

Abstract
Oncolytic viruses are an emerging class of cancer therapeutics that couple cytotoxicity with the induction of an anti-tumor immune response. Host-virus interactions are complex and modulated by a tumor microenvironment whose immunosuppressive activities can limit the effectiveness of cancer immunotherapies. In an effort to improve this aspect of oncolytic virotherapy, we combined the oncolytic herpes virus HSV1716 with the transforming growth factor beta receptor 1 (TGF-βR1) inhibitor A8301 to treat syngeneic models of murine rhabdomyosarcoma. Mice that received HSV1716 or A8301 alone showed little to no benefit in efficacy and survival over controls. Conversely, mice given combination therapy exhibited tumor stabilization throughout the treatment regimen, which was reflected in significantly prolonged survival times including some complete responses. In vitro cell viability and virus replication assays showed that the rhabdomyosarcoma cell lines were generally insensitive to HSV1716 and A8301. Likewise, in vivo virus replication assays showed that HSV1716 titers moderately decreased in the presence of A8301. The enhanced efficacy instead appears to be dependent on the generation of an improved anti-tumor T cell response as determined by its loss in athymic nude mice and following in vivo depletion of either CD4+ or CD8+ cells. These data suggest TGF-β inhibition can augment the immunotherapeutic efficacy of oncolytic herpes virotherapy.
Keywords: oncolytic virotherapy, oncolytic virus, herpes virus, TGF-β, smad signaling, rhabdomyosarcoma, childhood sarcoma, immunotherapy, T cell
Graphical Abstract
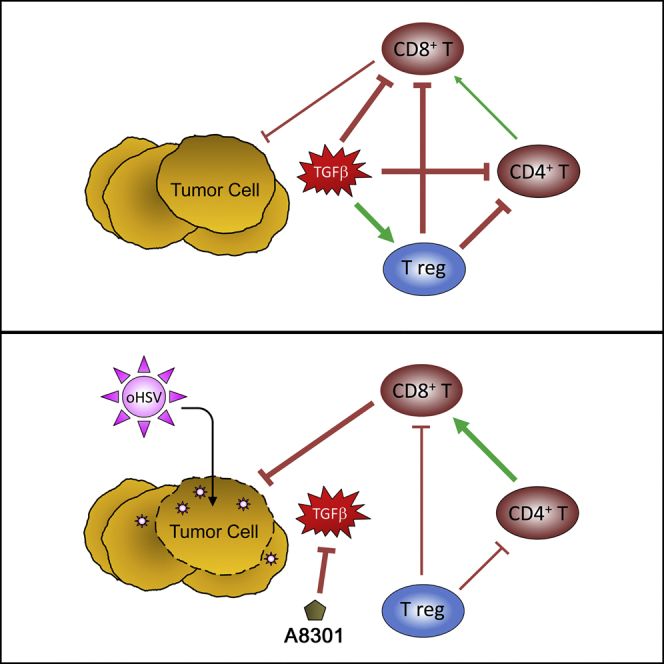
Introduction
Rhabdomyosarcoma (RMS) is the most common soft tissue sarcoma in children. With current standards of care composed of surgery, chemotherapy, and radiation, cure rates can be achieved in greater than 70% of newly diagnosed patients with localized disease.1 The prognosis for patients at relapse or with metastatic disease is decidedly less favorable, however, with overall survival rates of less than 30% despite aggressive multimodal therapy. New and more effective therapies for the treatment of RMS are therefore clearly needed.
We previously reported that human xenograft and immunocompetent transplantable mouse models of RMS are susceptible to oncolytic herpes virotherapy.2, 3, 4, 5, 6 Oncolytic herpes viruses (oHSVs) are members of an emerging class of cancer therapeutics that can selectively infect, replicate in, and lyse tumor cells without causing significant damage to the surrounding normal stroma.7 Although these direct lytic effects are important in themselves, they can also promote an adaptive antitumor immune response that can help eradicate local tumors and distant metastases alike.8, 9 The most pertinent example of this immunologic activity is currently found in Imlygic (Talimogene Laherparepvec), an oHSV that encodes the gene for granulocyte-macrophage colony-stimulating factor and the first oncolytic virus to be approved by the US Food and Drug Administration (FDA) as a cancer therapeutic. In a seminal phase III trial for unresectable recurrent melanoma, Imlygic treatment not only shrank 77.5% of virus-injected lesions, but also 52.3% of non-injected nonvisceral lesions and 29.9% of non-injected visceral lesions.10 Solid tumors have numerous mechanisms to evade antitumor immunity, however, and it has been well established that the immunosuppressive microenvironment they foster can impair the activities of infiltrating immune cells. Modulating the tumor microenvironment to better support virus-induced antitumor immunity thus marks a logical next step toward further improving oncolytic virotherapy. Several preclinical and clinical studies combining oncolytic virotherapy with immune checkpoint inhibitors have since been initiated with this goal in mind.11, 12, 13, 14
Transforming growth factor beta (TGF-β) comprises a family of pleiotropic cytokines whose activities vary by cell type and developmental or disease stage.15 Overexpression of TGF-β is a common occurrence and negative prognostic indicator in many cancers, because it can promote cell migration and invasion, epithelial-to-mesenchymal transition, remodeling of the extracellular matrix, and suppression of both the innate and adaptive arms of the immune system.16, 17, 18 TGF-β is also a known inhibitor of myogenic differentiation, and its overexpression is implicated in RMS tumorigenesis.19, 20 Attempts to “normalize” the tumor microenvironment through inhibition of the TGF-β signaling axis have shown promise as a cancer therapy, and a variety of TGF-β inhibitors are now being investigated in the preclinical setting and early-phase clinical trials.21
In the present study, we examined whether a small-molecule inhibitor of TGF-β receptor 1 (TGF-βR1), known as A8301,22 could potentiate the immunologic activities of HSV1716 (trade name Seprehvir). HSV1716 is an oHSV that is attenuated through deletion of the herpes neurovirulence factor ICP34.5. The loss of this factor, which normally counteracts the interferon and protein kinase R (PKR)-mediated shutdown of virus gene translation, restricts HSV1716 replication to cells in which these pathways are disabled (i.e., tumor cells).23 HSV1716 exhibits lytic activity against a wide variety of cancers and has been safely administered in multiple clinical trials, including an on-site trial for pediatric patients.24, 25 HSV1716 has also been shown to induce multiple immunostimulatory activities, such as the production of pro-inflammatory chemokines, enhanced dendritic cell phagocytosis and antigen presentation, and increased T cell recruitment to the site of the tumor.6, 26, 27 Using immunocompetent models of murine rhabdomyosarcoma (mRMS), we found that A8301 and HSV1716 had only a modest impact on tumor growth and animal survival when administered as single agents. Their combination had a profound antitumor effect, however, culminating in significantly prolonged survival times (and some durable complete responses) despite a precipitous drop in the amount of infectious virus that could be recovered from the injected tumors. We found these antitumor activities to be T cell dependent, because efficacy was nearly abolished when we repeated the studies in athymic nude mice and in C57BL/6 mice following CD4+ or CD8+ T cell depletion. Taken together, our results suggest that TGF-β inhibition can complement oncolytic herpes virotherapy of RMS by promoting an improved antitumor immunological response.
Results
A8301 Inhibits TGF-β-Induced Phosphorylation of SMAD2 In Vitro and In Vivo
We initially confirmed the TGF-β signaling inhibitory activities of A8301 in two established mRMS cell lines. The first cell line, M3-9-M, was originally derived from a male C57BL/6 mouse transgenic for hepatocyte growth factor and heterozygous for mutated p53.28 The second cell line, 76-9, was derived from a methylcholanthrene-induced mRMS tumor in a female C57BL/6 mouse.2 Flow cytometry analysis revealed that each mRMS cell line expressed low but otherwise comparable levels of TGF-βR1 (Figure 1A). We also noted that neither cell line showed detectable levels of TGF-β signaling activity when grown in culture short term, as evidenced by the lack of phosphorylation of SMAD2 (a TGF-β receptor signal transducer). We could induce robust SMAD2 phosphorylation by adding recombinant TGF-β (1 ng/mL, 30 min) to the cell media, however, and subsequently reverse it with the addition of 1 μM A8301 (Figure 1A). We next evaluated the ability of A8301 to inhibit TGF-β signaling in vivo. For these experiments, we injected C57BL/6 mice bearing M3-9-M or 76-9 tumors (gender-matched to tumor origin) with 6 mg/kg A8301 or vehicle control every other day and harvested their tumors for immunoblot analysis on day 7. SMAD2 phosphorylation was evident in the vehicle control-treated tumors of each mRMS model, but was substantially diminished in the tumors treated with A8301 (Figure 1B).
Figure 1.

A8301 Inhibits Smad2 Phosphorylation In Vitro and In Vivo
(A) Cultured M3-9-M and 76-9 cells were exposed to 1 ng/mL recombinant TGF-β for 30 min alone or in the presence of 1 μM A8301. FACS analysis for TGF-βR1 expression was performed with unstimulated cells. (B) Immunocompetent C57BL/6 mice bearing M3-9-M or 76-9 tumors were given intratumoral injections of vehicle control (DMSO in PBS) or 6 mg/kg A8301 every other day for a total of three doses and then sacrificed 24 hr after the final dose.
TGF-β Inhibition Augments Oncolytic Herpes Virotherapy in the M3-9-M and 76-9 Models of mRMS
We next proceeded with in vivo efficacy and survival studies to gauge the effects of TGF-β inhibition on oncolytic herpes virotherapy within the context of a tumor microenvironment. We subcutaneously implanted C57BL/6 mice with M3-9-M or 76-9 mRMS cells and initiated treatment once the tumors had grown to a volume of 200–300 mm3. The mice were then given intratumoral injections of 6 mg/kg A8301 or vehicle control every other day up until day 20, when these treatments were withdrawn. These mice were also given three intratumoral injections of HSV1716 or an equivalent volume of PBS during the first week of their treatment regimen on alternating days with A8301 or vehicle control (Figure 2A).
Figure 2.

Combining A8301 Treatment with HSV1716 Oncolytic Virotherapy Enhances Anti-tumor Efficacy
(A) A schematic of the treatment regimen. Mice were give intratumoral vehicle control or 6 mg/kg A8301 every other day for 20 days. During the first week of treatment, these mice also received three intratumoral 1 × 108 PFU doses of HSV1716 or an equivalent volume of PBS. (B) Average tumor volumes for each treatment group plotted until the first mouse reached endpoint criteria. (C) Kaplan-Meier survival curves demonstrating potency of combined therapy. A log rank test was used to determine statistical significance. n = 5 mice per group for M3-9-M studies; for 76-9 studies, n = 8 mice for vehicle and A8301 groups, and n = 7 for HSV1716 and combination therapy groups. *p < 0.05. Error bars represent SD.
Antitumor efficacy for each treatment group, presented as average tumor volume until the first animal reached endpoint criteria, is shown in Figure 2B. A8301 or HSV1716 treatment only slightly delayed tumor growth compared to the vehicle controls. Their combination, however, resulted in tumor stabilization or shrinkage up until A8301 treatment was withdrawn on day 20, an observation that was consistent for both the M3-9-M and 76-9 models. We found that treating M3-9-M tumors with A8301 or HSV1716 produced a modest, but statistically significant improvement in overall survival relative to the vehicle control (log rank test p values of 0.03 and 0.003, respectively; n = 5 mice per group) (Figure 2C, left panel). Combination therapy was decidedly more efficacious than either treatment alone, resulting in overall survival times that were significantly longer than the next best-performing group (p = 0.01 versus HSV1716 alone). This included one complete response that we subsequently found to be resistant to M3-9-M tumor re-challenge (tumor volume = 0 mm3 at day 200; data not shown). Mice bearing 76-9 tumors responded similarly, exhibiting modest improvements in survival following HSV1716 treatment (p = 0.05 versus vehicle) and striking improvements following combination therapy (p = 0.0004 versus HSV1716). Two animals in this treatment group achieved durable complete responses, resisting 76-9 re-challenge and remaining completely free of tumor until the end of the experiment on day 200 (Figure 2C, right panel; data not shown; n = 6–8 mice per group).
A8301 Does Not Enhance HSV1716-Mediated Cytotoxicity or Virus Replication
After finding that A8301 and HSV1716 combination therapy improved therapeutic outcomes in the M3-9-M and 76-9 models, we sought to further explore the mechanism(s) behind this enhanced efficacy. In order to determine whether A8301 contributed directly to mRMS cell cytotoxicity, we performed in vitro MTS cell viability studies with varying concentrations of A8301 in the presence or absence of HSV1716 infection, which was established by an MOI of 0.5 plaque-forming unit (PFU) per cell. After 96 hr of treatment and/or infection, we noted that A8301 treatment by itself had only a modest impact on M3-9-M and 76-9 cell viability at the concentrations tested. We also found that the efficacy of HSV1716 treatment was neither significantly enhanced nor impaired by the presence of A8301 (Figure 3A). We then conducted a series of plaque assays to determine whether TGF-β or A8301 treatment could in any way alter the kinetics of virus infection and reproduction in vitro. For these experiments, we infected each mRMS cell line with MOI 0.5 HSV1716 alone or in combination with 1 ng/mL TGF-β and/or a 500 nM concentration of A8301. We then collected and quantified the amount of infectious virus particles present at 3, 24, 48, and 72 hr following infection. There was little variance in the kinetics of virus replication between the two cell lines, and neither the addition of TGF-β nor A8301 appreciably altered the amount of recoverable infectious virus relative to their respective controls (Figure 3B). To see whether this observation also held true in vivo, we treated M3-9-M and 76-9 tumor-bearing mice as described in Figure 2A, but opted to follow the kinetics of a single 1 × 108 PFU dose of virus as opposed to the three intermittent doses delivered in the efficacy studies. We sacrificed subsets of mice at 3, 24, 48, 72, and 168 hr after virus injection and quantified the amount of virus recovered from their tumors using standard plaque assays. Contrary to our in vitro findings, we found that co-administration of A8301 significantly diminished HSV1716 replication in both the M3-9-M and 76-9 models, in some cases decreasing virus titers by as much as a full log relative to the HSV1716-only controls (Figure 3C). Taken together, these data suggest A8301-mediated enhancement of cytotoxicity or virus replication and/or persistence is not responsible for the improved efficacy of combination therapy.
Figure 3.

Efficacy of Combination Therapy Is Unlikely Due to A8301 Cytotoxic Effects or Enhanced Virolytic Activity
(A) MTS viability assays for M3-9-M or 76-9 cells treated with listed concentrations of A8301 with and without HSV1716 infection (MOI 0.5). These data are from a 96 hr time point postinfection. Samples were run in triplicate. (B) In vitro virus replication assays for M3-9-M and 76-9 cells treated with vehicle control, 1 ng/mL TGF-β, and/or 500 nM A8301. The cells were treated at the time of HSV1716 infection (MOI 0.5) and again at 48 hr. Neither exogenous TGF-β nor A8301 significantly altered the kinetics of virus replication. Samples were run in triplicate. (C) In vivo virus replication assays in mice bearing M3-9-M or 76-9 tumors. A8301 treatment significantly diminishes HSV1716 replication. Four tumors from each treatment group were collected and assayed in triplicate. Two-tailed t tests were used to determine statistical significance between treatment groups at each time point. *p < 0.05. Error bars represent SD.
Improved Efficacy of A8301 and HSV1716 Combination Therapy Is T Cell Dependent
Because the inhibitory properties of TGF-β on the adaptive immune system are well-known,16 we hypothesized that A8301-mediated reversal of T cell suppression could be a major contributing factor for the improved therapeutic response. To examine the importance of T cells in this response, we repeated our M3-9-M and 76-9 efficacy and survival studies in athymic nude mice. Contrary to our earlier observations in the immunocompetent C57BL/6 models, A8301 and HSV1716 combination therapy had only a marginal impact on slowing tumor growth and prolonging survival in the M3-9-M nude mouse model (Figures 4A and 4B, left column; p = 0.02 versus vehicle control; n = 5 mice per group) and no impact on the 76-9 model (Figures 4A and 4B, right column; n = 5 mice per group). To further dissect what population of T cells was mediating this effect, we used antibodies to specifically deplete CD4+ and/or CD8+ T cells in 76-9 tumor-bearing C57BL/6 mice undergoing combination therapy or given vehicle control (Figure 5A). Whereas the average tumor growth curves for mice injected with an isotype control antibody showed a clear separation between the vehicle and combination therapy groups, this distinction was lost or greatly diminished in mice that were depleted of CD4+ or CD8+ T cells, suggesting that both of these populations of T cells are necessary for the enhanced efficacy of our combination therapy (Figure 5B; n = 4 mice per group). Representative fluorescence-activated cell sorting (FACS) analyses confirming the specific depletion of each T cell subtype in the tumors are shown in Figure 5C.
Figure 4.

The Efficacy of Combination Therapy Is Largely Dependent on a T Cell Response
(A) Average tumor volumes for athymic nude mice implanted with M3-9-M or 76-9. These mice were treated with A8301, HSV1716, or the combination thereof as outlined in Figure 2. The data are plotted until the first mouse in each respective treatment group reached endpoint criteria. n = 5 mice per group. (B) Kaplan-Meier survival curves for the athymic nude mice in (A). Error bars represent SD.
Figure 5.

CD4 and CD8 T Cells Contribute to the Efficacy of Combination Therapy in the 76-9 mRMS Model
(A) Treatment regimen of C57Bl-6 mice bearing 76-9 tumors. Mice were given intratumoral vehicle control or 6 mg/kg A8301 every other day for 12 days along with intermittent doses of 1 × 108 PFU HSV1716 or PBS. Subsets of these vehicle and combination therapy mice were also i.p. administered CD4 or CD8 T cell-depleting or isotype control antibodies every 4 days (500 μg antibody per injection). (B) Average tumor growth curves. Statistical significance was determined with two-way ANOVA tests (n = 4 mice per group; *p ≤ 0.05). (C) Representative FACS analysis of tumors isolated from each treatment group demonstrating specificity of T cell-depleting antibodies. Numerical values represent percentage of CD4- or CD8-positive T cells out of CD3-expressing cells. Error bars represent SD.
HSV1716 Treatment Increases CD8 T Cell Recruitment and Lowers Incidence of Regulatory T Cells
After determining that T cells were critical mediators of the enhanced therapeutic effect, we next wanted to examine whether A8301 treatment could modulate their recruitment to the site of the tumor. For these studies, we once again established 76-9 tumors on the flanks of female C57BL/6 mice and treated them as described previously in Figure 2A. These mice were then sacrificed 72 hr after the third and final dose of HSV1716, upon which their tumors were collected and processed into single-cell suspensions for FACS analysis. Treatment groups receiving HSV1716, regardless of other therapy, exhibited a significantly higher influx of CD3ε+ T cells to the site of the tumor (Figure 6A). Although the percentage of total CD4+ T cells remained steady across groups (Figure 6B), the proportion of these cells displaying a regulatory phenotype (regulatory T cell [Treg]; defined here as CD4+/Foxp3+/CD25+) was significantly reduced in the virus-treated groups (Figure 6C). Likewise, the influx of total CD8+ T cells and CD44+ effector memory CD8+ T cells was significantly increased in these same treatment groups (Figures 6D and 6E, respectively). Additional staining with an HSV glycoprotein B (GB) tetramer suggests that many of these were anti-HSV CD8+ T cells (Figure 6F). Nonetheless, the overall ratio of CD8+ T cells to Tregs (which is associated with improved therapeutic outcomes29) remained significantly higher in the treatment groups that received HSV1716 (Figure 6G).
Figure 6.

HSV1716 Treatment Increases CD8+ T Cell Infiltration and Lowers Incidence of Regulatory T Cells
Mice bearing 76-9 tumors were treated as previously described and sacrificed 3 days after the final dose of HSV1716 or PBS control. Single-cell suspensions were obtained from isolated tumors, stained, and then analyzed via flow cytometry. (A) Total T cell infiltrates presented as a percentage of total living cells. (B) Percentage of CD4+ T cells. (C) Percentage of total CD4+ T cells displaying a T regulatory cell phenotype (Foxp3+/CD25+). (D) Percentage of CD8+ T cells. (E) Percentage of CD8+ cells displaying a memory T cell phenotype (CD44+). (F) Percentage of CD8+ T cells specific for HSV1716 glycoprotein B (GB). (G) The ratio of CD8+ T cells to T regulatory cells. n = 4 tumors were analyzed for each group, with the exception of A8301+HSV1716, which had n = 7. Statistical significance was determined with one-way ANOVA (*p = 0.05–0.005; **p = 0.005–0.0005; ***p < 0.0005). Error bars represent SD.
Discussion
In this manuscript, we demonstrated that localized TGF-β inhibition could potentiate oncolytic herpes viroimmunotherapy in murine models of RMS. The efficacy of this approach is most likely rooted in the reversal or alleviation of the immunosuppressive tumor microenvironment, which serves to strengthen virus-mediated immunological activities. Both oHSV effects and TGF-β inhibitory effects were necessary for this improved therapeutic activity to occur, because HSV1716 and A8301 given as a monotherapy showed only limited efficacy. Although the combination of these agents ultimately proved to be beneficial in our mRMS survival studies, the case for TGF-β inhibition in oncolytic virotherapy is presently somewhat controversial. In a recent report by Han and colleagues,30 TGF-β inhibition was shown to have a negative impact on oHSV treatment of a syngeneic mouse model glioblastoma because it reduced viral titers and overall survival times. Instead, mice that were given a single orthotopic injection of exogenous TGF-β1 prior to oHSV administration fared better than all other experimental groups, an observation the authors attributed to the attenuated abilities of natural killer cells and macrophages/microglia to clear oHSV-infected glioblastoma cells.30 Although we did not examine the functionality of innate immune cells in our tumor models following TGF-β inhibition, it seems reasonable that the de-repression of their activities could be responsible for the decreased titers of HSV1716 we obtained from tumors co-treated with A8301, especially as the early time points when these samples were collected would likely preclude the induction of an anti-virus T cell response (Figure 3C).
The notion that TGF-β signaling in the tumor microenvironment can be beneficial to oncolytic virotherapy is also supported by a recent publication from Ilkow et al.,31 who found that TGF-β1 mediates reciprocal communication between cancer cells and cancer-associated fibroblasts (CAFs) to promote oncolytic virus replication and cytolytic activity. Although TGF-β1 treatment had no direct impact on the ability of cancer cells to support virus replication, it could sensitize normal fibroblasts and CAFs to oncolytic virus (OV) infection, leading to increased secretion of fibroblast growth factor 2 (FGF2) in the tumor milieu. The authors subsequently showed that this increase in FGF2 was responsible for antagonizing the activities of RIG-1 (a double-stranded RNA helicase that activates interferon response pathways upon sensing cytosolic viruses32) in cancer cells, thereby further sensitizing them to oncolytic virus infection. Although the authors did not specifically inhibit or knockout TGF-β signaling activity within their model systems, it is likely that such an action would diminish OV infection of CAFs and the concomitant FGF2-mediated infection of the cancer cells themselves.
In addition to our present report, there is also other emerging evidence that suggests TGF-β inhibition can benefit oncolytic virotherapy. Hu et al.33, 34 showed that an oncolytic adenovirus expressing a TGF-β ligand trap (a soluble fusion protein of TGF-β receptor 2 and the Fc fragment of human immunoglobulin) was more efficient than the parental virus at inhibiting bone metastasis and osteolysis in nude mouse models of breast and prostate cancers. In a follow-up paper, Zhang et al.35 found that variants of this armed adenovirus were similarly effective in a syngeneic 4T1 mouse mammary bone metastasis model following their systemic administration. While the combination of both replicating virus and TGF-β signaling inhibition were critical for the antitumor effects, detailed mechanistic analysis on how these therapies complement one another remain forthcoming.
Taking these various studies into account, TGF-β inhibition can perhaps be considered somewhat of a double-edged sword for oncolytic virotherapy. It is possible that the efficacy of this approach will be tumor specific; inhibiting TGF-β in tumors that are more responsive to direct oncolysis could potentially be counterproductive, whereas tumors that are more susceptible to immune cell clearance may respond better in the less immunosuppressive microenvironment that TGF-β inhibition might provide. The challenge, then, would be to identify biomarkers to help determine which tumors stand to benefit the most from these treatments and devise means to enhance those specific aspects of the therapy accordingly. Another possibility to consider is that TGF-β signal activation and TGF-β signal inhibition can potentiate oncolytic virotherapy in tandem, provided the means of doing so are administered with proper staging. Although our combination therapy regimen resulted in significantly improved survival times (Figure 2), we could potentially optimize it further by delaying A8301 treatment or even administering exogenous TGF-β prior to HSV1716 injection. In theory, this could bolster early virolytic effects (or at least allow them to proceed relatively unhindered), setting the stage for perhaps even greater anti-tumor immunologic activity after the initiation of A8301 treatment.
One caveat of the present study is the generally non-permissive nature of mouse tumors to oHSV replication and persistence.2 Both mRMS models displayed minimal HSV1716 replication and rapid clearance of infectious virus regardless of other treatment (Figure 3C). As such, they may not fully recapitulate the effects and extent of direct oncolysis that can occur in more permissive human tumors, which in turn may impact the added utility of TGF-β inhibition. Previous studies have shown that human RMS tumors exhibit a range of sensitivity to oHSV, however, with the least susceptible models being roughly on par with the mRMS lines used here.2, 3 Although further study is clearly needed, we might surmise that our findings will be more reflective of less susceptible human tumors than those readily infected and lysed by oHSV. Going forward, we may be able to address this issue through the creation of a mouse-adapted oHSV or by substituting HSV1716 for a virus that displays a greater tropism for murine hosts.
Although we have demonstrated that the efficacy of our combination therapy in mRMS is T cell dependent (Figures 4 and 5), there are several unresolved questions as to how TGF-β inhibition and HSV1716 ultimately contribute to the therapeutic response. The inhibitory effects of TGF-β on cytotoxic T lymphocyte proliferation and function are well-documented,36 as is TGF-β’s ability to drive naive CD4+ T cells toward a regulatory T cell phenotype.37 In our own T cell recruitment studies, we observed no significant differences between the amount of CD8+ T cells or Tregs recruited to 76-9 tumors treated with HSV1716 alone or in combination with A8301 (Figure 6). One hypothesis to account for the disparity we observe between these groups in therapeutic outcomes is that the improved efficacy is not due to the absolute number of T cells recruited to the tumor, but rather their activation status once there. However, because these studies were performed at a single time point postinfection, it is also possible that we simply missed any differences before they had a chance to become more pronounced or sustained. Although we might speculate that A8301 is simply reversing factors that contribute to T cell immunosuppression, understanding what factors and cell populations are involved, when they are involved, and how they intersect with virus-associated immunologic events should yield valuable clues for further optimizing oncolytic viroimmunotherapy going forward. In the meantime, our present findings suggest that combining TGF-β inhibition with oHSV may be a novel and more effective treatment for RMS, which warrants further study.
Materials and Methods
Cell Lines and Viruses
Murine RMS cell lines M3-9-M and 76-9 were the kind gifts of Dr. Crystal Mackall (Stanford Cancer Institute) and Dr. Brenda Weigel (University of Minnesota), respectively. All mRMS cell lines were maintained in RPMI 1640 supplemented with 15% heat-inactivated fetal bovine serum (FBS), 1% L-glutamine, 1% nonessential amino acids, 50 μM 2-mercaptoethanol, and 100 IU/mL penicillin and 100 μg/mL streptomycin. Vero cells (ATCC) were cultured in DMEM supplemented with 10% FBS and penicillin/streptomycin. All cell lines were verified to be free of mycoplasma contamination by the MycoAlert Mycoplasma Detection Kit (LT07-318; Lonza, Allendale, NJ, USA) prior to use. The oHSV HSV1716 was provided by Virttu Biologics (Glasgow, UK).
TGF-β Inhibitor A8301
A8301 was purchased from Sigma-Aldrich (SML0788; St. Louis, MO, USA), reconstituted in DMSO to a final concentration of 5 mg/mL, and aliquoted and stored at −20°C. Recombinant TGF-β was purchased from PeproTech (100-21B; Rocky Hill, NJ, USA) and stored at −20°C.
Immunoblot Analysis
Whole-cell lysates and flash-frozen and pulverized tumor samples were prepared in cell lysis buffer (9803; Cell Signaling Technology, Danvers, MA, USA) supplemented with Protease Inhibitor Cocktail (78415; Fisher Scientific, Waltham, MA, USA) and Halt Phosphatase Inhibitor Cocktail (P-78420; Fisher Scientific). Protein concentrations were determined via the Micro BCA Protein Assay Kit (23235; Fisher Scientific), and 50 μg of total protein per sample was resolved on NuPAGE 4%–12% Bis-Tris pre-cast gels (NP0336BOX; Fisher Scientific). The proteins were then transferred to polyvinylidene fluoride (PVDF) membrane, blocked for 30 min with 5% bovine serum albumin in Tris-buffered saline with 0.2% Tween 20 (TBST), and probed overnight at 4°C with the following antibodies (1:1,000 dilution) purchased from Cell Signaling Technology: phospho-Smad2 Ser465/467 (3108S), Smad2 (3122S), and glyceraldehyde 3-phosphate dehydrogenase (GAPDH; 2118S). The membranes were washed three times in TBST and then probed with a secondary antibody conjugated to horseradish peroxidase (7074S). After another series of washes in TBST, the membranes were developed using Western Lightning Plus-ECL reagent (NEL103001EA; Perkin Elmer, Waltham, MA, USA) and exposed on X-ray film. Blots for phospho-Smad2 (60 kDa) and GAPDH (37 kDa) were probed and developed concurrently. The membranes were then stripped with OneMinute Western Blot Stripping Buffer (GM6001; GM Biosciences, Frederick, MD, USA) and re-probed for total Smad2.
Cell Viability (MTS) Assay
Cells were plated in 96-well dishes at a density of 3,000 cells/well and incubated overnight at 37°C prior to treatment and/or HSV1716 infection. Viability assays were performed 96 hr post-treatment using the Cell Titer 96 Aqueous Non-Radioactive Cell Proliferation Assay (G5421; Promega, Madison, WI, USA). Samples were run in triplicate, and the data shown are representative of three independent experiments. Results are presented as percent cell survival relative to uninfected and/or vehicle-treated controls. Error bars represent SD.
Animal Studies
All animal studies were approved by Nationwide Children’s Hospital Institutional Animal Care and Use Committee (protocol AR12-00074). Tumors were established by subcutaneously implanting 5 × 106 mRMS cells into the flanks of 6-week-old C57BL/6 mice or athymic nude mice (Envigo, Frederick, MD, USA). Tumor sizes were measured every other day, and volumes were calculated using length × width2 × π/6, as described previously.2 When tumors reached ∼200–300 mm3 in size, the mice were pooled and divided into treatment groups to have comparable average tumor burdens. Animals with tumor burdens that exceeded ∼200–300 mm3 in size, or who did not reach this threshold within a week of their cohorts, were excluded from further study.
Mice were given intratumoral injections of A8301 (6 mg/kg in DMSO + PBS, total volume = 50 μL) or a DMSO + PBS vehicle control every other day for a total of 10 treatments. During the first week, on alternating days, the mice were also given fractionated doses of HSV1716 intratumorally (1 × 108 PFU in 50 μL) or PBS (see Figure 2A). Tumor sizes were measured, unblinded, until they reached a volume of 2500 mm3 or a diameter of 2 cm upon which the animal was euthanized. Mice determined to be tumor free were subsequently re-challenged with implant of 5 × 106 mRMS cells in their contralateral flanks and observed until 200 days postinitiation of treatment.
Cell Depletion Studies
T cell depletion studies were conducted by intraperitoneally (i.p.) injecting 500 μg of anti-CD4 (GK1.5) and/or anti-CD8 (YTS169.4) antibody into C57BL/6 mice bearing bi-flank M3-9-M or 76-9 tumors every 96 hr. Isotype control mice were similarly injected with 500 μg of anti-Phytopthora IgG AFRC MAC 51 antibody. Specific depletion of the respective T cell subtypes was confirmed by flow cytometry analysis.
Flow Cytometry Analysis
Flow cytometric analyses were conducted as described previously.6 In brief, single-cell suspensions from tumors were prepared and lysed with ACK red blood cell lysis buffer (Lonza) and blocked with 5% mouse Fc blocking reagent (2.4G2; BD Biosciences, San Jose, CA, USA) in FACS buffer (1% FBS and 1 mM EDTA in PBS). These cells were then washed and labeled on ice for 30 min with one of the following antibody staining panels for analysis of infiltrating immune cells: Panel 1, CD4-allophycocyanin (APC) (GK1.5), CD25-phycoerythrin (PE) (7D4), CD8a-PE-Cy7 (53-6.7), CD3ε-Violet 421 (145-2C11), NK1.1-PerCP and B220-APC-Cy7 (RA3-6B2); Panel 2, CD4-fluorescein isothiocyanate (FITC), CD44-PE-Cy7 (IM7), CD8a-PE, CD3ε-Violet 421, NK1.1-PerCP, and HSV-GB-TETRAMER-APC. HSV glycoprotein B (GB) monomer (SSIEFARL) was obtained from the NIH tetramer core facility (Human B2M H-2Kb). After labeling, the cells were washed, fixed in 1% paraformaldehyde, and a minimum of 100,000 events were collected and analyzed on a BD FACS LSR II (BD Biosciences). Analysis was carried out using the FlowJo software, version 10.0.3 (Tree Star, Ashland, OR, USA).
For Foxp3 intracellular staining, mononuclear cells were enriched by Percoll (GE Healthcare Bio-Sciences, Pittsburgh, PA, USA) density gradient centrifugation and stained with cell surface markers including CD4-APC, CD8-PE-Cy7, CD25-PE, NK1.1-PerCP, B220-APC-Cy7, and CD3ε-Violet 421 followed by Foxp3-FITC (FJK-16 s) intracellular staining using a cell fixation and permeabilization kit (Invitrogen GAS001S100 and GAS002S100; Thermo Fisher Scientific). Stained cells were fixed in 0.5% paraformaldehyde but otherwise collected and analyzed as described above. All the staining antibodies were purchased from BioLegend (San Diego, CA, USA) except for anti-Foxp3 (eBioscience, San Diego, CA, USA), anti-CD25 (BD Biosciences), and TGF-βR1 (R&D Systems, Minneapolis, MN, USA).
Virus Replication Assays
Mouse RMS cells were seeded in 12-well plates at densities to ensure approximately 80% confluency the following day. They were then infected with MOI 0.5 HSV1716 in the presence or absence of 1 ng/mL TGF-β or 500 nM A8301 (1 mL total volume). Fresh TGF-β or A8301 was added after 48 hr. After 3, 24, 48, and 72 hr, the plates were freeze-thawed on dry ice three times, and the media and lysed cells from each well were transferred to 1.5 mL tubes and centrifuged at 1,500 × g for 10 min to pellet debris. Serial dilutions of these supernatants were then titrated on Vero cells by standard plaque assay. Each sample was run and titrated in triplicate.
For in vivo virus replication studies, mice were treated as described above but given only a single intratumoral dose of 1 × 108 PFU HSV1716. They were then sacrificed at 3, 24, 48, 72, and 168 hr post-infection. Tumors were isolated, sectioned, and transferred to 50 mL conical tubes containing 1 mL of DMEM, where they subsequently homogenized with a mechanical disruptor. These samples were then freeze-thawed on dry ice three times and centrifuged at 1,500 × g for 10 min to pellet debris. The supernatants were collected for titration on Vero cells per established protocol.2, 5 An n = 4 for each sample and time point was collected and titrated in triplicate. Error bars represent SD.
Statistical Analysis
All statistical analysis was performed with GraphPad Prism 7.0a for Mac OS X (GraphPad Software, La Jolla, CA, USA).
Author Contributions
Conceptualization, C.-Y.C. and T.P.C.; Methodology, B.H. and C.-Y.C.; Investigation, B.H., P.-Y.W., L.S., and H.M.S.; Visualization, B.H.; Writing – Original Draft, B.H.; Writing – Review & Editing, B.H., C.-Y.C., P.-Y.W., L.B., and T.P.C.; Resources, J.C., J.L., and L.B.; Supervision, C.-Y.C. and T.P.C.; Funding Acquisition, T.P.C.
Conflicts of Interest
The authors declare no conflicts of interest with the exception of J.C., who is an employee of Virttu Biologics, Ltd.
Acknowledgments
Funding for this project was provided by a start-up grant awarded to T.C. by the Research Institute at Nationwide Children’s Hospital.
References
- 1.Smith M.A., Altekruse S.F., Adamson P.C., Reaman G.H., Seibel N.L. Declining childhood and adolescent cancer mortality. Cancer. 2014;120:2497–2506. doi: 10.1002/cncr.28748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Leddon J.L., Chen C.Y., Currier M.A., Wang P.Y., Jung F.A., Denton N.L., Cripe K.M., Haworth K.B., Arnold M.A., Gross A.C. Oncolytic HSV virotherapy in murine sarcomas differentially triggers an antitumor T-cell response in the absence of virus permissivity. Mol Ther Oncolytics. 2015;1:14010. doi: 10.1038/mto.2014.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Currier M.A., Adams L.C., Mahller Y.Y., Cripe T.P. Widespread intratumoral virus distribution with fractionated injection enables local control of large human rhabdomyosarcoma xenografts by oncolytic herpes simplex viruses. Cancer Gene Ther. 2005;12:407–416. doi: 10.1038/sj.cgt.7700799. [DOI] [PubMed] [Google Scholar]
- 4.Eshun F.K., Currier M.A., Gillespie R.A., Fitzpatrick J.L., Baird W.H., Cripe T.P. VEGF blockade decreases the tumor uptake of systemic oncolytic herpes virus but enhances therapeutic efficacy when given after virotherapy. Gene Ther. 2010;17:922–929. doi: 10.1038/gt.2010.82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Currier M.A., Gillespie R.A., Sawtell N.M., Mahller Y.Y., Stroup G., Collins M.H., Kambara H., Chiocca E.A., Cripe T.P. Efficacy and safety of the oncolytic herpes simplex virus rRp450 alone and combined with cyclophosphamide. Mol. Ther. 2008;16:879–885. doi: 10.1038/mt.2008.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chen C.Y., Wang P.Y., Hutzen B., Sprague L., Swain H.M., Love J.K., Stanek J.R., Boon L., Conner J., Cripe T.P. Cooperation of oncolytic herpes virotherapy and PD-1 blockade in murine rhabdomyosarcoma models. Sci. Rep. 2017;7:2396. doi: 10.1038/s41598-017-02503-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Saha D., Wakimoto H., Rabkin S.D. Oncolytic herpes simplex virus interactions with the host immune system. Curr. Opin. Virol. 2016;21:26–34. doi: 10.1016/j.coviro.2016.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Keller B.A., Bell J.C. Oncolytic viruses-immunotherapeutics on the rise. J. Mol. Med. (Berl.) 2016;94:979–991. doi: 10.1007/s00109-016-1453-9. [DOI] [PubMed] [Google Scholar]
- 9.Andtbacka R.H., Ross M., Puzanov I., Milhem M., Collichio F., Delman K.A., Amatruda T., Zager J.S., Cranmer L., Hsueh E. Patterns of clinical response with talimogene laherparepvec (T-VEC) in patients with melanoma treated in the OPTiM phase III clinical trial. Ann. Surg. Oncol. 2016;23:4169–4177. doi: 10.1245/s10434-016-5286-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Andtbacka R.H., Kaufman H.L., Collichio F., Amatruda T., Senzer N., Chesney J., Delman K.A., Spitler L.E., Puzanov I., Agarwala S.S. Talimogene laherparepvec improves durable response rate in patients with advanced melanoma. J. Clin. Oncol. 2015;33:2780–2788. doi: 10.1200/JCO.2014.58.3377. [DOI] [PubMed] [Google Scholar]
- 11.Rajani K.R., Vile R.G. Harnessing the power of onco-immunotherapy with checkpoint inhibitors. Viruses. 2015;7:5889–5901. doi: 10.3390/v7112914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Woller N., Gürlevik E., Fleischmann-Mundt B., Schumacher A., Knocke S., Kloos A.M., Saborowski M., Geffers R., Manns M.P., Wirth T.C. Viral infection of tumors overcomes resistance to PD-1-immunotherapy by broadening neoantigenome-directed T-cell responses. Mol. Ther. 2015;23:1630–1640. doi: 10.1038/mt.2015.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cassady K.A., Haworth K.B., Jackson J., Markert J.M., Cripe T.P. To Infection and beyond: the multi-pronged anti-cancer mechanisms of oncolytic viruses. Viruses. 2016;8:8. doi: 10.3390/v8020043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Puzanov I., Milhem M.M., Minor D., Hamid O., Li A., Chen L., Chastain M., Gorski K.S., Anderson A., Chou J. Talimogene laherparepvec in combination with ipilimumab in previously untreated, unresectable stage IIIB-IV melanoma. J. Clin. Oncol. 2016;34:2619–2626. doi: 10.1200/JCO.2016.67.1529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Akhurst R.J., Hata A. Targeting the TGFβ signalling pathway in disease. Nat. Rev. Drug Discov. 2012;11:790–811. doi: 10.1038/nrd3810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yoshimura A., Muto G. TGF-β function in immune suppression. Curr. Top. Microbiol. Immunol. 2011;350:127–147. doi: 10.1007/82_2010_87. [DOI] [PubMed] [Google Scholar]
- 17.Pickup M., Novitskiy S., Moses H.L. The roles of TGFβ in the tumour microenvironment. Nat. Rev. Cancer. 2013;13:788–799. doi: 10.1038/nrc3603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Padua D., Massagué J. Roles of TGFbeta in metastasis. Cell Res. 2009;19:89–102. doi: 10.1038/cr.2008.316. [DOI] [PubMed] [Google Scholar]
- 19.Bouché M., Canipari R., Melchionna R., Willems D., Senni M.I., Molinaro M. TGF-beta autocrine loop regulates cell growth and myogenic differentiation in human rhabdomyosarcoma cells. FASEB J. 2000;14:1147–1158. doi: 10.1096/fasebj.14.9.1147. [DOI] [PubMed] [Google Scholar]
- 20.Wang S., Guo L., Dong L., Guo L., Li S., Zhang J., Sun M. TGF-beta1 signal pathway may contribute to rhabdomyosarcoma development by inhibiting differentiation. Cancer Sci. 2010;101:1108–1116. doi: 10.1111/j.1349-7006.2010.01512.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Neuzillet C., Tijeras-Raballand A., Cohen R., Cros J., Faivre S., Raymond E., de Gramont A. Targeting the TGFβ pathway for cancer therapy. Pharmacol. Ther. 2015;147:22–31. doi: 10.1016/j.pharmthera.2014.11.001. [DOI] [PubMed] [Google Scholar]
- 22.Tojo M., Hamashima Y., Hanyu A., Kajimoto T., Saitoh M., Miyazono K., Node M., Imamura T. The ALK-5 inhibitor A-83-01 inhibits Smad signaling and epithelial-to-mesenchymal transition by transforming growth factor-beta. Cancer Sci. 2005;96:791–800. doi: 10.1111/j.1349-7006.2005.00103.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.MacKie R.M., Stewart B., Brown S.M. Intralesional injection of herpes simplex virus 1716 in metastatic melanoma. Lancet. 2001;357:525–526. doi: 10.1016/S0140-6736(00)04048-4. [DOI] [PubMed] [Google Scholar]
- 24.Cripe, T. (2009). HSV1716 in patients with non-central nervous system (non-CNS) solid tumors. Report of the U.S. National Institutes of Health. May 2009, NCT00931931. http://clinicaltrials.gov/show/NCT00931931.
- 25.Streby K.A., Geller J.I., Currier M.A., Warren P.S., Racadio J.M., Towbin A.J., Vaughan M.R., Triplet M., Ott-Napier K., Dishman D.J. Intratumoral injection of HSV1716, an oncolytic herpes virus, is safe and shows evidence of immune response and viral replication in young cancer patients. Clin. Cancer Res. 2017;23:3566–3574. doi: 10.1158/1078-0432.CCR-16-2900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Benencia F., Courrèges M.C., Conejo-García J.R., Mohamed-Hadley A., Zhang L., Buckanovich R.J., Carroll R., Fraser N., Coukos G. HSV oncolytic therapy upregulates interferon-inducible chemokines and recruits immune effector cells in ovarian cancer. Mol. Ther. 2005;12:789–802. doi: 10.1016/j.ymthe.2005.03.026. [DOI] [PubMed] [Google Scholar]
- 27.Benencia F., Courrèges M.C., Fraser N.W., Coukos G. Herpes virus oncolytic therapy reverses tumor immune dysfunction and facilitates tumor antigen presentation. Cancer Biol. Ther. 2008;7:1194–1205. doi: 10.4161/cbt.7.8.6216. [DOI] [PubMed] [Google Scholar]
- 28.Meadors J.L., Cui Y., Chen Q.R., Song Y.K., Khan J., Merlino G., Tsokos M., Orentas R.J., Mackall C.L. Murine rhabdomyosarcoma is immunogenic and responsive to T-cell-based immunotherapy. Pediatr. Blood Cancer. 2011;57:921–929. doi: 10.1002/pbc.23048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mandl S.J., Rountree R.B., Dalpozzo K., Do L., Lombardo J.R., Schoonmaker P.L., Dirmeier U., Steigerwald R., Giffon T., Laus R., Delcayre A. Immunotherapy with MVA-BN®-HER2 induces HER-2-specific Th1 immunity and alters the intratumoral balance of effector and regulatory T cells. Cancer Immunol. Immunother. 2012;61:19–29. doi: 10.1007/s00262-011-1077-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Han J., Chen X., Chu J., Xu B., Meisen W.H., Chen L., Zhang L., Zhang J., He X., Wang Q.E. TGFβ treatment enhances glioblastoma virotherapy by inhibiting the innate immune response. Cancer Res. 2015;75:5273–5282. doi: 10.1158/0008-5472.CAN-15-0894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ilkow C.S., Marguerie M., Batenchuk C., Mayer J., Ben Neriah D., Cousineau S., Falls T., Jennings V.A., Boileau M., Bellamy D. Reciprocal cellular cross-talk within the tumor microenvironment promotes oncolytic virus activity. Nat. Med. 2015;21:530–536. doi: 10.1038/nm.3848. [DOI] [PubMed] [Google Scholar]
- 32.Yoneyama M., Kikuchi M., Natsukawa T., Shinobu N., Imaizumi T., Miyagishi M., Taira K., Akira S., Fujita T. The RNA helicase RIG-I has an essential function in double-stranded RNA-induced innate antiviral responses. Nat. Immunol. 2004;5:730–737. doi: 10.1038/ni1087. [DOI] [PubMed] [Google Scholar]
- 33.Hu Z., Zhang Z., Guise T., Seth P. Systemic delivery of an oncolytic adenovirus expressing soluble transforming growth factor-β receptor II-Fc fusion protein can inhibit breast cancer bone metastasis in a mouse model. Hum. Gene Ther. 2010;21:1623–1629. doi: 10.1089/hum.2010.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hu Z., Gupta J., Zhang Z., Gerseny H., Berg A., Chen Y.J., Zhang Z., Du H., Brendler C.B., Xiao X. Systemic delivery of oncolytic adenoviruses targeting transforming growth factor-β inhibits established bone metastasis in a prostate cancer mouse model. Hum. Gene Ther. 2012;23:871–882. doi: 10.1089/hum.2012.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhang Z., Hu Z., Gupta J., Krimmel J.D., Gerseny H.M., Berg A.F., Robbins J.S., Du H., Prabhakar B., Seth P. Intravenous administration of adenoviruses targeting transforming growth factor beta signaling inhibits established bone metastases in 4T1 mouse mammary tumor model in an immunocompetent syngeneic host. Cancer Gene Ther. 2012;19:630–636. doi: 10.1038/cgt.2012.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Thomas D.A., Massagué J. TGF-beta directly targets cytotoxic T cell functions during tumor evasion of immune surveillance. Cancer Cell. 2005;8:369–380. doi: 10.1016/j.ccr.2005.10.012. [DOI] [PubMed] [Google Scholar]
- 37.Wan Y.Y., Flavell R.A. ‘Yin-Yang’ functions of transforming growth factor-beta and T regulatory cells in immune regulation. Immunol. Rev. 2007;220:199–213. doi: 10.1111/j.1600-065X.2007.00565.x. [DOI] [PMC free article] [PubMed] [Google Scholar]