

Spinal muscular atrophy (SMA) is a genetic disorder which is clinically characterized by progressive muscle weakness and atrophy and is associated with the degeneration of spinal and lowers bulbar motor neurons. SMA is the most common genetic cause of infant mortality, and seems to be present in general populations. The clinical spectrum of SMA ranges from early infant death to normal adult life with only mild weakness. Approximately 81.2–95.0% of cases of SMA resulted from homozygous deletion of survival of motor neuron 1 (SMN1) and 5.0% were compound heterozygous patients.[1] SMA might manifest not only the dysfunction of pure motor neurons but also abnormalities in neuromuscular junction (NMJ), osteoporotic bone formation, cardiac abnormalities, and vascular defects.[2] These phenomena have been described in severe SMA (Type I, II) patients and in mouse models while data from SMA Type III individuals are not available. Patients with SMA Type III demonstrate progressive proximal weakness affecting the legs more severely than the arms, and might ultimately end up in the wheelchair. Herein, we report one patient with SMA Type III manifesting an atrial septal defect (ASD), NMJ defect, short stature, and thick toes.
A 28-year-old Han Chinese man presented with a past medical history of early-onset, slowly progressive proximal lower limb weakness beginning at 3-year-old, with development abdominal lordosis and a swaying back. His muscle weakness progressed slowly to involve the proximal muscles of his upper limbs at the age of 18. The patient continued to require assistance and became wheelchair bound at the age of 20. On evaluation, it was found that he had weakness in neck flexion (4/5 on a medical research council scale, Graded 0–5), reduced strength in the proximal muscles of his upper extremities (3/5), lower extremities (2/5), and distal muscle (4/5). Furthermore, hypomyotonia and atrophy of the proximal upper and lower limb muscles were noted, as well as the gastrocnemius muscle. Tendon reflexes were absent in all limbs. In addition, the patient had thick toes and talipes equinovarus [Figure 1a]. The patient measured 152 cm tall and reported that his father was 156 cm and mother was 165 cm tall.
Figure 1.
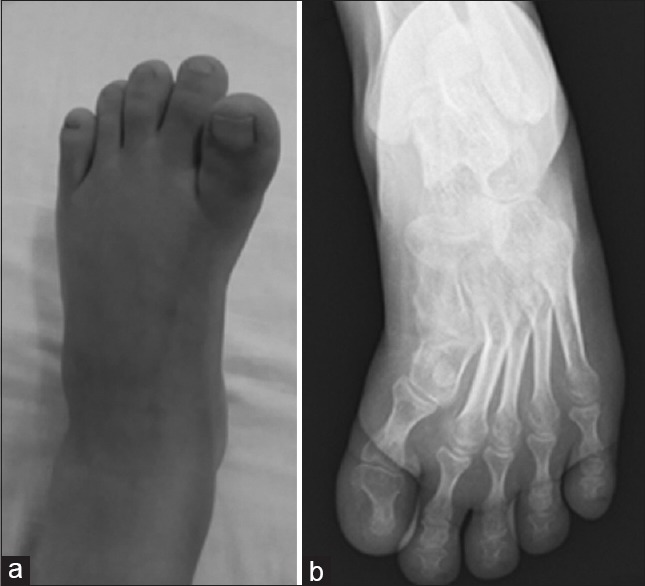
Representative images of the left foot. Thick toes and talipes equinovarus are noticed (a). The left foot X-ray shows osteoporosis and the first metatarsal is short and thick (b).
Auxiliary examination: serum creatine kinase, thyroid hormone, parathyroid hormone, and 25-hydroxyvitamin D were normal. Electromyography showed evidence of neurogenic discharge with a great amount of fasciculation and high amplitude of motor unit potentials. Pathologic decrement during repetitive nerve stimulation, 25.8–46.1% amplitude reduction of the fifth compound muscle action potential compared with the first during a train of supramaximal nerve stimulations at 3 Hz in the left abductor digiti minimi and right deltoid strongly suggests dysfunction of the NMJ. Nerve conduction study (NCS) was normal. Echocardiogram showed an ASD (20 mm) and pulmonary artery hypertension (45 mmHg). The left foot X-ray showed osteoporosis, and the first metatarsal was short and thick [Figure 1b]. The multiplex ligation-dependent probe amplification analysis showed that the patient had homozygous deletions of exons 7 and 8 in SMN1 gene.
The patient was found to possess exons 7 and 8 deletions of the SMN1 gene and manifested clinically after the age of 18 months with the onset of proximal muscle weakness. There was no fluctuating or fatigable weakness, creatine kinase, and NCS was normal. Myasthenia gravis, myopathy, muscular dystrophy, or peripheral neuropathies were excluded. Therefore, he was diagnosed as SMA Type III. Based on the information from this case, it seems that the patient clinically manifests with SMA Type III, ASD, defects at the NMJ and bone remodeling.
As previous case report demonstrated, SMA Type I patients were more likely to develop the cardiac disease while cardiac function and rhythm remain remarkably stable in SMA Type II and III, even severely handicapped patients at the end stage of disease.[3] Detailed classification of the reported abnormalities suggested convergence to specific pathologies in patients with SMA might be linked to downstream effects of SMN deficiency. As we know, SMA is the leading genetic cause of infant mortality with an incidence of approximately one in 10,000 newborns, whereas ASDs are detected in one per 1500 live births. In spite of an ASD in the SMA Type III patient, he also had a normal population risk to develop ASD. Whether ASD is associated with SMA Type III need more observation and research.
NMJ pathology has been reported in human SMA patients. SMN protein is localized at NMJs and evidence for the role in NMJ of SMA is provided by the failure of cultured patients’ muscle cells to cluster acetylcholine receptors (AChRs) and alterations in AChR clustering, misplacement, accumulation of synaptic vesicles in SMA Type I fetal samples.[4] Although it is currently unclear how low levels of SMN cause these defects at the NMJ, the available evidence indicates that these pathologies might be a consequence of failed synaptic maintenance due to impaired axonogenesis.
Short stature is defined as a height that is 2 standard deviations (2SDs) or more below the mean height for individuals of the same sex and chronologic age in a given population. This patient measured at 152 cm tall, which is 2SD below the mean height for same age Chinese males group (160.5 cm). In addition, osteoporosis and a short, thick metatarsal were also noticed. SMN protein interacts with osteoclast stimulatory factor, and SMN might play a role in skeletal development and bone remodeling.[5] Skeletal abnormity could be a complication of SMA.[5]
To date, more and more clinical phenomenon and basic study make it clear that additional cells and tissue types are selectively vulnerable to reduced levels of SMN.[3] The evidence suggests SMA goes beyond the motor system and might be a multi-organ disease, including muscle, brain, heart, vasculature, bone, pancreas, liver, lung, and intestine.[2,3] Some of the phenotypes are observed only in animal models while others in both SMA patients and mice, including the severe and mild types. SMA Type III patients could manifest with a mild limb weakness, defects in heart, NMJ and skeletal abnormalities. The further study is required to investigate and follow-up SMA Type III patients for improving survival and quality of life.
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent forms. In the form, the patient has given his consent for his images and other clinical information to be reported in the journal. The patient understands that his name and initial will not be published and due efforts will be made to conceal his identity, but anonymity cannot be guaranteed.
Financial support and sponsorship
This work was supported by grants from the National Natural Science Foundation of China (No. 81200965, No. 81071024, No. 81171202, and No. 81471287).
Conflicts of interest
There are no conflicts of interest.
Footnotes
Edited by: Peng Lyu
REFERENCES
- 1.Qu YJ, Song F, Yang YL, Jin YW, Bai JL. Compound heterozygous mutation in two unrelated cases of Chinese spinal muscular atrophy patients. Chin Med J. 2011;124:385–9. doi: 10.3760/cma.j.issn.0366-6999.2011.03.012. [PubMed] [Google Scholar]
- 2.Hamilton G, Gillingwater TH. Spinal muscular atrophy: Going beyond the motor neuron. Trends Mol Med. 2013;19:40–50. doi: 10.1016/j.molmed.2012.11.002. doi: 10.1016/j.molmed.2012.11.002. [DOI] [PubMed] [Google Scholar]
- 3.Shababi M, Lorson CL, Rudnik-Schöneborn SS. Spinal muscular atrophy: A motor neuron disorder or a multi-organ disease? J Anat. 2014;224:15–28. doi: 10.1111/joa.12083. doi: 10.1111/joa.12083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Martínez-Hernández R, Bernal S, Also-Rallo E, Alías L, Barceló MJ, Hereu M, et al. Synaptic defects in type I spinal muscular atrophy in human development. J Pathol. 2013;229:49–61. doi: 10.1002/path.4080. doi: 10.1002/path.4080. [DOI] [PubMed] [Google Scholar]
- 5.Shanmugarajan S, Swoboda KJ, Iannaccone ST, Ries WL, Maria BL, Reddy SV. Congenital bone fractures in spinal muscular atrophy: Functional role for SMN protein in bone remodeling. J Child Neurol. 2007;22:967–73. doi: 10.1177/0883073807305664. doi: 10.1177/0883073807305664. [DOI] [PMC free article] [PubMed] [Google Scholar]