

Abstract
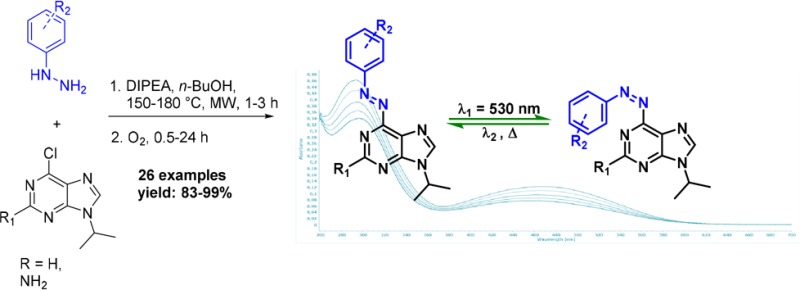
The first general two-step, one-pot synthetic route to 6-azopurines is presented. Microwave-assisted nucleophilic aromatic substitution of protected 6-chloropurines with hydrazines or hydrazides, followed by metal-free oxidation with oxygen, gives 6-azopurines in high to excellent yields. Photophysical studies revealed intensive n−π* absorption band that makes trans-to-cis photoswitching possible using visible light (λ = 530 nm).
Controlling biological processes with light is an emerging field of chemical biology.1 It offers high spatiotemporal resolution and bioorthogonality, and it generates no waste. Biological functions can be controlled with light using molecular photoswitches on the oligonucleotide,1a,1d,1e,2 small molecule,3 or genetic level.1a,1d,1e,2a,4 Photopharmacology represents the recently introduced concept of reversible photocontrol over the activity of small molecules. This type of regulation relies on the incorporation of a photoswitchable moiety into bioactive molecule to achieve a very precise control through on- and off-switching of their activity by light.3c−3e Similarly, photomanipulation of genetic processes can be achieved through reversible modulation of structure and function of DNA and RNA by incorporating a molecular photoswitch into structures of oligonucleotides.5 Translation and transcription are essential processes in the cell, and their photoregulation could therefore provide insight into many biological mechanisms. Through these applications, in the past decade both photopharmacology and the application of reversibly photocontrolled oligonucleotides provided important alternatives to optogenetics,4 which uses genetic manipulation to introduce photoresponsive ion channels to cells.
Nucleobases, as parts of nucleotides and nucleosides, are fundamental building blocks of DNA and RNA, as well as other biologically important molecules. For example, ATP and GTP are involved in many crucial cell activities, such as energy storage and signal transduction pathways.6 Furthermore, structures of many kinase inhibitors and other biologically relevant molecules are based on a purine scaffold (Figure 1, compounds A1–A5).7 Despite some side effects (nausea, vomiting, hyperglycemia, etc.), some of these compounds have already reached clinical phase research, illustrating the importance of purine moieties in drug discovery.8 In order to reduce side effects of purine-based drugs, one of the biggest challenges is precise spatiotemporal control of their activity in biological processes involving DNA, RNA, and kinases.
Figure 1.
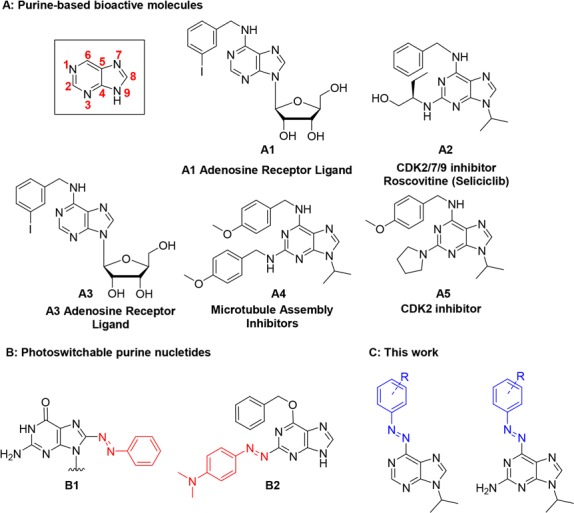
(A) Bioactive molecules with a purine scaffold; (B) examples of photoswitchable nucleotides; (C) structures enabled by this work.
Toward this goal, a photochromic unit needs to be introduced into the structure of a nucleobase to render it photoresponsive and thereby controllable by light. This must be achieved without compromising the activity and function of these molecules, which implies that the structural perturbation caused by the incorporation of the photochromic unit has to be as subtle as possible. Moreover, concerning the application of these photoresponsive molecules in biological systems, an additional and major challenge is to ensure control by visible light. For these purposes, azobenzenes are undoubtedly the most suitable and widely used organic chromophores, due to their photostability, high tunability, and low molecular weight.1a,1b,4,9
Examples of successful transformation of the purine scaffold into a photoswitchable unit are, however, scarce and mainly focused on positions 2 and 8 of the structure (for example B1 and B2; Figure 1B).5 To the best of our knowledge, position 6 has never been directly modified into a heterocyclic azobenzene, even though it appears to be the most promising position for incorporation of a photoswitch that allows to preserve the original molecular function (see examples A1–A5 in Figure 1).10
Constructing a heterocyclic azobenzene at the position 6 would impose a minor structural change in adenine and guanine while preserving a nitrogen atom that acts as a good hydrogen-bond acceptor. This, however, poses a synthetic challenge due to the low nucleophilicity of the 6-amino group of purines. The only existing example in the literature, which also highlights the difficulty to directly modify the 6-amino group, is the indirect attachment of the whole azobenzene scaffold through amide bond formation.11
With this in mind, we were interested in the development of a general synthetic approach for the incorporation of an azobenzene moiety to the pyrimidine ring of purines (Figure 1C). Our initial attempts to make 6-azopurines by commonly employed synthetic methods for azobenzenes, such as Mills reaction5c,12 and diazo coupling,13 were not successful in functionalizing the unreactive 6-amino group. We further attempted to apply Pd-catalyzed coupling reaction of aryl hydrazides with 6-chloro-9-isopropyladenine (compound 1, Figure 2), followed by NBS oxidation in pyridine.14 However, none of the reaction conditions that were screened afforded the desired coupling product (for details, see Scheme S1).
Figure 2.
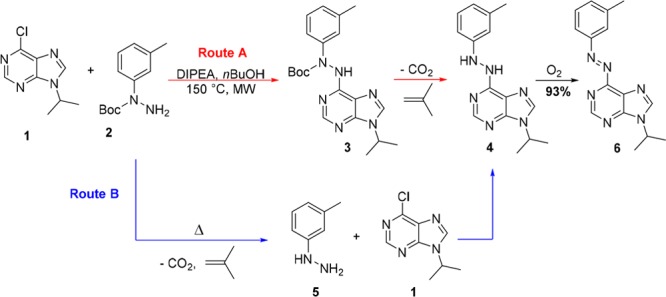
Representation of the two proposed pathways for the synthesis of azobenzene 6, which either involve a three-step approach (route A) or a two-step, one-pot synthesis (route B).
Finally, we focused on the easily accessible N-Boc-hydrazide 2 (Figure 2),15 that we subjected to a simple, uncatalyzed nucleophilic aromatic substitution with 6-chloro-9-isopropyladenine 1, aiming to synthesize N-Boc-N,N′-diaryl hydrazide 3 that could be further deprotected and oxidized to the desired azo compound. However, under the reaction conditions (equimolar 1 and 2, n-BuOH as a solvent, DIPEA as a base, rt to 60 °C), no product formation was observed and the starting material was quantitatively recovered. Then a reaction was submitted to microwave irradiation at 150 °C for 2 h, followed by 12 h of storage at room temperature. A red-brown layer was observed on the top of a yellow solution. NMR and MS analysis of precisely taken aliquots showed that the upper layer was consisted of pure heterocyclic azobenzene 6, whereas the yellow layer gave a pure spectrum of N,N′-diarylhydrazine 4 (Figure 2; see Figure S1).
From this unexpected observation, we hypothesized (Figure 2) that hydrazine 4 was formed either by deprotection of compound 3 (route A) or by deprotection of 2, followed by nucleophilic aromatic substitution with 5 (route B). Compound 2 was thus submitted to the same reaction conditions (without substrate 1 present), and the TLC analysis showed slow in situ conversion of N-Boc-3-methylphenyl hydrazide 2 to hydrazine 5. This led us to the conclusion that the synthesis of such heterocyclic azo-compounds can be performed both by using commercially available arylhydrazines and N-Boc-aryl hydrazides. Indeed, in reaction with compound 1, hydrazine 5 gave the same yield (93%) of compound 4 as starting from compound 2.
The second remarkable observation was the in situ formation of 6-azoadenine 6. The only available oxidant for this reaction was oxygen from the limited volume of air in the vessel. Slow diffusion of air through the solution could explain formation of the colored layer on the top of the reaction mixture (see Figure S1a). Therefore, two aliquots of the same microwave irradiated reaction were submitted to the different sources of oxygen–air and balloon filled with pure oxygen. After few minutes, it was already clear by color change that reaction under oxygen atmosphere proceeds much faster, and after only 2 h it gave pure oxidation product 6 in 93% yield. Reaction conducted under the air did not reach full conversion even after 16 h of vigorous stirring (Figure S1b). This result is highly promising since oxidation of diaryl hydrazines does not require use of transition metals,16 and it is fully compatible with initial reaction conditions of nucleophilic aromatic substitution. The significance of the newly introduced reaction sequence lies in the successful synthesis of important heterocyclic azobenzenes in a one-pot, two-step procedure, merging two consecutive reactions.
Further optimization of reaction conditions included a screening of the temperature (25, 60, and 150–180 °C with MW irradiation), base (K2CO3, Cs2CO3, K3PO4, and DIPEA) and solvent (DMSO, n-BuOH, CH3CN, and THF). We found that the reaction at 150 °C (for adenines) or 180 °C (for guanines) in n-BuOH showed the fastest rate for nucleophilic aromatic substitution in the presence of DIPEA as a base.
A library of 18 6-azoadenines and eight 6-azoguanines was prepared to study the substrate scope (Scheme 1) by using both electron-rich and electron-poor hydrazines while maintaining 6-chloro-9-isopropyladenine (1) and 6-chloro-9-isopropylguanine (8) scaffolds for nucleophilic aromatic substitution. The use of an N-isopropyl substituent on 6-chloropurines effectively mimicked the secondary carbon atom that is commonly attached to purines in nucleotides at position 9. Under the optimized conditions, all products were obtained in high to excellent yields using unprotected substrates in a simple experimental procedure (Scheme 1; see the SI).
Scheme 1. Synthetic Sequence for Obtaining Diverse 6-Azopurines.

Reaction conditions: (1) 1 or 8 (0.36 mmol), 7 (1.2 equiv), DIPEA (5 equiv), n-BuOH (2 mL), 150 or 180 °C, 1–3 h; (2) O2, rt.
Isolated yield after column chromatography.
2.8 mmol scale.
Nucleophilic aromatic substitution on substrate 1 was successfully performed upon MW heating at 150 °C in n-BuOH in the presence of DIPEA as base, while the reaction temperature for substitution on compound 8 had to be increased to 180 °C. This can be explained by the presence of an additional 2-amino group that decreases the electrophilicity of the guanine core, which affects the reaction time of nucleophilic aromatic substitution. The reaction time increased from 1 to 2 h, when substrate 1 was used, to 3 h in the case of 8 (Scheme 1). Moreover, it was found that the nature of the substituents on phenyl hydrazine precursors plays an important role regarding the reaction time of nucleophilic aromatic substitution. We have observed a slight increase of reaction time when hydrazines bearing electron-withdrawing substituents were employed. The reaction time of the oxidation also increased significantly in case of electron-poor diaryl hydrazines (from 1 h for 6b to 24 h for 6r; Scheme 1). On the other hand, the minute amounts of side products in the reaction with electron-poor hydrazines decreased, allowing pure products to be obtained after simple column chromatography. Otherwise, recrystallization from ethyl acetate/pentane was used to obtain pure target compounds.
This hydrazine substituent effect was further assessed by analyzing a series of hydrazines (7a–r) in the reaction with 1 (Scheme 1). At the specified reaction times, no obvious steric and electronic effects on the reaction yields were observed with a variety of meta- and para-substituted hydrazines (6a–m, 9a–g). To our satisfaction, optimized conditions also tolerated different ortho-substituents (F, Cl, Me), despite the reported challenges in the synthesis of recently developed highly desired ortho-substituted azobenzenes,17 yielding products in high yields (6n, 6o, and 6r). Nucleophilic substitution with sterically demanding 2-naphthyl hydrazine, an extended aromatic system, also worked (6p, 9h). Furthermore, the reaction was efficient even for highly electron-deficient substrates, such as 2,4,6-triflorophenylhydrazine, whereas the only tested hydrazine that did not provide the desired product was pentafluorophenylhydrazine.
The photophysical properties of the synthesized 6-azopurines were explored for representative cases of both adenine (6o) and guanine (9f) analogues. We examined a few characteristic parameters, including UV–vis absorption maxima (λmax), photoswitchability, photostationary state (PSS), and fatigue resistance upon long-term irradiation (for details, see the SI).
First, biological applications require red-shifted photoswitches that absorb in the visible spectral region, which enables lower energy inputs, deeper penetration, and lower toxicity.17,18 UV–vis absorption spectroscopy revealed that all of the representative examples of azopurines absorb in the visible spectral domain with absorption maxima (λmax) that are summarized in Figure 3. UV–vis spectra featured two characteristic signals, one of which can be assigned to a π–π* transition, whereas the other lower energy band from the n−π* transitions.
Figure 3.
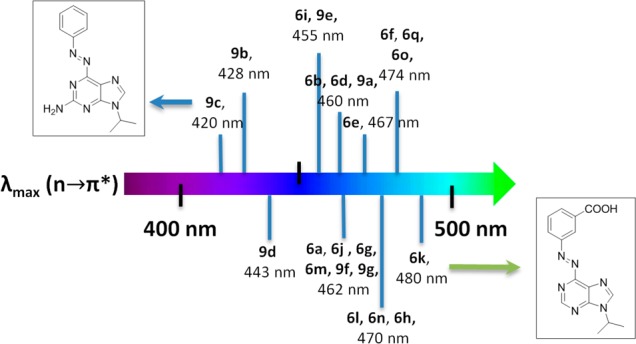
Spectral map that summarizes the absorption maxima (λmax) that correspond to a low energy n−π* transition for a series of 6-azopurines based on adenine (6a–r) and guanine (9a–h) scaffolds (structures are summarized in Scheme 1).
The absorption that corresponds to the n−π* transitions was found to be weak for azoadenines, whereas it presented a strong contribution for azoguanines reflected in their higher extinction coefficients. Furthermore, substituent effects on the spectral features were apparent. In this regard, electron-donating groups were found to induce a bathochromic shift of the π–π* transition, whereas para-substituted azopurines showed particularly good correlation between λmax and σpara Hammett parameter (for details, see Figure S50). This implies that para-substitution could act as an important design principle for azopurines, enabling red-shifted π–π* transition. Moreover, an interesting trend has been observed in red-shifting of n-π* transitions. Additional amino groups in guanines increase the electronic density and clearly lead to a hypsochromic shift (compounds 9a–g, Figure 3). Remarkably, despite the literature expectations on the ortho effect,17 substitution with carboxylic group in the meta position yielded the most bathochromically shifted 6-azopurine (6k). This surprising outcome is probably related to the presence of an acidic group that can protonate the purine core making it even more electron withdrawing.
Next, the photoswitchability of representative azopurines was investigated, and we found that green light (λ = 530 nm) can be used for trans-to-cis photoisomerization of both adenine and guanine analogues. This photoexcitation relied on promoting the low energy n-π* transition, which was effective for both adenine and guanine analogs. The photoswitchability was assessed for compounds 6o and 9f upon irradiation at 530 nm that resulted in a PSS characterized by trans/cis isomer ratios of 58:42 for 6o and 36:64 for 9f, as determined by 1H NMR spectroscopy (for details, see Figures S36–38). This finding thereby further supported the relevance of these systems in biological applications.
Finally, an important prerequisite for application of photoswitches is low switching fatigue. Remarkably, even after exposure to UV light at 365 nm (photon flux, Φ = 1.46 × 10–9 mol s–1) over a period of 12 h, the representative examples, such as 6a, showed very little fatigue (below 25% over >50 cycles) as exemplified by multiple cycles of switching in PBS buffer at physiological pH (see Figures S33–34). This analysis highlights desirable photophysical properties of a new generation of azopurines that make them promising candidates for further applications in biological systems.
In summary, we demonstrate an efficient and versatile synthetic route to a range of new heterocyclic azobenzenes based on the purine scaffold. A series of 6-azopurines from commercially available arylhydrazines and 6-chloro-9-isopropyladenine 1 or 6-chloro-9-isopropylguanine 8 were obtained in high yield. The generality of this method has been demonstrated through the nucleophilic aromatic substitution followed by oxidation of a wide variety of substituted—both electron-rich and electron-poor—phenylhydrazines. Final compounds are shown to have red-shifted absorption maxima featuring high fatigue resistance. Their synthetic accessibility, high stability, and visible-light-mediated isomerization make them ideal for biological applications. Further investigation of the photopharmacological utility of these systems is ongoing in our research group.
Acknowledgments
We gratefully acknowledge generous support from NanoNed, The Netherlands Organization for Scientific Research (NWO-CW, Top grant to B.L.F., and VIDI Grant No. 723.014.001 for W.S.), the Royal Netherlands Academy of Arts and Sciences Science (KNAW), the Ministry of Education, Culture and Science (Gravitation program 024.001.035), and the European Research Council (Advanced Investigator Grant No. 227897 to B.L.F. D.K. acknowledges the receipt of a fellowship from the Dositeja Fund for Young Talents for international studies. We thank. P. van der Meulen for help with the NMR studies, and T. Tiemersma-Wegman for ESI-MS analyses (both from Stratingh Institute for Chemistry, University of Groningen).
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.orglett.7b02361.
Experimental procedures, photochemistry, and characterization data for all new compounds PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- a Szymański W.; Beierle J. M.; Kistemaker H. A. V.; Velema W. A.; Feringa B. L. Chem. Rev. 2013, 113, 6114. 10.1021/cr300179f. [DOI] [PubMed] [Google Scholar]; b Chatterjee D. K.; Fong L. S.; Zhang Y. Adv. Drug Delivery Rev. 2008, 60, 1627. 10.1016/j.addr.2008.08.003. [DOI] [PubMed] [Google Scholar]; c Brieke C.; Rohrbach F.; Gottschalk A.; Mayer G.; Heckel A. Angew. Chem., Int. Ed. 2012, 51, 8446. 10.1002/anie.201202134. [DOI] [PubMed] [Google Scholar]; d Liu Q.; Deiters A. Acc. Chem. Res. 2014, 47, 45. 10.1021/ar400036a. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Wang F.; Liu X.; Willner I. Angew. Chem., Int. Ed. 2015, 54, 1098. 10.1002/anie.201404652. [DOI] [PubMed] [Google Scholar]
- a Lubbe A. S.; Szymanski W.; Feringa B. L. Chem. Soc. Rev. 2017, 46, 1052. 10.1039/C6CS00461J. [DOI] [PubMed] [Google Scholar]; b Nakasone Y.; Ooi H.; Kamiya Y.; Asanuma H.; Terazima M. J. Am. Chem. Soc. 2016, 138, 9001. 10.1021/jacs.6b02525. [DOI] [PubMed] [Google Scholar]
- a Lerch M. M.; Hansen M. J.; van Dam G. M.; Szymanski W.; Feringa B. L. Angew. Chem., Int. Ed. 2016, 55, 10978. 10.1002/anie.201601931. [DOI] [PubMed] [Google Scholar]; b Broichhagen J.; Frank J. A.; Trauner D. Acc. Chem. Res. 2015, 48, 1947. 10.1021/acs.accounts.5b00129. [DOI] [PubMed] [Google Scholar]; c Velema W. A.; van der Berg J. P.; Hansen M. J.; Szymanski W.; Driessen A. J. M.; Feringa B. L. Nat. Chem. 2013, 5, 924. 10.1038/nchem.1750. [DOI] [PubMed] [Google Scholar]; d Borowiak M.; Nahaboo W.; Reynders M.; Nekolla K.; Jalinot P.; Hasserodt J.; Rehberg M.; Delattre M.; Zahler S.; Vollmar A.; Trauner D.; Thorn-Seshold O. Cell 2015, 162, 403. 10.1016/j.cell.2015.06.049. [DOI] [PubMed] [Google Scholar]; e Ferreira R.; Nilsson J. R.; Solano C.; Andréasson J.; Grøtli M. Sci. Rep. 2015, 5, 9769. 10.1038/srep09769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Williams S. C. P.; Deisseroth K. Proc. Natl. Acad. Sci. U. S. A. 2013, 110, 16287. 10.1073/pnas.1317033110. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Deisseroth K. Nat. Methods 2011, 8, 26. 10.1038/nmeth.f.324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Ogasawara S.; Maeda M. Angew. Chem., Int. Ed. 2008, 47, 8839. 10.1002/anie.200803496. [DOI] [PubMed] [Google Scholar]; b Ogasawara S.; Ito S.; Miyasaka H.; Maeda M. Chem. Lett. 2010, 39, 956. 10.1246/cl.2010.956. [DOI] [Google Scholar]; c Zhu R.; Baumann R. P.; Penketh P. G.; Shyam K.; Sartorelli A. C. J. Med. Chem. 2013, 56, 1355. 10.1021/jm301804p. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Wang H.-X.; Xi D.-D.; Xie M.-S.; Wang H.-X.; Qu G.-R.; Guo H.-M. ChemBioChem 2016, 17, 1216. 10.1002/cbic.201600171. [DOI] [PubMed] [Google Scholar]; e Singer M.; Jäschke A. J. Am. Chem. Soc. 2010, 132, 8372. 10.1021/ja1024782. [DOI] [PubMed] [Google Scholar]
- Knowles J. R. Annu. Rev. Biochem. 1980, 49, 877. 10.1146/annurev.bi.49.070180.004305. [DOI] [PubMed] [Google Scholar]
- a Schow S. R.; Mackman R. L.; Blum C. L.; Brooks E.; Horsma A. G.; Joly A.; Kerwar S. S.; Lee G.; Shiffman D.; Nelson M. G.; Wang X.; Wick M. M.; Zhang X.; Lum R. T. Bioorg. Med. Chem. Lett. 1997, 7, 2697. 10.1016/S0960-894X(97)10076-2. [DOI] [Google Scholar]; b MacCallum D. E.; Melville J.; Frame S.; Watt K.; Anderson S.; Gianella-Borradori A.; Lane D. P.; Green S. R. Cancer Res. 2005, 65, 5399. 10.1158/0008-5472.CAN-05-0233. [DOI] [PubMed] [Google Scholar]; c Gao Z.-G.; Kim S.-K.; Biadatti T.; Chen W.; Lee K.; Barak D.; Kim S. G.; Johnson C. R.; Jacobson K. A. J. Med. Chem. 2002, 45, 4471. 10.1021/jm020211+. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Chang Y.-T.; Wignall S. M.; Rosania G. R.; Gray N. S.; Hanson S. R.; Su A. I.; Merlie J.; Moon H.-S.; Sangankar S. B.; Perez O.; Heald R.; Schultz P. G. J. Med. Chem. 2001, 44, 4497. 10.1021/jm010451+. [DOI] [PubMed] [Google Scholar]
- Benson C.; White J.; De Bono J.; O’Donnell A.; Raynaud F.; Cruickshank C.; McGrath H.; Walton M.; Workman P.; Kaye S.; Cassidy J.; Gianella-Borradori A.; Judson I.; Twelves C. Br. J. Cancer 2007, 96, 29. 10.1038/sj.bjc.6603509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Bandara H. M. D.; Burdette S. C.; Lee S.-N.; Itoh S.; Noda K.; Usui T.; Ishihara K.; Inamo M.; Takagi H. D.; Asano T. Chem. Soc. Rev. 2012, 41, 1809. 10.1039/C1CS15179G. [DOI] [PubMed] [Google Scholar]; b Flade S.; Jasper J.; Gieß M.; Juhasz M.; Dankers A.; Kubik G.; Koch O.; Weinhold E.; Summerer D. ACS Chem. Biol. 2017, 12, 1719. 10.1021/acschembio.7b00324. [DOI] [PubMed] [Google Scholar]
- Liu M.; Jinmei H.; Abe H.; Ito Y. Bioorg. Med. Chem. Lett. 2010, 20, 2964. 10.1016/j.bmcl.2010.02.109. [DOI] [PubMed] [Google Scholar]
- Davey M. H.; Lee V. Y.; Miller R. D.; Marks T. J. J. Org. Chem. 1999, 64, 4976. 10.1021/jo990235x. [DOI] [PubMed] [Google Scholar]
- Haghbeen K.; Tan E. W. J. Org. Chem. 1998, 63, 4503. 10.1021/jo972151z. [DOI] [Google Scholar]
- Lim Y.-K.; Lee K.-S.; Cho C.-G. Org. Lett. 2003, 5, 979. 10.1021/ol027311u. [DOI] [PubMed] [Google Scholar]
- Wang Z.; Skerlj R. T.; Bridger G. J. Tetrahedron Lett. 1999, 40, 3543. 10.1016/S0040-4039(99)00561-4. [DOI] [Google Scholar]
- Drug E.; Gozin M. J. Am. Chem. Soc. 2007, 129, 13784. 10.1021/ja074413c. [DOI] [PubMed] [Google Scholar]
- a Dutta B.; Biswas S.; Sharma V.; Savage N. O.; Alpay S. P.; Suib S. L. Angew. Chem., Int. Ed. 2016, 55, 2171. 10.1002/anie.201508223. [DOI] [PubMed] [Google Scholar]
- a Dong M.; Babalhavaeji A.; Samanta S.; Beharry A. A.; Woolley G. A. Acc. Chem. Res. 2015, 48, 2662. 10.1021/acs.accounts.5b00270. [DOI] [PubMed] [Google Scholar]; b Dong M.; Babalhavaeji A.; Hansen M. J.; Kálmán L.; Woolley G. A. Chem. Commun. 2015, 51, 12981. 10.1039/C5CC02804C. [DOI] [PubMed] [Google Scholar]; c Konrad D. B.; Frank J. A.; Trauner D. Chem. - Eur. J. 2016, 22, 4364. 10.1002/chem.201505061. [DOI] [PubMed] [Google Scholar]; d Bleger D.; Schwarz J.; Brouwer A.; Hecht S. J. Am. Chem. Soc. 2012, 134, 20597. 10.1021/ja310323y. [DOI] [PubMed] [Google Scholar]
- John A. A.; Ramil C. P.; Tian Y.; Cheng G.; Lin Q. Org. Lett. 2015, 17, 6258. 10.1021/acs.orglett.5b03268. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.