

Abstract
Background
Telomere defects are thought to play a role in cardiomyopathies, but the specific cell type affected by the disease in human hearts is not yet identified. The aim of this study was to systematically evaluate the cell type specificity of telomere shortening in patients with heart failure in relation to their cardiac disease, age, and sex.
Methods and Results
We studied cardiac tissues from patients with heart failure by utilizing telomere quantitative fluorescence in situ hybridization, a highly sensitive method with single‐cell resolution. In this study, total of 63 human left ventricular samples, including 37 diseased and 26 nonfailing donor hearts, were stained for telomeres in combination with cardiomyocyte‐ or α‐smooth muscle cell‐specific markers, cardiac troponin T, and smooth muscle actin, respectively, and assessed for telomere length. Patients with heart failure demonstrate shorter cardiomyocyte telomeres compared with nonfailing donors, which is specific only to cardiomyocytes within diseased human hearts and is associated with cardiomyocyte DNA damage. Our data further reveal that hypertrophic hearts with reduced ejection fraction exhibit the shortest telomeres. In contrast to other reported cell types, no difference in cardiomyocyte telomere length is evident with age. However, under the disease state, telomere attrition manifests in both young and older patients with cardiac hypertrophy. Finally, we demonstrate that cardiomyocyte‐telomere length is better sustained in women than men under diseased conditions.
Conclusions
This study provides the first evidence of cardiomyocyte‐specific telomere shortening in heart failure.
Keywords: cardiomyocyte, cardiomyopathy, telomere length
Subject Categories: Cardiomyopathy, Heart Failure, Hypertrophy
Clinical Perspective
What Is New?
Cardiac telomere attrition is a cell type‐specific, age‐independent phenomenon in heart failure.
Patients with reduced ejection fraction have cardiomyocytes with shorter telomeres.
Telomere shortening is associated with increased cardiomyocyte DNA damage.
What Are the Clinical Implications?
Additional studies should be undertaken to evaluate the clinical benefits of targeting telomeres for preventive or therapeutic purposes in heart failure.
Introduction
Hypertrophic cardiomyopathy (HCM) is a clinically heterogeneous disorder, commonly characterized by left ventricular hypertrophy with serious adverse outcomes, including heart failure (HF) and sudden cardiac death.1 HCM is caused by an increase in myocyte size, but not myocyte number, and by a greater amount of myocardial fibrosis, which is distributed both throughout the interstitium and in discrete foci.2 Although HCM is often caused by an identifiable mutation in a gene encoding for a sarcomeric protein and inherited in an autosomal‐dominant pattern,3 many patients do not have any relatives in whom the disease is manifested. Extensive studies are being performed to link phenotype with genotype in patients with cardiomyopathies; however, the mechanisms responsible for this progressive disease have yet to be elucidated.4 Further research is required to gain a full understanding of its pathogenesis and ultimately directed therapy.
During recent years, a few reports suggested a connection between telomere dysfunction and cardiovascular pathology.5, 6, 7, 8, 9 Telomeres are the distal ends of chromosomes consisting of (TTAGGG)n tandem repeats that cap the ends of chromosomes providing protection from structural degradation, DNA damage checkpoint responses, and end‐to‐end fusion.10, 11 In proliferative cells, telomere shortening occurs with each cell division as DNA polymerases fail to completely replicate telomeres. Further, newly replicated telomeres are naturally processed by exonucleases to generate the proper 3′ overhang.10 Oxidative stress has also been suggested to be a modulator of telomere loss.12, 13 The combination of these processes may lead to critical shortening and genomic instability. More importantly, when telomeres reach a critical length, cells can enter replicative senescence or become apoptotic.10, 11 Most studies measure telomere length (TL) either in noncardiac cells (such as isolated leukocytes)6, 7, 14, 15 or in total cardiac extracts,16 and use biochemical methods (eg, Southern‐based methods)17, 18, 19 that are unable to distinguish TL in a cell‐specific manner within the heart. Invaluable to our understanding of telomere dynamics are tissue‐ and cell‐specific analyses of telomeres in human diseased and nondiseased hearts using high‐resolution methods. Telomere quantitative fluorescence in situ hybridization (Q‐FISH) allows TL assessments in paraffin‐embedded archival material, while providing single‐cell resolution and maintaining intact tissue architecture.20, 21, 22 Although successfully used on a number of tissues, telomere Q‐FISH was only recently optimized for cardiac tissues.5 In order to better understand cardiac telomeric dynamics in human HF, we investigated cell type‐specific changes in TL of diseased hearts. Our results demonstrate that patients with HF exhibit shorter telomeres compared with nonfailing cohorts. Importantly, the telomere shortening is specific to cardiomyocytes and not to cardiac smooth muscle cells of the same hearts. Our findings indicate that telomere shortening in patients with HCM is associated with extensive DNA damage within cardiomyocytes. Furthermore, we found that aging does not alter TL in cardiomyocytes of nonfailing donor (NFD) hearts. However, under diseased conditions, telomere attrition occurs independently of age. Interestingly, we also show that women sustain their TL better than men in the presence of HF. Collectively, our results provide the first clear evidence that cardiomyocyte‐specific telomere shortening is a characteristic of HF in humans, highlights its potential significance in aging and sex, and suggests possible new scientific avenues important for cardiomyocyte function and survival in cardiac disease conditions.
Methods
Human Heart Procurement
Whole human hearts were procured from 2 separate patient groups: nonfailing brain‐dead organ donors with no history of HF (NFDs) and patients with end‐stage HF transplant (HCM, ischemic cardiomyopathy [ICM], and idiopathic dilated cardiomyopathy [IDCM]) that were obtained at the time of orthotopic heart transplantation. All hearts received cold cardioplegia in situ and were placed on wet ice in 4°C Krebs‐Henseleit buffer. Transmural left ventricular samples (epicardial fat removed) were snap‐frozen in liquid nitrogen and stored at −80°C or fixed overnight in 4% paraformaldehyde/PBS. Following progressive tissue dehydration with ethanol and xylene, the fixed samples were embedded in paraffin. All procedures were approved by the University of Pennsylvania institutional review board. Informed consent for research use of heart tissue was obtained prospectively from all transplant recipients. In the case of organ donors, informed consent for research use of explanted heart tissue was obtained from next of kin.
Patient Cohort
Our cases consist in total of 63 human left ventricular samples, including 37 diseased and 26 nonfailing hearts from organ donors (Table 1). Diseased hearts analyzed throughout the main figures were from patients with HCM (n=17), while diseased hearts analyzed for supplemental figures were from patients with IDCM (n=11) and ICM (n=9), with all manifesting signs of myocardial fibrosis (Figure S1). Transmural samples of the left ventricular myocardium were obtained from the free wall at the time of heart transplantation. Controls (NFDs) were selected from brain‐dead organ donors within the same age range of diseased patients as well as the same distribution of sex, history of diabetes mellitus, body mass index, and left ventricular ejection fraction (LVEF) >45%. Similarly, exclusion criteria for the failing cohort included myocarditis (viral, autoimmune), hepatitis B, hepatitis C, or HIV, severe renal disease, or prior left ventricular assist device support. All women were postmenopausal and no hormone therapy was reported. Left ventricular samples were collected from patients with HCM with LVEF ≥45% (HFpEF) and <45% (HFrEF). Ejection fraction values were available for 20 NFDs and 8 patients with HFrEF and 7 patients with HFpEF.
Table 1.
Demographic Data and Cardiac Parameters of Patients
| Etiology | NFD (n=26) | HCM (n=17) | IDCM (n=11) | ICM (n=9) |
|---|---|---|---|---|
| Sex |
62% Male 38% Female |
80% Male 20% Female |
100% Male | 100% Male |
| Age |
42% Young 58% Old |
24% Young 76% Old |
100% Old | 100% Old |
| BMI, kg/m2 | 29.15±1.26 | 27.66±1.21 | 26.01±1.32 | 27.94±1.25 |
| BSA, m2 | 2.02±0.06 | 1.99±0.06 | 2.06±0.03 | 2.02±0.02 |
| Heart Weight, g | 367.92±20.01 | 505.38±45.96 | 487.22±36.31 | 550±53.17 |
| HMI, g/m2 | 179.14±38.29 | 252.94±18.97 | 241.28±14.90 | 294.45±29.20 |
| Creatinine, mg/dL | 1.14±0.12 | 1.46±0.17 | 1.34±0.17 | 1.29±0.19 |
| Type 2 diabetes mellitus, No. (%) | 2 (8) | 1 (6) | 3 (27) | 1 (11) |
Values are expressed as mean±SD unless otherwise indicated. History and selected clinical parameters of patients selected for cardiac tissue analysis. Left ventricular samples were collected from nonfailing donors (NFDs) and patients with hypertrophic cardiomyopathy (HCM), ischemic cardiomyopathy (ICM), and idiopathic dilated cardiomyopathy (IDCM). NFDs were selected from brain‐dead organ donors to match diseased patients on age, sex, no history of diabetes mellitus, and nonobese status by body mass index (BMI) criteria (BMI <30), no left ventricular assist device, and with a left ventricular ejection fraction of >45%. BSA indicates body surface area; HMI, heart mass index.
Q‐FISH Analysis
Human cardiac paraffin sections (7 μm) were washed with xylene to remove paraffin, rehydrated through a graded ethanol series, followed by antigen unmasking in citrate buffer at pH 6.0 and dehydration. To quantify telomere fluorescence, slides were hybridized with a Cy3‐labeled peptide nucleic acid probe complementary to the mammalian telomere repeat sequence ([N‐terminus to C‐terminus] CCCTAACCCTAACCCTAA; synthesized by PNA Bio Inc.), denaturated for 5 minutes at 86°C, and further incubated at room temperature in the dark for 2.5 hours. Slides were blocked for 1 hour with blocking solution (DAKO) followed by primary antibody incubation (anti‐mouse α‐cardiac troponin T [already diluted; Abcam] or anti‐rabbit α‐smooth muscle [1:100; Abcam]). Slides were then incubated with secondary antibodies (anti‐mouse IgG fraction conjugated with Alexa Fluor 488 [Molecular Probes] or anti‐rabbit IgG fraction Alexa Fluor 488 [Molecular Probes] and counterstained with 4′,6‐diamidino‐2‐phenylindole (DAPI) solution. Between 13–51 different areas were analyzed per sample (see Tables 2 and 3 for details). Telomere FISH and nuclear DNA (DAPI) images were acquired at 100× using a Nikon Eclipse Ni‐U, using identical exposure settings for all telomere images and all DAPI images. The investigators were blinded during experimental procedures, imaging acquisition, and analysis. TL was measured using the Software for Telomere Counting (developed at Johns Hopkins, http://demarzolab.pathology.jhmi.edu/telometer/) as previously described.5, 20 Briefly, the nuclear area (using the DAPI staining) of the cell type of interest is identified. The software then takes into account only the red dots (telomere foci) within the nucleus for automatic analysis.
Table 2.
Number of Cardiac Cell Nuclei Scored for the Telomere Analysis
| Cell Type | Cell No. Per Group | No. of Different Areas Per Sample | Total No. of Cells |
|---|---|---|---|
| Nonfailing donors | |||
| Cardiomyocytes | 70 to 135 | 32 to 42 | 2725 |
| α‐SMA | 66 to 237 | 13 to 18 | 966 |
| HCM | |||
| Cardiomyocytes | 80 to 135 | 20 to 43 | 1326 |
| α‐SMA | 79 to 222 | 22 to 34 | 814 |
| ICM | |||
| Cardiomyocytes | 70 to 155 | 33 to 51 | 1030 |
| α‐SMA | 90 to 176 | 27 to 36 | 1188 |
| ICDM | |||
| Cardiomyocytes | 73 to 197 | 31 to 47 | 749 |
| α‐SMA | 77 to 159 | 29 to 41 | 1053 |
Number of cell nuclei scored for telomere analysis per etiology. α‐SMA indicates α‐smooth muscle actin; HCM, hypertrophic cardiomyopathy; ICDM, idiopathic dilated cardiomyopathy; ICM, ischemic cardiomyopathy.
Table 3.
Number of Cardiomyocyte Nuclei Scored in Subgroups
| Group | Cardiomyocyte No. Per Group | No. of Different Areas Per Sample | Total No. of Cells |
|---|---|---|---|
| Nonfailing donors | |||
| Men | 89 to 132 | 32 to 40 | 1369 |
| Women | 109 to 153 | 33 to 42 | 1356 |
| Young | 89 to 166 | 32 to 42 | 1096 |
| Old | 95 to 151 | 32 to 40 | 1629 |
| HCM | |||
| HFrEF | 80 to 135 | 32 to 43 | 819 |
| HFpEF | 78 to 128 | 20 to 40 | 507 |
| Men | 70 to 128 | 20 to 43 | 1093 |
| Women | 96 to 135 | 36 to 39 | 833 |
| Young | 70 to 128 | 32 to 40 | 716 |
| Old | 78 to 135 | 20 to 43 | 810 |
Number of cardiomyocyte nuclei and number of areas scored in each subgroup. HCM indicates hypertrophic cardiomyopathy; HFrEF, heart failure with reduced ejection fraction; HFpEF, heart failure with preserved ejection fraction.
Cardiac Morphology and Morphometric Analysis
Using paraformaldehyde‐fixed, paraffin‐embedded tissues, 7‐μm‐thick cross‐sections were subjected to trichrome staining (Sigma‐Aldrich) for nuclei, cytoplasm, and collagen visualization. Images were collected using a Nikon stage microscope Nikon Eclipse Ni‐U, and morphometric analysis for collagen tissue deposition was performed with ImageJ software.
Immunofluorescence Microscopy
Slides were de‐paraffinized in xylene and rehydrated in serial ethanol concentration washes. Antigen masking was performed in citrate buffer (10 mmol/L sodium citrate, pH 6.0) for 40 minutes in a steamer. Slides were blocked with 5% normal goat serum (DAKO) for 1 hour at room temperature. Incubation with primary antibodies was performed for 2h at RT. The primary antibodies used were: anti‐mouse α‐cardiac troponin T (Abcam), 53BP1 (1:500; Abcam), and γH2AX (1:300; EMD Millipore). After washing in PBS, the slides were incubated with the goat anti‐rabbit Alexa Fluor 488 (Invitrogen) secondary antibody for 53BP1. γH2AX secondary antibodies were biotin‐conjugated goat anti‐mouse (Jackson ImmunoResearch Laboratories, Inc) and streptavidin‐Alexa Fluor 488 (Jackson ImmunoResearch Laboratories, Inc.) for 1.5 hours in the dark at room temperature and nuclei were counterstained with DAPI. Images were collected using a Nikon stage microscope Nikon Eclipse Ni‐U.
Confocal Microscopy
Confocal images were acquired using a Leica TCS SP8 STED X system equipped with a tunable white light laser. A 63x/1.4 NA PlanApo objective was used for all images. Z stacks were acquired using a Z step size of 0.4 microns. Wavelength ranges of 500 to 545 nm, 561 to 631 nm, and 414 to 464 nm were used to excite green, red, and blue, respectively. Green (immunofluorescence) and red (telomere FISH) images were acquired using HyD detectors and DAPI images were acquired using a standard photomultiplier tube detector.
Statistical Analysis
Wilcoxon rank sum test was used to calculate the statistical significance of differences in cardiomyocyte TL distribution between patients (histogram figures). Mann–Whitney tests were performed for comparison between median values of identified groups in bar graph and box plot figures. Results from Kruskal‐Wallis tests for comparisons between average TL values of NFDs and patients are presented in Figures 4B, 5E, and 6E. Two‐tailed, unpaired Student t tests were performed for comparisons between mean values from the 2 groups and are shown in Figure 3; the difference was considered significant when P<0.05. Kruskal‐Wallis and Mann–Whitney tests were performed for comparison between median values of identified groups and shown in Figures S1 and S2, respectively. All statistical analyses were performed using GraphPad Prism 6 (GraphPad Software, Inc).
Results
Patients With HF Encounter Cardiomyocyte Telomere Shortening
Telomere shortening occurs when proliferating cells undergo each cell cycle, finally leading to cell senescence and apoptosis.23, 24 However, the role of telomere attrition in the setting of HF and specifically in human cardiomyocytes, classically known as post‐mitotic cells,25, 26 is undefined. To investigate TL in HCM cardiomyocytes, human cardiac tissues from patients with HCM and NFDs were subjected to Q‐FISH (Figure 1A). Our analysis demonstrated that the cardiomyocyte TL distribution significantly shifts towards a shorter TL range in HCM hearts compared with NFDs (P=0.016, Figure 1B). Similar results were obtained when we plotted the average TL between NFDs and patients with HCM (P=0.003, Figure 1C). Likewise, patients with ICM (P=0.041) exhibit cardiomyocytes with shorter telomeres than NFDs (Figure S2A), while analysis of TL in patients with idiopathic cardiomyopathy (IDCM) did not reveal the same result (P=0.101).
Figure 1.
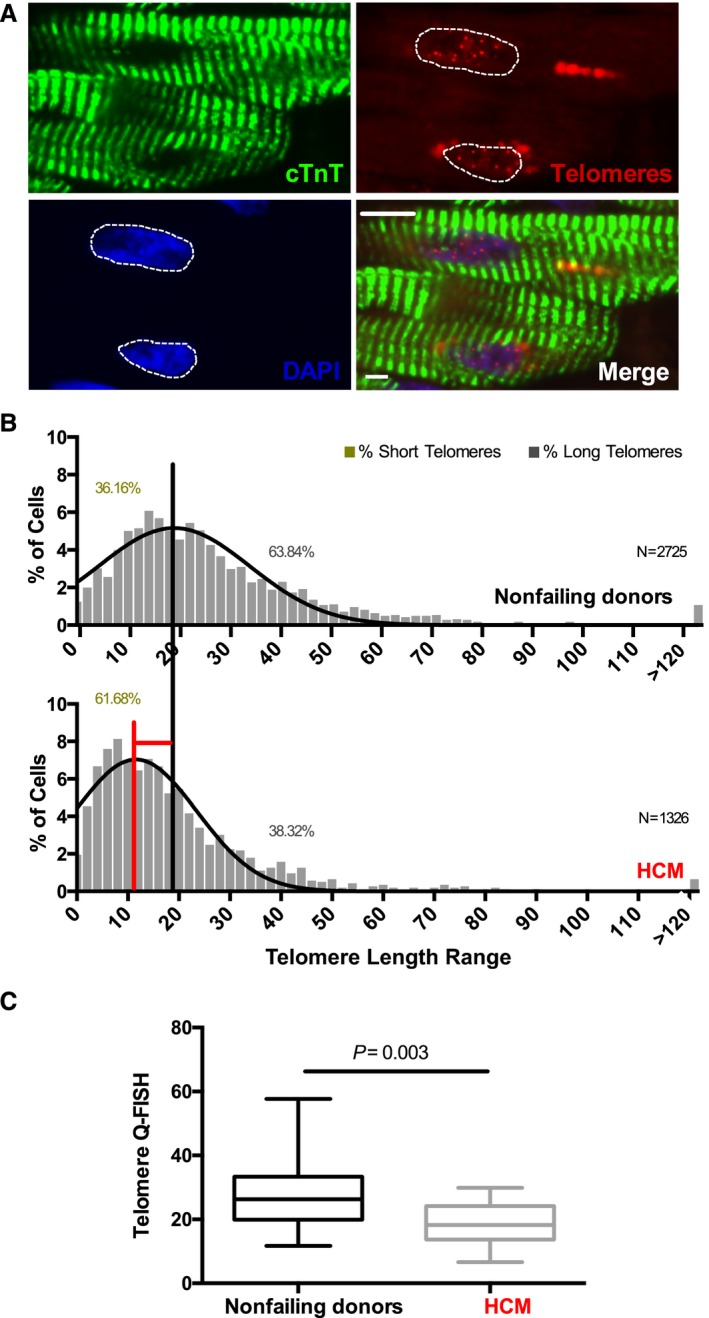
Patients with hypertrophic cardiomyopathy have short cardiomyocyte telomeres. A, Representative images of quantitative fluorescence in situ hybridization (Q‐FISH) analysis, immunostained for the cardiomyocyte‐specific marker, cardiac troponin T (cTnT, green), telomeric probe (red), and 4′,6‐diamidino‐2‐phenylindole (DAPI) for nuclei (blue). White dotted lines mark the area used for telomere analysis within the nucleus of each cardiomyocyte. Scale bar, 10 μm. B, Telomere length distribution histogram of individual cardiomyocytes from nonfailing donors (NFDs) and hypertrophic cardiomyopathy (HCM) patient cardiac samples. Data are presented as percentage of cells within the patients' spectrum of the telomeric length range. Black and red vertical lines were drawn at the median value of the histogram obtained for NFDs and patients with HCM, respectively. The shift in telomeric length from NFD to HCM histogram median value is shown by the red horizontal line (Wilcoxon rank sum test, P=0.016). N indicates the number of cardiomyocytes scored (see also Table 2). The percentage of cells with short and long telomeres is shown in the graph. C, Boxplot analysis shows average telomere length measurements in NFDs and patients with HCM (Mann–Whitney test, P=0.003). A total of 26 NFDs and 17 patients with HCM were analyzed.
To determine whether the observed cardiomyocyte‐telomere shortening is a general feature within the cardiac tissue of patients, we investigated whether vascular smooth muscle cells, which have been implicated in the pathogenesis of cardiovascular diseases,27 were also affected (Figure 2A). Remarkably, we found a comparable TL distribution in cardiac α‐smooth muscle cells between patients with HCM (Figure 2B and 2C), ICM (Figure S2C), and IDCM (Figure S2D) and NFDs (P=0.322 and 0.798 for Figure 2B and 2C, and P=0.578 and 0.921 for Figure S2C and S2D) arguing against a universal telomere shortening in diseased hearts. These data emphasize the selectivity of cardiomyocyte telomere shortening in human HF.
Figure 2.
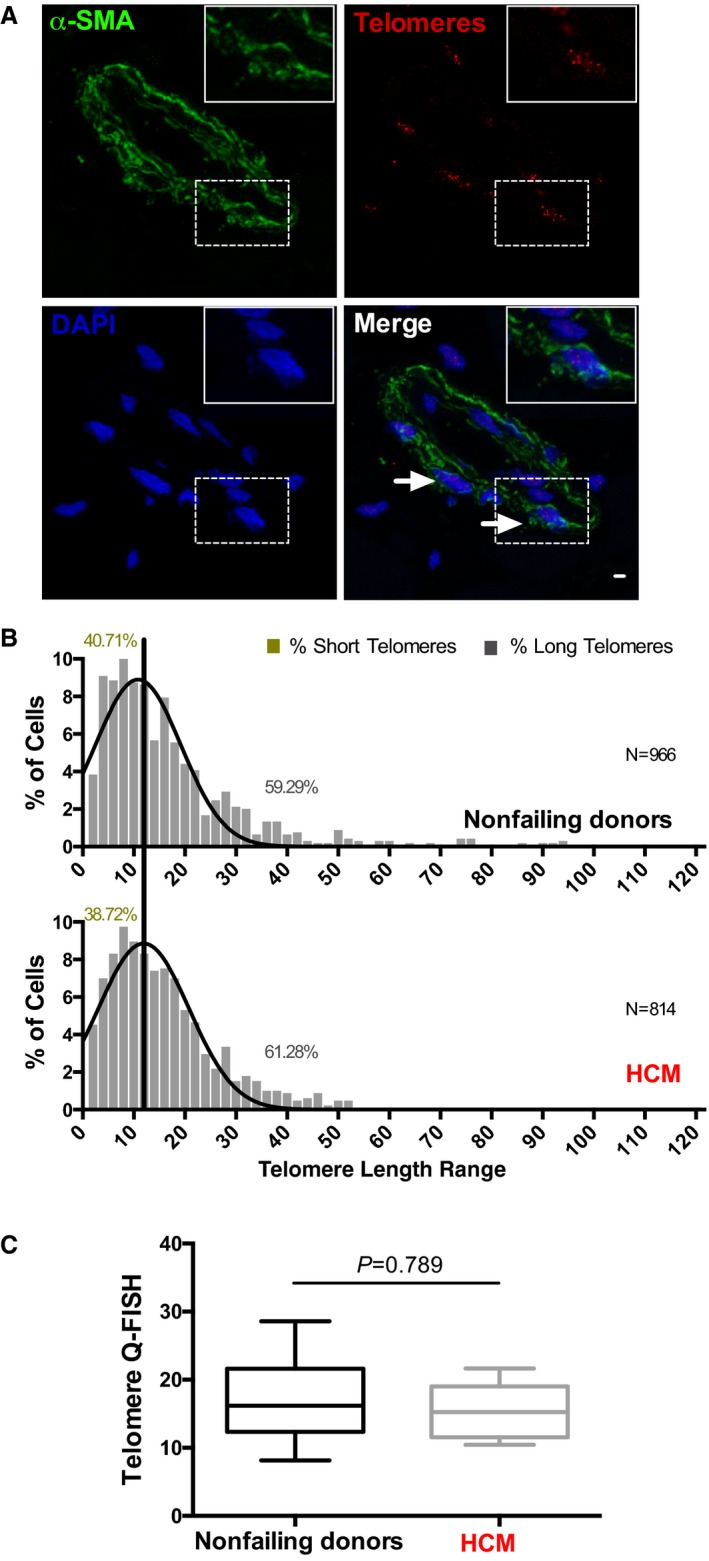
Cardiac smooth muscle cells have comparable telomere lengths in nonfailing donor (NFD) and hypertrophic cardiomyopathy (HCM) hearts. A, Representative picture of left ventricular tissues subjected to quantitative fluorescence in situ hybridization (Q‐FISH) analysis and immunostained for smooth muscle actin cell marker (α‐smooth muscle actin [α‐SMA]), telomeres, and 4′,6‐diamidino‐2‐phenylindole (DAPI) shown in green, red, and blue, respectively. Scale bar, 20 μm. Arrowheads indicate representative analyzed α‐SMA + cells. Insert shows a higher magnification of an α‐SMA + cell. B, Telomere length distribution histogram analysis of α‐SMA + cells from NFDs and HCM patient cardiac samples. Data are presented as percentage of cells within the patients' spectrum of telomeric length range. A black vertical line was drawn at the median value of the histogram obtained for NFDs. Statistical comparison of NFDs and patients with HCM, Wilcoxon rank sum test, P=0.322. N indicates the number of cardiomyocytes scored (see also Table 2). The percentage of cells with short and long telomeres is shown in the graph. C, Boxplot analysis shows average telomere length measurements between NFDs and patients with HCM (Mann–Whitney test, P=0.798). A total of 26 NFDs and 17 patients with HCM were analyzed.
TL is Associated With DNA Damage in Cardiomyocytes of Patients With HCM
It has been reported that telomere shortening and dysfunction are associated with activation of DNA damage pathways,28, 29 but such correlations have never been reported in adult human cardiomyocytes. To investigate whether HCM cardiomyocytes have persistent DNA damage, we performed immunofluorescence staining for 2 commonly used DNA damage markers, γH2AX and p53‐binding protein 1 (53BP1), in cardiac tissues from NFDs and HCM patients (Figure 3A and 3B). Our data showed that adult HCM cardiomyocytes exhibit a significantly higher DNA damage load compared with NFDs (P=0.016 [Figure 3C] and P<0.0001 [Figure 3D]). These results indicate that telomere shortening in human HCM cardiomyocytes is correlated with activation of DNA damage signaling.
Figure 3.
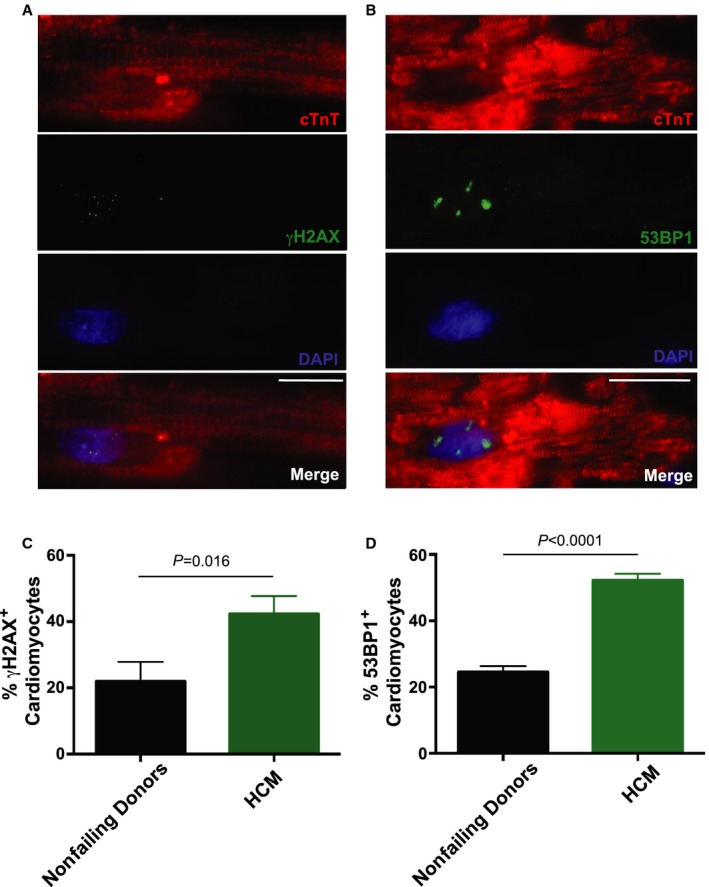
Activated DNA damage response in hypertrophic cardiomyopathy (HCM) cardiomyocytes. A, Representative immunofluorescence image: γH2AX in green, 4′,6‐diamidino‐2‐phenylindole (DAPI)‐stained nuclei in blue, and cardiac troponin T (cTnT) in red. Scale bar, 5 μm. B, Representative immunofluorescence image of 53BP1 in green, DAPI‐stained nuclei in blue, and cTnT in red. Scale bar, 5 μm. Quantification of DNA damage as detected by (C) γH2AX + and (D) by 53BP1+ cardiomyocytes. Data are represented as mean±SEM. P<0.05 were considered significant; statistical comparison of mean values (2‐tailed, unpaired Student t test) from nonfailing donor and HCM groups for γH2AX and p53‐binding protein 1 (53BP1) are P=0.016 and P<0.0001, respectively.
Patients With HCM With the Shortest TL Have Reduced Ejection Fraction
LVEF is a key parameter employed for identifying and tracking myocardial dysfunction. Virtually all transplanted hearts from patients with IDCM and ICM have reduced LVEF. In contrast, patients with HCM may progress to severe HF requiring transplant either with or without a reduction in LVEF, known as reduced systolic function (HFrEF) or preserved systolic function (HFpEF), respectively (discussed in Borlaug and Paulus30 and Udelson31). This provides an opportunity to investigate whether telomere dynamics correlate with cardiac EF. Notably, median values from TL distribution histogram analysis showed that both groups exhibited shorter telomeres compared with NFDs (NFDs versus HCM HFpEF and NFDs versus HCM HFrEF with P=0.004 and 0.034, respectively; Figure 4A). Plotting the average patient TL revealed that telomere shortening primarily affects HCM HFrEF patients (P=0.02, Figure 4B). Intriguingly, plotting the average TL per patient versus ejection fraction revealed that the majority of patients with HFrEF (62.5%) clustered together within the shorter telomere spectrum (Figure 4C, green triangles) compared with those with HFpEF (Figure 4C, brown squares) and the NFDs (Figure 4C, black circles).
Figure 4.

Correlation between telomere length and ejection fraction. A, Cardiomyocyte telomere length distribution of nonfailing donors (NFDs) and patients with hypertrophic cardiomyopathy (HCM): HCM heart failure with preserved ejection fraction (HFpEF) and HCM heart failure with reduced ejection fraction (HFrEF). Black and red vertical lines are drawn at median value of the histogram obtained for NFDs and patients with HCM, respectively. Shifts in patients' telomeric length distribution median values are shown in brown and green horizontal lines for HCM HFpEF and HCM HFrEF, respectively. Wilcoxon rank sum analysis between NFDs and patients with HCM HFpEF and HCM HFrEF: P=0.004 and P=0.034, respectively. N indicates the number of cardiomyocytes scored (see also Table 3). The percentage of cells with short and long telomeres is shown in graphs. B, Boxplot analysis shows average telomere length measurements (per patient) between NFDs and patients with HCM HFpEF and HCM HFrEF (Kruskal‐Wallis test, P=0.164 and P=0.02, respectively). C, Distribution of telomere length (per patient) and cardiac function as measured by ejection fraction in NFDs (black circles), patients with HCM HFrEF (green triangles), and patients with HCM HFpEF (brown squares). Patients with shorter average telomere length are presented below the horizontal black line. A total of 20 NFD, 8 HCM HFpEF, and 9 HCM HFrEF patient samples with available ejection fraction data were analyzed. Q‐FISH indicates quantitative fluorescence in situ hybridization.
Age‐Associated Cardiomyocyte Telomere Attrition Solely Occurs in Diseased Hearts
Age has been suggested to be a major determinant of telomere shortening (reviewed in Blackburn et al32). However, the effect of telomere shortening in adult cardiomyocytes remains equivocal. To address the effect of aging on cardiomyocyte telomeric attrition we compared cardiac tissues from young (21–46 years) and older (50–75 years) NFDs. Notably, no shift in TL distribution median value was evident in young and old hearts from NFDs (P=0.874, Figure 5A and 5B), suggesting that aging per se is not sufficient to induce cardiomyocyte telomere shortening in NFD hearts. However, when compared with corresponding NFDs, the cardiomyocyte median value of TL distribution significantly shifts in young patients with HCM (P=0.012, Figure 5C), shown with blue horizontal lines, as well as in older patients with HCM (P=0.036, Figure 5D), shown with green horizontal lines. Similar results were obtained by plotting the patients' average TL (Figure 5E). Together, these findings suggest that cardiomyocyte telomere attrition is likely to be caused by the disease environment rather than aging.
Figure 5.

Effect of age in telomere shortening in cardiac tissues. A through D, Histograms presenting distribution of the percentage of cells within the patients' range of cardiomyocyte telomeric length. A, young and (B) old nonfailing donors (NFDs) and (C) young (green, aged 21–46 years) and (D) old (blue, aged 50–75 years) patients with hypertrophic cardiomyopathy (HCM). N indicates the number of cardiomyocytes scored. The percentage of cells with short and long telomeres is shown in the graph. Black and red vertical lines were drawn at the median value of the histogram obtained for NFDs and patients with HCM, respectively. The shift in telomeric length distribution from young patients with HCM to young NFDs is shown with the horizontal line green (Wilcoxon rank sum test, P=0.012); the shift in telomeric length distribution from old patients with HCM to NFDs is shown with the blue horizontal line (Wilcoxon rank sum test, P=0.036); P=0.874 (Wilcoxon rank sum test) for comparison of young NFDs and old NFDs. E, Boxplot analysis shows average telomere length measurements (per patient) between young and old NFDs and young and old patients with HCM (Kruskal‐Wallis test, P=0.850, 0.004, and 0.102). A total of 9 young NFDs, 15 old NFDs, 5 young patients with HCM, and 11 old patients with HCM were analyzed. Q‐FISH indicates quantitative fluorescence in situ hybridization.
Cardiomyocyte‐Specific Telomere Attrition Is Attenuated in Women With HF
As the prevalence of HCM is higher in men compared with women,33 we were inclined to explore the role of sex in the scope of TL and HCM. Our analysis revealed no TL difference between men and women in NFDs (P=0.996, Figure 6A and 6B). However, men with HCM showed significant telomere shortening (P=0.007; Figure 6C, light green horizontal line) compared with their nonfailing counterparts (Figure 6A), while women with HCM did not show a significant shift in their TL distribution median value (P=0.132; Figure 6B, light blue horizontal line) compared with nonfailing women (Figure 6D). Similar results were obtained by plotting the patients' average TL of the different groups (Figure 6E).
Figure 6.
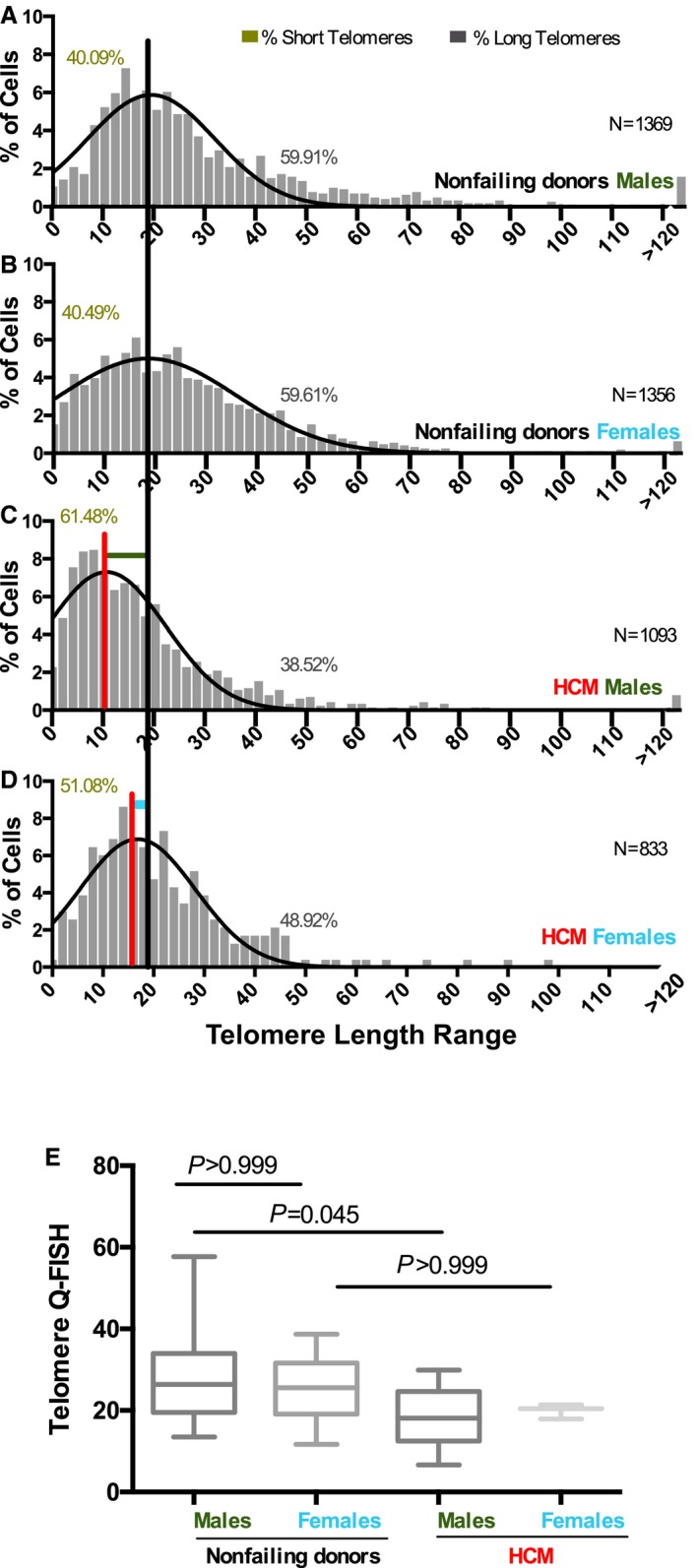
Sex‐dependent telomere shortening in response to hypertrophic cardiomyopathy (HCM) disease environment. A through D, Distribution of the percentage of cells within the patients' range of cardiomyocyte telomeric length is presented as histograms in nonfailing donor (NFD; A) males and (B) females as well as (C) male and (D) female patients with HCM. N indicates the number of cardiomyocytes scored. The percentage of cells with short and long telomeres is shown in the graph (see also Table 3). Black and red vertical lines are drawn at the median value of the histogram for NFDs and patients with HCM, respectively. The shift in telomeric length distribution in male patients with HCM from male NFDs is shown with horizontal dark green line (Wilcoxon rank sum test, P=0.007) for comparison of male NFDs and female NFDs; the shift in telomeric length distribution in female patients with HCM to female NFDs is shown with the light blue horizontal line (Wilcoxon rank sum test, P=0.132); P=0.996 (Wilcoxon rank sum test). E, Boxplot analysis shows average telomere length measurements (per patient) between male (dark green) and female (light blue) NFDs and male and female patients with HCM (Kruskal‐Wallis test, P>0.999, P=0.045 and P>0.999). A total of 14 male NFDs, 10 female NFDs, 3 male patients with HCM, and 13 female patients with HCM were analyzed. Q‐FISH indicates quantitative fluorescence in situ hybridization.
Conclusions
The findings of the current study demonstrate, for the first time, that post‐mitotic cardiomyocytes from patients with HF have significantly shorter TL compared with healthy individuals. We also found that the observed telomere shortening occurs in a cell type‐specific manner. Further, in‐depth analysis revealed that cardiomyocytes with the shortest telomeric lengths are mainly from patients with reduced ejection fraction. This discovery is compelling to consider in light of the fact that HCM is a familial disease caused by sarcomeric protein gene mutations primarily affecting cardiomyocytes.34 Interestingly, when the same sarcomeric mutations are introduced into mice, they poorly mimic the human spontaneous cardiac condition unless high levels of the mutation are introduced via transgene overexpression35, 36 or mice are placed under severe stress conditions.37, 38, 39 An intriguing interpretation of this phenotypic discrepancy could be that the significantly longer telomeres and generally higher levels of telomerase activity of inbred laboratory mice40, 41 protect their hearts from the degree of tissue damage and organ failure that is observed in human patients. In support of this notion, it was recently shown that shorter telomeres combined with dystrophin deficiency (a cardiac structural protein) in mice unmask the full pathologic spectrum of cardiac phenotype similar to the cardiac defect seen in patients.5
Recent studies highlight both causal and secondary roles for telomere attrition in various conditions, including cardiopulmonary diseases (discussed in Blackburn et al32). Further, the involvement of mitochondrial abnormalities and excessive reactive oxygen species is well established in cardiac dysfunction from patients of diverse etiologies.42, 43 These irregularities may also be key factors affecting TL in cardiac muscle cells of patients with HCM. In fact, we recently reported elevated oxidative stress, potentially exacerbated by oxidative DNA damage, in cardiac tissues that exhibit short telomeres.5 These findings are in accordance with previous reports suggesting oxidative stress as a contributor to telomere shortening independent of cell replication44 and that telomere shortening leads to activation of DNA damage signaling.28, 45, 46 The importance of DNA damage in human HCM is emphasized by our finding of increased DNA damage in adult diseased cardiomyocytes. Based on our results, we postulate that excessive oxidative damage of HCM hearts causes DNA damage in cardiomyocytes, leading to telomere uncapping and consequently telomere shortening. Further, as previously suggested,5 it is possible that telomere shortening may also further enhance oxidative stress leading to additional telomere erosion as a feedback loop. Although further analyses are necessary to clearly uncover whether our finding of cardiomyocyte‐specific telomere shortening is causative or a consequential effect, the results presented here suggest that the observed telomere shortening is associated with increased cardiomyocyte DNA damage in human patients with HF.
As a marker of “biological age” rather than “chronological age,” TL plays a critical role in maintaining genome integrity and consequently cell health.47 In support of previous observations, in contrast to highly proliferative tissues where TL decreases with chronological age,48, 49 our cardiac analysis demonstrated that cardiomyocyte telomere attrition is not an age‐dependent phenomenon in healthy human donors.16 However, we show that telomeric shortening occurs regardless of age in patients with HF, suggesting that cardiomyocyte “biological aging” is exacerbated under the disease state. We further found that women better sustain their cardiomyocyte TL in the presence of the disease, indicating that women may reach the TL required for the onset of HF later in life compared with men. Although individual patient characteristics are likely to further influence the observed telomere shortening of the subgroups, our finding is concurrent with previously documented sex‐related cardiac functional differences in patients with HCM as well as the reported lower occurrence of HCM in women.33
The clinical impact of reduction in cardiomyocyte TL remains uncertain. It could indicate chronic disease—specific markers and/or it may indicate the increase in susceptibility to clinical deterioration. As heart transplant recipients, all of the patients in our diseased heart groups had severe degrees of clinical disease. Within the HCM group, many had progressed to the phase of overt cardiac dysfunction as previously described50 and many had LVEFs <45%. Future studies in relevant animal models of progressive myocardial disease and human myocardium from individuals with earlier phases of disease should provide definitive clues to the causes of cardiomyocytes telomere shortening in human HF. Ultimately, studies using interventions that mitigate cardiomyocyte TL changes and gene alterations, including animal models engineered to affect these processes, will provide the greatest functional insight and might cultivate potential future therapeutic implications. The human findings presented here highlight the need to fully understand telomere dynamics by studying new disease models that will take cardiomyocyte telomere biology into account.
Sources of Funding
This work was supported by startup funds, a Pilot and Feasibility Grant from National Institutes of Health (P30 AR069619) and funds from the McCabe Foundation to Mourkioti. Procurement of human heart tissues were enabled by grants from the National Institutes of Health (HL089847 and HL105993) to Margulies.
Disclosures
None.
Supporting information
Figure S1. Increased fibrosis in diseased cardiac tissues. A, Representative trichrome staining of cardiac tissues from nonfailing donors and patients with hypertrophic cardiomyopathy (HCM), patients with ischemic cardiomyopathy (ICM), and patients with idiopathic dilated cardiomyopathy (IDCM). B, Calculated fibrosis boxplot graphs are presented as the percentage of blue area (fibrotic tissue) over whole tissue area. A total of 26 nonfailing donor, 17 HCM, 9 ICM, and 11 IDCM samples were analyzed. Statistical comparison between nonfailing donors and patients with HCM, ICM, and IDCM show significance of difference (Kruskal‐Wallis, P=0.0002, P<0.0001, and P=0.004, respectively).
Figure S2. Telomere length measurements in ischemic (ICM) and idiopathic dilated (IDCM) cardiomyopathies. Left ventricular tissues from nonfailing donors (black, n=9) and patients with ICM (grey, n=9, A and C) and IDCM (grey, n=11, B and D) were subjected to quantitative fluorescence in situ hybridization (Q‐FISH) analysis. Significant telomere shortening occurs in patient cardiomyocytes (A) ICM (Mann–Whitney, *P=0.041) but not in (B) IDCM (Mann–Whitney, P=0.101), while α‐smooth muscle‐positive cells from (C) ICM (Mann–Whitney, P=0.578) and (D) IDCM (Mann–Whitney, P=0.922) show comparable telomere lengths with nonfailing donors. The number of nuclei (N) scored per group is shown in Table 2.
Acknowledgments
We would like to thank Jeffrey Brandimarto for assistance with human cardiac tissue processing and data quantification; all of the patients with cardiomyopathy and their families who contribute to UPenn's tissue repository; Dr Robert Pignolo and Mr Robert Caron at the Penn Center Musculoskeletal Disorders Histology Core Facility for assistance with histological sections; Cell Development Biology Histology Microscopy Core at University of Pennsylvania specifically Dr Andrea Stout for help with confocal microscopy; and Dr Alan Meeker (Johns Hopkins) for protocol advice and for sharing the software for telomere analysis. We are also grateful to telomere expert Dr Brad Johnson (UPenn) and histology expert Dr Lachlan Smith (UPenn) for critical comments and helpful discussions on the manuscript.
(J Am Heart Assoc. 2017;6:e005086 DOI: 10.1161/JAHA.116.005086.)28882819
References
- 1. Fatkin D, Seidman CE, Seidman JG. Genetics and disease of ventricular muscle. Cold Spring Harb Perspect Med. 2014;4:a021063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. McNally EM, Barefield DY, Puckelwartz MJ. The genetic landscape of cardiomyopathy and its role in heart failure. Cell Metab. 2015;21:174–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Refaat MM, Fahed AC, Hassanieh S, Hotait M, Arabi M, Skouri H, Seidman JG, Seidman CE, Bitar FF, Nemer G. The muscle‐bound heart. Card Electrophysiol Clin. 2016;8:223–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Semsarian C, Ingles J, Maron MS, Maron BJ. New perspectives on the prevalence of hypertrophic cardiomyopathy. J Am Coll Cardiol. 2015;65:1249–1254. [DOI] [PubMed] [Google Scholar]
- 5. Mourkioti F, Kustan J, Kraft P, Day JW, Zhao MM, Kost‐Alimova M, Protopopov A, DePinho RA, Bernstein D, Meeker AK, Blau HM. Role of telomere dysfunction in cardiac failure in Duchenne muscular dystrophy. Nat Cell Biol. 2013;15:895–904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Raymond AR, Norton GR, Sareli P, Woodiwiss AJ, Brooksbank RL. Relationship between average leucocyte telomere length and the presence or severity of idiopathic dilated cardiomyopathy in black Africans. Eur J Heart Fail. 2013;15:54–60. [DOI] [PubMed] [Google Scholar]
- 7. van der Harst P, van der Steege G, de Boer RA, Voors AA, Hall AS, Mulder MJ, van Gilst WH, van Veldhuisen DJ; Group M‐HS . Telomere length of circulating leukocytes is decreased in patients with chronic heart failure. J Am Coll Cardiol. 2007;49:1459–1464. [DOI] [PubMed] [Google Scholar]
- 8. Leri A, Franco S, Zacheo A, Barlucchi L, Chimenti S, Limana F, Nadal‐Ginard B, Kajstura J, Anversa P, Blasco MA. Ablation of telomerase and telomere loss leads to cardiac dilatation and heart failure associated with p53 upregulation. EMBO J. 2003;22:131–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Chimenti C, Kajstura J, Torella D, Urbanek K, Heleniak H, Colussi C, Di Meglio F, Nadal‐Ginard B, Frustaci A, Leri A, Maseri A, Anversa P. Senescence and death of primitive cells and myocytes lead to premature cardiac aging and heart failure. Circ Res. 2003;93:604–613. [DOI] [PubMed] [Google Scholar]
- 10. Blackburn EH. Switching and signaling at the telomere. Cell. 2001;106:661–673. [DOI] [PubMed] [Google Scholar]
- 11. Wong JM, Collins K. Telomere maintenance and disease. Lancet. 2003;362:983–988. [DOI] [PubMed] [Google Scholar]
- 12. Serra V, von Zglinicki T, Lorenz M, Saretzki G. Extracellular superoxide dismutase is a major antioxidant in human fibroblasts and slows telomere shortening. J Biol Chem. 2003;278:6824–6830. [DOI] [PubMed] [Google Scholar]
- 13. von Zglinicki T. Oxidative stress shortens telomeres. Trends Biochem Sci. 2002;27:339–344. [DOI] [PubMed] [Google Scholar]
- 14. Farzaneh‐Far R, Cawthon RM, Na B, Browner WS, Schiller NB, Whooley MA. Prognostic value of leukocyte telomere length in patients with stable coronary artery disease: data from the Heart and Soul Study. Arterioscler Thromb Vasc Biol. 2008;28:1379–1384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Muezzinler A, Zaineddin AK, Brenner H. A systematic review of leukocyte telomere length and age in adults. Ageing Res Rev. 2013;12:509–519. [DOI] [PubMed] [Google Scholar]
- 16. Takubo K, Izumiyama‐Shimomura N, Honma N, Sawabe M, Arai T, Kato M, Oshimura M, Nakamura K. Telomere lengths are characteristic in each human individual. Exp Gerontol. 2002;37:523–531. [DOI] [PubMed] [Google Scholar]
- 17. Kimura M, Stone RC, Hunt SC, Skurnick J, Lu X, Cao X, Harley CB, Aviv A. Measurement of telomere length by the Southern blot analysis of terminal restriction fragment lengths. Nat Protoc. 2010;5:1596–1607. [DOI] [PubMed] [Google Scholar]
- 18. Oh H, Wang SC, Prahash A, Sano M, Moravec CS, Taffet GE, Michael LH, Youker KA, Entman ML, Schneider MD. Telomere attrition and Chk2 activation in human heart failure. Proc Natl Acad Sci USA. 2003;100:5378–5383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Aviv A, Valdes AM, Spector TD. Human telomere biology: pitfalls of moving from the laboratory to epidemiology. Int J Epidemiol. 2006;35:1424–1429. [DOI] [PubMed] [Google Scholar]
- 20. Shekhani MT, Barber JR, Bezerra SM, Heaphy CM, Roibon ND, Taheri D, Reis LO, Guner G, Joshu CE, Netto GJ, Meeker AK. High‐resolution telomere fluorescence in situ hybridization reveals intriguing anomalies in germ cell tumors. Hum Pathol. 2016;54:106–112. [DOI] [PubMed] [Google Scholar]
- 21. Heaphy CM, Gaonkar G, Peskoe SB, Joshu CE, De Marzo AM, Lucia MS, Goodman PJ, Lippman SM, Thompson IM Jr, Platz EA, Meeker AK. Prostate stromal cell telomere shortening is associated with risk of prostate cancer in the placebo arm of the Prostate Cancer Prevention Trial. Prostate. 2015;75:1160–1166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Flores I, Canela A, Vera E, Tejera A, Cotsarelis G, Blasco MA. The longest telomeres: a general signature of adult stem cell compartments. Genes Dev. 2008;22:654–667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Chin L, Artandi SE, Shen Q, Tam A, Lee SL, Gottlieb GJ, Greider CW, DePinho RA. p53 deficiency rescues the adverse effects of telomere loss and cooperates with telomere dysfunction to accelerate carcinogenesis. Cell. 1999;97:527–538. [DOI] [PubMed] [Google Scholar]
- 24. Takai H, Smogorzewska A, de Lange T. DNA damage foci at dysfunctional telomeres. Curr Biol. 2003;13:1549–1556. [DOI] [PubMed] [Google Scholar]
- 25. Bergmann O, Bhardwaj RD, Bernard S, Zdunek S, Barnabe‐Heider F, Walsh S, Zupicich J, Alkass K, Buchholz BA, Druid H, Jovinge S, Frisen J. Evidence for cardiomyocyte renewal in humans. Science. 2009;324:98–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Elser JA, Margulies KB. Hybrid mathematical model of cardiomyocyte turnover in the adult human heart. PLoS One. 2012;7:e51683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Rivard A, Andres V. Vascular smooth muscle cell proliferation in the pathogenesis of atherosclerotic cardiovascular diseases. Histol Histopathol. 2000;15:557–571. [DOI] [PubMed] [Google Scholar]
- 28. Aix E, Gutierrez‐Gutierrez O, Sanchez‐Ferrer C, Aguado T, Flores I. Postnatal telomere dysfunction induces cardiomyocyte cell‐cycle arrest through p21 activation. J Cell Biol. 2016;213:571–583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Lazzerini‐Denchi E, Sfeir A. Stop pulling my strings—what telomeres taught us about the DNA damage response. Nat Rev Mol Cell Biol. 2016;17:364–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Borlaug BA, Paulus WJ. Heart failure with preserved ejection fraction: pathophysiology, diagnosis, and treatment. Eur Heart J. 2011;32:670–679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Udelson JE. Heart failure with preserved ejection fraction. Circulation. 2011;124:e540–e543. [DOI] [PubMed] [Google Scholar]
- 32. Blackburn EH, Epel ES, Lin J. Human telomere biology: a contributory and interactive factor in aging, disease risks, and protection. Science. 2015;350:1193–1198. [DOI] [PubMed] [Google Scholar]
- 33. Olivotto I, Maron MS, Adabag AS, Casey SA, Vargiu D, Link MS, Udelson JE, Cecchi F, Maron BJ. Gender‐related differences in the clinical presentation and outcome of hypertrophic cardiomyopathy. J Am Coll Cardiol. 2005;46:480–487. [DOI] [PubMed] [Google Scholar]
- 34. Morita H, Nagai R, Seidman JG, Seidman CE. Sarcomere gene mutations in hypertrophy and heart failure. J Cardiovasc Transl Res. 2010;3:297–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Robbins J. Remodeling the cardiac sarcomere using transgenesis. Annu Rev Physiol. 2000;62:261–287. [DOI] [PubMed] [Google Scholar]
- 36. James J, Zhang Y, Osinska H, Sanbe A, Klevitsky R, Hewett TE, Robbins J. Transgenic modeling of a cardiac troponin I mutation linked to familial hypertrophic cardiomyopathy. Circ Res. 2000;87:805–811. [DOI] [PubMed] [Google Scholar]
- 37. Danialou G, Comtois AS, Dudley R, Karpati G, Vincent G; Des Rosiers C; Petrof BJ. Dystrophin‐deficient cardiomyocytes are abnormally vulnerable to mechanical stress‐induced contractile failure and injury. FASEB J. 2001;15:1655–1657. [DOI] [PubMed] [Google Scholar]
- 38. Houser SR, Margulies KB, Murphy AM, Spinale FG, Francis GS, Prabhu SD, Rockman HA, Kass DA, Molkentin JD, Sussman MA, Koch WJ; American Heart Association Council on Basic Cardiovascular Sciences CoCC, Council on Functional G and Translational B . Animal models of heart failure: a scientific statement from the American Heart Association. Circ Res. 2012;111:131–150. [DOI] [PubMed] [Google Scholar]
- 39. Patten RD, Hall‐Porter MR. Small animal models of heart failure: development of novel therapies, past and present. Circ Heart Fail. 2009;2:138–144. [DOI] [PubMed] [Google Scholar]
- 40. Kipling D, Cooke HJ. Hypervariable ultra‐long telomeres in mice. Nature. 1990;347:400–402. [DOI] [PubMed] [Google Scholar]
- 41. Calado RT, Dumitriu B. Telomere dynamics in mice and humans. Semin Hematol. 2013;50:165–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Lesnefsky EJ, Chen Q, Hoppel CL. Mitochondrial metabolism in aging heart. Circ Res. 2016;118:1593–1611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Shirakabe A, Ikeda Y, Sciarretta S, Zablocki DK, Sadoshima J. Aging and autophagy in the heart. Circ Res. 2016;118:1563–1576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Passos JF, Saretzki G, von Zglinicki T. DNA damage in telomeres and mitochondria during cellular senescence: is there a connection? Nucleic Acids Res. 2007;35:7505–7513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Denchi EL. Give me a break: how telomeres suppress the DNA damage response. DNA Repair (Amst). 2009;8:1118–1126. [DOI] [PubMed] [Google Scholar]
- 46. Denchi EL, de Lange T. Protection of telomeres through independent control of ATM and ATR by TRF2 and POT1. Nature. 2007;448:1068–1071. [DOI] [PubMed] [Google Scholar]
- 47. Aviv A. Chronology versus biology: telomeres, essential hypertension, and vascular aging. Hypertension. 2002;40:229–232. [DOI] [PubMed] [Google Scholar]
- 48. Blasco MA. Telomere length, stem cells and aging. Nat Chem Biol. 2007;3:640–649. [DOI] [PubMed] [Google Scholar]
- 49. Bar C, Blasco MA. Telomeres and telomerase as therapeutic targets to prevent and treat age‐related diseases. F1000Res. 2016;5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Olivotto I, Cecchi F, Poggesi C, Yacoub MH. Patterns of disease progression in hypertrophic cardiomyopathy: an individualized approach to clinical staging. Circ Heart Fail. 2012;5:535–546. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Increased fibrosis in diseased cardiac tissues. A, Representative trichrome staining of cardiac tissues from nonfailing donors and patients with hypertrophic cardiomyopathy (HCM), patients with ischemic cardiomyopathy (ICM), and patients with idiopathic dilated cardiomyopathy (IDCM). B, Calculated fibrosis boxplot graphs are presented as the percentage of blue area (fibrotic tissue) over whole tissue area. A total of 26 nonfailing donor, 17 HCM, 9 ICM, and 11 IDCM samples were analyzed. Statistical comparison between nonfailing donors and patients with HCM, ICM, and IDCM show significance of difference (Kruskal‐Wallis, P=0.0002, P<0.0001, and P=0.004, respectively).
Figure S2. Telomere length measurements in ischemic (ICM) and idiopathic dilated (IDCM) cardiomyopathies. Left ventricular tissues from nonfailing donors (black, n=9) and patients with ICM (grey, n=9, A and C) and IDCM (grey, n=11, B and D) were subjected to quantitative fluorescence in situ hybridization (Q‐FISH) analysis. Significant telomere shortening occurs in patient cardiomyocytes (A) ICM (Mann–Whitney, *P=0.041) but not in (B) IDCM (Mann–Whitney, P=0.101), while α‐smooth muscle‐positive cells from (C) ICM (Mann–Whitney, P=0.578) and (D) IDCM (Mann–Whitney, P=0.922) show comparable telomere lengths with nonfailing donors. The number of nuclei (N) scored per group is shown in Table 2.