

Abstract
Background:
Congenital myasthenic syndromes (CMSs) are a group of clinically and genetically heterogeneous disorders caused by impaired neuromuscular transmission. The defect of AGRN was one of the causes of CMS through influencing the development and maintenance of neuromuscular transmission. However, CMS reports about this gene mutation were rare. Here, we report a novel homozygous missense mutation (c.5302G>C) of AGRN in a Chinese CMS pedigree.
Methods:
We performed a detailed clinical assessment of a Chinese family with three affected members. We screened for pathogenic mutations using a disease-related gene panel containing 519 genes associated with genetic myopathy (including 17 CMS genes).
Results:
In the family, the proband showed limb-girdle pattern of weakness with sparing of ocular, facial, bulbar, and respiratory muscles. Repetitive nerve stimulation showed a clear decrement of the compound muscle action potentials at 3 Hz only. Pathological analysis of the left tibialis anterior muscle showed predominance of type I fiber and the presence of scattered small angular fibers. The proband's two elder sisters shared a similar but more severe phenotype. By gene analysis, the same novel homozygous mutation (c.5302G>C, p. A1768P) of AGRN was identified in all three affected members, whereas the same heterozygous mutation was found in both parents, revealing an autosomal recessive transmission pattern. All patients showed beneficial responses to adrenergic agonists.
Conclusions:
This study reports a Chinese pedigree in which all three children carried the same novel AGRN mutation have CMS only affecting limb-girdle muscle. These findings might expand the spectrum of mutation in AGRN and enrich the phenotype of CMS.
Keywords: AGRN, Congenital Myasthenic Syndrome, Gene Mutation
INTRODUCTION
Congenital myasthenic syndromes (CMSs [MIM 608931]) represents a group of clinically and genetically heterogeneous disorders caused by impaired neuromuscular junction (NMJ) transmission leading to fatigable weakness.[1] Conventionally, CMS were classified on the basis of the location of a mutated protein as presynaptic, synaptic basal lamina-associated, or postsynaptic. Currently, gene defects that influence the development and maintenance of NMJ are assigned to a separate group of the CMS and rank second in the disease causes following defects of the acetylcholine receptors (AChRs).[2] These genes include RAPSN, DOK7, LRP4, MUSK, and AGRN.[3,4] Agrin, encoded by AGRN, is a cell-specific heparan sulfate proteoglycan generated by alternative splicing. Motoneuron-derived agrin is secreted from nerve terminals into the synaptic cleft and leads to clustering and synthesis of postsynaptic AChRs through activation of the postsynaptic LRP4-MuSK-Dok-7 complex.[5] There are only a few cases reported about this gene mutation so far.[6,7,8,9] Here, we report a novel homozygous missense mutation (c.5302G>C) of AGRN in a Chinese CMS pedigree.
METHODS
Ethical approval
The study was conducted in accordance with the Declaration of Helsinki and was approved by the Ethics Committee of Chinese People's Liberation Army General Hospital. Informed consent was obtained from all subjects.
Clinical assessment
A detailed history was taken, and a thorough neurological examination was performed. Electrophysiological studies and muscle pathology studies were performed to determine the location and nature of the impairment. Auxiliary examinations included muscular magnetic resonance imaging (MRI), creatine kinase levels and anti-AChR and anti-MuSK antibody tests. The diagnosis of CMS can be suspected when there are clinical symptoms of early onset fatigable muscle weakness, a positive family history, and a decremental response of repetitive nerve stimulation (RNS). Genetic studies are needed to confirm the diagnosis.
Genetic and bioinformatics analyses
Venous blood samples were obtained from the pedigree. Genomic DNA was extracted from peripheral blood using a standard procedure. The amplified DNA of the proband was captured with a disease-related gene panel containing 519 genes associated with genetic myopathy including 17 CMS genes [Supplementary Table S1] using biotinylated oligoprobes (MyGenostics GenCap Enrichment technologies) and sequenced on an Illumina HiSeq 2000. The candidate variant was confirmed by Sanger's sequencing and was evaluated the pathogenicity by three algorithms, namely, SIFT (http://sift.jcvi.org/), PolyPhen (http://genetics.bwh.harvard.edu/pph2/index.shtml) and Mutation Taster (http://mutationtaster.org/) as described previously. Sanger's sequencing was then conducted across the family.
Supplementary Table S1.
The list of 519 genes related with genetic myopathy contained in the panel
| ABAT | ABCB1 | ABCC2 | ABCC8 | ACOX1 | ACY1 | ADCK3 | ADSL |
| AGA | AHI1 | AKT2 | AKT3 | ALDH4A1 | ALDH5A1 | ALDH7A1 | ALG1 |
| ALG3 | ALG6 | ALG8 | ALG9 | ALG11 | ALG12 | ALG13 | AMT |
| APIS2 | APTX | ARFGEF2 | ARG1 | ARHGEF15 | ARHGEF9 | ARL13B | ARSA |
| ARSB | ARX | ASAH1 | ASPA | ATIC | ATN1 | ATP13A2 | ATP1A2 |
| ATP1A3 | ATP2A2 | ATP5A1 | ATP6AP2 | ATP6VOA2 | ATP7A | ATPAF2 | ATR |
| ATRX | AVPR1A | B4GALT1 | BCKDHA | BCKDHB | BCKDK | BCS1L | BDNF |
| BLK | BRAF | BRAT1 | BRD2 | BTD | BUB1B | C12orf57 | C12orf65 |
| C12orf12 | CACNA1A | CACNA1C | CACNA1H | CACNB4 | CASK | CASR | CBL |
| CC2D2A | CCL2 | CDK5RAP2 | CDKL5 | CDON | CEL | CENPJ | CEP152 |
| CEP290 | CHD2 | CHD4 | CHD7 | CHD8 | CHRNA2 | CHRNA4 | CHRNB2 |
| CISD2 | CLCN2 | CLCN4 | CLCNKA | CLCNKB | CLN3 | CLN5 | CLN6 |
| CLN8 | CNTN2 | CNTNAP2 | COG1 | COH4 | COG5 | COG6 | COG7 |
| COG8 | COL18A1 | COL4A1 | COQ2 | COQ9 | COX15 | CP | CPT1A |
| CPT2 | CREBBP | CSTB | CTSA | CTSD | CTSF | CUL4B | CYP1B1 |
| CYP2A6 | CYP2B6 | CYP2C19 | CYP2C9 | CYP2D6 | CYP2R1 | CYP2U1 | CYP3A5 |
| DBT | DCAF17 | DCX | DDC | DDOST | DEPDC5 | DHCR7 | DLD |
| DOLK | DPM1 | DPM2 | DPM3 | DPYD | DYRK1A | EEF1A2 | EFHC1 |
| EFHC2 | EHMT1 | EIF2AK3 | EIF2B1 | EIF2B2 | EIF2B3 | EIF2B4 | EIF2B5 |
| EMX2 | EPM2A | ERCC6 | ERCC8 | ETFA | ETFB | ETFDH | FA2H |
| FAAH | FAM126A | FDG1 | FGF8 | FGFR1 | FGFR2 | FGFR3 | FH |
| FKRP | FKTN | FLVCR2 | FMR1 | FOLR1 | FOXR1 | FOXH1 | FOXP1 |
| FOXP2 | FOXP3 | FTL | FUCA1 | GABBR2 | GABRA1 | GABRA2 | GABRA3 |
| GABRD | GABRG2 | GALC | GALNS | GAMT | GATA6 | GATM | GCDH |
| GCK | GCSH | GFAP | GLB1 | GLDC | GLI2 | GLT3 | GLIS3 |
| GLRA1 | GLRB | GLUD1 | GLUL | GNAO1 | GNE | GNPTAB | GNPTG |
| GNS | GOSR2 | GPC3 | GPHN | GPR56 | GRIA3 | GRIN1 | GRIN2A |
| GRIN2B | GU2B | HADH | HCN1 | HCN4 | HDAC8 | HEXA | HEXB |
| HGSNAT | HNF1A | HNF1B | HNF4A | HNRNPU | HOXA1 | HPD | HPRT1 |
| HGAS | HSD17B10 | HSD17B4 | HYAL1 | IBA57 | IDH2 | IDS | IDUA |
| IER3IP1 | INPP5E | INS | INSR | IQSEC2 | KAT6B | KCNA1 | KCNV2 |
| KCDH7 | KDM5C | KIAA1279 | KLF11 | KRAS | L1CAM | L2HGDH | LARGE |
| LRB | LGI1 | LIAS | LIG4 | LRPPRC | MAGI2 | MAGT1 | MAP2K1 |
| MAP2K2 | MAPK10 | MBD5 | MCOLN1 | MCPH 1 | ME2 | MECP2 | MED12 |
| MED17 | MEF2C | MET | MFSD8 | MGAT2 | MID1 | MKKS | MLC1 |
| MMACHC | MOCS1 | MOCS2 | MOGS | MPDU1 | MPI | MTHFR | MTR |
| MTRR | MYBPC1 | NAGLU | NDE1 | NDUFA1 | NDUFA2 | NDUFS1 | NDUFS3 |
| NDUFS4 | NDUFS7 | NDUFS8 | NDUFV1 | NEU1 | NEUROD1 | NEUROG3 | NF1 |
| NGLY1 | NHEJ1 | NHLRC1 | NHS | NIPBL | NKX2-2 | NLGN3 | NLGN4X |
| NODAL | NOTCH3 | NPC1 | NPC2 | NPHP1 | NRAS | NRXN1 | NSD1 |
| OFD1 | OPA1 | OPHN1 | PAFAH1B1 | PAK3 | PANK2 | PAX4 | PAX6 |
| PC | PCDH19 | PDGFRB | PDHA1 | PDHX | PDSS1 | PDSS2 | PDX1 |
| PEX1 | PEX10 | PEX12 | PEX13 | PEX14 | PEX16 | PEX19 | PEX2 |
| PEX26 | PEX3 | PEX5 | PEX6 | PEX7 | PGK1 | PGM1 | PHF6 |
| PHFDH | PIGV | PIK3CA | PIK3R2 | PLA2G6 | PLAGL1 | PLCB1 | PLP1 |
| PMM2 | PNKP | PNPO | POLG | POMGNT1 | POMT1 | POMT2 | PPT1 |
| PQBP1 | PRICKLE1 | PRICKLE2 | PRODH | PRRT2 | PSAP | PSAT1 | PTCH2 |
| PTEN | PTF1A | PTPN11 | QDPR | RAB39B | RAB3GAP1 | RAD21 | RAF1 |
| RAI1 | RARS2 | RFT1 | RFX6 | RNASEH2A | RNASEH2B | RNASEH2C | RPGRIP1L |
| RPS6KA3 | RRP1B | RTTN | SAMHD1 | SCARB2 | SCN10A | SCN11A | SCN1A |
| SCN1B | SCN2A | SCN2B | SCN3A | SCN3B | SCN4B | SCN5A | SCN8A |
| SCN9A | SCO2 | SDHA | SERPINI1 | SETBP1 | SGCE | SGSH | SHANK2 |
| SHANK3 | SHH | SHOC2 | SIX3 | SLC16A2 | SLC17A5 | SCL19A2 | SLC19A3 |
| SLC1A3 | SLC2OA2 | SLC25A15 | SLC25A19 | SLC25A22 | SLC2A1 | SLC35A1 | SLC35A2 |
| SLC35C1 | SLC46A1 | SLC6A4 | SLC6A5 | SLC6A8 | SLC9A6 | SMC1A | SMC3 |
| SMN1 | SMPD1 | SMS | SNAP29 | SNIP1 | SOS1 | SPRED1 | SPTAN1 |
| SRD5A3 | SRPX2 | ST3GAL5 | STIL | STRADA | STXBP1 | SUCLA2 | SUMF1 |
| SUOX | SURF1 | SYN1 | SYNGAP1 | SYP | TACO1 | TBC1D24 | TBX1 |
| TCF4 | TGIF1 | TMEM165 | TMEM216 | TMEM67 | TMEM70 | TPP1 | TREX1 |
| TRPM6 | TSC1 | TSC2 | TSEN2 | TSEN34 | TSEN54 | TUBA1A | TUBA8 |
| YUBB2B | TUSC3 | UBE3A | UCP2 | VANGL1 | VPS13A | VPS13B | VPK1 |
| WDR45 | WDR62 | WFS1 | ZEB2 | ZFP57 | ZIC2 | CHAT* | COLQ* |
| LAMB2* | CHRNA1* | CHRNB1* | CHRND* | CHRNE* | CHRNG* | AGRN* | DOK7* |
| MUSK* | RAPSN* | GFPT1* | DPAGT1* | ALG2* | PLEC* | SCN4A* |
*The 17 genes are congenital myasthenic syndrome related genes screened in the study.
RESULTS
Clinical features
The proband (II-3, the pedigree shown in Figure 1) was a 27-year-old man who had an apparently normal childhood and adolescence except failing to pass the physical examination of high jump and running. At 21 years old, he began to suffer from fatigable weakness of lower limbs. Gradually, he had difficulty standing up from a squat position, jumping, and running. During the cause of the disease, he had no ptosis, bulbar or facial weakness. Neurological examination at the age of 25 years revealed normal cranial nerves and mild muscle atrophy of lower legs. Muscle strength of lower limbs was Medical Research Council (MRC) Grade 4−/5 in proximal and Grade 4+/5 in distal. Tendon reflexes were preserved except bilateral Achilles reflexes. Ocular, facial, bulbar, and respiratory muscles were not involved. Creatine kinase level was normal and anti-AChR, and anti-MuSK antibody tests were negative. The MRI of lower extremities was normal. The nerve conduction study and needle electromyography were within normal limits. RNS at 3 Hz evoked from common peroneal nerves showed a clear decrement of the compound muscle action potentials, with 16% and 18% decline in left and right tibialis anterior, respectively. No significant changes were recorded of RNS at 10 Hz or 20 Hz. Pathological analysis of the left tibialis anterior muscle under light microscopy showed a predominance of type I fiber and the presence of scattered small angular fibers [Figure 2].
Figure 1.
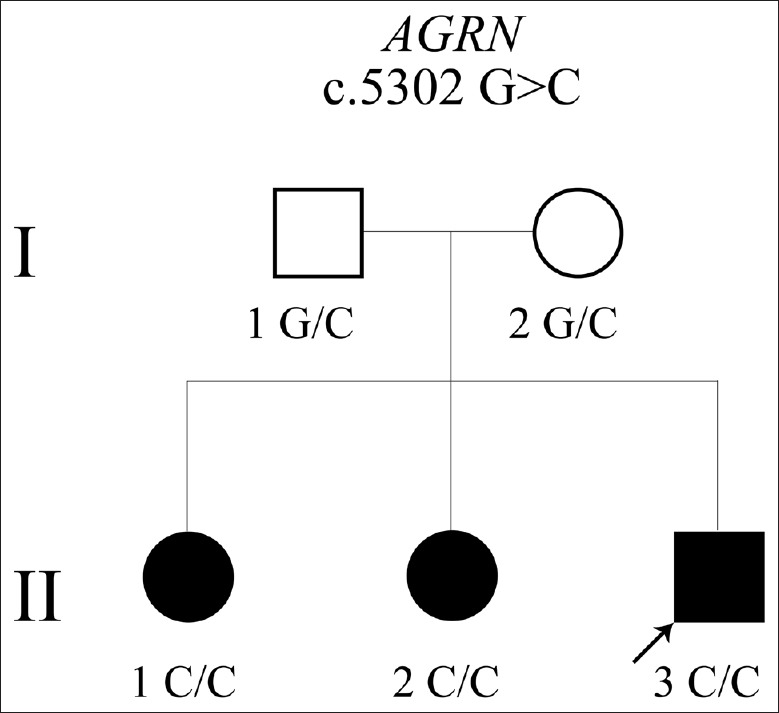
A Chinese congenital myasthenic syndrome pedigree with a novel AGRN mutation only affecting limb-girdle muscle. Arrow indicates the proband. The homozygous AGRN mutation (c.5302G>C) was inherited from parents.
Figure 2.

Pathological results of left tibialis anterior muscle from the proband (original magnification, ×100). (a) Presence of scattered small angular fibers (H & E staining). (b) ATPase staining showed predominance of type I fiber (dark).
The other two elder sisters shared a similar but more severe phenotype. The 29-year-old sister (II-2) suffered from lower limb weakness at the age of 7 years. She complained of walking slowly, difficulty in climbing and a tendency to fall. Upper limbs became involved from the age of 9 years. Neurological assessment at 12 years old showed normal cranial nerve function except trapezius muscles weakness (MRC Grade 4/5). Muscle strength of limbs was Grade 4/5 in proximal and Grade 5−/5 in distal. Deep tendon reflexes were decreased. Muscle enzyme levels were normal. Needle electromyography of distal muscles in four extremities showed short duration and low amplitude motor unit potentials with a few abnormal spontaneous potentials. Nerve conduction studies were normal. Pathological analysis of muscle biopsy under light microscopy revealed type II muscle fiber atrophy. Another sibling, a 31-year-old female (II-1), showed a similar manifestation, but she did not undergo evaluation.
Genetic analysis
We identified a novel homozygous missense mutation (c.5302G>C) in exon 31 of AGRN leading to the substitution of alanine to proline in the C-terminal LG2 domain of agrin (p. A1768P; RefSeq: NM_198576). All three siblings were homozygous for the mutation while both parents were heterozygous [Figure 3]. This variation is not found in ExAC population database. SIFT predicted the substitution to affect protein function with a score of 0.03. Polyphen revealed the mutation to be probably damaging with a score of 1.0 and Mutation Taster predicted that this mutation was disease-causing. Therefore, we made the diagnosis of CMS caused by a novel homozygous mutation in AGRN (c.5302G>C) (we have submitted the variant to Leiden Open Variation Database http://databases.lovd.nl/shared/variants/0000128826).
Figure 3.

Sanger sequences of AGRN mutation (c.5302G>C) across the family. The red arrow indicated the mutation site.
Treatment and follow-up
First treatment with pyridostigmine only showed a beneficial response during the 1st month, then, the symptoms were aggravated, so we tried ephedrine and acquired an evident symptomatic improvement after only 3 days of treatment. Due to the difficulty in obtaining ephedrine, we changed the treatment to salbutamol and observed a similar therapeutic effect as ephedrine. After treatment, the more severely affected sister (II-1) could walk a much longer distance, improving from <50 m to more than 500 m. All three patients are still receiving treatment and have taken salbutamol (2 mg tid) for more than 1 year, and the movement status is sustained.
DISCUSSION
We report a Chinese pedigree with all three CMS patients harboring the same novel missense pathogenic mutation (c.5302G>C p. A1768P) of AGRN. Genetic analysis revealed both parents were heterozygous carrying one single mutated allele that had been transmitted to their three affected children. The parents denied that they were consanguineous, but both of them were from a small village. To the best of our knowledge, previously, only four reports described CMS caused by defects in AGRN, which displayed heterogeneous clinical features. In 2009, Huzé et al.[6] first reported two siblings from a consanguineous family carrying a homozygous missense mutation (G1709R) and presented with ptosis, mild facial and limb-girdle muscles weakness. The second report described a severe CMS patient who required continuous respiratory support caused by two compound heterozygous mutations (V1727F, Q353X).[7] The third article reported five patients from three unrelated families who shared different phenotypes of distal muscle weakness and atrophy.[8] The latest case reported a 17-month-old boy harboring a homozygous mutation (G1765S) who presented with dropped head in addition to proximal muscle weakness, ptosis, and ophthalmoplegia.[9] Acetylcholinesterase inhibitors were not helpful in most of the cases, while adrenergic agonists provided a positive effect for some of the patients. More detailed, there are three mutations located in the LG2 domain as well as our report. As we know, agrin includes three globular, C-terminal LG domains, an N-terminal (NtA) domain and follistatin-like domains.[10] The NtA domain is responsible for binding to basal laminae. The C-terminal LG3 domain is critical for the aggregation of AChRs and other molecules at the NMJ, whereas LG1 and LG2 domain of agrin are involved in interacting with α-dystroglycan, which is a multimeric transmembrane protein complex and is thought to be associated with structural stability of muscle cell membrane.[11] The interaction seems to promote the binding of agrin to the surface of muscle cells, and hence increase the potency of agrin in inducing AChRs clustering, which is an important event in NMJ development.[12] The way in which the interaction affects neuromuscular transmission remains unclear. Studied about the G1709R substitution in LG2 domain showed that the mutation did not affect agrin's ability to activate MuSK or cluster AChRs, nor does it affect the interaction with a-dystroglycan, it seemed to perturb the endplate maintenance.[6] On the contrary, another analysis showed that V1727Fmutation in LG2 domain significantly reduced AChRs clustering activity by impairing MuSK activation and increased affinity to α-dystroglycan, which mimics non-neural isoform agrin.[7] In our report, the patients showed a typical electrophysiological change in the RNS test. The pathology demonstrated the predominance of type I fiber and a slight myopathic change. The therapeutic effects of adrenergic agonists on all three patients are evident. All these features are in accordance with congenital muscular dystrophy caused by AGRN mutation. However, the clinical manifestations of our patients were somewhat different from those of previously reported cases. They showed a limb-girdle pattern weakness without the involvement of ocular, facial, bulbar, and respiratory muscles. Although bearing the same mutation, the three siblings showed variations in age of onset and in symptom severity. The missense mutation we identified were predicted to affect the function of the protein. However, future investigations are needed to pin down the detailed molecular mechanism how a defect in the C-terminal LG2 domain of agrin influence NMJ.
In conclusion, we report a Chinese CMS pedigree with a novel AGRN mutation only affecting limb-girdle muscle. The study findings might expand the spectrum of mutation in AGRN and enrich the phenotype of CMS.
Supplementary information is linked to the online version of the paper on the Chinese Medical Journal website.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
Acknowledgments
We would like to thank all the patients and clinicians who took part in this study and Beijing MyGenostics for technical assistance.
Footnotes
Edited by: Qiang Shi
REFERENCES
- 1.Rodríguez Cruz PM, Palace J, Beeson D. Congenital myasthenic syndromes and the neuromuscular junction. Curr Opin Neurol. 2014;27:566–75. doi: 10.1097/WCO.0000000000000134. doi: 10.1097/WCO.0000000000000134. [DOI] [PubMed] [Google Scholar]
- 2.Engel AG, Shen XM, Selcen D, Sine SM. Congenital myasthenic syndromes: Pathogenesis, diagnosis, and treatment. Lancet Neurol. 2015;14:420–34. doi: 10.1016/S1474-4422(14)70201-7. doi: 10.1016/S1474-4422(14)70201-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Singhal N, Martin PT. Role of extracellular matrix proteins and their receptors in the development of the vertebrate neuromuscular junction. Dev Neurobiol. 2011;71:982–1005. doi: 10.1002/dneu.20953. doi: 10.1002/dneu.20953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tezuka T, Inoue A, Hoshi T, Weatherbee SD, Burgess RW, Ueta R, et al. The MuSK activator agrin has a separate role essential for postnatal maintenance of neuromuscular synapses. Proc Natl Acad Sci U S A. 2014;111:16556–61. doi: 10.1073/pnas.1408409111. doi: 10.1073/pnas.1408409111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Burden SJ, Yumoto N, Zhang W. The role of muSK in synapse formation and neuromuscular disease. Cold Spring Harb Perspect Biol. 2013;5:a009167. doi: 10.1101/cshperspect.a009167. doi: 10.1101/cshperspect. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Huzé C, Bauché S, Richard P, Chevessier F, Goillot E, Gaudon K, et al. Identification of an agrin mutation that causes congenital myasthenia and affects synapse function. Am J Hum Genet. 2009;85:155–67. doi: 10.1016/j.ajhg.2009.06.015. doi: 10.1016/j.ajhg.2009.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Maselli RA, Fernandez JM, Arredondo J, Navarro C, Ngo M, Beeson D, et al. LG2 agrin mutation causing severe congenital myasthenic syndrome mimics functional characteristics of non-neural (z-) agrin. Hum Genet. 2012;131:1123–35. doi: 10.1007/s00439-011-1132-4. doi: 10.1007/s00439-011-1132-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nicole S, Chaouch A, Torbergsen T, Bauché S, de Bruyckere E, Fontenille MJ, et al. Agrin mutations lead to a congenital myasthenic syndrome with distal muscle weakness and atrophy. Brain. 2014;137:2429–43. doi: 10.1093/brain/awu160. doi: 10.1093/brain/awu160. [DOI] [PubMed] [Google Scholar]
- 9.Karakaya M, Ceyhan-Birsoy O, Beggs AH, Topaloglu H. A novel missense variant in the AGRN gene; congenital myasthenic syndrome presenting with head drop. J Clin Neuromuscul Dis. 2017;18:147–51. doi: 10.1097/CND.0000000000000132. doi: 10.1097/CND.0000000000000132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Burgess RW, Skarnes WC, Sanes JR. Agrin isoforms with distinct amino termini: Differential expression, localization, and function. J Cell Biol. 2000;151:41–52. doi: 10.1083/jcb.151.1.41. doi: 10.1083/jcb.151.1.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sciandra F, Bozzi M, Bianchi M, Pavoni E, Giardina B, Brancaccio A, et al. Dystroglycan and muscular dystrophies related to the dystrophin-glycoprotein complex. Ann Ist Super Sanita. 2003;39:173–81. [PubMed] [Google Scholar]
- 12.Gesemann M, Cavalli V, Denzer AJ, Brancaccio A, Schumacher B, Ruegg MA, et al. Alternative splicing of agrin alters its binding to heparin, dystroglycan, and the putative agrin receptor. Neuron. 1996;16:755–67. doi: 10.1016/s0896-6273(00)80096-3. doi: 10.1016/S0896-6273(00)80096-3. [DOI] [PubMed] [Google Scholar]