

Abstract
Background
In the BARI 2D (Bypass Angioplasty Revascularization Investigation 2 Diabetes) trial, randomization of diabetic patients with stable ischemic heart disease to insulin provision (IP) therapy, as opposed to insulin sensitization (IS) therapy, resulted in biochemical evidence of impaired fibrinolysis but no increase in adverse clinical outcomes. We hypothesized that the prothrombotic effect of IP therapy in combination with the hypercoagulable state induced by active smoking would result in an increased risk of myocardial infarction (MI).
Methods and Results
We analyzed BARI 2D patients who were active smokers randomized to IP or IS therapy. The primary end point was fatal or nonfatal MI. PAI‐1 (plasminogen activator inhibitor 1) activity was analyzed at 1, 3, and 5 years. Of 295 active smokers, MI occurred in 15.4% randomized to IP and in 6.8% randomized to IS over the 5.3 years (P=0.023). IP therapy was associated with a 3.2‐fold increase in the hazard of MI compared with IS therapy (hazard ratio: 3.23; 95% confidence interval, 1.43–7.28; P=0.005). Baseline PAI‐1 activity (19.0 versus 17.5 Au/mL, P=0.70) was similar in actively smoking patients randomized to IP or IS therapy. However, IP therapy resulted in significantly increased PAI‐1 activity at 1 year (23.0 versus 16.0 Au/mL, P=0.001), 3 years (24.0 versus 18.0 Au/mL, P=0.049), and 5 years (29.0 versus 15.0 Au/mL, P=0.004) compared with IS therapy.
Conclusions
Among diabetic patients with stable ischemic heart disease who were actively smoking, IP therapy was independently associated with a significantly increased hazard of MI. This finding may be explained by higher PAI‐1 activity in active smokers treated with IP therapy.
Clinical Trial Registration
URL: http://www.clinicaltrials.gov. Unique identifier: NCT00006305.
Keywords: coronary artery disease, diabetes mellitus, insulin, myocardial infarction, smoking
Subject Categories: Diabetes, Type 2; Cardiovascular Disease; Chronic Ischemic Heart Disease; Myocardial Infarction
Clinical Perspective
What Is New?
Diabetic patients with stable ischemic heart disease who smoke and are treated with insulin provision therapy as opposed to insulin sensitization therapy may be at increased risk of myocardial infarction.
What Are the Clinical Implications?
Preferential treatment with insulin sensitization therapy rather than insulin provision therapy should be considered for the care of diabetic patients with stable ischemic heart disease who smoke.
Introduction
Patients with type 2 diabetes mellitus and stable ischemic heart disease (SIHD) are at increased risk of myocardial infarction (MI) compared with their nondiabetic counterparts.1, 2, 3, 4, 5 The increased risk is thought to be mediated by a multifactorial diabetic vasculopathy resulting in a prothrombotic coronary vascular milieu.6 Potentially compounding this prothrombotic tendency, treatment of diabetes mellitus with insulin provision pharmacotherapy impairs fibrinolysis. In the BARI 2D (Bypass Angioplasty Revascularization Investigation 2 Diabetes) trial of patients with diabetes mellitus and SIHD, PAI‐1 (plasminogen activator inhibitor 1) antigen and activity levels increased in those randomized to an insulin provision (IP) strategy compared with those randomized to an insulin sensitization (IS) strategy. Despite the significant differences in fibrinolytic biomarker profiles, no difference was observed in death or major cardiovascular events between the 2 treatment strategies.7, 8 Active cigarette smoking also induces a multifactorial hypercoagulable state through increased synthesis of PAI‐1, elevation in the blood fibrinogen concentration, increased platelet activity, and increased expression of tissue factor and is associated with an increased risk of MI in both diabetic and nondiabetic patients with SIHD.9 We hypothesized that in diabetic patients with SIHD, the prothrombotic effects of IP therapy in combination with the hypercoagulable state induced by active smoking would result in an increased hazard of coronary thrombosis manifested by MI.
Methods
Data Source
The BARI 2D data set was obtained on request from the Biologic Specimen and Data Repository Information Coordinating Center of the National Heart, Lung, and Blood Institute under a data use agreement. The Washington University Human Research Protection Office granted this study an exemption from institutional review board oversight.
Study Design
The rationale and design of the BARI 2D trial have been described previously.10, 11 Briefly, the BARI 2D trial randomized 2368 patients with both type 2 diabetes mellitus and SIHD to receive either prompt revascularization with percutaneous coronary intervention (PCI) or coronary artery bypass grafting (CABG) in addition to intensive medical therapy or to intensive medical therapy alone and to receive either IS or IP therapy. Randomization was stratified according to the method of revascularization, PCI or CABG, selected by the treating physician to be the optimal treatment for each patient.
Study Patients
Participants in the BARI 2D trial were recruited between January 1, 2001, and March 31, 2005, at 49 clinical sites in the United States, Brazil, Canada, Mexico, Austria, and the Czech Republic. Participants had to be ≥25 years old with a diagnosis of type 2 diabetes mellitus and coronary artery disease (≥50% stenosis of a major epicardial coronary artery associated with a positive stress test or ≥70% stenosis of a major epicardial coronary artery and classic angina). All patients had to be eligible for either elective PCI or CABG. The main exclusion criteria were the need for immediate revascularization, left main coronary disease, coronary revascularization within the 12 months before randomization, class III or IV heart failure, hepatic dysfunction, serum creatinine >2.0 mg/dL (177 μmol/L), and glycosylated hemoglobin level (HbA1c) >13.0%. The protocol was approved by each institution's review board or ethics committee. All participants provided written informed consent.
Treatment and Follow‐up
Patients were randomized to either an IS strategy for treatment of hyperglycemia using primarily metformin and/or a thiazolidinedione drug or an IP strategy using primarily a sulfonylurea and/or a meglitinide drug and insulin itself. Patients randomized to the prompt revascularization strategy were to undergo PCI or CABG within 4 weeks of randomization. Patients in the intensive medical therapy group were permitted to receive revascularization only if they developed worsening angina, severe ischemia, or an acute coronary syndrome. Intensive medical therapy in both strategies consisted of lifestyle management targeting smoking cessation, weight loss, and regular exercise as well as pharmacological therapy according to contemporary guidelines to maintain HbA1c <7.0%, low‐density lipoprotein cholesterol <100 mg/dL (2.6 mmol/l), and blood pressure ≤130/80 mm Hg. Patients were evaluated monthly for 6 months and every 3 months thereafter for a mean follow‐up of 5.3 years. Smoking status (active, former or never) was ascertained for all patients at baseline; 6 months; and 1, 2, 3, 4, and 5 years.
Outcomes
The primary end point for the present analysis was fatal or nonfatal MI. All myocardial ischemic events requiring hospitalization were adjudicated based on source documents including emergency department records, admission history and physical examination, ECGs and biomarkers, discharge summaries, and records of revascularization procedures.2 The BARI 2D criteria for MI required that an abnormal biomarker profile exceed at least twice the upper limits of normal for the local laboratory. When cardiac troponin and creatine kinase‐MB were acquired simultaneously, cardiac troponin took precedence in establishing the diagnosis of MI. MI was confirmed in the presence of abnormal cardiac biomarkers and evidence of angina or angina‐equivalent symptoms, ECG or imaging evidence of new myocardial ischemia, or autopsy evidence of new MI. All ECGs were interpreted at the core laboratory with the use of the Minnesota code with an adaptation of the NOVACODE for serial ECG comparisons.12, 13 A Q‐wave MI required the development of new pathological Q waves13 or the new occurrence of a left bundle‐branch block in addition to abnormal biomarkers. Non–Q‐wave MI required the aforementioned MI criteria without new pathological Q waves. Fatal MIs were defined as death occurring within 30 days after the event with a causal relationship to the death.
Laboratory Measurements
Insulin, tissue plasminogen activator (tPA) antigen, and PAI‐1 activity samples were drawn by peripheral venipuncture, placed on ice within 15 minutes, centrifuged for separation of plasma, frozen, and shipped in batches to the core laboratory. Insulin, tPA antigen, and PAI‐1 assays were performed by ELISA. The tPA activity was not measured because it complexes with excess PAI‐1 in plasma resulting in activity that is undetectable.7 PAI‐1 activity was assayed with a chromogenic substrate kinetic procedure.
Statistical Analyses
Because we were interested in the prothrombotic effects of active smoking, we compared patients who were actively smoking to those who were not smoking at the time of assessment (never and former smokers). Continuous variables were compared using the Student t test for independent groups, and categorical variables were compared with the χ2 or Fisher exact test, as appropriate. Non‐normal data were summarized by the median (first, third quartiles) and compared using the Mann–Whitney U test. No adjustment was made for multiple comparisons. Kaplan–Meier curves were created by insulin therapy strategy to examine the rates of fatal and nonfatal MI by smoking status as defined at baseline and were compared using the log‐rank test. Risk‐factor levels and medication use during follow‐up were compared based on concurrent smoking status. We evaluated the independent effect of IP (compared with IS) therapy on MI among smokers and nonsmokers by including smoking status, therapy, and the interaction between smoking status and therapy in a multivariable Cox regression model. In addition, variables with univariate P<0.10 in baseline comparisons of smokers randomized to IS versus IP were included in the model. These included race, hypercholesterolemia, HbA1c level, prior revascularization, aspirin use, statin use, angiotensin‐converting enzyme inhibitor or angiotensin receptor blocker use, and biguanide use. In addition, age, sex, and beta blocker use were forced into the model given their relation to MI. The risk of MI for IP therapy (versus IS) was summarized via the adjusted hazard ratios (HRs) and 95% confidence intervals from the multivariable model for both smokers and nonsmokers. The difference in HRs between smokers and nonsmokers was evaluated by smoking status and therapy interaction term. Smoking status was treated as an updated time‐dependent exposure variable. In the event that smoking status was missing for a subject at any time point from study entry to the last follow‐up, the smoking status of the previous time point was used. Statistical significance was assessed using 2‐sided P values, with P<0.05 considered statistically significant. All analyses were conducted in SAS v9.4 (SAS Institute Inc).
Results
Patients
Of 2360 patients with known smoking status, 295 were active smokers at baseline. Among active smokers, the mean age was 57 years, 76% were male, and 65% were white. At baseline, 33.1% of patients had never smoked, 54.4% were former smokers, and 12.5% were current smokers. Among the 295 patients who smoked at study entry, the mean (±SD) daily number of cigarettes smoked was 15±12. At 1, 3, and 5 years, 9.9%, 9.8%, and 9.9%, respectively, were active smokers. Of those who smoked at baseline, 53% had a change in smoking status at any of the follow‐up visits. Conversely, of the 2065 nonsmokers at baseline, 4% smoked at some time during the study.
Of the 295 active smokers at baseline, 143 were randomized to IP therapy and 152 were randomized to IS therapy. Patients in the IP arm were more likely to have hypercholesterolemia, more often to be taking a biguanide and a statin, and less likely to have a prior revascularization (Tables 1 and 2).
Table 1.
Baseline Characteristics of Active Smokers
| Total | IS | IP | P Value | |
|---|---|---|---|---|
| No. of patients | 295 | 152 | 143 | |
| Age, y, mean±SD | 57±7 | 57±8 | 57±7 | 0.76 |
| Female, % | 24 | 28 | 20 | 0.14 |
| Nonwhite, % | 35 | 40 | 30 | 0.09 |
| Clinical characteristics, % | ||||
| History of MI | 59 | 58 | 60 | 0.81 |
| History of CHF | 6 | 4 | 9 | 0.01 |
| Hypertension | 77 | 75 | 79 | 0.40 |
| Hypercholesterolemia | 76 | 70 | 83 | 0.013 |
| Cerebrovascular accident | 12 | 10 | 13 | 0.37 |
| Prior revascularization | 22 | 17 | 27 | 0.049 |
| ABI ≤0.9 | 34 | 35 | 33 | 0.80 |
| Insulin use | 28 | 28 | 28 | 0.90 |
| Cardiovascular risk factors | ||||
| BMI, mean±SD | 31±6 | 31±7 | 31±5 | 0.71 |
| HDL‐C, mg/dL, mean±SD | 37±11 | 37±10 | 38±11 | 0.44 |
| LDL‐C, mg/dL, mean±SD | 99±33 | 99±31 | 98±36 | 0.94 |
| HbA1c, %, mean±SD | 7.8±1.7 | 8.1±1.8 | 7.5±1.6 | 0.006 |
| Regular exercise, % | 18 | 21 | 15 | 0.22 |
| Treatment randomization | ||||
| SIHD treatment, % | 0.91 | |||
| Medical therapy | 48 | 48 | 49 | |
| Prompt revascularization | 52 | 52 | 51 | |
ABI indicates ankle brachial index; BMI, body mass index; CHF, congestive heart failure; HbA1c, glycosylated hemoglobin; HDL‐C, high‐density lipoprotein cholesterol; IP indicates insulin provision; IS, insulin sensitization; LDL‐C, low‐density lipoprotein cholesterol; MI, myocardial infarction; SIHD, stable ischemic heart disease.
Table 2.
Use of Recommended Medications Among Active Smokers Over Time by Diabetes Mellitus Treatment Strategy
| Treatment | Baseline | Year 1 | Year 3 | Year 5 | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| IS | IP | P Value | IS | IP | P Value | IS | IP | P Value | IS | IP | P Value | |
| No. of patients | 152 | 143 | 114 | 103 | 112 | 83 | 60 | 39 | ||||
| Aspirin, % | 81 | 89 | 0.07 | 90 | 93 | 0.62 | 89 | 94 | 0.40 | 88 | 97 | 0.24 |
| Statin, % | 67 | 80 | 0.012 | 96 | 93 | 0.35 | 93 | 97 | 0.47 | 94 | 93 | 1.00 |
| ACEI/ARB, % | 70 | 79 | 0.08 | 83 | 90 | 0.23 | 90 | 88 | 0.80 | 98 | 93 | 0.55 |
| Beta blocker, % | 70 | 74 | 0.52 | 84 | 85 | 0.85 | 91 | 76 | 0.012 | 92 | 77 | 0.09 |
| All 4 medication classes, % | 36 | 48 | 0.044 | 66 | 68 | 0.77 | 72 | 66 | 0.49 | 76 | 77 | 1.00 |
ACEI indicates angiotensin‐converting enzyme inhibitor; ARB, angiotensin receptor blocker; IP, insulin provision; IS, insulin sensitization.
Medication Compliance and Risk Factor Control
Compliance with the 4 pharmacologic components of intensive medical therapy increased significantly from baseline to 5 years in IP and IS groups (Table 2). At 5‐year follow‐up, 76% of IS patients and 77% of IP patients were taking aspirin, a statin, an angiotensin‐converting enzyme inhibitor or angiotensin receptor blocker, and a beta blocker (P=1.00). Control of individual risk factors for active smokers was similar throughout the study between IP and IS groups (Table 3). Overall, risk factor control increased over the 5 years of the study for smokers in both treatment arms. At year 5, the mean number of the 5 targeted risk factors controlled (exclusive of smoking) for IP patients was 3.25±1.07, whereas the mean number of risk factors controlled for IS patients was 2.91±1.52 (P=0.39).
Table 3.
Achievement of Risk Factor Goals (Exclusive of Smoking) Over Time Among Active Smokers by Diabetes Mellitus Treatment Strategy
| Treatment | Baseline | Year 1 | Year 3 | Year 5 | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| IS | IP | P Value | IS | IP | P Value | IS | IP | P Value | IS | IP | P Value | |
| No. of patients | 152 | 143 | 114 | 103 | 112 | 83 | 60 | 39 | ||||
| Risk factors | ||||||||||||
| LDL‐C <100 mg/dL, % | 54 | 56 | 0.9 | 74 | 69 | 0.49 | 86 | 76 | 0.24 | 75 | 86 | 0.50 |
| Triglycerides <150 mg/dL, % | 45 | 45 | 1.0 | 44 | 49 | 0.55 | 58 | 50 | 0.39 | 53 | 44 | 0.51 |
| SBP ≤130 mm Hg, % | 52 | 49 | 0.64 | 65 | 70 | 0.46 | 69 | 68 | 1.00 | 59 | 71 | 0.34 |
| DBP ≤80 g/dL, % | 68 | 64 | 0.46 | 72 | 80 | 0.19 | 81 | 80 | 1.00 | 71 | 87 | 0.17 |
| HbA1c <7%, % | 33 | 44 | 0.09 | 47 | 36 | 0.18 | 41 | 26 | 0.08 | 42 | 21 | 0.10 |
| Number of risk factors at goal, mean±SD | 2.63±1.25 | 2.60±1.33 | 0.87 | 3.07±1.13 | 3.08±1.32 | 0.99 | 3.42±1.15 | 3.14±1.29 | 0.22 | 2.91±1.52 | 3.25±1.07 | 0.39 |
DBP, diastolic blood pressure; HbA1c, glycosylated hemoglobin; IP indicates insulin provision; IS, insulin sensitization; LDL‐C, low‐density lipoprotein cholesterol; SBP, systolic blood pressure.
Outcomes
Fatal or nonfatal MI occurred in 15.4% of smokers randomized to IP and in 6.8% randomized to IS over the 5‐year follow‐up period (P=0.023). Among nonsmokers, fatal or nonfatal MI occurred in 12% of patients in both IP and IS groups (P=0.84). Event‐free survival curves for smokers and nonsmokers are presented in Figure 1A and 1B, respectively. Randomization of smokers to IP therapy was independently associated with a 3.2‐fold increase in the hazard of MI compared with randomization to IS (HR: 3.23; 95% CI, 1.42–7.28; P=0.005; Table 4), whereas there was no difference in the rate of fatal or nonfatal MI among nonsmokers randomized to IP versus IS therapy (HR: 1.00; 95% CI, 0.7–1.29; P=0.98). Compared with nonsmokers, the effect of insulin treatment strategy in active smokers was significantly different with regard to risk of fatal or nonfatal MI (P=0.007 for interaction). In active smokers, fatal or nonfatal MI was not influenced by assignment to the PCI or CABG strata (P=0.63 for interaction) or by randomization to prompt revascularization or intensive medical therapy (P=0.49 for interaction).
Figure 1.
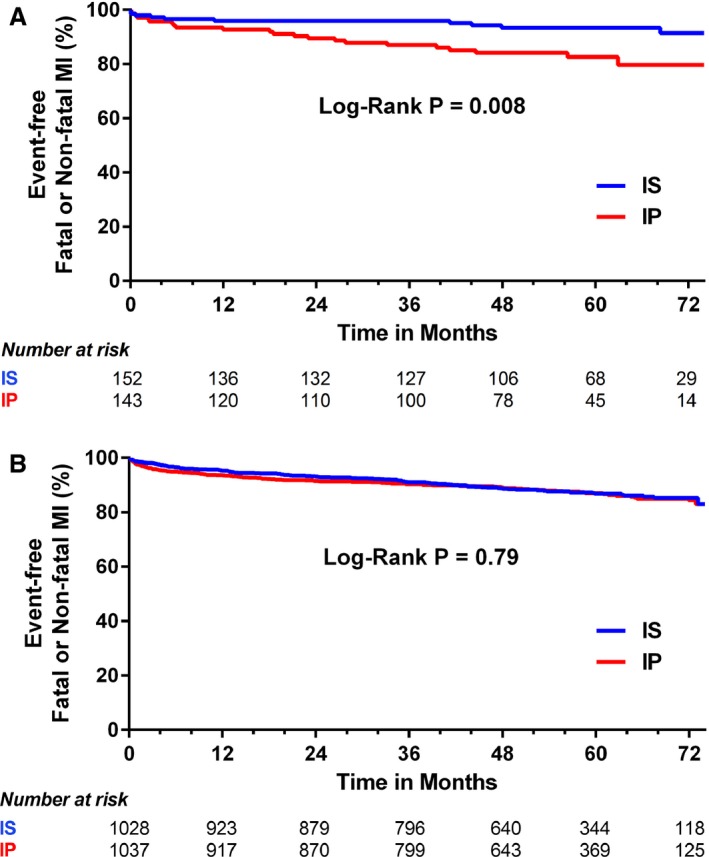
A, Rates of freedom from fatal or nonfatal MI in active smokers (smoking status as a time‐dependent variable) with IS vs IP therapy. B, Rates of freedom from fatal or nonfatal MI in nonsmokers (smoking status as a time‐dependent variable) with IS vs IP therapy. IP indicates insulin provision; IS, insulin sensitization; MI, myocardial infarction.
Table 4.
Cox Proportional Hazards Model for MI
| Variables | HR | 95% CI | P Value | P Value for Interaction |
|---|---|---|---|---|
| Insulin providing (vs insulin sensitizing) | ||||
| Current smokers | 3.228 | 1.431–7.281 | 0.005 | 0.007 |
| Former/never smokers | 0.997 | 0.772–1.287 | 0.98 | |
| White | 0.793 | 0.610–1.030 | 0.08 | |
| Male sex | 0.872 | 0.671–1.135 | 0.31 | |
| Age (per 5 y) | 1.051 | 0.977–1.131 | 0.18 | |
| Hypercholesterolemia | 0.761 | 0.534–1.084 | 0.13 | |
| Prior revascularization | 1.657 | 1.273–2.156 | 0.002 | |
| HbA1c (per 1 U) | 1.102 | 1.023–1.188 | 0.011 | |
| Baseline biguanide treatment | 0.702 | 0.550–0.895 | 0.004 | |
| Baseline aspirin treatment | 0.993 | 0.670–1.471 | 0.97 | |
| Baseline statin treatment | 0.943 | 0.672–1.324 | 0.73 | |
| Baseline ACEI/ARB treatment | 1.508 | 1.089–2.089 | 0.014 | |
| Baseline beta blocker treatment | 1.086 | 0.816–1.446 | 0.57 | |
ACEI indicates angiotensin‐converting enzyme inhibitor; ARB, angiotensin receptor blocker; CI, confidence interval; HbA1c, glycosylated hemoglobin; HR, hazard ratio; MI, myocardial infarction.
Fibrinolytic Factors
Median baseline PAI‐1 activity (17.5 versus 19.0 Au/mL, P=0.70) and antigen values (26.0 versus 25.5 ng/mL, P=0.76) were not found to be significantly different in active smokers randomized to IS or IP therapy, respectively (Table 5). However, active smokers randomized to IP therapy were found to have significantly increased mean PAI‐1 activity and antigen levels at 1 year (23.0 versus 16.0 Au/mL [P=0.001] and 33.0 versus 22.5 ng/mL [P=0.003]), 3 years (24.0 versus 18.0 Au/mL [P=0.049] and 31.0 versus 24.0 ng/mL [P=0.008]), and 5 years (29.0 versus 15.0 Au/mL [P=0.004] and 37.0 versus 23.0 ng/mL [P=0.003]) compared with IS therapy (Figure 2A and 2B). For nonsmokers (Table 6), mean baseline PAI‐1 activity (15.0 versus 16.0 Au/mL, P=0.85) and antigen values (23.0 versus 22.0 Au/mL, P=0.91) were also not found to be significantly different in those randomized to IS or IP therapy (Figure 2A and 2B). At follow‐up, however, nonsmokers randomized to IP therapy were found to have significantly increased PAI‐1 activity and antigen levels at 1 year (20.0 versus 13.0 Au/mL [P<0.001] and 27.0 versus 19 ng/mL [P<0.001]), 3 years (19.0 versus 12.0 Au/mL [P<0.001] and 27.0 versus 19.0 ng/mL [P<0.001]), and 5 years (20.0 versus 15.0 Au/mL [P<0.001] and 27.0 versus 20.0 ng/mL [P<0.001]) compared with IS therapy (Figure 2A and 2B). PAI‐1 activity and antigen levels were significantly greater in active smokers than in nonsmokers at baseline (19.0 versus 16.0 [P=0.004] and 26.0 versus 23.0 [P<0.001]), 1 year (19.0 versus 16.0 [P=0.03] and 25.0 versus 22.0 [P<0.001]), and 3 years (20.5 versus 15.0 [P<0.001] and 26.0 versus 23.0 [P=0.004]). By year 5, those differences were no longer found to be significant (22.5 versus 17.0 [P=0.06] and 28.0 versus 23.0 [P=0.13]).
Table 5.
Biochemical Measures in Active Smokers
| Variable | IS | IP | P Valuea |
|---|---|---|---|
| Baseline | |||
| No. of patients | 152 | 143 | |
| PAI‐1 activity, AU/mL, median (IQR) | 17.5 (10.5–31.0) | 19.0 (9.8–29.0) | 0.70 |
| PAI‐1 antigen, ng/mL, median (IQR) | 26.0 (17.0–43.0) | 25.5 (17.0–37.0) | 0.76 |
| tPA, ng/mL, median (IQR) | 10.0 (7.6–13.0) | 9.7 (7.2–13.0) | 0.48 |
| Insulin, IU/mL, median (IQR) | 9.5 (4.8–17.0) | 10.0 (6.5–19.0) | 0.14 |
| HbA1c, %, mean (SD) | 8.09±1.78 | 7.53±1.59 | 0.006 |
| Year 1 | |||
| No. of patients | 114 | 103 | |
| PAI‐1 activity, AU/mL, median (IQR) | 16.0 (7.9–27.0) | 23.0 (13.0–34.5) | 0.001 |
| PAI‐1 antigen, ng/mL, median (IQR) | 22.5 (18.0–36.0) | 33.0 (22.0–48.0) | 0.003 |
| tPA, ng/mL, median (IQR) | 9.0 (6.9–12.0) | 11.0 (8.2–13.0) | 0.004 |
| Insulin, IU/mL, median (IQR) | 6.8 (4.1–13.0) | 10.0 (4.9–17.0) | 0.023 |
| HbA1c, %, mean (SD) | 7.18±1.34 | 7.52±1.46 | 0.10 |
| Year 3 | |||
| No. of patients | 112 | 83 | |
| PAI‐1 activity, AU/mL, median (IQR) | 18.0 (9.6–32.0) | 24.0 (13.0–41.0) | 0.049 |
| PAI‐1 antigen, ng/mL, median (IQR) | 24.0 (15.0–37.0) | 31.0 (23.0–49.0) | 0.008 |
| tPA, ng/mL, median (IQR) | 8.8 (5.9–11.0) | 11.0 (8.0–13.0) | 0.010 |
| Insulin, IU/mL, median (IQR) | 7.8 (4.4–11.0) | 10.2 (7.0–20.0) | 0.038 |
| HbA1c, %, mean (SD) | 7.46±1.75 | 7.90±1.52 | 0.13 |
| Year 5 | |||
| No. of patients | 60 | 39 | |
| PAI‐1 activity, AU/mL, median (IQR) | 15.0 (9.4–27.0) | 29.0 (22.0–35.0) | 0.004 |
| PAI‐1 antigen, ng/mL, median (IQR) | 23.0 (14.0–31.0) | 37.0 (26.0–55.0) | 0.003 |
| tPA, ng/mL, median (IQR) | 9.3 (6.0–11.5) | 12.0 (9.6–13.0) | 0.009 |
| Insulin, IU/mL, median (IQR) | 7.0 (4.1–9.7) | 15.0 (8.7–30.0) | 0.002 |
| HbA1c, %, mean (SD) | 7.78±2.00 | 8.06±1.77 | 0.59 |
HbA1c indicates glycosylated hemoglobin; IP indicates insulin provision; IQR, interquartile range; IS, insulin sensitization; PAI‐1, plasminogen activator inhibitor‐1; tPA, tissue plasminogen activator.
Kruskal–Wallis test.
Figure 2.
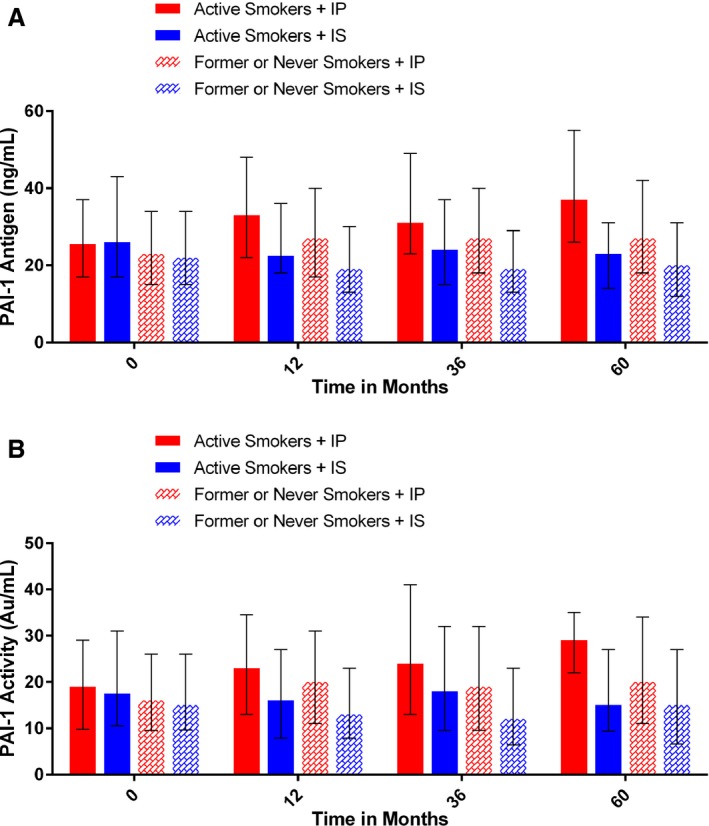
A, Median (first, third quartiles) PAI‐1 antigen levels in active smokers and nonsmokers randomized to IP or IS therapy at baseline and at 12, 36, and 60 mo following randomization. B, Median (first, third quartiles) PAI‐1 activity levels in active smokers and nonsmokers randomized to IP or IS therapy at baseline and at 12, 36, and 60 mo following randomization. IP indicates insulin provision; IS, insulin sensitization; PAI‐1, plasminogen activator inhibitor 1.
Table 6.
Biochemical Measures in Nonsmokers
| Variable | IS | IP | P Valuea |
|---|---|---|---|
| Baseline | |||
| No. of patients | 1028 | 1037 | |
| PAI‐1 activity, AU/mL, median (IQR) | 15.0 (9.7–26.0) | 16.0 (9.6–26.0) | 0.85 |
| PAI‐1 antigen, ng/mL, median (IQR) | 22.0 (15.0–34.0) | 23.0 (15.0–34.0) | 0.91 |
| tPA, ng/mL, median (IQR) | 9.7 (7.4–12.0) | 9.7 (7.3–12.0) | 0.71 |
| Insulin, IU/mL, median (IQR) | 10.0 (5.6–17.0) | 9.7 (5.8–17.0) | 0.80 |
| HbA1c, %, mean (SD) | 7.57±1.58 | 7.68±1.61 | 0.10 |
| Year 1 | |||
| No. of patients | 986 | 996 | |
| PAI‐1activity, AU/mL, median (IQR) | 13.0 (7.9–23.0) | 20.0 (11.0–31.0) | <0.001 |
| PAI‐1 antigen, ng/mL, median (IQR) | 19.0 (13.0–30.0) | 27.0 (17.0–40.0) | <0.001 |
| tPA, ng/mL, median (IQR) | 7.5 (5.4–9.7) | 11.0 (8.3–13.0) | <0.001 |
| Insulin, IU/mL, median (IQR) | 6.3 (3.6–11.0) | 9.5 (5.5–18.0) | <0.001 |
| HbA1c, %, mean (SD) | 6.98±1.26 | 7.29±1.37 | <0.001 |
| Year 3 | |||
| No. of patients | 886 | 911 | |
| PAI‐1 activity, AU/mL, median (IQR) | 12.0 (6.4–23.0) | 19.0 (9.6–32.0) | <0.001 |
| PAI‐1 antigen, ng/mL, median (IQR) | 19.0 (13.0–29.0) | 27.0 (18.0–40.0) | <0.001 |
| tPA, ng/mL, median (IQR) | 7.7 (5.5–10.0) | 11.0 (8.5–13.0) | <0.001 |
| Insulin, IU/mL, median (IQR) | 6.2 (3.8–10.0) | 9.7 (5.2–18.0) | <0.001 |
| HbA1c, %, mean (SD) | 6.97±1.15 | 7.46±1.41 | <0.001 |
| Year 5 | |||
| No. of patients | 434 | 463 | |
| PAI‐1 activity (AU/mL), median (IQR) | 15.0 (6.6–27.0) | 20.0 (11.0–34.0) | <0.001 |
| PAI‐1 antigen (ng/mL), median (IQR) | 20.0 (12.0–31.0) | 27.0 (18.0–42.0) | <0.001 |
| tPA (ng/mL), median (IQR) | 8.0 (5.9–10.5) | 11.0 (8.7–13.0) | <0.001 |
| Insulin (IU/mL), median (IQR) | 6.3 (4.4–11.0) | 9.5 (6.2–19.0) | <0.001 |
| HbA1c, mean (SD), % | 7.12±1.22 | 7.41±1.31 | 0.003 |
HbA1c indicates glycosylated hemoglobin; IP indicates insulin provision; IQR, interquartile range; IS, insulin sensitization; PAI‐1, plasminogen activator inhibitor‐1; tPA, tissue plasminogen activator.
Kruskal–Wallis test.
In active smokers, tPA levels were not found to be significantly different at baseline for IS or IP therapies (10.0 versus 9.7 ng/mL, P=0.48; Table 5). However, IP‐treated patients had significantly increased tPA levels at 1 year (11.0 versus 9.0 ng/mL, P=0.004), 3 years (11.0 versus 8.8 ng/mL, P=0.01), and 5 years (12.0 versus 9.3 ng/mL, P=0.009) compared with IS‐treated patients. For nonsmokers (Table 6), no difference was found in tPA levels between IS or IP at baseline (9.7 versus 9.7 ng/mL, P=0.71). However, IP therapy was associated with significantly increased tPA levels at 1 year (11.0 versus 7.5 ng/mL, P<0.001), 3 years (11.0 versus 7.7 ng/mL, P<0.001), and 5 years (11.0 versus 8.0 ng/mL, P<0.001) compared with IS therapy.
Insulin levels in active smokers were not found to be significantly different at baseline for IS or IP (9.5 versus 10.0 IU/mL, P=0.14). However, IP therapy was found to have significantly increased insulin levels at 1 year (10.0 versus 6.8 IU/mL, P=0.023), 3 years (10.2 versus 7.8 IU/mL, P=0.038), and 5 years (15.0 versus 7.0 IU/mL, P=0.002) compared with IS therapy (Table 5). In smokers, HbA1c levels were greater at baseline in patients receiving IS therapy; however, at 1, 3, and 5 years of follow‐up, no significant differences were found in HbA1c levels between IS and IP treatment arms (Table 5). For nonsmokers at baseline, no difference was found in insulin levels (10.0 versus 9.7 IU/mL, P=0.80) between IS or IP. However, IP was associated with significantly increased insulin levels at 1 year (9.5 versus 6.3 IU/mL, P<0.001), 3 years (9.7 versus 6.2 IU/mL, P<0.001), and 5 years (9.5 versus 6.3 IU/mL, P<0.001) compared with IS (Table 6). At baseline, HbA1c levels were not found to be significantly different between IS and IP groups. However, at 1, 3, and 5 years of follow‐up, HbA1c levels were found to be significantly lower in patients treated with IS (Table 6).
Discussion
The significant findings of this retrospective analysis of a randomized clinical trial are 3‐fold. First, the combination of IP therapy and active smoking in diabetic patients with SIHD was associated was a 323% increase in the hazard of fatal or nonfatal MI over 5.3 years of follow‐up. Second, this association may be mediated through a significant increase in PAI‐1 activity in smokers treated with IP therapy compared with smokers treated with IS therapy. Third, IP therapy also increased PAI‐1 activity in nonsmokers but to a lesser degree than in active smokers and was not associated with an increased rate or hazard of MI compared with nonsmokers treated with IS therapy.
The increased rate of macrovascular complications in patients with type 2 diabetes mellitus compared with nondiabetic patients has been attributed to a specific diabetic vasculopathy.6 Dysglycemia caused by insulin resistance initiates structural changes of the vessel wall that culminate in diabetic vascular complications. These alterations in vascular homeostasis due to endothelial and smooth muscle dysfunction are the main features of diabetic vasculopathy that promote a proinflammatory/thrombotic state that ultimately leads to atherothrombosis. Insulin resistance promotes atherothrombosis through increased cellular synthesis of PAI‐1 that binds to and inhibits tPA, resulting in impaired fibrinolysis.6
Aggressive treatment of diabetic patients to specific glucose levels as end points of therapy has not resulted in a reduction in cardiovascular complications14, 15; therefore, targeting the mechanism of hyperglycemia has received increased attention. Improving insulin sensitivity with drugs such as metformin, rather than increasing insulin levels with insulinotropic sulfonylureas, has resulted in improved cardiovascular outcomes. In the 10‐year follow‐up of the UK Prospective Diabetes Study (UKPDS), metformin‐treated patients had a 33% reduction in MI; this reduction was significantly greater than observed in the sulfonylurea arm, despite a lack of difference in HbA1c between the metformin and sulfonylurea arms in the 5 years after the initial study ended.16 A case–control study found that IS drugs significantly reduced the risk of MI compared with sulfonylurea treatment.17 An analysis of BARI 2D by Chaitman et al found that in the CABG‐stratum patients who had greater atherosclerotic burden, the reduction of MI seen in patients randomized to revascularization was found to be significant only in those randomized to an IS treatment strategy.2 Sobel and colleagues subsequently evaluated the impact of IS and IP treatment strategies on fibrinolytic biomarker profiles in the BARI 2D trial.7 Although IP therapy was associated with greater PAI‐1 antigen and activity, these changes did not translate into any difference in outcomes in the overall trial. We hypothesized that the failure to detect a difference in outcomes between patients randomized to IS or IP in BARI 2D was due to the intensive treatment of other risk factors such as hypertension and hyperlipidemia, along with aggressive lifestyle modification that mitigated the clinical consequences of prothrombotic IP therapy on outcomes. We further hypothesized that a second prothrombotic condition might tip the hemostatic balance toward a prothrombotic phenotype.
Active smoking is a major risk factor for acute coronary thrombosis leading to MI.18 Exposure to cigarette smoke alters the balance of antithrombotic/prothrombotic factors by affecting the function of endothelial cells, platelets, fibrinogen, and coagulation factors along with elevation of PAI‐1 activity.18 The dramatic increase in the rate and hazard of fatal and nonfatal MI in active smokers randomized to IP therapy in BARI 2D supports a “2‐hit” theory for the development of coronary thrombotic events. That the increase in MI may be mediated, at least in part, through inhibition of fibrinolysis is suggested by both the significant increase in PAI‐1 activity levels among active smokers in the IP group at all time points following randomization and the fact that, although IP therapy also increased PAI activity in nonsmokers, the magnitude of increase was less in nonsmokers than active smokers.
Limitations
Our study has several potential limitations. First, there was crossover between the IS and IP groups. In patients randomized to IP, 92% of patients were on IP therapy at 5 years, whereas 18% were receiving IS therapy. In patients randomized to IS, 80% were still taking IS drugs at 5 years, and 54% were taking IP drugs.8 Second, other mediators of hypercoagulability such as platelet function were not measured. Third, although we adjusted for all observed differences by smoking status, it is not possible to adjust for unmeasured confounders between active smokers and nonsmokers. Fourth, smoking status was self‐reported with no biochemical validation of smoking status. Fifth, these results were achieved in a selected population treated by specific protocols in the context of a clinical trial and thus may not be generalizable to other populations and settings. Finally, this study was a post hoc subgroup analysis and, as such, the results can only be considered hypothesis generating.
Conclusion
In summary, in this post hoc analysis of the BARI 2D trial, we found that after 5.3 years of follow‐up, the combination of smoking and IP therapy in patients with type 2 diabetes mellitus and SIHD was associated with a significantly increased hazard of MI. This result may be explained by higher PAI‐1 activity in smokers treated with IP. If confirmed in future studies, our findings suggest that diabetic smokers with SIHD should be preferentially treated with IS rather than IP therapy.
Disclosures
None.
(J Am Heart Assoc. 2017;6:e005946 DOI: 10.1161/JAHA.117.005946.)28903941
References
- 1. Beckman JA, Creager MA, Libby P. Diabetes and atherosclerosis: epidemiology, pathophysiology, and management. JAMA. 2002;287:2570–2581. [DOI] [PubMed] [Google Scholar]
- 2. Chaitman BR, Hardison RM, Adler D, Gebhart S, Grogan M, Ocampo S, Sopko G, Ramires JA, Schneider D, Frye RL. The Bypass Angioplasty Revascularization Investigation 2 Diabetes randomized trial of different treatment strategies in type 2 diabetes mellitus with stable ischemic heart disease: impact of treatment strategy on cardiac mortality and myocardial infarction. Circulation. 2009;120:2529–2540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Leal J, Gray AM, Clarke PM. Development of life‐expectancy tables for people with type 2 diabetes. Eur Heart J. 2009;30:834–839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Stevens RJ, Kothari V, Adler AI, Stratton IM. The UKPDS risk engine: a model for the risk of coronary heart disease in type II diabetes (UKPDS 56). Clin Sci (Lond). 2001;101:671–679. [PubMed] [Google Scholar]
- 5. Franco OH, Steyerberg EW, Hu FB, Mackenbach J, Nusselder W. Associations of diabetes mellitus with total life expectancy and life expectancy with and without cardiovascular disease. Arch Intern Med. 2007;167:1145–1151. [DOI] [PubMed] [Google Scholar]
- 6. Paneni F, Beckman JA, Creager MA, Cosentino F. Diabetes and vascular disease: pathophysiology, clinical consequences, and medical therapy: part 1. Eur Heart J. 2013;34:2436–2443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Sobel BE, Hardison RM, Genuth S, Brooks MM, McBane RD III, Schneider DJ, Pratley RE, Huber K, Wolk R, Krishnaswami A, Frye RL. Profibrinolytic, antithrombotic, and antiinflammatory effects of an insulin‐sensitizing strategy in patients in the Bypass Angioplasty Revascularization Investigation 2 Diabetes (BARI 2D) trial. Circulation. 2011;124:695–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Frye RL, August P, Brooks MM, Hardison RM, Kelsey SF, MacGregor JM, Orchard TJ, Chaitman BR, Genuth SM, Goldberg SH, Hlatky MA, Jones TL, Molitch ME, Nesto RW, Sako EY, Sobel BE. A randomized trial of therapies for type 2 diabetes and coronary artery disease. N Engl J Med. 2009;360:2503–2515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Barua RS, Ambrose JA. Mechanisms of coronary thrombosis in cigarette smoke exposure. Arterioscler Thromb Vasc Biol. 2013;33:1460–1467. [DOI] [PubMed] [Google Scholar]
- 10. Sobel BE, Frye R, Detre KM. Burgeoning dilemmas in the management of diabetes and cardiovascular disease: rationale for the Bypass Angioplasty Revascularization Investigation 2 Diabetes (BARI 2D) trial. Circulation. 2003;107:636–642. [DOI] [PubMed] [Google Scholar]
- 11. Brooks MM, Frye RL, Genuth S, Detre KM, Nesto R, Sobel BE, Kelsey SF, Orchard TJ. Hypotheses, design, and methods for the Bypass Angioplasty Revascularization Investigation 2 Diabetes (BARI 2D) trial. Am J Cardiol. 2006;97:9 g–19 g. [DOI] [PubMed] [Google Scholar]
- 12. Rautaharju PM, Calhoun HP, Chaitman BR. Novacode serial ECG classification system for clinical trials and epidemiologic studies. J Electrocardiol. 1992;24(suppl):179–187. [DOI] [PubMed] [Google Scholar]
- 13. Chaitman BR, Zhou SH, Tamesis B, Rosen A, Terry AB, Zumbehl KM, Stocke K, Takase B, Gussak I, Rautaharju PM. Methodology of serial ECG classification using an adaptation of the Novacode for Q wave myocardial infarction in the Bypass Angioplasty revascularization Investigation (BARI). J Electrocardiol. 1996;29:265–277. [DOI] [PubMed] [Google Scholar]
- 14. Gerstein HC, Miller ME, Byington RP, Goff DC Jr, Bigger JT, Buse JB, Cushman WC, Genuth S, Ismail‐Beigi F, Grimm RH Jr, Probstfield JL, Simons‐Morton DG, Friedewald WT. Effects of intensive glucose lowering in type 2 diabetes. N Engl J Med. 2008;358:2545–2559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Turnbull FM, Abraira C, Anderson RJ, Byington RP, Chalmers JP, Duckworth WC, Evans GW, Gerstein HC, Holman RR, Moritz TE, Neal BC, Ninomiya T, Patel AA, Paul SK, Travert F, Woodward M. Intensive glucose control and macrovascular outcomes in type 2 diabetes. Diabetologia. 2009;52:2288–2298. [DOI] [PubMed] [Google Scholar]
- 16. Holman RR, Paul SK, Bethel MA, Matthews DR, Neil HA. 10‐year follow‐up of intensive glucose control in type 2 diabetes. N Engl J Med. 2008;359:1577–1589. [DOI] [PubMed] [Google Scholar]
- 17. Sauer WH, Cappola AR, Berlin JA, Kimmel SE. Insulin sensitizing pharmacotherapy for prevention of myocardial infarction in patients with diabetes mellitus. Am J Cardiol. 2006;97:651–654. [DOI] [PubMed] [Google Scholar]
- 18. Ambrose JA, Barua RS. The pathophysiology of cigarette smoking and cardiovascular disease: an update. J Am Coll Cardiol. 2004;43:1731–1737. [DOI] [PubMed] [Google Scholar]