

Abstract
The majority of characterised ferrochelatase enzymes catalyse the final step of classical haem synthesis, inserting ferrous iron into protoporphyrin IX. However, for the recently discovered coproporphyrin-dependent pathway, ferrochelatase catalyses the penultimate reaction where ferrous iron is inserted into coproporphyrin III. Ferrochelatase enzymes from the bacterial phyla Firmicutes and Actinobacteria have previously been shown to insert iron into coproporphyrin, and those from Bacillus subtilis and Staphylococcus aureus are known to be inhibited by elevated iron concentrations. The work herein reports a Km (coproporphyrin III) for S. aureus ferrochelatase of 1.5 µM and it is shown that elevating the iron concentration increases the Km for coproporphyrin III, providing a potential explanation for the observed iron-mediated substrate inhibition. Together, structural modelling, site-directed mutagenesis, and kinetic analyses confirm residue Glu271 as being essential for the binding of iron to the inhibitory regulatory site on S. aureus ferrochelatase, providing a molecular explanation for the observed substrate inhibition patterns. This work therefore has implications for how haem biosynthesis in S. aureus is regulated by iron availability.
Keywords: coproporphyrinogen, ferrochelatase, haem biosynthesis
Introduction
Ferrochelatase enzymes catalyse the insertion of metal ions into porphyrin rings. The insertion of ferrous iron (Fe2+) into protoporphyrin IX is the final step in the well-characterised classical haem synthesis pathway. However, bacterial species in the Firmicutes and Actinobacteria Phyla (Bacillus subtilis, Mycobacterium tuberculosis, Propionibacterium acnes, S. aureus) have been shown to utilise a ‘coproporphyrin-dependent’ pathway [1,2], where Fe2+ insertion into coproporphyrin III is the penultimate step (Figure 1). Branching and regulation of these bacterial haem biosynthetic pathways are discussed in a recent review that suggests a series of new abbreviations based on enzyme activities [3]. These new abbreviations provide a useful way of discriminating between protoporphyrin ferrochelatase (PpfC) and coproporphyrin ferrochelatase (CpfC), and this nomenclature is adopted herein in place of the traditional HemH abbreviation.
Figure 1. The coproporphyrin-dependent pathway of haem synthesis.

This figure depicts a recently discovered route for haem synthesis in Firmicutes and Actinobacteria catalysed by coproporphyrinogen oxidase (CgoX), CpfC, and coprohaem decarboxylase (ChdC). These new abbreviations are introduced in a recent review [3].
Ferrochelatases are membrane-bound proteins in eukaryotes and many prokaryotes, but Gram-positive bacteria have been shown to encode a soluble form of the protein [4–6]. All of these forms of ferrochelatase have been shown to insert a variety of divalent metals into the centre of porphyrin molecules, with zinc being the most common metal used in in vitro assays due to being more stable in an aerobic environment than the natural Fe2+ substrate [7] and allows for fluorescence-based assays to be utilised. Catalytic rates for ferrochelatases vary with kcat values typically ranging from 0.11 to 15.3 min−1, although S. aureus CpfC was recently reported to have a kcat of 165 min−1 using coproporphyrin III and Fe2+ as substrates [1]. Parallel experiments performed for the B. subtilis CpfC gave a kcat of 78 min−1, which compares to another report of 0.11 min−1 [2]. This recent work also identified 0.8 µM as the threshold concentration of Fe2+ above which inhibition occurs for the B. subtilis and S. aureus enzymes, which represents an interesting feature unique to coproporphyrin-dependent ferrochelatases.
While bacterial and mammalian ferrochelatase enzymes generally exhibit poor sequence identity (e.g. <10% between human and B. subtilis), the secondary and tertiary structures are similar in the core region [8]. However, human and yeast PpfC enzymes have an extended C-terminal region compared with B. subtilis CpfC. The human enzyme ligates a [2Fe–2S] iron–sulphur cluster in this region, which is not present in the S. cerevisiae enzyme. This cluster is ligated by one residue in the core of the protein (Cys 196) and three in the extended C-terminal region (Cys 403, 406, 411), and at the time was thought to be responsible for anchoring the extended C-terminal to the rest of the monomer, and was also suggested to be indirectly involved in stabilising the dimer form of the protein [8]. S. cerevisiae does not contain the necessary residues in the same location to form a cluster and the dimer stability is reportedly not affected [9]. Some ferrochelatases from Gram-positive bacteria have been shown to ligate [2Fe–2S] clusters via an extra insertion, although loss of this cluster did not result in a total loss of activity [10] in contrast with the [2Fe–2S] cluster of the human enzyme [11]. The in vivo role for these cofactors remains poorly understood.
Ferrochelatase structures exhibit a degree of variability with respect to the residues present in the substrate binding cleft. For eukaryotic PpfC enzymes a series of leucine residues form part of the hydrophobic lip that covers the active site pocket, which is not present in the B. subtilis enzyme. The Tyr13 residue in B. subtilis CpfC, which has been determined to aid the conserved His183 residue in stabilisation of the B ring of the porphyrin macrocycle, is not present in the membrane-associated ferrochelatases [12]. The residue in this position in the other enzymes is a methionine whose role is not entirely understood [8,9]. The tyrosine residue in B. subtilis has been implicated in a metal-binding function in the catalytic metal-binding site along with the conserved histidine and glutamic acid residues [12,13] and it is possible that the methionine residue in this position aids in the insertion of metals into the porphyrin ring in a similar manner to the tyrosine residue found in the soluble form of ferrochelatase. The amino acid at this position has also been linked to the specificity of metal chelation, with an Y13M mutation of the B. subtilis enzyme resulting in a more rapid insertion of Co2+ and a complete loss of Cu2+ utilisation [14].
Soluble ferrochelatases have been crystallised with a variety of metals present in the metal-binding cleft, and have been shown to have two separate sites for metal binding: one close to the porphyrin-binding site stabilised by the conserved His183 and one closer to the surface of the protein ∼7 Å from the H183 residue [12]. In B. subtilis CpfC, this outer site has been shown to bind Mg2+ ions, which are co-ordinated by the residues Arg46, Glu268, and Glu272 [12]. This particular metal site has been reported to provide a Mg2+-mediated stimulation of zinc insertion into deuteroporphyrin IX, with mutation of the Glu272 residue resulting in abolition of the stimulatory effects of Mg2+ [13]. The mechanism for this stimulation was proposed to be due to repulsion between metal ions present at both sites pushing the inserted metal at the inner (catalytic)-binding site closer to the porphyrin ring [13]. Such a regulatory metal site has not been investigated in the human enzyme, and it seems unlikely that S. cerevisiae PpfC could bind metals at this site due to the presence of a glycine residue instead of a glutamate residue at position 322 [9]. However, it has been suggested that S. cerevisiae PpfC can bind the inhibitor Cd2+ at two sites, with one site slightly further away from the active site than the outer metal-binding site for B. subtilis CpfC. Human PpfC is susceptible to substrate inhibition when zinc is the metal ion substrate. This zinc inhibition is more pronounced at higher concentrations of the porphyrin substrate, suggesting that an inhibitory metal ion binds to the enzyme–product complex [15]. Residues that are predicted to be predominantly involved in metal binding in B. subtilis, where Zn2+ and Cd2+ have been observed, are the conserved residues H183 and E264 and the unique Y13 residue that resides on the other side of the porphyrin-binding site [13]. Cd2+ has been shown to also bind to the nearby H262, which is located underneath the porphyrin substrate and is proposed to inhibit CpfC by hampering the binding of porphyrin substrate [13].
The current work investigates the kinetic properties of CpfC from the human pathogen S. aureus using the native substrates Fe2+ and coproporphyrin III. Since iron has dramatic inhibitory properties for this class of CpfCs [1], this study aims to test the hypothesis that iron inhibition is mediated by the regulatory metal-binding site previously shown to bind Mg2+ in the B. subtilis enzyme [12,13]. This work has implications for how staphylococci respond to nutritional immunity (e.g. metal sequestration) encountered during infection.
Experimental procedures
Cloning, overexpression, and purification of S. aureus ferrochelatase
The cpfC (hemH) gene was amplified from S. aureus strain USA300 [16] via colony PCR using Q5 polymerase (NEB) and cloned into the NheI/HindIII sites of plasmid pTrcHis (ThermoFischer), and the correct sequence of the resultant pTrcHis-Sa-cpfC vector was confirmed by Sanger sequencing. Escherichia coli JM109 cells (Sigma–Aldrich) were transformed with pTrcHis-Sa-cpfC and a 10 ml LB overnight culture of this expression strain was used to inoculate 1 l of Circlegrow medium in a 2-l baffled flask (125 µg ml−1 ampicillin was included throughout for plasmid selection). Cultures were grown for 24 h at 37°C and 160 rpm and cells were harvested (4 k rpm, 4°C, 20 min) and stored at −20°C. Cell pellets from 1 l cultures were re-suspended in protein storage buffer (30 ml) containing 50 mM Tris/MOPS pH 8.0, 100 mM KCl, 1% sodium cholate. Cell suspensions were sonicated (6 × 30 s on ice at 10 µ, with 30 s intervals between bursts), and cell debris was removed via centrifugation (18 k rpm, 4°C, 20 min). The supernatant was applied to a column containing Talon metal affinity resin (2.5 ml bed volume) equilibrated with 5 column volumes of protein storage buffer. The column was washed with 20 column volumes of protein storage buffer followed by 5 column volumes of wash buffer (protein storage buffer including 15 mM imidazole), and purified protein was eluted with 5 column volumes of elution buffer (protein storage buffer including 300 mM imidazole). Purified protein was concentrated to <1 ml using a 10 kDa spin concentrator (Millipore) and imidazole was removed using a PD10 desalting column (GE Healthcare) equilibrated with protein storage buffer.
Assay of S. aureus ferrochelatase activity
Purified CpfC was assayed using coproporphyrin III (Frontier Scientific) as the porphyrin substrate and ferrous iron as the inserted metal ion. Coproprophyrin III powder was solubilised using a few drops of ammonium hydroxide and was then diluted in assay buffer (0.1 M Tris–HCl pH 8.1, 0.5% sodium cholate, 0.5% Tween 20, 1 mM β-mercaptoethanol). Coproporphyrin III stock solutions were quantified in 0.1 M HCl spectrophotometrically using an extinction coefficient of ε399.5 = 489 mM−1 cm−1 [17]. Ferrous iron solutions were quantified spectrophotometrically using ferrozine solution and an extinction coefficient of ε562 = 27.9 mM−1 cm−1 [18]. Iron chelation was measured using a continuous assay essentially, as previously described [10]. Activity of 50 nM CpfC was monitored using a Cary 60 spectrophotometer at 30°C. The reactions were initiated by the addition of enzyme, which was preincubated at 30°C, and the depletion of coproporphyrin III was monitored at 392 nm for 3 min (ε392 = 115 mM−1 cm−1, [1]). All assays contained 0.1 M Tris–HCl pH 8.1, 0.5% sodium cholate, 0.5% Tween 20, 1 mM β-mercaptoethanol. The steady-state rates were estimated using linear regression of the timecourse at the start of the reaction. Michaelis plots (v vs. [S]) were fitted to the Michaelis–Menten equation using non-linear regression. Ki,app values were calculated by fitting v vs. [inhibitor] data to a single-binding site model described by eqn (1) [19], where no mechanistic assumptions are made to perform a quantitative assessment of the key features of inhibition
| 1 |
where v is the enzymatic rate, vcontrol is the rate in the absence of inhibitor, vmin is the rate in the presence of saturating inhibitor, [I] is the inhibitor concentration, Ki,app is the apparent inhibition constant defining the inhibitor concentration that elicits 50% of the total inhibition, and n is the Hill coefficient.
Structural modelling of S. aureus ferrochelatase
A structural model for S. aureus CpfC was generated using the RaptorX server [20,21] with B. subtilis CpfC as a template (PDBid = 1AK1, α-carbon RMSD = 0.23 Å). The S. aureus CpfC model was superposed (α-carbon RMSD = 0.66 Å) onto a Mg2+-bound and porphyrin-bound structure of B. subtilis CpfC (PDBid = 1C1H), and the native substrate coproporphyrin III was modelled into the active site in place of N-methyl mesoporphyrin via superposition using the CCP4MG software.
Results
Measurement of kinetic constants for S. aureus ferrochelatase
Since much of the previous work on CpfCs has been performed with non-native substrates (i.e. Zn2+, protoporphyrin IX, and deuteroporphyrin IX), it was of interest to define the kinetic constants for the in vivo substrates Fe2+ and coproporphyrin III. Kinetic assays where [Fe2+] was varied (Figure 2A) demonstrate substrate inhibition above 0.8 µM, and the data below this threshold value were fitted to the Michaelis equation to provide an estimate of 0.27 ± 0.04 µM for an apparent Km for iron binding at the inner catalytic metal site. Varying [coproprophyrin III] at a fixed concentration of 0.7 µM Fe2+ (Figure 2B) provides the first estimate of an apparent Km for coproprophyrin III as 1.5 ± 0.2 µM, and kcat was fitted as 10.5 ± 0.4 min−1 at this sub-inhibitory Fe2+ concentration.
Figure 2. Kinetic analysis of S. aureus ferrochelatase.

(A) 50 nM CpfC was assayed in the presence of varying concentrations of iron and a fixed concentration of 1 µM of coproporphyrin. Data collected at 0–0.8 µM iron were fitted to a single rectangular hyperbola to provide an estimate of the apparent Km for iron (Kapp = 0.27 ± 0.04). (B) 50 nM CpfC was assayed in the presence of varying concentrations of coproprophyrin III and a fixed iron concentration of 0.7 µM (below the 0.8 µM inhibitory iron concentration). Data were fitted to a single rectangular hyperbola via non-linear regression: kcat = 10.5 ± 0.4 min−1, Kapp = 1.5 ± 0.2 µM.
Elevated iron increases the Km for coproporphyin III
To gain an insight into the influence of iron upon coproporphyrin III binding, kinetic plots were produced for each substrate at fixed concentrations of the other substrate (Figure 3). Firstly, varying the [coproporphyrin III] in the presence of fixed (sub-inhibitory) concentrations of Fe2+ revealed that increasing [Fe2+] increases the apparent Km for coproporphyrin (Figure 3A). Performing the converse experiment with variation of [Fe2+] in the presence of fixed concentrations of coproporphyrin III revealed a more conventional pattern of increasing the concentration of coproporphyrin III also decreases the apparent Km for Fe2+ (Figure 3B).
Figure 3. Opposing kinetic patterns for the two ferrochelatase substrates.

Primary Michaelis–Menten plots were generated with fixed concentrations of iron (A) and coprorporphyrin III (B), and data were fitted to single rectangular hyperbolae via non-linear regression. Lineweaver–Burk plots (insets) are shown to highlight differences in kinetic patterns between panels (A) and (B). To avoid well-known problems with these secondary plots, trend lines were generated using the kinetic constants obtained from non-linear regression of the untransformed data.
Structural modelling of S. aureus ferrochelatase reveals a regulatory metal-binding site
Based on the data in Figure 3, it was hypothesised that S. aureus CpfC has a regulatory metal-binding site, and the most simple explanation would be that this metal site is at the same location as the Mg2+-binding site in B. subtilis CpfC. The only difference is that this site would be inhibitory when occupied by iron, as opposed to stimulatory when occupied by Mg2+ [13]. To investigate the local environment of this putative regulatory metal site, a structural model for S. aureus CpfC was generated using the B. subtilis CpfC crystal structure as a template (Figure 4). The model and the template were superposed and the Mg2+ ion from the B. subtilis structure was shown in the same spatial location in the S. aureus model (Figure 4A). The metal-binding residues were fully conserved between species, with residues Glu271, Glu267, and Arg45 in the S. aureus model all mapping adjacent to the outer regulatory metal site (Figure 4B).
Figure 4. Structural modelling of bound coproprophyrin and a regulatory metal-binding site for S. aureus ferrochelatase.

(A) Surface electrostatics view of a S. aureus CpfC structural model generated using the RaptorX server with B. subtilis CpfC as a template (PDBid = 1AK1, α-carbon RMSD = 0.3 Å). The S. aureus CpfC model was superposed (α-carbon RMSD = 0.66 Å) onto an Mg2+-bound (cyan) and porphyrin-bound structure of B. subtilis CpfC (PDBid = 1C1H), and the native substrate coproporphyrin III (yellow) was modelled into the active site in place of N-methyl mesoporphyrin. (B) Cartoon representation of the active site of S. aureus CpfC showing conserved residues implicated in metal binding at an outer regulatory site (E271, E267, R45) and an inner catalytic site (E263, Y12, H181).
Magnesium potentiates Fe2+-mediated inhibition of S. aureus ferrochelatase
Since Mg2+ has previously been used to investigate the regulatory metal site in B. subtilis CpfC and was shown to stimulate chelatase activity [13], it was of interest to determine whether Mg2+ could also be used as a molecular probe to study S. aureus CpfC. To investigate the effect of Mg2+ upon the Fe2+ kinetics, iron was varied in the presence and absence of Mg2+ (Figure 5). Surprisingly, the addition of Mg2+ potentiates chelatase inhibition, lowering the threshold concentration of Fe2+ required for substrate inhibition.
Figure 5. Impact of magnesium upon ferrochelatase kinetics.

v vs. [Fe2+] plots were generated for S. aureus CpfC assayed in the presence (○) and absence (●) of 4 mM Mg2+. Fixed concentrations of 50 nM enzyme and 2 µM coproporphyrin III were used, with the Fe2+ concentration being varied.
Loss of the regulatory-binding site abolishes inhibition by Mg2+ or Fe2+
To test the hypothesis that the metal-binding site shown in Figure 4 is involved in the Mg2+-mediated inhibition kinetics shown in Figure 5, Glu271 was mutated to serine and the resultant E271S mutant protein was purified and assayed in the presence and absence of Mg2+ (Figure 6). Figure 6A shows that Mg2+-potentiated inhibition was dramatically diminished in the E271S mutant, and that inhibition by Fe2+ in the absence of Mg2+ was also dramatically diminished. Figure 6B displays Ki,app(Mg2+) values for wild-type and E271S CpfC as 66 µM and 11 mM, respectively, highlighting a dramatic loss of sensitivity to Mg2+ for the mutant enzyme. Together, these data strongly indicate that in wild-type S. aureus CpfC, the binding of Mg2+ or Fe2+ at this metal site results in enzyme inhibition. In addition, the data in Figures 4–6 suggest that loss of the inhibitory metal site should reverse the inhibitory effects of increasing iron concentrations seen in Figure 3A, so a similar kinetic study was carried out with the E271S variant of S. aureus CpfC including iron levels well above the inhibitory threshold for the wild-type enzyme (Figure 7). These data confirm that loss of the E271 metal site abolishes the iron-mediated inhibition with respect to coproporphyrin III (Figure 7A), and the effect of varying [coproporphyrin III] upon the kinetic constants for iron (Figure 7B) remains similar to the wild-type data (Figure 3B).
Figure 6. Loss of the metal-binding residue Glu271 abrogates the inhibition of S. aureus ferrochelatase by iron and magnesium.

(A) v vs. [Fe2+] plots were generated for wild-type (red) and an E271S variant of S. aureus CpfC (blue) assayed in the presence (squares) and absence (circles) of 4 mM Mg2+. Fixed concentrations of 50 nM enzyme and 2 µM coproporphyrin III were used. The fitted blue line provides estimates of apparent kinetic constants when the inhibition by Fe2+ is removed: kcat = 24.1 ± 2.0 min−1, Kapp(Fe2+) = 1.2 ± 0.2 µM. (B) Inhibition dose responses for wild-type (red) and an E271S variant of S. aureus CpfC (blue) assayed in the presence of increasing concentrations of Mg2+. Fixed concentrations of 50 nM enzyme, 2 µM coproporphyrin III, and 0.7 µM Fe2+ were used. To estimate Ki,app values, the data were fitted to a four parameter Hill equation (eqn 1, [19]) with the minimum activity (i.e. vmin) constrained to a positive value.
Figure 7. Loss of inhibitory metal site restores conventional Michaelis kinetics.

Primary Michaelis–Menten plots for an E271S variant of S. aureus CpfC were generated with fixed concentrations of iron (A) and coproporphyrin III (B), and data were fitted to single rectangular hyperbolae via non-linear regression. Lineweaver–Burk plots (insets) are shown to highlight differences in kinetic patterns between panels (A) and (B). To avoid well-known problems with these secondary plots, trend lines were generated using the kinetic constants obtained from non-linear regression of the untransformed data.
Discussion
Preliminary kinetic analysis of S. aureus CpfC revealed a kcat of 10.5 ± 0.4 min−1. This rate is comparable to previous measurements for other bacterial PpfCs [10], and slightly higher than previous measurements of 1.8 and 0.11 min−1 for CpfCs from M. tuberculosis and B. subtilis, respectively [2]. While a kcat for S. aureus CpfC was recently calculated to be 165 min−1 via extrapolation of a substrate inhibition curve [1], the maximum rates measured (∼28 min−1) were of a similar magnitude to those presented herein. We report the first measurement of Kapp (coproporphyrin III) for S. aureus CpfC as 1.5 ± 0.2 µM, which is slightly lower than previous measurements of 10.5 and 7.8 µM for CpfC enzymes from M. tuberculosis and B. subtilis, respectively [2]. Our estimate of 0.27 ± 0.04 µM for Kapp (Fe2+) is comparable with a previous estimate of 0.6 ± 0.1 µM for this enzyme [1], although both of these measurements were hampered by substrate inhibition by iron.
Primary and secondary kinetic plots clearly demonstrate that elevating the iron concentration increases the Km for coproporphyrin III (Figure 3A), which is consistent with Fe2+ causing inhibition at higher concentrations. It was therefore hypothesised that Fe2+ is inserted into the porphyrin substrate following binding to a well-known catalytic site adjacent to the tetrapyrrole substrate, and higher concentrations of Fe2+ inhibit chelation by binding at a second regulatory site. Structural modelling (Figure 4) provides convincing evidence that S. aureus possesses a second metal-binding site similar to the Mg2+-binding site in B. subtilis CpfC [12], although Mg2+ was found to potentiate Fe2+-mediated inhibition in the current study (Figure 5) compared with having a stimulatory role in previous reports [13]. This could be due to differences in substrates used, as the current study uses the native substrates, whereas the previous work has used Zn2+ and deuteroporphyrin IX as substrates [13], which could influence the behaviour of metals in the active site. Indeed, previous measurements of S. aureus CpfC kinetics demonstrate a lack of Fe2+-mediated inhibition when protoporphyrin IX is used as a substrate [1].
To confirm the involvement of the regulatory metal site (Figure 4) in allosteric modulation of S. aureus CpfC, the E271S mutant enzyme was purified and kinetic analyses were performed in the presence and absence of Mg2+. A high concentration of 4 mM Mg2+ was used initially to saturate metal binding (i.e. Fe2+ and Mg2+) at the inhibitory site. Substitution of this glutamate residue with serine resulted in two dramatic outcomes: substrate inhibition by Fe2+ was lost and Mg2+ was no longer a potent inhibitor of metal chelation (Figure 6A). This strongly suggests that the binding of Fe2+ or Mg2+ is stabilised by Glu271. To add weight to this conclusion, Mg2+ dose–response data confirm that Mg2+ is a potent inhibitor of the wild-type enzyme but not the E271S mutant (Figure 6B). Steady-state kinetics for the E271S mutant revealed that loss of metal binding at this residue reverses the Fe2+-mediated increase in Km(coproporphyrin III) (Figure 7A) that is seen for the wild-type enzyme in Figure 3A. The effect of varying [coproporphyrin III] upon the kinetic constants for iron elicits the same pattern for the wild-type (Figure 3B) and E271S mutant (Figure 7B), which strongly suggests that metal binding at the E271S site largely inhibits coproprophyrin binding and/or chelation rather than iron binding at the catalytic site. Taken together, all data presented herein support the model for wild-type S. aureus CpfC in Figure 8 where iron binding to a lower affinity regulatory site at E271 (blue) diminishes the rate of iron chelation at an inner catalytic site (green) via increasing the Km for the coproporphyrin III substrate.
Figure 8. Kinetic model for iron-dependent inhibition of S. aureus ferrochelatase.
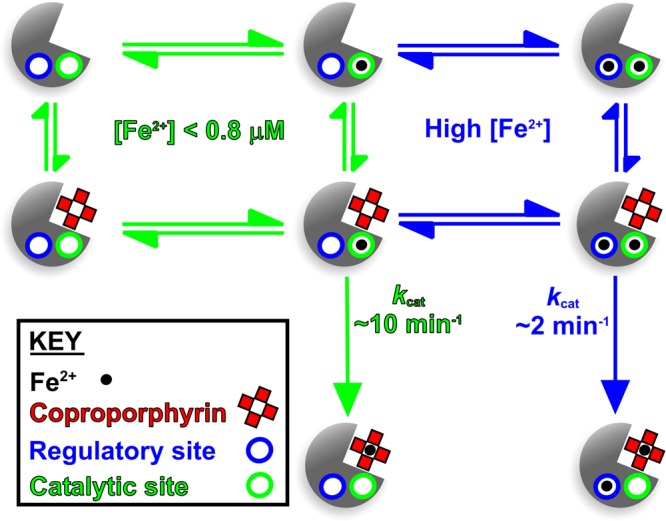
Processes shown in blue and green depict conditions of high- and low-iron concentrations, where the inhibitory regulatory site is occupied or empty, respectively. Since inhibition is only observed at higher iron concentrations, it is unlikely that the regulatory site would be occupied when the catalytic site is empty, so these intermediates are not shown.
It is well-known that there is fierce competition for iron both during infection resulting from nutritional immunity [22], and during growth in mixed microbial communities mediated by microbial siderophores. Therefore, where iron is scarce, it is important for S. aureus to tightly regulate how this nutrient is used within the cell. The current work is consistent with a model whereby at very low Fe2+ concentrations ferrochelatase is able to insert iron into coproporphyrin III, whereas at higher iron concentrations flux through this step of the pathway is down-regulated. There may be many reasons for this substrate inhibition, including avoiding accumulation of Fe-coproporphyrin III, or perhaps once the haem requirements of the cell are satisfied Fe2+ is made available for other processes (e.g. Fe–S cluster assembly). Future studies on bacteria that perform coproporphyrin-dependent haem biosynthesis are required to reveal the hierarchical channelling of iron for different uses in the cell, which will be important during infection (S. aureus, M. tuberculosis, and P. acnes) and growth in microbial communities (B. subtilis).
Acknowledgements
We thank the laboratory of Prof. Harry Dailey (University of Georgia) for supplying expression plasmids for ferrochelatases.
Abbreviations
- CgoX
coproporphyrinogen oxidase
- ChdC
coprohaem decarboxylase
- CpfC
coproporphyrin ferrochelatase
- PpfC
protoporphyrin ferrochelatase
Author Contribution
C.H. performed the experiments and M.S. drove the experimental design. C.H. and M.S. analysed the data. M.S., J.D.R., and C.H. wrote the paper.
Funding
This work was funded by a Research Grant from the Royal Society [RG110528 to M.S.].
Competing Interests
The Authors declare that there are no competing interests associated with the manuscript.
References
- 1.Lobo S.A.L., Scott, A., Videira, M.A.M., Winpenny, D., Gardner, M., Palmer, M.J. et al. (2015) Staphylococcus aureus haem biosynthesis: characterisation of the enzymes involved in final steps of the pathway. Mol. Microbiol. 97, 472–487 10.1111/mmi.13041 [DOI] [PubMed] [Google Scholar]
- 2.Dailey H.A., Gerdes, S., Dailey, T.A., Burch, J.S. and Phillips, J.D. (2015) Noncanonical coproporphyrin-dependent bacterial heme biosynthesis pathway that does not use protoporphyrin. Proc. Natl Acad. Sci. U.S.A. 112, 2210–2215 10.1073/pnas.1416285112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dailey H.A., Dailey, T.A., Gerdes, S., Jahn, D., Jahn, M., O'Brian, M.R. et al. (2017) Prokaryotic heme biosynthesis: multiple pathways to a common essential product. Microbiol. Mol. Biol. Rev. 81, e00048-16 10.1128/MMBR.00048-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hansson M. and Hederstedt, L. (1992) Cloning and characterization of the Bacillus subtilis hemEHY gene cluster, which encodes protoheme IX biosynthetic enzymes. J. Bacteriol. 174, 8081–8093 10.1128/jb.174.24.8081-8093.1992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hansson M. and Hederstedt, L. (1994) Purification and characterisation of a water-soluble ferrochelatase from Bacillus subtilis. Eur. J. Biochem. 220, 201–208 10.1111/j.1432-1033.1994.tb18615.x [DOI] [PubMed] [Google Scholar]
- 6.Dailey T.A. and Dailey, H.A. (2002) Identification of [2Fe-2S] clusters in microbial ferrochelatases. J. Bacteriol. 184, 2460–2464 10.1128/JB.184.9.2460-2464.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Camadro J.M., Ibraham, N.G. and Levere, R.D. (1984) Kinetic studies of human liver ferrochelatase - role of endogenous metals. J. Biol. Chem. 259, 5678–5682 PMID: [PubMed] [Google Scholar]
- 8.Wu C.-K., Dailey, H.A., Rose, J.P., Burden, A., Sellers, V.M. and Wang, B.-C. (2001) The 2.0 angstrom structure of human ferrochelatase, the terminal enzyme of heme biosynthesis. Nat. Struct. Biol. 8, 156–160 10.1038/84152 [DOI] [PubMed] [Google Scholar]
- 9.Karlberg T., Lecerof, D., Gora, M., Silvegren, G., Labbe-Bois, R., Hansson, M. et al. (2002) Metal binding to Saccharomyces cerevisiae ferrochelatase. Biochemistry 41, 13499–13506 10.1021/bi0260785 [DOI] [PubMed] [Google Scholar]
- 10.Shepherd M., Dailey, T.A. and Dailey, H.A. (2006) A new class of [2Fe-2S]-cluster-containing protoporphyrin (IX) ferrochelatases. Biochem. J. 397, 47–52 10.1042/BJ20051967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dailey H.A., Sellers, V.M. and Dailey, T.A. (1994) Mammalian ferrochelatase - expression and characterization of normal and two human protoporphyric ferrochelatases. J. Biol. Chem. 269, 390–395 PMID: [PubMed] [Google Scholar]
- 12.Lecerof D., Fodje, M., Hansson, A., Hansson, M. and Al-Karadaghi, S. (2000) Structural and mechanistic basis of porphyrin metallation by ferrochelatase. J. Mol. Biol. 297, 221–232 10.1006/jmbi.2000.3569 [DOI] [PubMed] [Google Scholar]
- 13.Lecerof D., Fodje, M.N., Alvarez León, R., Olsson, U., Hansson, A., Sigfridsson, E. et al. (2003) Metal binding to Bacillus subtilis ferrochelatase and interaction between metal sites. J. Biol. Inorg. Chem. 8, 452–458 10.1007/s00775-002-0436-1 [DOI] [PubMed] [Google Scholar]
- 14.Hansson M.D., Karlberg, T., Söderberg, C.A.G., Rajan, S., Warren, M.J., Al-Karadaghi, S. et al. (2011) Bacterial ferrochelatase turns human: Tyr13 determines the apparent metal specificity of Bacillus subtilis ferrochelatase. J. Biol. Inorg. Chem. 16, 235–242 10.1007/s00775-010-0720-4 [DOI] [PubMed] [Google Scholar]
- 15.Davidson R.E., Chesters, C.J. and Reid, J.D. (2009) Metal ion selectivity and substrate inhibition in the metal ion chelation catalyzed by human ferrochelatase. J. Biol. Chem. 284, 33795–33799 10.1074/jbc.M109.030205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kazakova S.V., Hageman, J.C., Matava, M., Srinivasan, A., Phelan, L., Garfinkel, B. et al. (2005) A clone of methicillin-resistant Staphylococcus aureus among professional football players. N. Engl. J. Med. 352, 468–475 10.1056/NEJMoa042859 [DOI] [PubMed] [Google Scholar]
- 17.Smith K.M. (1975) Porphyrins and Metalloporphyrins, Elsevier, New York [Google Scholar]
- 18.Berlett B.S., Levine, R.L., Chock, P.B., Chevion, M. and Stadtman, E.R. (2001) Antioxidant activity of ferrozine-iron-amino acid complexes. Proc. Natl Acad. Sci. U.S.A. 98, 451–456 10.1073/pnas.98.2.451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Stein R.L. (2011) in Kinetics of Enzyme Action, pp. 73–87. John Wiley & Sons, Inc., Hoboken, New Jersey, U.S.A. [Google Scholar]
- 20.Käellberg M., Wang, H., Wang, S., Peng, J., Wang, Z., Lu, H. et al. (2012) Template-based protein structure modeling using the RaptorX web server. Nat. Protoc. 7, 1511–1522 10.1038/nprot.2012.085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Peng J. and Xu, J. (2011) Raptorx: Exploiting structure information for protein alignment by statistical inference. Proteins 79, 161–171 10.1002/prot.23175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hammer N.D. and Skaar, E.P. (2012) The impact of metal sequestration on Staphylococcus aureus metabolism. Curr. Opin. Microbiol. 15, 10–14 10.1016/j.mib.2011.11.004 [DOI] [PMC free article] [PubMed] [Google Scholar]