49E‐03 | 1.46E‐03 | −1.26E+00 | 1.69E‐03 |
| Deoxycytidine monophosphate (dCMP) | 21.3202 | 7.62E‐04 | 1.82E‐04 | −2.07E+00 | 7.87E‐03 |
| Fructose | 17.8171 | 9.71E‐01 | 3.30E+00 | 1.76E+00 | 2.45E‐02 |
| Fructose 1,6‐bisphosphate (fructose‐1, 6p2) | 11.5276 | 6.02E+00 | 1.30E+01 | 1.11E+00 | 1.65E‐02 |
| Fructose 6‐phosphate (fructose‐6p) | 21.2896 | 3.78E‐03 | 5.70E‐03 | 5.93E‐01 | 4.18E‐02 |
| Glucose 6‐phosphate (glucose‐6p) | 21.4070 | 7.04E‐02 | 7.72E‐01 | 3.45E+00 | 9.13E‐04 |
| Lactate | 8.8119 | 2.20E‐02 | 7.75E‐03 | −1.51E+00 | 4.97E‐02 |
| Malate | 13.8313 | 2.25E+00 | 5.46E+00 | 1.28E+00 | 4.02E‐02 |
| Oxaloacetate | 13.5829 | 1.22E‐02 | 2.51E‐02 | 1.04E+00 | 4.88E‐02 |
| Pyruvate | 18.9793 | 2.10E‐01 | 4.21E‐01 | 1.00E+00 | 3.40E‐02 |
| Succinate | 11.8994 | 1.40E‐01 | 6.21E‐02 | −1.17E+00 | 2.41E‐03 |
| Thymine | 13.0700 | 5.41E‐03 | 1.82E‐02 | 1.75E+00 | 4.35E‐03 |
| Uridine diphosphate (UDP) | 21.5776 | 2.73E‐03 | 6.28E‐03 | 1.20E+00 | 1.15E‐02 |
| 2‐hydroxybutanoic acid | 9.5741 | 4.89E‐02 | 2.55E‐02 | −9.36E‐01 | 1.81E‐03 |
| Aspartate | 14.2188 | 1.36E‐01 | 2.61E‐01 | 9.40E‐01 | 4.35E‐02 |
| Fructose 2,6‐bisphosphate (fructose‐2, 6p2) | 20.6541 | 4.81E‐01 | 3.42E‐01 | −4.92E‐01 | 5.43E‐03 |
| Fucose | 16.4754 | 4.34E‐03 | 7.03E‐04 | −2.63E+00 | 7.12E‐06 |
| Fumarate | 12.2976 | 1.56E‐01 | 2.29E‐01 | 5.54E‐01 | 5.66E‐04 |
| Xylitol | 16.1919 | 3.77E‐01 | 1.08E‐01 | −1.81E+00 | 7.84E‐05 |
| Uridine monophosphate (UMP) | 24.2838 | 4.53E‐02 | 1.26E‐01 | 1.48E+00 | 1.97E‐02 |
| Uracil | 12.2391 | 1.57E‐02 | 7.11E‐02 | 2.18E+00 | 2.94E‐03 |
| Uridine | 22.6655 | 7.55E‐04 | 1.53E‐03 | 1.02E+00 | 1.06E‐02 |
| Xanthosine | 24.2838 | 1.07E‐03 | 1.82E‐04 | −2.56E+00 | 4.95E‐02 |
| β‐alanine | 13.2484 | 3.79E‐02 | 1.86E‐02 | −1.03E+00 | 7.11E‐05 |
| Nicotinamide adenine dinucleotide phosphate (NADP+) | 4.7442 | 4.03E‐01 | 2.53E‐01 | −6.72E‐01 | 1.22E‐04 |
| Glutathione metabolism | |||||
| L‐glutamate | 15.2758 | 6.10E‐03 | 1.55E‐02 | 1.35E+00 | 2.02E‐02 |
| 5‐oxoproline | 14.4595 | 3.84E‐01 | 7.26E‐01 | 9.19E‐01 | 1.17E‐02 |
| Glycine | 11.8509 | 2.22E+00 | 1.63E+00 | −4.46E‐01 | 2.26E‐02 |
| Nicotinamide adenine dinucleotide phosphate (NADP+) | 4.7442 | 4.03E‐01 | 2.53E‐01 | −6.72E‐01 | 1.22E‐04 |
| Glutathione (GSH) | 17.7547 | 2.23E‐01 | 2.95E‐01 | 4.04E‐01 | 4.52E‐02 |
| γ‐aminobutyric acid (GABA) | 10.0654 | 6.19E‐03 | 1.53E‐02 | 1.31E+00 | 4.15E‐02 |
| Succinate | 11.8994 | 1.40E‐01 | 6.21E‐02 | −1.17E+00 | 2.41E‐03 |
| Cholesterol and bile acid biosynthesis | |||||
| Glycine | 11.8509 | 2.22E+00 | 1.63E+00 | −4.46E‐01 | 2.26E‐02 |
| Cholesterol | 10.7894 | 5.41E‐03 | 1.82E‐02 | 1.75E+00 | 4.35E‐03 |
| Cholate | 24.7399 | 2.30E+00 | 3.60E‐01 | −2.68E+00 | 2.68E‐04 |
The different cluster of metabolites identified by PCA analysis between wild‐type liver tissues (n = 8) and HCC (n = 8) was shown. The metabolites were classified into corresponding pathways analyzed in Figure 4. P < 0.05 means the significant difference. W, wild‐type liver; T, hepatocellular carcinoma (HCC); Fold change: log2 (T mean/W mean).
Transcriptional differences between HCC and wild‐type liver tissues screened by NGS
Transcriptional data for 28225 well‐identified transcripts in HCC and wild‐type liver tissues were analyzed by NGS. Among them, 3747 transcripts were significantly and differentially expressed between the two groups based on P‐values (P < 0.05) (Table S1). Values for differentially expressed enzymes involved in the metabolic pathways of glycolysis, TCA cycle, PPP, lipid, glutathione, and cholesterol are shown in Table 2.
Table 2.
Differently expressed genes identified by Next‐Generation Sequencing (NGS) between HCC and wild‐type liver tissues related to the pathways analyzed in Figure 4
| mRNA accession NO. | Gene symbol | Full name | W value | T value | Fold change | P‐value |
|---|---|---|---|---|---|---|
| Glycolysis; TCA cycle; Pentose phosphate pathway; Lipid biosynthesis | ||||||
| NM_133904 | Acacb | Acetyl‐Coenzyme A carboxylase beta | 5.65E+00 | 2.03E+01 | 1.28E+00 | 1.27E‐02 |
| NM_001199296 | Acly | ATP‐citrate synthase isoform 1 | 1.43E+01 | 1.23E+02 | 2.15E+00 | 2.47E‐02 |
| NM_019477 | Acsl4 | Long‐chain fatty‐acid–CoA ligase 4 isoform 2 | 7.41E+00 | 5.07E+01 | 1.92E+00 | 6.63E‐04 |
| NM_028176 | Cda | Cytidine deaminase | 2.51E+00 | 1.40E+01 | 1.72E+00 | 7.28E‐03 |
| NM_023203 | Dctpp1 | dCTP pyrophosphatase 1 | 5.87E+01 | 2.50E+01 | −8.54E‐01 | 3.99E‐02 |
| NM_007861 | Dld | Dihydrolipoyl dehydrogenase | 9.25E+01 | 3.90E+01 | −8.64E‐01 | 4.69E‐02 |
| NM_170778 | Dpyd | Dihydropyrimidine dehydrogenase | 1.30E+02 | 2.91E+01 | −1.50E+00 | 3.64E‐03 |
| NM_001164466 | Dpys | Dihydropyrimidinase | 7.85E+01 | 9.94E+00 | −2.07E+00 | 1.39E‐07 |
| NM_007988 | Fasn | Fatty acid synthase | 7.90E+01 | 2.58E+02 | 1.18E+00 | 5.74E‐03 |
| NM_010209 | Fh | Fumarate hydratase 1 | 1.37E+02 | 5.65E+01 | −8.86E‐01 | 6.23E‐05 |
| NM_008061 | G6pc | Glucose‐6‐phosphatase | 1.99E+02 | 1.47E+01 | −2.61E+00 | 2.28E‐09 |
| NM_019468 | G6pd2 | Glucose‐6‐phosphate 1‐dehydrogenase 2 | 2.66E‐01 | 6.06E+00 | 3.13E+00 | 2.63E‐03 |
| NM_011829 | Impdh1 | Inosine‐5′‐monophosphate dehydrogenase 1 | 8.62E‐01 | 3.44E+00 | 1.38E+00 | 2.29E‐02 |
| NM_008797 | Pcx | Pyruvate carboxylase | 6.31E+00 | 5.19E+01 | 2.11E+00 | 1.20E‐04 |
| NM_008826 | Pfkl | 6‐phosphofructokinase | 4.00E+00 | 1.30E+01 | 1.18E+00 | 9.30E‐03 |
| NM_001081274 | Pgd | 6‐phosphogluconate dehydrogenase | 2.31E+01 | 6.10E+01 | 9.71E‐01 | 3.58E‐02 |
| NM_011099 | Pkm2 | Pyruvate kinase isozymes M1/M2 | 1.09E+01 | 2.96E+01 | 9.99E‐01 | 2.30E‐02 |
| NM_011506 | Sucla2 | Succinyl‐CoA ligase [ADP‐forming] subunit beta | 8.10E+01 | 3.23E+01 | −9.19E‐01 | 2.64E‐02 |
| NM_133995 | Upb1 | Beta‐ureidopropionase | 2.14E+02 | 8.74E+01 | −8.95E‐01 | 3.23E‐05 |
| NM_146006 | Lss | Lanosterol synthase | 1.18E+01 | 6.96E+01 | 1.77E+00 | 1.02E‐03 |
| NM_007856 | Dhcr7 | 7‐dehydrocholesterol reductase | 2.68E+01 | 9.47E+01 | 1.26E+00 | 2.58E‐02 |
| NM_138656 | Mvd | Mevalonate (diphospho) decarboxylase | 3.42E+00 | 2.57E+01 | 2.02E+00 | 6.45E‐05 |
| NM_145927 | Fntb | Protein farnesyltransferase subunit beta | 1.61E‐01 | 1.63E+00 | 2.31E+00 | 2.82E‐02 |
| Glutathione metabolism | ||||||
| NM_172961 | Abat | 4‐aminobutyrate aminotransferase | 3.77E+01 | 1.14E+01 | −1.20E+00 | 3.64E‐03 |
| NM_019468 | G6pd2 | Glucose‐6‐phosphate 1‐dehydrogenase 2 | 2.66E‐01 | 6.06E+00 | 3.13E+00 | 2.63E‐03 |
| NM_001081274 | Pgd | 6‐phosphogluconate dehydrogenase | 2.31E+01 | 6.10E+01 | 9.71E‐01 | 3.58E‐02 |
| NM_008160 | Gpx1 | Glutathione peroxidase 1 | 1.38E+03 | 1.28E+02 | −2.38E+00 | 1.35E‐04 |
| NM_177410 | Bcl2 | B cell leukemia/lymphoma 2 | 2.16E‐01 | 1.03E+00 | 1.56E+00 | 1.01E‐02 |
| NM_011671 | Ucp2 | Uncoupling protein 2 | 3.94E+00 | 1.89E+01 | 1.57E+00 | 1.79E‐04 |
| Cholesterol and bile acid biosynthesis | ||||||
| NM_145364 | Akr1d1 | 3‐oxo‐5‐beta‐steroid 4‐dehydrogenase | 1.13E+02 | 9.67E+00 | −2.46E+00 | 2.21E‐09 |
| NM_008537 | Amacr | Alpha‐methylacyl‐CoA racemase | 4.97E+01 | 2.17E+01 | −8.29E‐01 | 4.15E‐02 |
| NM_007519 | Baat | Bile acid‐CoA:amino acid N‐acyltransferase | 1.44E+02 | 3.16E+01 | −1.52E+00 | 3.42E‐04 |
| NM_016668 | Bhmt | Betaine–homocysteine S‐methyltransferase 1 | 1.57E+03 | 5.90E+01 | −3.28E+00 | 1.29E‐03 |
| NM_024264 | Cyp27a1 | Sterol 26‐hydroxylase | 1.82E+02 | 2.49E+01 | −1.99E+00 | 3.36E‐06 |
| NM_010010 | Cyp46a1 | Cholesterol 24‐hydroxylase | 1.59E+00 | 1.28E‐01 | −2.52E+00 | 6.88E‐04 |
| NM_007824 | Cyp7a1 | Cholesterol 7‐alpha‐monooxygenase | 6.62E+01 | 3.18E+00 | −3.04E+00 | 1.64E‐13 |
| NM_007825 | Cyp7b1 | 25‐hydroxycholesterol 7‐alpha‐hydroxylase | 1.74E+02 | 9.14E+00 | −2.95E+00 | 3.37E‐12 |
| NM_010012 | Cyp8b1 | 7‐alpha‐hydroxycholest‐4‐en‐3‐one | 6.44E+01 | 1.31E+00 | −3.90E+00 | 0.00E+00 |
| NM_028772 | Dmgdh | Dimethylglycine dehydrogenase | 2.08E+02 | 2.70E+01 | −2.04E+00 | 2.72E‐05 |
| NM_010321 | Gnmt | Glycine N‐methyltransferase | 1.44E+03 | 8.06E+01 | −2.88E+00 | 2.01E‐06 |
| NM_133943 | Hsd3b7 | 3 beta‐hydroxysteroid dehydrogenase type 7 | 7.31E+01 | 2.82E+01 | −9.53E‐01 | 1.79E‐02 |
| NM_008952 | Pipox | Peroxisomal sarcosine oxidase | 1.88E+02 | 5.13E+01 | −1.30E+00 | 3.65E‐03 |
| NM_146006 | Lss | Lanosterol synthase | 1.18E+01 | 6.96E+01 | 1.77E+00 | 1.02E‐03 |
| NM_007856 | Dhcr7 | 7‐dehydrocholesterol reductase | 2.68E+01 | 9.47E+01 | 1.26E+00 | 2.58E‐02 |
| NM_138656 | Mvd | Mevalonate (diphospho) decarboxylase | 3.42E+00 | 2.57E+01 | 2.02E+00 | 6.45E‐05 |
| NM_145927 | Fntb | Protein farnesyltransferase subunit beta | 1.61E‐01 | 1.63E+00 | 2.31E+00 | 2.82E‐02 |
The mRNA levels of differently expressed genes related to metabolism pathways of glycolysis, TCA cycle, pentose phosphate, lipid, glutathione, and cholesterol are shown. HCC (n = 5) and wild‐type liver (n = 5) and tissues were analyzed. P values were calculated using Student's t‐test and P < 0.05 means the significant difference. W, wild‐type liver; T, hepatocellular carcinoma (HCC); Fold change: ln (T mean/W mean).
Pathway enrichment and metabolite set enrichment analysis
Although we cannot conclusively determine which of the potential metabolic pathways are relevant to a phenotype, KEGG pathway enrichment (Fig. 3A, B; Tables [Link], [Link], [Link]) and metabolite set enrichment analysis (MSEA) (Fig. 3C; Tables [Link], [Link]) identified several key metabolic pathways. The following pathways were selected from the outputs of the KEGG pathway analysis and MSEA based on their biological relevance to HCC.
Figure 3.
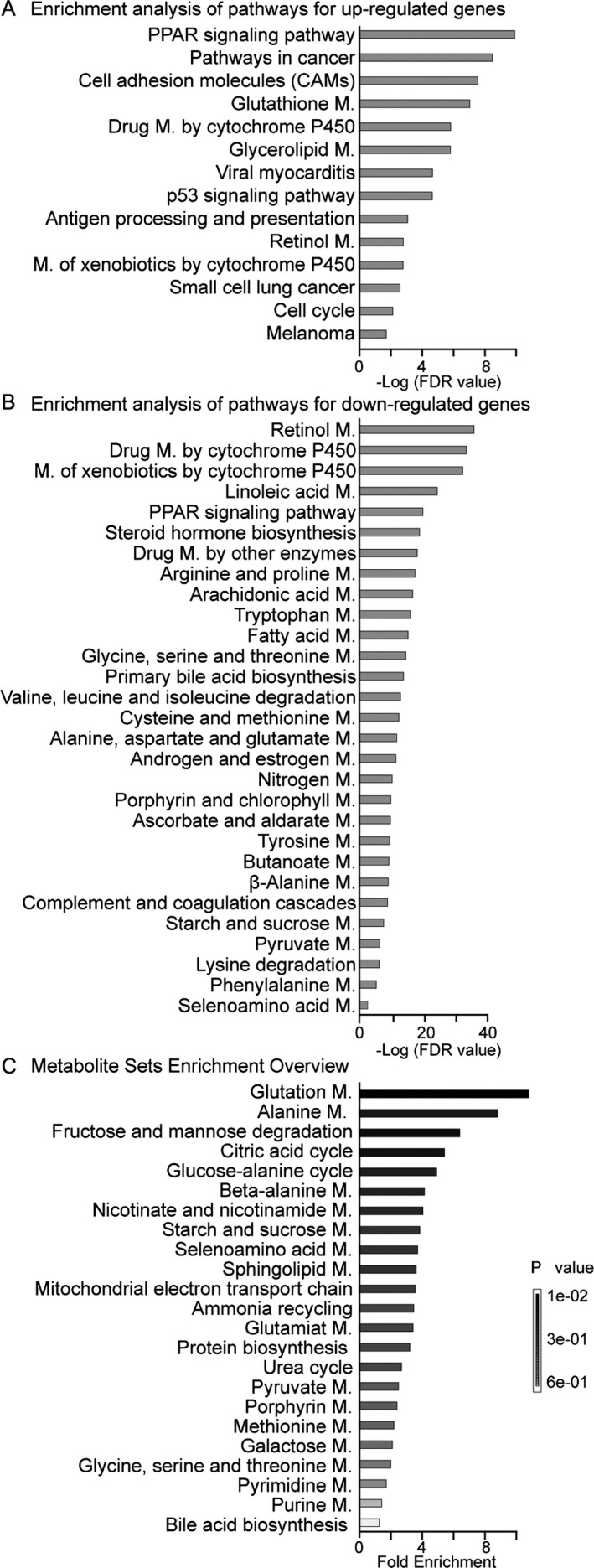
Pathway enrichment analysis for significantly changed genes and metabolites. (A) The significantly upregulated genes and (B) downregulated genes in HCC compared to wild‐type liver tissues were analyzed by KEGG pathway enrichment assays. (C) Significantly changed metabolites in HCC compared to wild‐type liver tissues were analyzed by MetaboAnalyst version 3.0 for metabolite set enrichment. The p values for the metabolic pathways are color coded, with dark black representing the most significant values and white representing the least significant values. Detailed information is shown in Tables [Link], [Link], [Link], [Link]. M.: metabolism.
Alterations of glycolysis, PPP, TCA cycle, and lipid biosynthesis in HCC
Disordered lipid metabolism has been widely reported in cancer cells. Because we observed a large quantity of lipid droplets in HCC cells based on gross anatomy and histopathological diagnosis (Fig. 1B, D), lipid biosynthesis and its closely related pathways, including glycolysis, PPP, and the TCA cycle, were comprehensively analyzed based on the metabolomics and transcriptomics database.
For the glycolysis metabolism pathway, we found that mainstream metabolites, such as glucose‐1‐phosphate (glucose‐1p), glucose‐6‐phosphate (glucose‐6p), fructose‐6‐phosphase (frucotose‐6p), fructose‐1,6‐bisphosphage (fructose‐1,6p2), and pyruvate, and the related enzymes Pfk1 and Pkm2 were significantly increased. In addition, the increased fructose and downregulated G6PC enzyme ensured a sufficient supply of raw materials. This outcome indicates that the enhanced glycolysis may not only maintain the excessive energy need for tumor growth but also provide a large quantity of the final metabolite, pyruvate.
The pyruvate from the glycolysis pathway enters the mitochondria and participates in the TCA cycle. Interestingly, in the TCA cycle, parts of metabolites, such as malate, oxaloacetate, and citrate, and the related enzymes Pcx and Acly were significantly increased, while parts of other metabolites, such as succinate, and the related enzymes Dld, Fh, and Sucl were significantly decreased. The results indicate that the shuttling of citrate and malate across the membrane of mitochondria may be enhanced, and therefore, the flow of pyruvate to lipid biosynthesis is enhanced by combining TCA and the citrate shuttle, in which pyruvate is taken from the cytosol, converted into acetyl‐CoA, condensed with oxaloacetate to citrate, and transferred back to the cytosol (Fig. 4A, Tables 1, 2).
Figure 4.
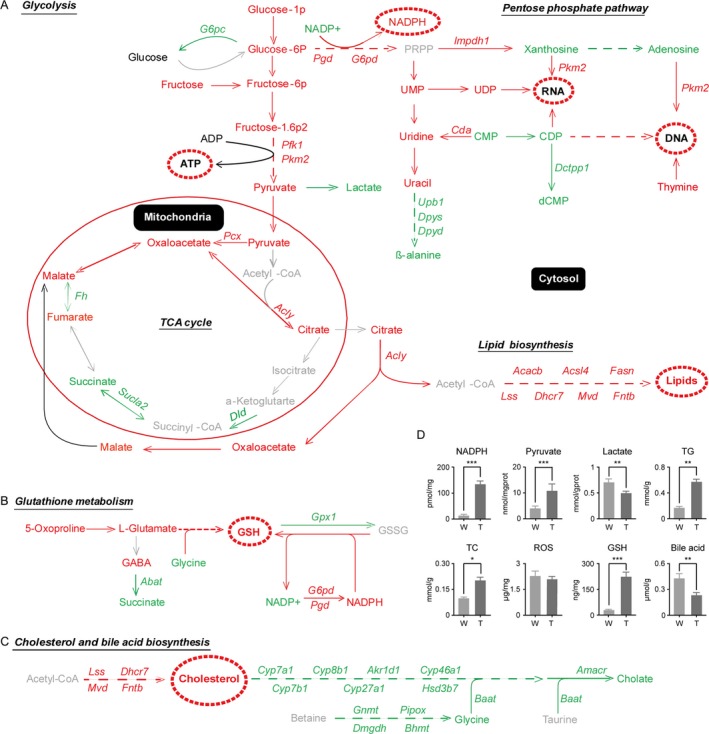
Schematic representations of the most relevant metabolic and transcriptional differences between HCC and wild‐type liver tissues. Metabolic pathways related to glycolysis, the TCA cycle, PPP, and lipid biosynthesis. (B) Metabolic pathways related to glutathione. (C) Metabolic pathways related to cholesterol and bile acid synthesis. Red indicates significantly higher concentrations of metabolites, expression levels of enzymes, or enhanced pathways in HCC; green indicates significantly lower concentrations of metabolites, expression levels of enzymes, or attenuated pathways in HCC; and gray indicates unchanged or undetermined results. The standard letters indicate metabolites, the italic letters indicate enzymes, and the underlined italic letters indicate pathways. The metabolites with broken circles indicate the central metabolites in HCC. (D) The reduced form of nicotinamide adenine dinucleotide phosphate (NADPH), pyruvate, lactate, cholesterol (TC), triglyceride (TG), ROS, GSH, and bile acid (BA) was detected in wild‐type liver (W) and HCC (T) tissues using the methods described in the Materials and Methods section. The data are expressed as the mean ± SEM (n = 8). *P < 0.05, **P < 0.01, and ***P < 0.001.
Another main raw material, hydrogen (H), is dominantly supplied by NADPH from the pentose phosphate pathway (PPP). In PPP, the levels of enzymes Pgd and G6pd and the concentration of NADPH were significantly elevated. Following the PPP, metabolites such as UMP, UDP, uridine, uracil, and thymine were significantly increased, while xanthosine, adenosine, CMP, CDP, beta‐alanine, and dCMP were significantly decreased. In addition, related enzymes, such as Impdh1, Pkm2, and Cda, were significantly increased, while Upb1, Dpys, Dpyd, and Dctpp1 were significantly decreased (Fig. 4A; Tables 1, 2). This outcome indicates that the enhanced PPP may supply not only the NADPH but also the raw materials for nucleotide synthesis needed for the rapid growth of tumor cells.
Finally, the enhanced glycolysis pathway and the citrate shuttle offered sufficient acetyl‐CoA, and the enhanced PPP offered sufficient NADPH. Additionally, increased expression levels of enzymes such as Acacb, Acsl, Fasn, Lss, Dhcr7, Mvd, and Fntb stimulated lipid synthesis and the accumulation of lipids to form large quantities of lipid droplets in HCC cells (Fig. 4A; Tables 1, 2; Fig. 1B, D).
Alterations of the glutathione metabolism pathway in HCC
Glutathione (GSH), especially in hepatocytes, plays important roles not only as an antioxidant but also in integrated detoxification. The increase in GSX (GSH+GSSG) in cancer cells indicates that enhanced de novo glutathione synthesis is occurring, which is crucial in cancer biology 23. In this study, GSH and the substrates for its synthesis, such as 5‐oxoproline and L‐glutamate, were significantly increased in HCC compared to wild‐type liver tissues (Table 1). The accumulated GABA resulted from significantly downregulated expression of Abat enzymes and subsequently decreased succinate, which may contribute to a sufficient L‐glutamate supply (Tables 1, 2). However, glycine, which is a widely used substrate for synthesis in cells, was significantly decreased, which may be due to its excessive consumption (Table 1). In addition, the significantly increased NADPH resulted from enhanced PPP via significant upregulation of the Pgd and G6pd enzymes, which may contribute to the enhanced reduction processes from GSSG to GSH (Tables 1, 2). The decreased Gpx1 level may attenuate the oxidation processes from GSH to GSSG. These changes may play crucial roles in maintaining the high concentration of GSH and low ROS levels in HCC cells. The summary of metabolites and enzymes related to the GSH metabolism pathway based on metabolomics and transcriptomics is shown in Figure 4B.
Alterations of cholesterol and the bile acid metabolism pathway in HCC
Cholesterol plays an important role not only in the formation of the cell membrane but also the synthesis of bile acids, vitamin D, and steroid hormones 24. Compared to wild‐type liver tissues, in HCC, the concentration of cholesterol was significantly increased, while glycine and cholate were significantly decreased (Table 1). Consistently, the transcriptional analysis showed that the enzymes related to cholesterol synthesis, such as Lss, Dhcr7, Mvd, and Fntb, were significantly upregulated in HCC compared to wild‐type liver tissues. However, the enzymes associated with bile acid synthesis from cholesterol, such as Cyp7a1, Cyp27a1, Baat, and Cyp8b1, were significantly downregulated in HCC (Table 2). The summary of metabolites and enzymes related to the cholesterol metabolism pathway based on metabolomics and transcriptomics is shown in Figure 4C.
Discussion
One of the hallmarks of cancers and precancerous lesions is the increase in de novo fatty acid synthesis regardless of the serum lipid levels 25. The liver is the major organ for regulating lipid metabolism, and changes in lipid homeostasis are observed in various liver diseases 26. Clinically, fat deposition is often (36%) seen in well‐differentiated HCCs between 1.1 and 1.5 cm in size 27. Recent evidence showed that both de novo synthesized and exogenous fatty acids support the growth of HCC cells 28. Although fatty acids are recognized as an important risk factor in hepatocarcinogenesis, the detailed mechanisms related to fatty acid accumulation remain unknown.
In this study, we showed that abundant quantities of lipid droplets (Fig. 1) are a histopathological characteristic of HCC and detected significantly elevated levels of triglyceride and cholesterol in HCC of Ras‐Tg mice compared to the liver tissues of wild‐type mice (Fig. 4A). Further integrative analysis of the metabolomic and transcriptomic data showed that the key enzymes associated with lipid biosynthesis were significantly enhanced at the mRNA level. In addition, acetyl‐CoA, which is a basic raw material for lipid de novo biosynthesis, is continuously supplied by (1) the enhanced glycolysis pathway, which produces pyruvate, and (2) the enhanced citrate shuttle, which converts and transfers mitochondrial pyruvate into cytosol acetyl‐CoA for further lipid synthesis. Additionally, another raw material for lipid synthesis, NADPH, was sufficiently supplied by enhanced PPP (Fig. 4A, Tables 1, 2). This evidence suggests that there are underlying molecular mechanisms for the increase in de novo lipid synthesis in HCC cells. Importantly, the combination of TCA and the citrate shuttle to switch mitochondrial pyruvate to cytosol acetyl‐CoA may be the key point for lipid synthesis and offers possible targets for the treatment of steatosis and HCC.
The roles of cholesterol in hepatic tumorigenesis and development are obscure. Because the liver is the dominant de novo synthesis organ for cholesterol 29, cholesterol synthesis in the liver is reduced, and the serum cholesterol content is gradually depressed along with the progression of liver diseases from chronic hepatitis to cirrhosis 30. Interestingly, several clinical reports showed that the serum cholesterol levels increased in cirrhosis patients with HCC compared to cirrhosis patients without HCC 31, 32, 33. This evidence indicates that the cholesterol levels were elevated in HCC cells 34, and the relatively lower serum level of cholesterol in HCC patients may result from the severity of cirrhosis, which occurred for 80% of HCCs 35. Therefore, the negative correlation of the serum cholesterol level to the mortality of HCC patients 36 may reflect the functional reserve for liver damage 37. Furthermore, several studies have suggested an important role for cholesterol in liver cancer 38, 39. Intriguingly, this study showed that cholesterol was significantly elevated in the HCC cells of Ras‐Tg mice (Fig. 4A). Apart from the sufficient supply of acetyl‐CoA and NADPH and the elevated levels of enzymes related to cholesterol biosynthesis, we found that bile acid synthesis pathways were attenuated via downregulation of the expression of multiple key enzymes that may play important roles in the accumulation of cholesterol in HCC of Ras‐Tg mice (Fig. 4A, C, Tables 1, 2). However, the detailed mechanisms related to cholesterol accumulation and its roles in hepatic tumorigenesis and development need to be investigated further.
Bile acid homeostasis is disrupted in multiple liver diseases, and the association between elevated BA levels and liver cancer has been demonstrated in both human and animal studies. Cell death and inflammation induced by BAs are suggested to be the underlying mechanisms 40. However, other investigations indicate that bile acid tends to be downregulated in clinical human HCC 41, and a significant negative correlation between bile acid levels and tumor burden was also observed 42. Consistent with the latter, we find that bile acid biosynthesis was substantially attenuated and that bile acid levels were significantly decreased in hepatic tumors. ERK is especially suggested to play important roles in suppressing Cyp7a1 and Cyp8b1, which are the rate‐limiting enzymes for bile acid biosynthesis 43. Consistently, activated Ras/ERK is a feature of hepatic tumors in Ras‐Tg mice, and the expression levels of Cyp7a1 and Cyp8b1 were significantly downregulated (Table 2). Because the Ras/ERK signaling pathway is frequently hyperactivated in human HCC 13, we suggest that the Ras/ERK pathway may contribute to the accumulation of cholesterol in HCC cells by suppressing bile acid biosynthesis.
High levels of ROS play dual functions not only in damaging biological macromolecules and destroying cells but also in promoting tumorigenesis 44. However, recent evidence indicates that in vivo tumor cells showed lower ROS levels, which contributes to tumorigenesis 45, 46. This evidence questions the role of ROS in tumorigenesis and development. The expression of the ras oncogene leads to the dysfunction of mitochondria and elevation of ROS levels 47. This phenomenon was also observed in nontumor hepatocytes from Ras‐Tg mice, which showed significantly higher ROS levels compared to the hepatocytes from wild‐type mice 11. However, there was no difference in ROS levels between the HCC cells of Ras‐Tg mice and hepatocytes of wild‐type mice (Fig. 4B). This evidence indicates that the roles of high levels of ROS in ras oncogene‐expressing hepatocytes in tumorigenesis remain unclear, yet the relatively low levels of ROS are essential for HCC development. However, the strategies employed by tumor cells to downregulate ROS levels are still unknown. Glutathione, which is one of the main ROS‐eliminating molecules, has been shown to be elevated in tumor cells 48. In HCC of Ras‐Tg mice, a significantly elevated quantity of GSH was also observed (Fig. 4B, Table 1). However, Gpx1, which is the main enzyme that eliminates ROS by converting GSH into GSSG, was significantly decreased in HCC cells (Fig. 4B, Table 2). Therefore, whether the high ratio of GSH/GSSG contributes to low ROS levels has yet to be investigated. In addition, Bcl2 and Ucp2, which are two molecules that have been recognized to play important roles in eliminating ROS 49, 50, were also significantly overexpressed in HCC cells (Table 2). Taken together, although the mechanisms associated with a normal level of ROS in HCC cells have yet to be elucidated, the high ratio of GSH/GSSG and high expression levels of Bcl2 and Ucp2 may be involved.
The interpretation of our findings may be limited to the mouse HCC induced by the Hras12V oncogene and the analysis based on static metabolic phenotypes. In human HCC, although hyperactivation of Ras/MAPK is a frequent event, a portion of patients do not show signs of hyperactivation 51. In addition, various metabolic profiles have been reported due to the complicated causes, different grades, or subgroups of human HCC, and there have been different study designs 41, 52, 53, 54. Similar to the findings on HCC of Ras‐Tg mice, it was suggested that there was a high glycolytic conversion of glucose into pyruvate in the cytosol followed by conversion into acetyl‐CoA and entry into the mitochondrial citric acid cycle with little conversion of pyruvate into lactate or alanine based on a panel of 31 pairs of human HCC tumors and corresponding nontumor liver tissues 52. Further ascertaining the metabolic fluxes in HCC of Ras‐Tg mice by using dynamic data through isotopic substrate labeling experiments will contribute to generalizable knowledge of HCC.
In addition, several limitations of this study must be mentioned. (1) We only presented a snapshot of differences in metabolic pools. Without dynamic 13C‐isotopic labeling experiments, it is hard to ascertain metabolic fluxes and rates. Therefore, the interpretation offered in this study needs further verification. (2) The metabolic profile of the nontumor liver harboring the ras oncogene of Ras‐Tg mice was not investigated in this study. It will be of great significance to detect differences between cells that transformed into malignant tumors and those that did not transform while expressing the same ras oncogene were detected. (3) The metabolic profile of female HCC was not investigated in this study. It will be interesting to compare metabolic profiles of hepatic tumorigenesis between males and females, which should offer valuable information for determining the source of male prevalence in HCC. The investigations addressing these issues are ongoing in our laboratory.
In summary, the mouse HCC induced by the Hras12V oncogene was analyzed by integrative analysis of metabolomics and transcriptomics. The accumulation of triglycerides and cholesterol in HCC is related to the overexpression of lipid biosynthesis‐related enzymes and sufficient supply of acetyl‐CoA and NADPH by accelerated glycolysis, the citrate shuttle, and PPP. Specifically, the downregulation of bile acid synthesis‐associated enzymes may play an important role in cholesterol accumulation. In addition, the elevated quantities of GSH, the GSH/GSSG ratio, and Bcl2 and Ucp2 may contribute to a normal level of ROS in HCC.
Conflict of Interest
The authors declare no conflicts of interest.
Supporting information
Figure S1. Typical GC/MS total ion current (TIC) chromatograms of HCC of Ras‐Tg mice and liver tissues of wild‐type mice.
Figure S2. Tissue lipids detected by oil red staining.
Table S1. Differentially expressed transcripts between HCC of Ras‐Tg mice and liver tissues of wild‐type mice.
Table S2. KEGG pathway enrichment analysis for upregulated genes in HCC of Ras‐Tg mice compared to liver tissues of wild‐type mice.
Table S3. KEGG pathway enrichment analysis for downregulated genes in HCC of Ras‐Tg mice compared to liver tissues of wild‐type mice.
Table S4. The number of metabolites in the pathways output by MSEA.
Table S5. Significant metabolites identified in the pathways output by MSEA.
Table S6. Overrepresentation analysis of the metabolic pathways output by MSEA.
Cancer Medicine 2017; 6(10):2370–2384
Contributor Information
Jingyu Wang, Email: wangjingyus@163.com.
Aiguo Wang, Email: wangaiguotl@hotmail.com.
References
- 1. Parkin, D. M. , Bray F., Ferlay J., and Pisani P.. 2005. Global cancer statistics, 2002. CA Cancer J. Clin. 55:74–108. [DOI] [PubMed] [Google Scholar]
- 2. Boguski, M. S. , and McCormick F.. 1993. Proteins regulating Ras and its relatives. Nature 366:643–654. [DOI] [PubMed] [Google Scholar]
- 3. Hunter, T. 1997. Oncoprotein networks. Cell 88:333–346. [DOI] [PubMed] [Google Scholar]
- 4. Newell, P. , Toffanin S., Villanueva A., Chiang D. Y., Minguez B., Cabellos L., et al. 2009. Ras pathway activation in hepatocellular carcinoma and anti‐tumoral effect of combined sorafenib and rapamycin in vivo. J. Hepatol. 51:725–733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Wang, A. G. , Moon H. B., Lee M. R., Hwang C. Y., Kwon K. S., Yu S. L., et al. 2005. Gender‐dependent hepatic alterations in H‐ras12V transgenic mice. J. Hepatol. 43:836–844. [DOI] [PubMed] [Google Scholar]
- 6. Rong, Z. , Fan T., Li H., Li J., Wang K., Wang X., et al. 2017. Differential proteomic analysis of gender‐dependent hepatic tumorigenesis in Hras12V transgenic mice. Mol. Cell Proteomics 16:1475–1490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Wang, K. , Nie X., Rong Z., Fan T., Li J., Wang X., et al. 2017. B lymphocytes repress hepatic tumorigenesis but not development in Hras12V transgenic mice. Int. J. Cancer 141:1201–1214. [DOI] [PubMed] [Google Scholar]
- 8. Kim, J. H. , Shin H. J., Ha H. L., Park Y. H., Kwon T. H., Jung M. R., et al. 2014. Methylsulfonylmethane suppresses hepatic tumor development through activation of apoptosis. World J. Hepatol. 6:98–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kopanja, D. , Pandey A., Kiefer M., Wang Z., Chandan N., Carr J. R., et al. 2015. Essential roles of FoxM1 in Ras‐induced liver cancer progression and in cancer cells with stem cell features. J. Hepatol. 63:429–436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Wang, A. G. , Moon H. B., Chae J. I., Kim J. M., Kim Y. E., Yu D. Y., et al. 2011. Steatosis induced by the accumulation of apolipoprotein A‐I and elevated ROS levels in H‐ras12V transgenic mice contributes to hepatic lesions. Biochem. Biophys. Res. Commun. 409:532–538. [DOI] [PubMed] [Google Scholar]
- 11. Kim, S. K. , Kim H., Koh G. Y., Lim D. S., Yu D. Y., Kim M. D., et al. 2015. Mouse hepatic tumor vascular imaging by experimental selective angiography. PLoS ONE 10:e0131687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Bae, H. J. , Jung K. H., Eun J. W., Shen Q., Kim H. S., Park S. J., et al. 2015. MicroRNA‐221 governs tumor suppressor HDAC6 to potentiate malignant progression of liver cancer. J. Hepatol. 63:408–419. [DOI] [PubMed] [Google Scholar]
- 13. Wang, A. G. , Song Y. N., Chen J., Li H. L., Dong J. Y., Cui H. P., et al. 2014. Activation of RAS/ERK alone is insufficient to inhibit RXRalpha function and deplete retinoic acid in hepatocytes. Biochem. Biophys. Res. Commun. 452:801–807. [DOI] [PubMed] [Google Scholar]
- 14. Kim, Y. K. , Minai‐Tehrani A., Lee J. H., Cho C. S., Cho M. H., and Jiang H. L.. 2013. Therapeutic efficiency of folated poly(ethylene glycol)‐chitosan‐graft‐polyethylenimine‐Pdcd4 complexes in H‐ras12V mice with liver cancer. Int. J. Nanomedicine 8:1489–1498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lee, H. S. , Lee S. Y., Ha H. L., Han D. C., Han J. M., Jeong T. S., et al. 2009. 2′‐Benzoyloxycinnamaldehyde inhibits tumor growth in H‐ras12V transgenic mice via downregulation of metallothionein. Nutr. Cancer 61:723–734. [DOI] [PubMed] [Google Scholar]
- 16. Wang, A. G. , Moon H. B., Chun S. Y., Lee T. H., Yu D. Y., and Lee D. S.. 2006. Orchiectomy reduces hepatotumorigenesis of H‐ras12V transgenic mice via the MAPK pathway. Life Sci. 79:1974–1980. [DOI] [PubMed] [Google Scholar]
- 17. Moon, E. Y. , Lee M. R., Wang A. G., Lee J. H., Kim H. C., Kim H. M., et al. 2006. Delayed occurrence of H‐ras12V‐induced hepatocellular carcinoma with long‐term treatment with cinnamaldehydes. Eur. J. Pharmacol. 530:270–275. [DOI] [PubMed] [Google Scholar]
- 18. Heo, C. K. , Woo M. K., Yu D. Y., Lee J. Y., Yoo J. S., Yoo H. S., et al. 2010. Identification of autoantibody against fatty acid synthase in hepatocellular carcinoma mouse model and its application to diagnosis of HCC. Int. J. Oncol. 36:1453–1459. [DOI] [PubMed] [Google Scholar]
- 19. Nicholson, J. K. , Holmes E., Kinross J. M., Darzi A. W., Takats Z., and Lindon J. C.. 2012. Metabolic phenotyping in clinical and surgical environments. Nature 491:384–392. [DOI] [PubMed] [Google Scholar]
- 20. Sung, W. K. , Zheng H., Li S., Chen R., Liu X., Li Y., et al. 2012. Genome‐wide survey of recurrent HBV integration in hepatocellular carcinoma. Nat. Genet. 44:765–769. [DOI] [PubMed] [Google Scholar]
- 21. Cuperlovic‐Culf, M. , Belacel N., and Culf A.. 2008. Integrated analysis of transcriptomics and metabolomics profiles. Expert. Opin. Med. Diagn. 2:497–509. [DOI] [PubMed] [Google Scholar]
- 22. Chen, K. , Zhu C., Cai M., Fu D., Cheng B., Cai Z., et al. 2014. Integrative metabolome and transcriptome profiling reveals discordant glycolysis process between osteosarcoma and normal osteoblastic cells. J. Cancer Res. Clin. Oncol. 140:1715–1721. [DOI] [PubMed] [Google Scholar]
- 23. Wise, D. R. , and Thompson C. B.. 2010. Glutamine addiction: a new therapeutic target in cancer. Trends Biochem. Sci. 35:427–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ikonen, E. 2008. Cellular cholesterol trafficking and compartmentalization. Nat. Rev. Mol. Cell Biol. 9:125–138. [DOI] [PubMed] [Google Scholar]
- 25. Menendez, J. A. , and Lupu R.. 2007. Fatty acid synthase and the lipogenic phenotype in cancer pathogenesis. Nat. Rev. Cancer 7:763–777. [DOI] [PubMed] [Google Scholar]
- 26. Jiang, J. T. , Xu N., Zhang X. Y., and Wu C. P.. 2007. Lipids changes in liver cancer. J. Zhejiang Univ. Sci. B 8:398–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kutami, R. , Nakashima Y., Nakashima O., Shiota K., and Kojiro M.. 2000. Pathomorphologic study on the mechanism of fatty change in small hepatocellular carcinoma of humans. J. Hepatol. 33:282–289. [DOI] [PubMed] [Google Scholar]
- 28. Cao, D. , Song X., Che L., Li X., Pilo M. G., Vidili G., et al. 2017. Both de novo synthetized and exogenous fatty acids support the growth of hepatocellular carcinoma cells. Liver Int. 37:80–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Lewis, G. F. 2006. Determinants of plasma HDL concentrations and reverse cholesterol transport. Curr. Opin. Cardiol. 21:345–352. [DOI] [PubMed] [Google Scholar]
- 30. Jiang, M. , Liu F., Xiong W. J., Zhong L., Xu W., Xu F., et al. 2010. Combined MELD and blood lipid level in evaluating the prognosis of decompensated cirrhosis. World J. Gastroenterol. 16:1397–1401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Alsabti, E. A. 1979. Serum lipids in hepatoma. Oncology 36:11–14. [DOI] [PubMed] [Google Scholar]
- 32. Venturini, I. , Amedei R., Modonesi G., Cosenza R., Miglioli L., Cioni G., et al. 1999. May plasma cholesterol level be considered a neoplastic marker in liver disease from cirrhosis to hepatocellular carcinoma? Ital. J. Gastroenterol. Hepatol. 31:61–65. [PubMed] [Google Scholar]
- 33. Venturini, I. , Zeneroli M. L., Corsi L., Baraldi C., Ferrarese C., Pecora N., et al. 1998. Diazepam binding inhibitor and total cholesterol plasma levels in cirrhosis and hepatocellular carcinoma. Regul. Pept. 74:31–34. [DOI] [PubMed] [Google Scholar]
- 34. Kawata, S. , Takaishi K., Nagase T., Ito N., Matsuda Y., Tamura S., et al. 1990. Increase in the active form of 3‐hydroxy‐3‐methylglutaryl coenzyme A reductase in human hepatocellular carcinoma: possible mechanism for alteration of cholesterol biosynthesis. Cancer Res. 50:3270–3273. [PubMed] [Google Scholar]
- 35. Llovet, J. M. , Burroughs A., and Bruix J.. 2003. Hepatocellular carcinoma. Lancet 362:1907–1917. [DOI] [PubMed] [Google Scholar]
- 36. Kitahara, C. M. , Berrington de Gonzalez A., Freedman N. D., Huxley R., Mok Y., Jee S. H., et al. 2011. Total cholesterol and cancer risk in a large prospective study in Korea. J. Clin. Oncol. 29:1592–1598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Wang, Q. , Lau W. Y., Zhang B., Zhang Z., Huang Z., Luo H., et al. 2014. Preoperative total cholesterol predicts postoperative outcomes after partial hepatectomy in patients with chronic hepatitis B‐ or C‐related hepatocellular carcinoma. Surgery 155:263–270. [DOI] [PubMed] [Google Scholar]
- 38. Montero, J. , Morales A., Llacuna L., Lluis J. M., Terrones O., Basanez G., et al. 2008. Mitochondrial cholesterol contributes to chemotherapy resistance in hepatocellular carcinoma. Cancer Res. 68:5246–5256. [DOI] [PubMed] [Google Scholar]
- 39. Morioka, S. , Sai K., Omori E., Ikeda Y., Matsumoto K., and Ninomiya‐Tsuji J.. 2016. TAK1 regulates hepatic lipid homeostasis through SREBP. Oncogene 35:3829–3838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Wang, X. , Fu X., Van Ness C., Meng Z., Ma X., and Huang W.. 2013. Bile Acid Receptors and Liver Cancer. Curr. Pathobiol. Rep. 1:29–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Fitian, A. I. , and Cabrera R.. 2017. Disease monitoring of hepatocellular carcinoma through metabolomics. World J. Hepatol. 9:1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Xiao, J. F. , Varghese R. S., Zhou B., Nezami Ranjbar M. R., Zhao Y., Tsai T. H., et al. 2012. LC‐MS based serum metabolomics for identification of hepatocellular carcinoma biomarkers in Egyptian cohort. J. Proteome Res. 11:5914–5923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Kong, B. , Wang L., Chiang J. Y., Zhang Y., Klaassen C. D., and Guo G. L.. 2012. Mechanism of tissue‐specific farnesoid X receptor in suppressing the expression of genes in bile‐acid synthesis in mice. Hepatology 56:1034–1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Panieri, E. , and Santoro M. M.. 2016. ROS homeostasis and metabolism: a dangerous liason in cancer cells. Cell Death Dis. 7:e2253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. DeNicola, G. M. , Karreth F. A., Humpton T. J., Gopinathan A., Wei C., Frese K., et al. 2011. Oncogene‐induced Nrf2 transcription promotes ROS detoxification and tumorigenesis. Nature 475:106–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Sayin, V. I. , Ibrahim M. X., Larsson E., Nilsson J. A., Lindahl P., and Bergo M. O.. 2014. Antioxidants accelerate lung cancer progression in mice. Sci. Transl. Med. 6:221ra15. [DOI] [PubMed] [Google Scholar]
- 47. Bellot, G. L. , Liu D., and Pervaiz S.. 2013. ROS, autophagy, mitochondria and cancer: ras, the hidden master? Mitochondrion 13:155–162. [DOI] [PubMed] [Google Scholar]
- 48. Marengo, B. , Nitti M., Furfaro A. L., Colla R., Ciucis C. D., Marinari U. M., et al. 2016. Redox homeostasis and cellular antioxidant systems: crucial players in cancer growth and therapy. Oxid. Med. Cell. Longev. 2016:6235641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Asher, G. , Lotem J., Kama R., Sachs L., and Shaul Y.. 2002. NQO1 stabilizes p53 through a distinct pathway. Proc. Natl. Acad. Sci. U S A 99:3099–3104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Bae, Y. S. , Oh H., Rhee S. G., and Yoo Y. D.. 2011. Regulation of reactive oxygen species generation in cell signaling. Mol. Cells 32:491–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Delire, B. , and Starkel P.. 2015. The Ras/MAPK pathway and hepatocarcinoma: pathogenesis and therapeutic implications. Eur. J. Clin. Invest. 45:609–623. [DOI] [PubMed] [Google Scholar]
- 52. Beyoglu, D. , Imbeaud S., Maurhofer O., Bioulac‐Sage P., Zucman‐Rossi J., Dufour J. F., et al. 2013. Tissue metabolomics of hepatocellular carcinoma: tumor energy metabolism and the role of transcriptomic classification. Hepatology 58:229–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Darpolor, M. M. , Basu S. S., Worth A., Nelson D. S., Clarke‐Katzenberg R. H., Glickson J. D., et al. 2014. The aspartate metabolism pathway is differentiable in human hepatocellular carcinoma: transcriptomics and (13) C‐isotope based metabolomics. NMR Biomed. 27:381–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Zhang, L. , Huang Y., Lian M., Fan Z., Tian Y., Wang Y., et al. 2017. Metabolic profiling of hepatitis B virus‐related hepatocellular carcinoma with diverse differentiation grades. Oncol. Lett. 13:1204–1210. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Typical GC/MS total ion current (TIC) chromatograms of HCC of Ras‐Tg mice and liver tissues of wild‐type mice.
Figure S2. Tissue lipids detected by oil red staining.
Table S1. Differentially expressed transcripts between HCC of Ras‐Tg mice and liver tissues of wild‐type mice.
Table S2. KEGG pathway enrichment analysis for upregulated genes in HCC of Ras‐Tg mice compared to liver tissues of wild‐type mice.
Table S3. KEGG pathway enrichment analysis for downregulated genes in HCC of Ras‐Tg mice compared to liver tissues of wild‐type mice.
Table S4. The number of metabolites in the pathways output by MSEA.
Table S5. Significant metabolites identified in the pathways output by MSEA.
Table S6. Overrepresentation analysis of the metabolic pathways output by MSEA.