

Abstract
The unicellular green alga Dunaliella salina is well adapted to salt stress and contains compounds (including β-carotene and vitamins) with potential commercial value. A large transcriptome database of D. salina during the adjustment, exponential and stationary growth phases was generated using a high throughput sequencing platform. We characterized the metabolic processes in D. salina with a focus on valuable metabolites, with the aim of manipulating D. salina to achieve greater economic value in large-scale production through a bioengineering strategy. Gene expression profiles under salt stress verified using quantitative polymerase chain reaction (qPCR) implied that salt can regulate the expression of key genes. This study generated a substantial fraction of D. salina transcriptional sequences for the entire growth cycle, providing a basis for the discovery of novel genes. This first full-scale transcriptome study of D. salina establishes a foundation for further comparative genomic studies.
Keywords: Dunaliella salina, Transcriptome profile, Metabolic processes and adjustment, Regulatory metabolism, Salt stress
1. Introduction
The unicellular green alga Dunaliella salina is well-known for its exceptional capacity to survive in hyper-saline environments due to the rapid accumulation of various osmolytes. As the main producer of natural β-carotene, D. salina is widely used for food, nutritional supplements, and cosmetics (Sathasivam et al., 2012). Large amounts of β-carotene are produced by modifying environmental conditions (Mogedas et al., 2009; Tian and Yu, 2009). However, under stress, the cell division of D. salina is retarded and cell growth is restricted (Rad et al., 2011), making it difficult to cultivate D. salina on a large-scale.
Several thousand expressed sequence tags (ESTs) of D. salina generated by traditional sequencing methods have been included in GenBank (National Center for Biotechnology Information (NCBI)) (Alkayala et al., 2010; Zhao et al., 2011). However, the metabolic pathways of prospective commercial compounds in D. salina are not well known. Due to the insufficient coverage of less-abundant transcripts (Wang et al., 2010), it is necessary to explore the development and evolution of this organism using genomic, transcriptomic, and proteomic approaches. With a better understanding of the metabolic processes and genetic information of the species, genetic manipulation may potentially enhance the quality and quantity of feedstock. Using a transcriptomic approach, the oleaginous marine alga D. tertiolecta was investigated to identify the pathways and genes involved in lipid synthesis, which has potential application in biofuel production (Rismani-Yazdi et al., 2011). The complete organelle (chloroplast and mitochondrial) genomes of D. salina were reported recently (Smith et al., 2010). Using complementary DNA (cDNA) microarrays, Kim(2010) investigated the genes (accession number GSE10271) which are important for the growth for D. salina under extremely low or high salinities. However, the genome annotation of D. salina is far from being completely understood.
For species without a reference genome, mRNA-sequencing technology can detect transcripts corresponding to the existing genomic sequences (Xu et al., 2012) and provide abundant information for a wide range of biological studies (Surget-Groba and Montoya-Burgos, 2010; Liu et al., 2015). In this study, we first characterized the transcriptomic database of D. salina during the adjustment, exponential and stationary growth phases, and performed transcriptome annotation with a special focus on stress-related compounds. Gene expression under salt stress was verified using relative real-time polymerase chain reaction (PCR).
2. Materials and methods
2.1. Plant materials and mRNA isolation
D. salina strain 435 was obtained from the Freshwater Algae Culture Collection at the Institute of Hydrobiology (FACHB-collection), Chinese Academy of Sciences, Wuhan, China. The cells were cultured in Dunaliella medium (DM) in a GXZ-1000A type incubator (Dongnan Instrument Co., Ltd., Ningbo, Zhejiang, China) (Liu et al., 2014).
For mRNA isolation, cells were harvested at different phases of their growth cycle: 30 d (adjustment phase), 80 d (exponential growth phase), and 120 d (stationary phase), using a Legend Micro 17R micro-centrifuge (Thermo Electron, Pittsburgh, PA, USA) at 1500g and 4 ○C for 5 min. The sediments were lysed using TRIzol reagent (Invitrogen Life Technologies Inc., Carlsbad, CA, USA) to isolate total RNA. Following the instructions of NEBNext Poly(A) mRNA Magnetic Isolation Module Dynabeads® Oligo(dT)25 (New England Biolabs, MA, USA), mRNA was isolated and then randomly fragmented by incubation at an elevated temperature (94 °C) in the presence of divalent cations (Mg2+).
2.2. Construction of a strand-specific cDNA library and transcriptome sequencing
A strand-specific cDNA library was constructed and the paired-end transcriptome was sequenced by the NovoGene Biological Information Technology Co., Ltd. (Beijing, China). All sequencing reads were submitted to the Short Read Archive (SRA) of the NCBI, and can be accessed under accession number SRR1036662.
2.3. Raw read analysis and de novo assembly of transcripts
The raw data (raw reads) obtained by sequencing the cDNA library were filtered for reads containing adapters, reads containing poly-N (N ≥10%), and low-quality reads, using Perl scripts (Novogene, Beijing, China). At the same time, the quality 20 (Q20), quality 30 (Q30), GC-content, and duplication sequence level of the clean data were calculated. All the downstream analyses were based on the clean, high-quality data.
The clean reads were first assembled into gene clusters using the Trinity v2012-10-05 program (Grabherr et al., 2011) with the settings: min_kmer_cov set to 2 and all other parameters set to default. Then, all long sequences generated by the Trinity program were assembled into contigs and singletons using CodonCode Aligner 5.0.2 with all parameters set to default.
2.4. Functional annotation of unique sequences
For sequence similarity alignment, the D. salina unique sequences (uniseqs; contigs and singletons of the CodonCode Aligner results) were annotated using BLAST 2.2.27+with a cutoff E-value of ≤10−5. The hmmscan of the HMMER 3.0 package was also used to predict the homologous conserved domains by searching the PFAM (protein families) database. Furthermore, uniseqs were functionally assigned into euKaryotic Orthologous Groups (KOGs) (Jensen et al., 2008). Using BLAST hit gene identifiers (GIs) and gene accessions, Blast2GO v2.5 software was used to process the BLAST matches and annotate them with gene ontology (GO) terms describing biological processes, molecular functions, and cellular components.
Uniseqs with corresponding enzyme commission (EC) numbers with a cutoff E-value of ≤10−5 were mapped into the Kyoto encyclopedia of genes and genomes (KEGG) database using KEGG Mapper. Then, uniseqs processed by the KEGG automatic annotation server (KAAS) were used to reconstruct the KEGG pathways with KAAS results containing KEGG Orthology (KO) assignments, and the basic rate interface transmission extension (BRITE) hierarchies (Moriya et al., 2007). Hence, uniseqs were categorized according to their role in the KEGG pathways.
2.5. Analysis of gene expression level under osmotic stress
To evaluate the effects of osmotic stress on uniseqs, the expression level of a transcript was quantified as reads per kilobase (kb) of the transcript per million mapped reads of the transcriptome (RPKM) (Mortazavi et al., 2008). For each uniseq, the RPKM was calculated based on the formula: RPKM=a/(b×c), where a represents the number of reads in each de Brujin graph, b represents the number of the total reads (millions), and c represents the uniseq length (kb).
To test the expression levels of key genes involved in the metabolic processes under salt stress, relative quantitative PCR (qPCR) was used to assess the mRNA amount in D. salina. We chose algae in the logarithmic phase of growth in DM medium (1.5 mol/L NaCl) as normal cells. The cells were treated by hypo salinity (0.5 mol/L NaCl) and hyper salinity (4.5 mol/L NaCl) for 2 h, and then lysed using TRIzol reagent (Invitrogen Life Technologies Inc., Carlsbad, CA, USA) to isolate total RNA. The co-isolated residual genomic DNA within the total RNA was removed using an RNase-freed DNase 1 enzyme (Promega Co., Madison, WI, USA). The integrity of the total RNA was assessed using formaldehyde agarose gel electrophoresis, and RNA quantity was determined by NanoDrop 2000 spectrophotometer measurements (Thermo Scientific, DE, USA). Relative qPCR was performed with the ABI PRISM® 7900 Real-Time PCR System (Applied Biosystems, CA, USA) using the FastStart Universal SYBR Green Master (ROX; Roche, Basel, Switzerland). The following actin primers were used for endogenous control: 5'-ACCACACCTTCTTCAACGA-3' and 5'-GG ATGGCTACATACATGGCA-3' (Chen et al., 2011). Other selected genes involved in the metabolic processes in D. salina were analyzed using the primers shown in Table S1. Three experiments were carried out in parallel and the results were calculated based on the mean of the three results.
2.6. Statistical analysis
Data are expressed as mean±standard deviation (SD). Statistical analysis of the results was performed using the Student’s t-test algorithm. P<0.05 was set as the level of significant difference.
3. Results
3.1. Sequencing and de novo assembly of the D. salina transcriptome
We constructed a strand-specific cDNA library of pooled RNA of D. salina at different growth phases (adjustment phase, logarithmic phase, and stationary phase) to analyze the metabolic processes. The paired-end in-depth sequencing of the cDNA library generated 30 445 912 (about 5.86 gigabases (Gb)) raw reads with an average length of 200 bp. The statistics of sequencing data quality is presented in Table 1. A total of 29 326 716 (96.32%) high-quality reads were obtained after filtering the 82 128 adapters, removing the 4085 sequences with unknown bases (N >10%) and trimming 1 032 983 low-quality reads (reads containing more than 50% of bases with a Phred quality score of ≤5).
Table 1.
Statistics of sequencing data quality from D. salina
| Raw read | Base number (Gb) | Length (bp) | Error (%) | Q20 (%)1 | Q30 (%)2 | GC content (%) |
| 30445912 | 5.86 | 200 | 0.04 | 96.83 | 90.89 | 52.77 |
The error rate of bases with Phred quality score of ≤20 less than 0.01;
The error rate of bases with Phred quality score of ≤30 less than 0.001
The high-quality clean reads were assembled de novo into 73 443 gene clusters using the Trinity v2012-10-05 program. About 75.4% (55 412) of the reads were found to comprise only one sequence (Fig. 1a) and the remaining 24.6% (18 031) covered 2 to 156 sequences, with 3286 gene clusters having more than five sequences (Fig. 1b). The longest sequence in each de Brujin graph representing the corresponding gene cluster was retrieved and considered as a Trinity unique sequence (Trinity uniseq). Using this criterion, 73 443 Trinity uniseqs were generated, with a mean length of 664 bp (ranging from 201 to 15 085 bp; Fig. 1c).
Fig. 1.

Overview of the sequencing and assembly of D. salina transcriptome
(a) Distribution of clean read coverage; (b) Distribution of Trinity-assembled gene cluster coverage; (c) Size distribution of Trinity uniseqs; (d) Distribution of contig coverage; (e) Size distribution of the contig length
The 50 406 (68.63%) overlapping sequences of 73 443 Trinity uniseqs were further assembled into 16 783 contigs using CodonCode Aligner 5.0.2 (CodonCode Co., USA) with parameters set to default.
The coverage depth for contigs (Fig. 1d) ranged from 2 to 29 bp with an average length of 1409 bp (Fig. 1e). The N50 indicates that 50% of the assembled bases were incorporated into contigs longer than 1727 bp (N50=1727 bp). The remaining 23 037 Trinity uniseqs could not be assembled into contigs and were defined as singletons, of which 18 240 (79.18%) were less than 500 bp. The contigs and singletons represent the transcriptome of D. salina with a total of 39 820 unique sequences (uniseqs).
3.2. Functional annotation of the D. salina transcriptome
Based on the similarity of sequence alignment, a total of 12 981 (32.57%) uniseqs (9143 contigs and 3838 singletons) had significant BLASTx matches with the non-redundant protein (NR) databases and Swiss-Prot (SP) database. In addition, about 1901 (4.77%) uniseqs had significant BLASTn matches with the nucleotide (NT) database (Table S2), of which 1739 uniseqs were found with BLASTx matches. In brief, a total of 13 143 (33.01 %) uniseqs (9206 contigs and 3937 singletons) were identified through BLAST matches (Table S2).
According to the species taxonomy, the top hits of the BLAST annotated uniseqs were further clustered into five categories including “Plants”, “Animals”, “Fungi”, “Prokaryotes”, and “Viruses” (Fig. 2a). Since there were only five virus species in the “Viruses” category, that category is not shown in Fig. 2a. Most of the 131 plant species had 10 599 (80.68%) genes similar to those of D. salina (Table S3). Among them, 33.59% species belonged to green algae, predominantly Volvox carteri and Chlamydomonas (Fig. 2b). In addition, 1326 (10.09%) uniseqs of D. salina were shared by 369 prokaryotes, of which 57.32% were shared with Halomonas, 894 uniseqs were shared with 264 animal species, and 313 (2.38%) with 138 fungal species.
Fig. 2.

Top-hit species distribution of BLAST results of D. salina uniseqs
(a) Number of top-hit species of BLAST results assigned with the “Plant”, “Animal”, “Fungi”, and “Prokaryote” categories. (b) Percent distribution of the top seven green algae (Volvox carteri, Chlamydomona, Coccomyxa, Chlorella, Dunaliella, Auxenochlorella, and Micromonas), remaining algae and higher plants in the plant category
The uniseqs were further annotated using the hmmscan program to find the conserved domain in the PFAM database. Finally, 18 182 uniseqs (45.66%, Table S2) were found, which had 4180 conserved domains. To further evaluate the completeness of the D. salina transcriptome, 8908 (22.37%) of the above uniseqs were assigned into 11 353 KOG functions with 26 clusters (Fig. S1). A total of 22 111 uniseqs (13 455 contigs and 8656 singletons) were annotated by the BLAST, PFAM, and KOG databases (Table S2).
Using Blast2GO v2.5 software, a total of 18 963 uniseqs (47.62%) were assigned into GO terms describing biological processes (BP), molecular functions (MF), and cellular components (CC) as shown in Fig. S2. Of the GO terms for annotated uniseqs, 5260 uniseqs (3837 contigs and 1423 singletons) were assigned EC numbers (Table S2). Apart from 1872 of the 5260 uniseqs that could not be mapped into any pathway, 6.4% were annotated with biological processes (Fig. S3) using the KEGG mapper (Huson et al., 2011), and were analyzed further for metabolic processes of D. salina, with detailed descriptions of the stress-related metabolic compounds (osmolytes and polyamines) and carotenoid metabolic compounds, as described below.
3.3. Metabolic processes in D. salina
3.3.1. Osmolyte metabolism in D. salina
Using the de novo assembly and functional annotation of the D. salina transcriptome, we completed the metabolic pathway of glycerol (Fig. 3a). The identified enzymes are shown in Table S4 and the sequences in Data S1. The best hits of their BLAST results are shown in Table S5. An important three-carbon metabolic intermediate for glycerol metabolism is dihydroxyacetone phosphate (DHAP), a product of glycolysis. Nicotinamide adenine dinucleotide (NAD+)-dependent glycerol-3-phosphate dehydrogenase 1 (GPD1) catalyzes glycerol metabolism, which converts DHAP to glycerol-3-phosphate (GLP). Hyperosmotic stress can relieve the inhibition of GPD1 function and activate GPD1 to synthesize GLP (Brewster and Gustin, 2014). GLP is then dephosphorylated by GLP phosphatase 1 (GPPase 1, EC: 3.1.3.21) to form glycerol. Glycerol can also be synthesized by the reduction of dihydroxyacetone (DHA) through catalysis by dihydroxyacetone reductase (DHR, EC: 1.1.1.156). Excess glycerol in cells exposed to hypo-saline stress is degraded to maintain lower osmotic pressure. This reaction is catalyzed by DHR to produce DHA (Chen and Jiang, 2009), followed by phosphorylation with dihydroxyacetone kinase (DAK, EC: 2.7.1.29) to regenerate the three-carbon metabolic intermediate DHAP.
Fig. 3.
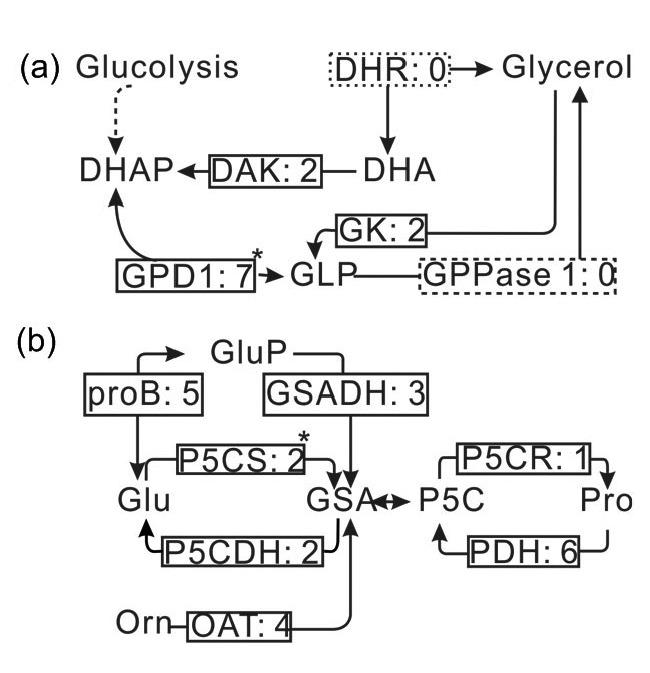
Osmolyte metabolic pathways reconstructed based on the de novo assembly and annotation of D. salina transcriptome
(a) Reconstruction of the glycerol metabolic pathway. Enzymes identified and unidentified are shown in the solid and dashed boxes, respectively, and include: DAK (EC: 1.7.1.29), dihydroxyacetone kinase; DHR (EC: 1.1.1.156), dihydroxyacetone reductase; GPD1 (EC: 1.1.1.8), nicotinamide adenine dinucleotide (NAD+)-dependent glycerol-3-phosphate dehydrogenase 1; GK (EC: 2.1.1.30), glycerol kinase; GPPase 1 (EC: 3.1.3.21), glycerol-3-phosphate phosphatase 1. (b) Reconstruction of proline metabolic pathway. Enzymes identified are shown in the solid boxes and include: proB (EC: 2.7.2.11), glutamate 5-kinase; GSADH (EC: 1.2.1.41), glutamate-5-semialdehyde dehydrogenase; P5CS (EC: 2.7.2.1, 1.2.1.41), δ-1-pyrroline-5-carboxylate synthetase; P5CDH (EC: 1.5.1.12), 1-pyrroline-5-carboxylate dehydrogenase; P5CR (EC: 1.5.1.2), pyrroline-5-carboxylate reductase; PDH (EC: 1.5.99.8), proline dehydrogenase; OAT (EC: 2.6.1.13), ornithine-oxo-acid transaminase. The digits behind the colon (:) in each rectangle represent the number of assigned transcripts of enzymes. Key enzymes are shown with a superscript symbol (*) next to the box. DHA, dihydroxyacetone; DHAP, dihydroxyacetone phosphate; GLP, glycerol-3-phosphate; Glu, glutamine; GluP, glutamate-phosphate; Orn, ornithine; GSA, glutamate-5-semialdehyde; P5C, δ-1-pyrroline-5-carboxylate; Pro, proline
In addition to glycerol, proline (Pro) is another osmolyte accumulated in D. salina during the process of adaptation. Pro acts both as a protective agent and a free-radical scavenger in the cytosol (Venekamp, 2006); however, its metabolism has not been analyzed. According to the functional annotation of the D.salina transcriptome, the metabolic pathway of Pro was reconstructed (Fig. 3b). The identified enzymes are shown in Table S4, with average transcript levels of 3.29. Their sequences and the best hit of their BLAST results are shown in Data S1 and Table S5. Pro can be synthesized from either glutamine (Glu) or ornithine via the molecule δ-1-pyrroline-5-carboxylate (P5C). Glu is phosphorylated by the enzyme glutamate 5-kinase (proB, EC: 2.7.2.11) to form glutamate-phosphate (GluP) products. Subsequently, it is reduced by glutamate-5-semialdehyde dehydrogenase (GSADH, EC: 1.2.1.41) into glutamate-5-semialdehyde (GSA). These two reactions can also be catalyzed directly by an enzyme, δ-1-pyrroline-5-carboxylate synthetase (P5CS, EC: 2.7.2.1, 1.2.1.41), which is the rate-limiting step for Pro biosynthesis (Deng et al., 2013). This enzyme has both the kinase and γ-glutamyl phosphate reductase domains. One uniseq with two transcripts was identified in the D. salina transcriptome. GSA can also be synthesized by transferring the NH2 groups from ornithine to 2-oxo acid. This reaction is catalyzed by ornithine-oxo-acid transaminase (OAT, EC: 2.6.1.13) which was expressed with four transcripts (Table S4). GSA is then spontaneously cyclized to form P5C. Finally, P5C is reduced to Pro by the enzyme pyrroline-5-carboxylate reductase (P5CR, EC: 1.5.1.2), which dehydrogenates H+ from nicotinamide adenine dinucleotide phosphate (NAPDH). In reverse, enzymes related to Pro catabolism were also identified (Table S4), including proline dehydrogenase (PDH, EC: 1.5.99.8) and 1-pyrroline-5-carboxylate dehydrogenase (P5CDH, EC: 1.5.1.12). PDH catalyzes the dehydrogenation of Pro to form P5C, whereas P5CDH converts GSA to Glu.
3.3.2. Polyamine biosynthetic pathway in D. salina
Polyamines, including putrescine (Put), spermidine (Spd), and spermine (Spm), are low molecular aliphatic nitrogen compounds found in all living organisms. There are two routes (arginine and ornithine routes) to synthesize putrescine (Put) in Chlamydomonas reinhardtii, a relative of Dunaliella. These have been investigated intensively and the results show that the ornithine route controls this process with the key enzyme ornithine decarboxylase (ODC, EC: 4.1.1.17) (Voigt et al., 2000). Through the functional annotation of the transcriptome, two uniseqs coding for N-carbamoylputrescine amidase (aguB, EC: 3.5.1.53) and ODC were identified (Table S4). Although arginine decarboxylase (speA, EC: 4.1.1.19) and agmatine deiminase (Adase, EC: 3.5.5.12) were not found in the D. salina transcriptome, the identification of aguB implies that D. salina might possess these two routes. Using the information from the assembly and annotation of the D. salina transcriptome, the polyamine metabolic pathway was reconstructed (Fig. 4). Enzymes involved in the polyamine biosynthetic pathway are shown in Table S4. Their sequences and the best hits of their BLAST results are shown in Data S1 and Table S5 individually. Arginine is decarboxylated by speA to form agmatine (Agm) products. Then, with the catalyzation of the speB enzyme or the aguB and Adase to synthesize Put. Then, Put is successively attached to the aminopropyl groups from the molecule decarboxylated S-adenosylmethionine (dcAdoMet) in two-step reactions. They are catalyzed by spermidine synthase (SPDS, EC: 2.5.1.16) and spermine synthase (SPS, EC: 2.5.1.22), with the production of Spd and Spm. Aminopropyl is produced by the enzyme S-adenosylmethionine decarboxylase (dsAM, EC: 4.1.1.50) from S-adenosyl-L-methionine (AdoMet). Heterologous expression of SPDS from D. salina may confer tolerance to high salt stress in Escherichia coli (Liu et al., 2014).
Fig. 4.
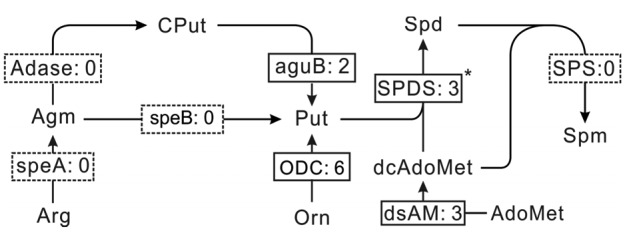
Polyamine biosynthetic pathway reconstructed based on the de novo assembly and annotation of D. salina transcriptome
Enzymes identified and unidentified are shown in the solid and dashed boxes, respectively, and include: speA (EC: 4.1.1.19), arginine decarboxylase; Adase (EC: 3.5.3.12), agmatine deiminase; speB (EC: 3.5.3.11), agmatinase; aguB (EC: 3.5.1.53), N-carbamoylputrescine amidase; ODC (EC: 4.1.1.17), ornithine decarboxylase; dsAM (EC: 4.1.1.50), S-adenosylmethionine decarboxylase; SPDS (EC: 2.5.1.16), spermidine synthase; SPS (EC: 2.5.1.22), spermine synthase. The digits behind the colon (:) in each box represent the number of assigned transcripts of enzymes. Key enzyme is shown with a superscript symbol (*) next to the box. Arg, arginine; Agm, agmatine; CPut, carbamoyl-putrescine; Put, putrescine; AdoMet, S-adenosyl-L-methionine; DcAdoMet, S-adenosyl-metioninamine; Orn, ornithine; Spd, spermidine; Spm, spermine
3.3.3. Carotenoid biosynthetic pathway in D. salina
Based on the annotation of the D. salina transcriptome, carotenogenesis-relevant enzymes were identified (Table S4). Their sequences and the best hits of the BLAST results are shown in Data S1 and Table S5 individually. The carotenogenesis pathway was reconstructed (Fig. 5). Four precursors, geranylgeranyl-phosphates (GGPPs), are condensed to form phytoene, the basic precursor of all carotenoids, and catalyzed by the enzyme phytoene synthase (PSY, EC: 2.5.1.32). Usually, this rate-limiting step is regulated through heterologous overexpression of this gene or environmental modification to enhance carotenoid production (Steinbrenner and Linden, 2001; Couso et al., 2011). Previous studies indicated that this gene is duplicated in the genome sequences in D. salina (Tran et al., 2009). Through the de novo assembly of the transcriptome, PSY with three transcripts was identified in D. salina with an RPKM of 31.53. Next, phytoene is converted to lycopene with step-wise desaturations by four enzymes: 15-cis-phytoene desaturase (PSD, EC: 1.3.5.5), δ-carotene isomerase (ZCI, EC: 5.2.1.12), δ-carotene desaturase (ZDS, EC: 1.3.5.6), and prolycopene isomerase (PLI, EC: 5.2.1.13). Lycopene is then cyclized to carotenes and their derivatives. Three lycopene cyclases were identified, including lycopene β-cyclase (LcyB, EC: 5.5.1.19), lycopene cyclase CruP (CruP), and lycopene ε-cyclase (LcyE, EC: 1.14.13.129). LcyB and CruP catalyze the biosynthesis of α-carotene, β-carotene, and γ-carotene, while LcyE catalyzes the formations of δ-carotene and ε-cyclase. As the main source for natural β-carotene production, Dunaliella has been developed to accumulate β-carotene through environmental modification (Mogedas et al., 2009). CruP cannot affect the production of cyclized carotenoids under photo-inhibitory stress and protects against reactive oxygen species (ROS) in oxygenic photosynthetic organisms, providing a unique target to develop algae for food and cosmetics (Bradbury et al., 2012). There were four transcripts coding the CruP enzyme (Table S4).
Fig. 5.
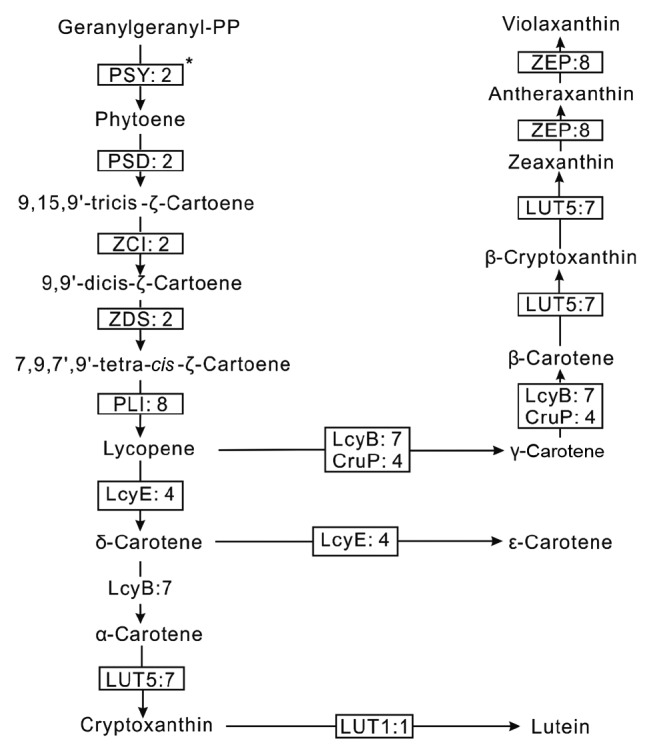
Carotenoid biosynthetic pathway reconstructed based on the de novo assembly and the functional annotation of D. salina transcriptome
Enzymes identified are shown in the solid boxes, and include: PSY (EC: 2.5.1.32), phytoene synthase; PSD (EC: 1.3.5.5), 15-cis-phytoene desaturase; ZCI (EC: 5.2.1.12), δ-carotene isomerase; ZDS (EC: 1.3.5.6), δ-carotene desaturase; PLI (EC: 5.2.1.13), prolycopene isomerase; LcyB (EC: 5.5.1.19), lycopene β-cyclase; CruP, lycopene cyclase CruP; LUT5 (EC: 1.14.−.−), cytochrome P450, family 97, subfamily A (β-ring hydroxylase); LcyE (EC: 5.5.1.18), lycopene ε-cyclase; ZEP (EC: 1.14.13.90), zeaxanthin epoxidase; LUT1 (EC:1.14.99.45), carotene ε-monooxygenase. The digits behind the colon (:) in each rectangle represent the number of assigned transcripts of enzymes. Key enzyme is shown with a superscript symbol (*) next to the box
Carotene derivatives are synthesized by hydroxyl-lation or epoxidation in D. salina. There were three hydroxylases identified in the D. salina transcriptome, including cytochrome P450 enzyme LUT5 (EC: 1.14.−.−), β-carotene hydroxylase (CrtZ, EC: 1.14.13), and carotene ε-monooxygenase (LUT1, EC: 1.14.13. 90). They catalyze the formations of zeaxanthin, zeinnxanthin, α-cryptoxanthin, β-cryptoxanthin, and lutein. Finally, the zeaxanthin epoxidase (ZEP, EC: 1.14.13.90) catalyzes antheraxanthin and violaxanthin biosynthesis with the substrate zeaxanthin.
The D. salina transcriptome presented here includes all of the enzymes required for carotenoid biosynthesis. The reconstructed pathway is consistent with those proposed for plants (Kim et al., 2009) and algae (Steinbrenner and Linden, 2001; Ramos et al., 2011).
3.4. Protective responses of Dunaliella: metabolic adjustments
Metabolite adjustment is a protective response of Dunaliella under high salinity resulting in a large accumulation of osmolytes and other organic solutes (Mishra et al., 2008). To clarify how it impacts metabolic patterns, relative qPCR was used to assess the mRNA abundance of key genes involved in the metabolic processes of osmolytes (gpd1, p5cs, and spds). In our test, gpd1 had a relatively high expression under salt stress. Genes (p5cs and spds) from Pro and polyamine metabolism were found with higher expression under hyper-than under hypo-salinity stress (Fig. 6). Therefore, we conclude that environmental shifts could modify this alga through metabolic adjustments by regulation of the corresponding key genes.
Fig. 6.
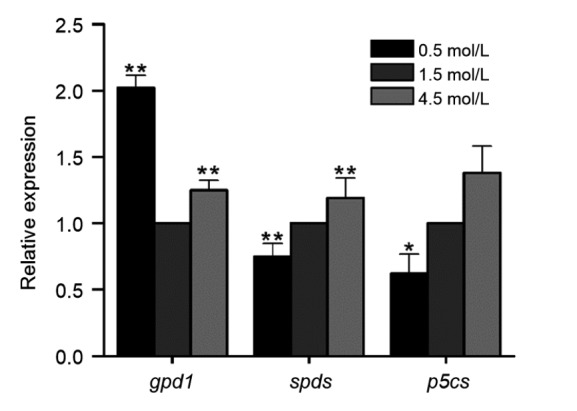
Analyses of relative expression levels of key genes in D. salina metabolic processes
Student’s t-test was used to check the significant differences. * indicates significant difference between salinity stress and control (P<0.05). ** indicates extremely significant difference between them (P<0.01)
4. Discussion
As an osmotic solute, glycerol is an essential molecule to counterbalance environmental osmotic pressure. An increase in salinity promotes intracellular glycerol production within two hours to maintain osmotic homeostasis. Cells recover with osmotic stress-induced gene expression and accumulation of proteins 24 h later (Chen and Jiang, 2009). The carbon for glycerol biosynthesis is obtained mainly from photosynthetic products or the degradation of stored polysaccharides with the latter dominating under increasing salt stress (Goyal, 2007a; 2007b). The key enzyme, GPD1, has two independent functional domains, glycerol-3-phosphate dehydrogenase (G3PDH) and phosphoserine phosphatase (SerB), which localizes in the chloroplast (Cai et al., 2013) and is highly expressed following hyper-or hypo-salinity stress for two hours, same with other results (Chen et al., 2011). Although the enzymes GPD1, DAK, DHR, and GPPase 1 in the glycerol pathway are regulated by salt stress, GPD1 drives the pathway and controls glycerol synthesis (Chen et al., 2012).
As the most diverse and widespread pigments in the chloroplast, carotenoids, including carotene and lutein, are indispensable in light harvesting and energy transfer during photosynthesis and in the protection of the photosynthetic apparatus against photo-oxidative damage (Varela et al., 2015). Dunaliella have an ability to accumulate carotenoids, especially β-carotene located within the chloroplast, under unfavourable conditions (salinity or nutritional deficiency). The new accumulated β-carotene in the plastids is found within newly formed triacylglycerol droplets. It was reported that the formations of these sequestering structures and β-carotene are interdependent (Rabbani et al., 1998). The biosynthesis of carotenoid and triacylglycerol needs the same precursor acyl-CoA. Two acyl-CoA molecules are condensed to generate mevalonate followed by decarboxylation with mevalonate 5-diphosphate decarboxylase to generate isopentenyl diphosphate (IPP). Farnesyl pyrophosphate synthase catalyzes the head-to-tail condensation reaction of dimethylallyl pyrophosphate with two molecules of IPP to form farnesyl pyrophosphate (FPP), which is the important precursor of all sesquiterpenes (Ferriols et al., 2015) such as carotenoids, ergosterol, and coenzyme Q.
The metabolic process of secondary metabolites is on the dynamic balance and metabolic pathways work under precise control (Wang et al., 2012). There are many ways to increase carotenoid production, e.g. by regulation of expression of the key genes, modification of the growth conditions, or screening for mutants with high carotenoid production. Polyamines are involved in stress signaling through intricate crosstalk with abscisic acid (ABA), Ca2+ signaling, and other hormonal pathways in plant defense and development (Marco et al., 2011). With regard to Spd and Spm syntheses, three transcripts coding for SPDS were identified, but none was found for SPS. In C. reinhardtii cells and other green algae (Vovles and Chlorella), no intracellular Spm or only trace amounts were detected (Hamana and Matsuzaki, 1982). Excessive Spm uptake blocks the cell cycle, which can be rescued by subsequent addition of spermidine or putrescine (Theiss et al., 2002). In general, Dunaleilla greatly accumulates valuable metabolic products, such as β-carotene, but shows a restricted capacity to grow under stress (high light, high salinity, or chilling) (García et al., 2007), which makes large-scale farming of Dunaliella challenging. The excessive Spm accumulated in Dunaliella under stress may be one of the causes of growth inhibition. A reduction in the substrates for spermine biosynthesis using inhibitors of S-adenosylmethionine decarboxylase (dsAM, EC: 4.1.1.50) and SPDS (Theiss et al., 2002) would allow Dunaliella to achieve greater economic value in large-scale production under environmental stress.
Our target was to characterize carotenoid metabolism and identify the corresponding enzymes, not to explore the best way to induce carotenogenesis. Although the pathway in D. salina may be the same as in other plants and bacteria, even some algae, this is the first time that a metabolic pathway has been constructed in D. salina through transcriptomic analysis.
In conclusion, the characterization of these metabolic pathways and identification of the relevant key enzymes may enable the manipulation of Dunaliella to produce these valuable metabolites through bioengineering. Most importantly, the first full-scale transcriptome of D. salina at a high resolution lays a foundation for further comparative genomic study.
List of electronic supplementary materials
Data S1 Sequences of the genes identified in D. salina transcriptome
Table S1 Primers of those selective genes involved in the metabolic processes in D. salina
Table S2 Summary of annotation of D. salina transcriptome
Table S3 Top-hit species (viridiplantae) list of D. salina BLAST-annotated uniseqs
Table S4 Enzymes identified in metabolism of osmolytes (glycerol and proline), polyamines, and carotenoid through annotation of D. salina transcriptome
Table S5 The best hit of the highlighted enzymes in the metabolic processes of D. salina
Fig. S1 KOG (euKaryotic Ortholog Groups) functional classification of D. salina uniseqs
Fig. S2 Gene ontology (GO) annotation of D. salina transcriptome
Fig. S3 KEGG functional analyses of D. salina uniseqs
Footnotes
Project supported by the National High-Tech R&D Program (863) of China (No. 2007AA09Z449)
Electronic supplementary materials: The online version of this article (http://dx.doi.org/10.1631/jzus.B1700088) contains supplementary materials, which are available to authorized users
Compliance with ethics guidelines: Ling HONG, Jun-li LIU, Samira Z. MIDOUN, and Philip C. MILLER declare that they have no conflict of interest.
This article does not contain any studies with human or animal subjects performed by any of the authors.
References
- 1.Alkayala F, Albionb RL, Tillettb RL, et al. Expressed sequence tag (EST) profiling in hyper saline shocked Dunaliella salina reveals high expression of protein synthetic apparatus components. Plant Sci. 2010;179(5):437–449. doi: 10.1016/j.plantsci.2010.07.001. [DOI] [PubMed] [Google Scholar]
- 2.Bradbury LMT, Shumskaya M, Tzfadia O, et al. Lycopene cyclase paralog CruP protects against reactive oxygen species in oxygenic photosynthetic organisms. Proc Natl Acad Sci USA. 2012;109:E1888–E1897. doi: 10.1073/pnas.1206002109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Brewster JL, Gustin MC. Hog1: 20 years of discovery and impact. Sci Signal. 2014;7(343):re7. doi: 10.1126/scisignal.2005458. [DOI] [PubMed] [Google Scholar]
- 4.Cai M, He LH, Yu TY. Molecular clone and expression of a NAD+-dependent glycerol-3-phosphate dehydrogenase isozyme gene from the halotolerant alga Dunaliella salina . PLoS ONE. 2013;8(4):e62287. doi: 10.1371/journal.pone.0062287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chen H, Jiang J. Osmotic responses of Dunaliella to the changes of salinity. J Cell Physiol. 2009;219(2):251–258. doi: 10.1002/jcp.21715. [DOI] [PubMed] [Google Scholar]
- 6.Chen H, Lao YM, Jiang JG. Effects of salinities on the gene expression of a (NAD+)-dependent glycerol-3-phosphate dehydrogenase in Dunaliella salina . Sci Total Environ. 2011;409(7):1291–1297. doi: 10.1016/j.scitotenv.2010.12.038. [DOI] [PubMed] [Google Scholar]
- 7.Chen H, Lu Y, Jiang JG. Comparative analysis on the key enzymes of the glycerol cycle metabolic pathway in Dunaliella salina under osmotic stresses. PLoS ONE. 2012;7(6):e37578. doi: 10.1371/journal.pone.0037578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Conesa A, Götz S, García-Gomez JM, et al. Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics. 2005;21(18):3674–3676. doi: 10.1093/bioinformatics/bti610. [DOI] [PubMed] [Google Scholar]
- 9.Couso I, Vila M, Rodriguez H, et al. Overexpression of an exogenous phytoene synthase gene in the unicellular alga Chlamydomonas reinhardtii leads to an increase in the content of carotenoids. Biotechnol Prog. 2011;27(1):54–60. doi: 10.1002/btpr.527. [DOI] [PubMed] [Google Scholar]
- 10.Deng G, Liang J, Xu D, et al. The relationship between proline content, the expression level of P5CS (Δ1-pyrroline-5-carboxylate synthetase), and drought tolerance in tibetan hulless barley (Hordeum vulgare var. nudum) Russ J Plant Physiol. 2013;60(5):693–700. doi: 10.1134/S1021443713050038. [DOI] [Google Scholar]
- 11.Ferriols VMEN, Yaginuma R, Adachi M, et al. Cloning and characterization of farnesyl pyrophosphate synthase from the highly branched isoprenoid producing diatom Rhizosolenia setigera . Sci Rep. 2015;5:10246. doi: 10.1038/srep10246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.García F, Freile-Pelegrin Y, Robledo D. Physiological characterization of Dunaliella sp. (Chlorophyta, Volvocales) from Yucatan, Mexico. Bioresour Technol. 2007;98(7):1359–1365. doi: 10.1016/j.biortech.2006.05.051. [DOI] [PubMed] [Google Scholar]
- 13.Goyal A. Osmoregulation in Dunaliella, part I: effects of osmotic stress on photosynthesis, dark respiration and glycerol metabolism in Dunaliella tertiolecta and its salt-sensitive mutant (HL 25/8) Plant Physiol Biochem. 2007;45(9):696–704. doi: 10.1016/j.plaphy.2007.05.008. [DOI] [PubMed] [Google Scholar]
- 14.Goyal A. Osmoregulation in Dunaliella, Part II: photosynthesis and starch contribute carbon for glycerol synthesis during a salt stress in Dunaliella tertiolecta . Plant Physiol Biochem. 2007;45(9):705–710. doi: 10.1016/j.plaphy.2007.05.009. [DOI] [PubMed] [Google Scholar]
- 15.Grabherr MG, Haas BJ, Yassour M, et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat Biotechnol. 2011;29(7):644–654. doi: 10.1038/nbt.1883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hamana K, Matsuzaki S. Widespread occurrence of norspermidine and norspermine in eukaryotic algae. J Biochem. 1982;91(4):1321–1328. doi: 10.1093/oxfordjournals.jbchem.a133818. [DOI] [PubMed] [Google Scholar]
- 17.Huson DH, Mitra S, Ruscheweyh HJ, et al. Integrative analysis of environmental sequences using MEGAN4. Genome Res. 2011;21(9):1552–1560. doi: 10.1101/gr.120618.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jensen LJ, Julien P, Kuhn M, et al. eggNOG: automated construction and annotation of orthologous groups of genes. Nucleic Acids Res. 2008;36(Databse issue):D250–D254. doi: 10.1093/nar/gkm796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kim J, Smith JJ, Tian L, et al. The evolution and function of carotenoid hydroxylases in Arabidopsis . Plant Cell Physiol. 2009;50(3):463–479. doi: 10.1093/pcp/pcp005. [DOI] [PubMed] [Google Scholar]
- 20.Liu H, Wu W, Hou K, et al. Transcriptome changes in Polygonum multiflorum Thunb. roots induced by methyl jasmonate. J Zhejiang Univ-Sci B (Biomed & Biotechnol) 2015;16(12):1027–1041. doi: 10.1631/jzus.B1500150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liu J, Zhang D, Hong L. Isolation, characterization and functional annotation of the salt tolerance genes through screening the high-quality cDNA library of the halophytic green alga Dunaliella salina (Chlorophyta) Ann Microbiol. 2014;24(3):1293–1302. doi: 10.1007/s13213-014-0967-z. [DOI] [Google Scholar]
- 22.Marco F, Alcázar RN, Tiburcio AF, et al. Interactions between polyamines and abiotic stress pathway responses unraveled by transcriptome analysis of polyamine overproducers. OMICS. 2011;15(11):775–782. doi: 10.1089/omi.2011.0084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mishra A, Mandoli A, Jha B. Physiological characterization and stress-induced metabolic responses of Dunaliella salina isolated from salt pan. J Ind Microbiol Biot. 2008;35(10):1093–1101. doi: 10.1007/s10295-008-0387-9. [DOI] [PubMed] [Google Scholar]
- 24.Mogedas B, Casal C, Forján E, et al. β-Carotene production enhancement by UV-A radiation in Dunaliella bardawil cultivated in laboratory reactors. J Biosci Bioeng. 2009;108(1):47–51. doi: 10.1016/j.jbiosc.2009.02.022. [DOI] [PubMed] [Google Scholar]
- 25.Moriya Y, Itoh M, Okuda S, et al. KAAS: an automatic genome annotation and pathway reconstruction server. Nucleic Acids Res. 2007;35(Suppl. 2):W182–W185. doi: 10.1093/nar/gkm321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mortazavi A, Williams BA, McCue K, et al. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat Methods. 2008;5(7):621–628. doi: 10.1038/nmeth.1226. [DOI] [PubMed] [Google Scholar]
- 27.Rabbani S, Beyer P, Lintig JV, et al. Induced β-carotene synthesis driven by triacylglycerol deposition in the unicellular alga Dunaliella bardawil . Plant Physiol. 1998;116:1239–1248. doi: 10.1104/pp.116.4.1239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rad FA, Aksoz N, Hejazi MA. Effect of salinity on cell growth and β-carotene production in Dunaliella sp. isolates from Urmia Lake in northwest of Ira. Afr J Biotechnol. 2011;10(12):2282–2289. [Google Scholar]
- 29.Ramos AA, Polle J, Tran D, et al. The unicellular green alga Dunaliella salina Teod. as a model for abiotic stress tolerance: genetic advances and future perspectives. Harmful Algae. 2011;26(1):3–20. doi: 10.4490/algae.2011.26.1.003. [DOI] [Google Scholar]
- 30.Rismani-Yazdi H, Haznedaroglu BZ, Bibby K, et al. Transcriptome sequencing and annotation of the microalgae Dunaliella tertiolecta: pathway description and gene discovery for production of next-generation biofuels. BMC Genomics. 2011;12:148. doi: 10.1186/1471-2164-12-148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sathasivam R, Kermanee P, Roytrakul S, et al. Isolation and molecular identification of β-carotene producing strains of Dunaliella salina and Dunaliella bardawil from salt soil samples by using species-specific primers and internal transcribed spacer (ITS) primers. Afr J Biotechnol. 2012;11(102):16677–16687. [Google Scholar]
- 32.Smith DR, Lee RW, Cushman JC, et al. The Dunaliella salina organelle genomes: large sequences, inflated with intronic and intergenic DNA. BMC Plant Biol. 2010;10:14. doi: 10.1186/1471-2229-10-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Steinbrenner J, Linden H. Regulation of two carotenoid biosynthesis genes coding for phytoene synthase and carotenoid hydroxylase during stress-induced astaxanthin formation in the green alga Haematococcus pluvialis . Plant Physiol. 2001;125(2):810–817. doi: 10.1104/pp.125.2.810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Surget-Groba Y, Montoya-Burgos JI. Optimization of de novo transcriptome assembly from next-generation sequencing data. Genome Res. 2010;20:1432–1440. doi: 10.1101/gr.103846.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Theiss C, Bohley P, Voigt J. Regulation by polyamines of ornithine decarboxylase activity and cell division in the unicellular green alga Chlamydomonas reinhardtii . Plant Physiol. 2002;128(4):1470–1479. doi: 10.1104/pp.010896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tian J, Yu J. Changes in ultrastructure and responses of antioxidant systems of algae (Dunaliella salina) during acclimation to enhanced ultraviolet-B radiation. J Photochem Photobiol B. 2009;97(3):152–160. doi: 10.1016/j.jphotobiol.2009.09.003. [DOI] [PubMed] [Google Scholar]
- 37.Tran D, Haven J, Qiu WG, et al. An update on carotenoid biosynthesis in algae: phylogenetic evidence for the existence of two classes of phytoene synthase. Planta. 2009;229(3):723–729. doi: 10.1007/s00425-008-0866-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Varela JC, Pereira H, Vila M, et al. Production of carotenoids by microalgae: achievements and challenges. Photosynth Res. 2015;125:423–436. doi: 10.1007/s11120-015-0149-2. [DOI] [PubMed] [Google Scholar]
- 39.Venekamp JH. Regulation of cytosol acidity in plants under conditions of drought. Physiol Plantarum. 2006;76(1):112–117. doi: 10.1111/j.1399-3054.1989.tb05461.x. [DOI] [Google Scholar]
- 40.Voigt J, Deinert B, Bohley P. Subcellular localization and light-dark control of ornithine decarboxylase in the unicellular green alga Chlamydomonas reinhardtii . Physiol Plant. 2000;108(2000):353–360. doi: 10.1034/j.1399-3054.2000.108004353.x. [DOI] [Google Scholar]
- 41.Wang X, Xia X, Huang F, et al. Genetic modification of secondary metabolite biosynthesis in higher plants: a review. J Biotechnol. 2012;28(10):1151–1163. (in Chinese) [PubMed] [Google Scholar]
- 42.Wang Z, Fang B, Chen J, et al. De novo assembly and characterization of root transcriptome using Illumina paired-end sequencing and development of cSSR markers in sweetpotato (Ipomoea batatas) BMC Genomics. 2010;11:726–739. doi: 10.1186/1471-2164-11-726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Xu DL, Long H, Liang JJ, et al. De novo assembly and characterization of the root transcriptome of Aegilops variabilis during an interaction with the cereal cyst nematode. BMC Genomics. 2012;13:133–141. doi: 10.1186/1471-2164-13-133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhao R, Cao Y, Xu H, et al. Analysis of espressed sequence tags from the green alga Dunaliella salina (Chalrophyta) J Phycol. 2011;47(6):1454–1460. doi: 10.1111/j.1529-8817.2011.01071.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1 Sequences of the genes identified in D. salina transcriptome
Table S1 Primers of those selective genes involved in the metabolic processes in D. salina
Table S2 Summary of annotation of D. salina transcriptome
Table S3 Top-hit species (viridiplantae) list of D. salina BLAST-annotated uniseqs
Table S4 Enzymes identified in metabolism of osmolytes (glycerol and proline), polyamines, and carotenoid through annotation of D. salina transcriptome
Table S5 The best hit of the highlighted enzymes in the metabolic processes of D. salina
Fig. S1 KOG (euKaryotic Ortholog Groups) functional classification of D. salina uniseqs
Fig. S2 Gene ontology (GO) annotation of D. salina transcriptome
Fig. S3 KEGG functional analyses of D. salina uniseqs