

Abstract
Background
Left ventricular noncompaction (LVNC) has since been classified as a primary genetic cardiomyopathy, but the genetic basis is not fully evaluated. The aim of the present study was to identify the genetic spectrum using next‐generation sequencing and to evaluate genotype–phenotype correlations in LVNC patients.
Methods and Results
Using next‐generation sequencing, we targeted and sequenced 73 genes related to cardiomyopathy in 102 unrelated LVNC patients. We identified 43 pathogenic variants in 16 genes in 39 patients (38%); 28 were novel variants. Sarcomere gene variants accounted for 63%, and variants in genes associated with channelopathies accounted for 12%. MYH7 and TAZ pathogenic variants were the most common, and rare variant collapsing analysis showed variants in these genes contributed to the risk of LVNC, although patients carrying MYH7 and TAZ pathogenic variants displayed different phenotypes. Patients with pathogenic variants had early age of onset and more severely decreased left ventricular ejection fractions. Survival analysis showed poorer prognosis in patients with pathogenic variants, especially those with multiple variants: All died before their first birthdays. Adverse events were noted in 17 patients, including 13 deaths, 3 heart transplants, and 1 implantable cardioverter‐defibrillator insertion. Congestive heart failure at diagnosis and pathogenic variants were independent risk factors for these adverse events.
Conclusions
Next‐generation sequencing revealed a wide spectrum of genetic variations and a high incidence of pathogenic variants in LVNC patients. These pathogenic variants were independent risk factors for adverse events. Patients harboring pathogenic variants showed poor prognosis and should be followed closely.
Keywords: genetics, noncompaction cardiomyopathy, prognosis
Subject Categories: Cardiomyopathy, Congenital Heart Disease
Clinical Perspective
What Is New?
This research revealed a wide spectrum of genetic variants and high incidence of novel pathogenic variants using a focused next‐generation sequencing strategy in a cohort of 102 patients with left ventricular noncompaction.
What Are the Clinical Implications?
The presence of a pathogenic variant was an independent risk factor for death, heart transplantation, or implantable cardioverter‐defibrillator insertion in patients with left ventricular noncompaction, and the prognosis was even worse in patients with double pathogenic variants or TAZ variants.
Introduction
Left ventricular noncompaction (LVNC) was originally described as cross‐linked infantile cardiomyopathy with poor prognosis1 but has since been classified as a primary genetic cardiomyopathy by the American Heart Association.2 LVNC is characterized by a pattern of prominent trabecular meshwork and deep intertrabecular recesses communicating with the left ventricular cavity. LVNC is postulated to be caused by an arrest of the normal process of intrauterine endomyocardial morphogenesis. LVNC may be a distinct disorder but also may be associated with other cardiomyopathies.2, 3, 4, 5, 6, 7 With the development of sequencing technologies, multiple gene variants have been found related to LVNC, but the genetics of LVNC have not been fully evaluated. Previous studies have shown that sarcomere gene variants likely play an important role in patients with LVNC8 but do not predict clinical phenotype.9 Next‐generation sequencing (NGS) was used recently because of the ability to investigate multiple genes at reasonable cost. The aim of this study was to investigate the genetic landscape of LVNC and to identify genotype–phenotype correlations in the largest cohort of well‐phenotyped Japanese LVNC patients.
Methods
Clinical Evaluation
Unrelated childhood patients were recruited from 2001 to 2016 from 61 Japanese hospitals with divisions of pediatric cardiology. A total of 102 patients with LVNC were included in this study. Three patients had Barth syndrome; none had neuromuscular disorders. In addition, patients with congenital heart disease that induced significant hemodynamic changes or with insufficient clinical information were excluded. Clinical evaluation consisted of clinical presentation and symptoms; a personal and family history (patient's biological family members showed existence of any cardiomyopathy disease, not only LVNC but also other cardiomyopathy or family members [parents or brother sisters]), arrhythmia, thromboembolism, ECG, 2‐dimensional Doppler, and color Doppler echocardiography. The diagnosis of heart failure was based on clinical symptoms of feeding difficulty, tachypnea, and cyanosis and findings of decreased left ventricular ejection fraction (LVEF) in the left ventricle on echocardiography and cardiomegaly on chest x‐ray. A diagnosis of LVNC was made according to (1) the characteristic 2‐layered appearance of the myocardium, with an increased N/C ratio (N/C>2.0) at end‐diastole and the disease process observed in ≥1 ventricular wall segment and (2) multiple deep intertrabecular recesses communicating with the ventricular cavity, as demonstrated by color Doppler imaging.3
Informed consent was obtained from all patients’ parents, according to institutional guidelines. This study protocol conforms to the ethics guidelines of the 1975 Declaration of Helsinki, as reflected in a priori approval by the research ethics committee of University of Toyama, Japan.
Mutation Screening
Genomic DNA was extracted from whole blood using a QuickGene DNA whole blood kit S (Kurabo). NGS of 73 cardiac disorder–related genes associated with cardiomyopathies and channelopathies (Table S1) was performed using an IonPGM system (Life Technologies). This custom panel utilized 2 separate polymerase chain reaction primer pools, yielding a total of 1870 amplicons and used to generate target amplicon libraries. Genomic DNA samples were polymerase chain reaction–amplified using the custom panel and an Ion AmpliSeq Library Kit v2.0 (Life Technologies, Carlsbad, CA). Individual samples were labeled using an Ion Xpress Barcode Adapters Kit (Life Technologies) and then pooled at equimolar concentrations. Emulsion polymerase chain reaction and ion sphere particle enrichment were performed using the Ion PGM HiQ OT2 Kit (Life Technologies), according to the manufacturer's instructions. Ion sphere particles were loaded onto a 316 chip and sequenced using an Ion PGM HiQ Sequencing Kit (Life Technologies).
Data Analysis and Variant Classification
Torrent Suite and Ion Reporter software version 5.0 (Life Technologies) were used to perform primary, secondary, and tertiary analyses, including optimized signal processing, base calling, sequence alignment, and variant analysis. The allelic frequency of all detected variants was determined using the Exome Aggregation Consortium (ExAC) East Asian database and the Human Genetic Variation Database (HGVD), which contains data for 1208 Japanese persons.10 Rare variants such as those single‐nucleotide polymorphisms with a minor allele frequency (MAF) below some threshold in the combined set of cases and controls were selected.11 All variants with a MAF ≥0.05% among the ExAC East Asian and HGVD populations were filtered out.12, 13 We utilized 7 different in silico predictive algorithms to improve the accuracy of evaluating the pathogenicity of the remaining variants: FATHMM, SIFT, PROVEAN, Align GVGD, MutationTaster2, PolyPhen2, and CADD (URLs listed in Table S2). Variants predicted to be deleterious or pathogenic by at least 5 of the 7 in silico algorithms were considered likely pathogenic. The pathogenicity of the detected variant was based on the guidelines of the American College of Medical Genetics and Genomics.13
Sanger Sequencing
For all candidate pathogenic variants that passed these selection criteria, Sanger sequencing was used to validate the NGS results. The nucleotide sequences of amplified fragments were analyzed by direct sequencing in both directions using the BigDye Terminator v3.1 Cycle Sequencing Kit (Applied Biosystems), and sequence analysis was performed using an ABI 3130xl automated sequencer (Applied Biosystems).
Assessment of the Frequency of Rare Variants in Control Population Data
Differences in proportions of rare variants versus controls from the ExAC East Asian and HGVD data were assessed using the Fisher exact test, with P<0.05 considered statistically significant. Potential pathogenicity of the variants was evaluated based on allele frequency, as recommended by recent guidelines for interpreting sequence variants.13
Gene‐Based Collapsing Test
We used a genic collapsing test to confer risk genes of LVNC.14, 15 Each gene was indicated as carrying or not carrying a “qualifying” variant. A qualifying variant was defined as a variant with an MAF cutoff of <0.05% among the ExAC East Asian population. Qualified variants were defined as nonsynonymous, frameshift, and splice‐site variants.
Statistical Analysis
Statistical analysis was performed with SPSS (version 24; IBM Corp) software and R software. The unpaired t test or the χ2 test was used to compare variables. P<0.05 was considered statistically significant. Important prognostic factors were used in the univariate analysis and then in Firth regression using R software.16 The event‐free rate for the combined end point of death, heart transplantation (HT), or implantable cardioverter‐defibrillator (ICD) insertion was calculated by the Kaplan‐Meier method and compared using the log‐rank test. The Fisher exact test was performed for each gene in collapsing analysis with a nominal significance level <1.37×10−4 according to Bonferroni correction for the number of assessable genes.
Results
Baseline Clinical Characteristics
A total of 102 patients were enrolled in this study; 54 were male and 48 were female, with an age range from fetus to 12 years (mean age: 1.8±0.4 years; Table 1). Pathogenic variants were identified in 39 patients (38%) who presented with a much earlier age of onset and lower LVEF (P<0.05) than those without pathogenic variants. The majority (76.9%) of patients with pathogenic variants presented with congestive heart failure at diagnosis. We divided the LVNC patients into 2 types: those with systolic dysfunction (n=63) and those without systolic dysfunction (n=39). Pathogenic variants were more commonly detected in patients with systolic dysfunction (31/63, 49%) than in those without (9/39, 23%; P=0.012). Family history was more common in patients with pathogenic variants but did not reach statistical significance. Survival analysis showed that patients with pathogenic variants had worse prognosis than patients without; 26% of the patients with pathogenic variants died or underwent HT or ICD insertion (Figure 1).
Table 1.
Characteristics of Patients With and Without Pathogenic Mutations
| P+ (n=39) | P− (n=63) | P Value | |
|---|---|---|---|
| Sex, male:female | 18:21 | 34:27 | 0.54 |
| Age at onset, y | 0.45±0.2 | 2.7±0.6 | 0.003 |
| CHF at diagnosis, n (%) | 30 (76.9) | 32 (50.8) | 0.01 |
| Family history, n (%) | 12 (30.8) | 12 (19) | 0.81 |
| LVEF, % | 37±2.0 | 46.3±3.0 | 0.01 |
| LVDD z score | 1.59±0.18 | 1.44±0.56 | 0.79 |
CHF indicates congestive heart failure; LVEF, left ventricular ejection fraction; LVDD, left ventricular end‐diastolic dimension; P+, patients with pathogenic mutations; P−, patients with no or nonpathogenic mutations.
Figure 1.
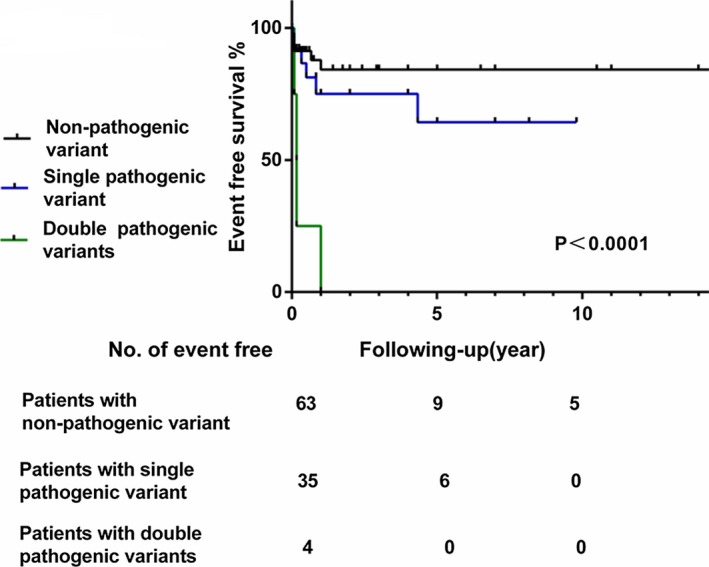
Event‐free survival to the combined end point of death, heart transplantation, and implantable cardioverter‐defibrillator insertion of patients with double pathogenic, pathogenic, and nonpathogenic mutations.
Genetic analysis
NGS of samples from the 102 patients yielded 540 830±11 986 sequence reads per person. The mean read length per sample was 163.6±1.1 base pairs, and the mean depth of base coverage was 247.0±5.8 reads; 95.23% had >10‐fold coverage, and 92.5% had >20‐fold coverage.
The distribution of pathogenic variants is shown in Figure 2. There were 43 pathogenic variants: 39 missense, 1 deletion, 1 nonsense, and 2 splice site variants. Sarcomere gene variants accounted for 63%, and variants in genes associated with channelopathies accounted for 12%. Overall, MYH7 was most commonly mutated (n=19, 44%), followed by TAZ (n=6, 14%). There was only 1 pathogenic variant in each of the following genes: MYBPC3, TNNC1, LMNA, ANK2, KCNH2, KCNE3, JUP, HCN4, BMPR1A, and TBX5. Notably, this is the first report of pathogenic variants in BMPR1A, ANK2, and TBX5 in LVNC patients. Ten missense variants were identified in MYBPC3, but 9 of them were filtered out because of their frequent occurrence (MAF >0.5%) in the ExAC East Asian or HGVD (Japanese) populations. Consequently, there is a significant difference in the prevalence of variants in MYH7 and MYBPC3 in this study, unlike other forms of cardiomyopathy (Table S3).
Figure 2.
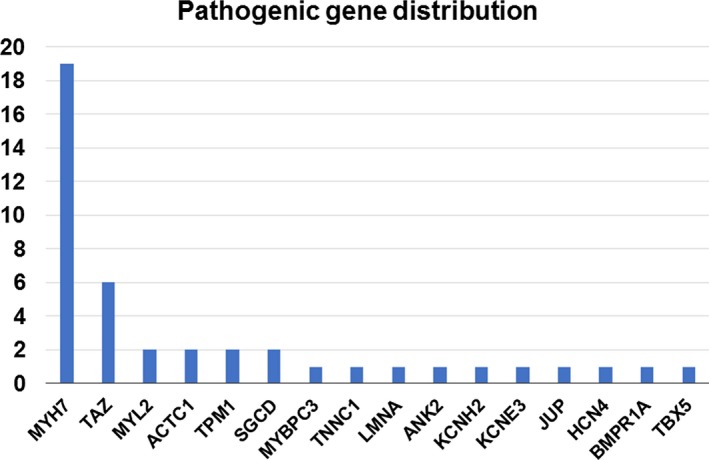
Pathogenic gene distribution of left ventricular noncompaction. The number of pathogenic mutations identified in each gene in which at least 1 mutation was identified.
Twenty‐nine novel variants (not detected in 60 706 persons of any race/ethnicity in the ExAC and HGVD databases) were identified in 12 genes: 19 novel variants in sarcomere genes (66%), including 12 MYH7 variants, and 4 novel variants in TAZ. Novel pathogenic variants were also identified in BMPR1A, HCN4, LMNA, SGCD, and TBX5 (Table S4).
In addition, 14 rare variants with MAF<0.05% in the 2 reference databases were identified in 7 genes (ANK2, JUP, KCNE3, KCNH2, MYH7, MYL2 and TAZ; Table 2). None of them had been reported previously in East Asian controls in ExAC or HGVD. The odds ratios for the association between the variant and the risk of disease were all significantly >1.0, and the Fisher exact P values were all <0.05 (Table 2). The genic collapsing test revealed that MYH7 (P=1.29E‐17, ranked first) and TAZ (P=3.48E‐9, ranked second) reached significance (adjusted a or P<1.37×10−4), strongly suggesting that variants in these genes contribute to an increased risk of LVNC. All other genes, including MYBPC3, ANK2, TPM1 and ACTC1, did not reach the adjusted α (Table S5).
Table 2.
The Frequency of Rare Variants in the Control Population Databases
| Gene | Variant | dbSNP | ExAC (All Individuals), % | HGVD, % | Genotype, Case (n=102) | ExAC (East Asian, n=4327) | Risk, OR | Frequency, 95% CI | P Value | Classification |
|---|---|---|---|---|---|---|---|---|---|---|
| ANK2 | R321W | rs753032598 | 0.0025 | ··· | 1 | 0 | 127.9 | 1.08 to + ∞ | 0.0230 | Likely pathogenic |
| JUP | E146K | rs146581757 | 0.002 | ··· | 1 | 0 | 127.9 | 1.08 to + ∞ | 0.0230 | Likely pathogenic |
| KCNE3 | R99H | rs121908441 | 0.0086 | ··· | 1 | 0 | 127.9 | 1.08 to + ∞ | 0.0230 | Pathogenic |
| KCNH2 | A561T | rs199472921 | ··· | ··· | 1 | 0 | 127.9 | 1.08 to + ∞ | 0.0230 | Pathogenic |
| MYH7 | R23W | rs730880828 | 0.0025 | ··· | 1 | 0 | 127.9 | 1.08 to + ∞ | 0.0230 | Likely pathogenic |
| L620P | rs199862338 | ··· | ··· | 1 | 0 | 127.9 | 1.08 to + ∞ | 0.0230 | Likely pathogenic | |
| P838L | rs397516153 | ··· | ··· | 1 | 0 | 127.9 | 1.08 to + ∞ | 0.0230 | Likely pathogenic | |
| R904C | rs727503253 | 0.00082 | ··· | 1 | 0 | 127.9 | 1.08 to + ∞ | 0.0230 | Likely pathogenic | |
| E1801K | rs397516248 | ··· | ··· | 2 | 0 | 215.3 | 8.0 to + ∞ | 0.0005 | Likely pathogenic | |
| E1914K | rs397516254 | ··· | ··· | 1 | 0 | 127.9 | 1.08 to + ∞ | 0.0230 | Likely pathogenic | |
| MYL2 | P144fs | rs199567559 | 0.00082 | ··· | 1 | 0 | 127.9 | 1.08 to + ∞ | 0.0230 | Likely pathogenic |
| TAZ | G197R | rs132630277 | ··· | ··· | 1 | 0 | 127.9 | 1.08 to + ∞ | 0.0230 | Likely pathogenic |
| c.109+1G>C | ··· | ··· | ··· | 1 | 0 | 127.9 | 1.08 to + ∞ | 0.0230 | Pathogenic |
CI indicates confidence interval; ExAC, Exome Aggregation Consortium database; HGVD, Human Genetic Variation Database; OR, odds ratio.
The Characteristics of Patients With Single or Double Pathogenic Variants
Double heterozygous variants were identified in 4 patients, all of whom presented with congestive heart failure during the fetal or neonatal periods and died before their first birthdays. Of note, none had family history of cardiomyopathy (Table 3). Survival analysis revealed that patients with double variants showed the worst prognosis compared with patients with a single variant and without variants (Figure 1). There were no differences in age of onset, heart failure at diagnosis, LVEF, and family history between the 2 groups (Table 3).
Table 3.
Characteristics of Patients With Single and Double Mutations
| Single Variant (n=35) | Double Variant (n=4) | P Value | |
|---|---|---|---|
| Sex, male:female | 15:20 | 3:1 | 0.32 |
| Age of onset, y | 0.5±0.2 | 0.001±0.001 | 0.43 |
| CHF at diagnosis, n (%) | 26 (74.3) | 4 (100) | 0.56 |
| Family history, n (%) | 12 (34.2) | 0 | 0.29 |
| LVEF, % | 36.9±2.2 | 37.5±3.8 | 0.93 |
| LVDD z score | 1.51±0.19 | 2.31±0.34 | 0.19 |
Double heterozygous variants: MYH7 and JUP, MYH7 and BMPR1A, TPM1 and SGCD, and TAZ and KCNE3. CHF indicates congestive heart failure; LVDD, left ventricular end‐diastolic dimension; LVEF, left ventricular ejection fraction.
The characteristics of patients with adverse events
Adverse events were noted in 16 patients: 12 died, 3 underwent HT, and 1 underwent ICD insertion. Among those 16, double heterozygous variants were identified in 4 patients, and single variants were noted in 6, including variants in TAZ in 2. No pathogenic variants were identified in the remaining 6 patients (Table 4). The majority of patients with adverse events were boys (76%). All of these patients were diagnosed before their first birthday, except 1 who was diagnosed at age 4 years and underwent ICD insertion after 9 months of follow‐up. Five patients were diagnosed during the fetal period, because of severe heart failure and hydrops fetalis, and died soon after birth. The multivariable proportional hazards model showed that congestive heart failure at diagnosis and pathogenic variant were independent risk factors for death, HT, or ICD insertion in all LVNC patients (Table 5).
Table 4.
Characteristics of Patients With Adverse Events
| ID | Gene and Variant | Age at Onset | Sex | Family History | CHF at Diagnosis | Outcome | Cause of Death |
|---|---|---|---|---|---|---|---|
| 234 | SGCD N99H; TPM1 D14G | 15 d | M | No | Yes | Death | CHF |
| 274 | TAZ H176Y; KCNE3 R99H | Fetus | M | No | Yes | Death | CHF |
| 280 | MYH7 K542N; JUP E146K | Fetus (30 WG) | M | No | Yes | Death | CHF |
| 342 | MYH7 P838L; BMPR1A R284L | 1 d | F | No | Yes | Death | CHF |
| 159 | TAZ splice donor c.109+1G>C | 2 mo | M | Yes | Yes | Death | CHF |
| 247 | MYH7 R712H | Fetus (32 WG) | F | No | Yes | HT | |
| 312 | ACTC1 T231R | 4 y | M | No | Yes | ICD insertion | |
| 313 | TAZ M185V | 1 mo | M | Yes | Yes | HT | |
| 233 | KCNH2 A561T | Fetus (25 WG) | M | No | Yes | Death | CHF |
| 321 | TNNC1 E94A | 4 mo | F | No | No | HT | |
| 193 | ··· | 1 d | M | No | Yes | Death | CHF |
| 275 | ··· | 1 d | M | No | Yes | Death | CHF |
| 294 | ··· | 1 y | M | No | Yes | Death | CHF |
| 356 | ··· | 15 d | M | Yes | Yes | Death | VF |
| 367 | ··· | Fetus | F | Yes | Yes | Death | CHF |
| 416 | ··· | 1 mo | M | No | Yes | Death | CHF |
CHF indicates congestive heart failure; F, female; HT, heart transplantation; ICD, implantable cardioverter‐defibrillator; M, male; VF, ventricular fibrillation; WG, weeks of gestation.
Table 5.
Multivariate Analysis of Risk Factors for LVNC
| Variable | Univariable Survival Analysis | Multivariable Survival Analysis | ||
|---|---|---|---|---|
| HR (95% CI) | P Value | HR (95% CI) | P Value | |
| Age at onset, y | 3.14 (1.17–8.42) | 0.03 | 0.47 (0.12–2.61) | 0.34 |
| Family history | 1.42 (0.46–4.43) | 0.16 | 2.08 (0.65–5.97) | 0.20 |
| CHF at diagnosis | 19.30 (2.98–20.31) | 0.0003 | 46.24 (5.39–6097.7) | 0.00002 |
| Genotype positive | 3.61 (1.27–10.20) | 0.01 | 3.22 (1.12–11.22) | 0.03 |
CHF indicates congestive heart failure; CI, confidence interval; HR, hazard ratio; LVNC, left ventricular noncompaction.
Genotype–phenotype correlations
Variants found in participants with systolic dysfunction and details of for each participant are shown in Tables S6 and S7. Single sarcomere variants were identified in 24 patients, single nonsarcomere variants were found in 11, and double variants were noted in 4 patients (MYH7 and JUP, MYH7 and BMPR1A, TPM1 and SGCD, TAZ and KCNE3; Table 4). There were no differences in age at onset, heart failure onset, LVEF, and family history between the sarcomere and nonsarcomere groups (Table 6). Survival analysis showed that the prognosis of patients with nonsarcomere variants was worse than that of patients with sarcomere variants (Figure 3).
Table 6.
Characteristics of Patients With Sarcomere and Nonsarcomere Mutations
| Sarcomere Variant (n=24) | Nonsarcomere Variant (n=11) | P Value | |
|---|---|---|---|
| Sex male:female | 8:16 | 3:8 | 0.99 |
| Age of onset, y | 0.7±0.3 | 0.15±0.07 | 0.26 |
| CHF at diagnosis, n (%) | 15 (62.5) | 10 (91) | 0.12 |
| Family history, n (%) | 6 (34.8) | 6 (54.5) | 0.13 |
| LVEF, % | 39.4±2.3 | 31.8±4.7 | 0.11 |
| LVDD z score | 1.24±0.2 | 2.1±0.4 | 0.04 |
CHF indicates congestive heart failure; LVDD, left ventricular end‐diastolic dimension; LVEF, left ventricular ejection fraction.
Figure 3.
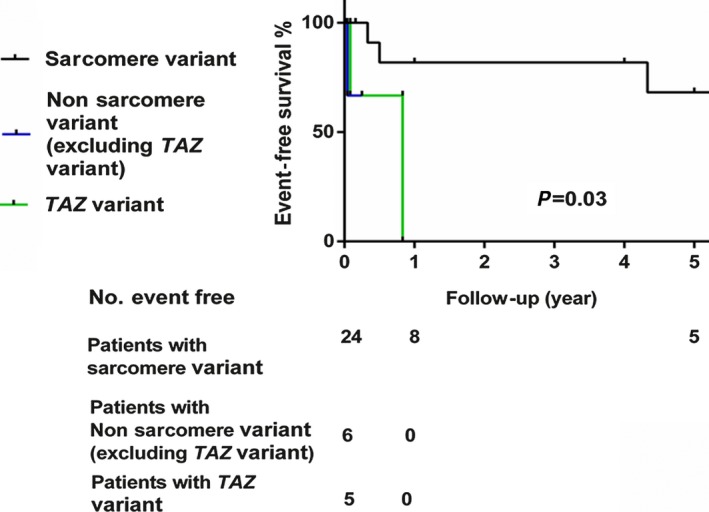
Event‐free survival to the combined end point of death, heart transplantation, and implantable cardioverter‐defibrillator insertion of patients with sarcomere, nonsarcomere (excluding TAZ mutations), or TAZ mutations.
Because MYH7 and TAZ were predicted to significantly contribute to the risk of LVNC, we compared the characteristics of patients with variants in these genes (Table 7). The patients carrying TAZ variants displayed a distinct phenotype; all were male infants who presented with congestive heart failure and had worse prognoses. Three had Barth syndrome, 1 with double variants. Overall, 80% of the TAZ group had family history of cardiomyopathy; this was much higher than the MYH7 group. The TAZ group presented with higher LVDD z scores and lower LVEF than the MYH7 group. There were no differences in age at onset between the groups. In our study, we found that the clinical manifestation varied significantly in the patients with MYH7 variants, from no symptoms to severe heart failure. Two patients with double variants of MYH7 and another gene and 1 patient with TAZ and another variant were excluded from the analysis (Table 7). Survival analysis showed that the prognosis was significantly worse for patients with TAZ variants compared with patients with sarcomere gene variants (P=0.03; Figure 3).
Table 7.
Characteristics of Patients With MYH7 or TAZ Mutations
| MYH7 (N=17) | TAZ (N=5) | P Value | |
|---|---|---|---|
| Sex, male:female | 5:12 | 5:0 | 0.01 |
| Age at onset, y | 0.5±0.4 | 0.3±0.1 | 0.71 |
| CHF at diagnosis, n (%) | 10 (58.8) | 5 (100) | 0.13 |
| Family history, n (%) | 4 (23.5) | 4 (80) | 0.039 |
| LVEF, % | 39.8±3.2 | 20.4±5.6 | 0.008 |
| LVDD z score | 1.07±0.27 | 3.13±0.36 | 0.001 |
Three patients with double mutation of MYH7 and another gene and 1 patient with TAZ and another mutation were excluded. CHF indicates congestive heart failure; LVDD, left ventricular end‐diastolic dimension; LVEF, left ventricular ejection fraction.
Among the patients with nonsarcomere gene variants, 5 carried variants in channelopathy‐related genes: ANK2, KCNE3, KCNH2, HCN4, and JUP. The ECG of the patient with the KCNE3 variant showed left bundle‐branch block. ECGs of the patients with ANK2, HCN4 and LMNA variants showed normal or nonspecific changes. The patient with the KCNH2 variant died at 2 weeks after birth due to severe congestive heart failure; however, no specific changes were identified on ECG.
One patient who carried both MYH7 and BMPR1A variants was diagnosed during the fetal period and died after 1 year of follow‐up. We extracted DNA from her postmortem heart and found the same variants in MYH7 and BMPR1A that were detected previously in blood samples (Figure 4).
Figure 4.
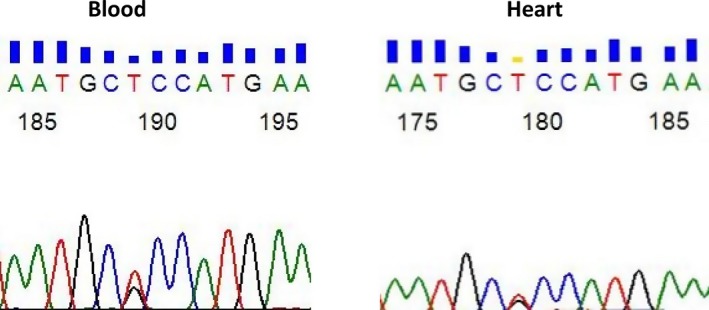
Detection of the BMPR1A c.851G>T (p. R284L) variant in DNA isolated from blood and heart samples of a patient with left ventricular noncompaction.
The variant in TPM1 appeared de novo (Figure 5A), as neither parent nor a brother carried this variant.
Figure 5.
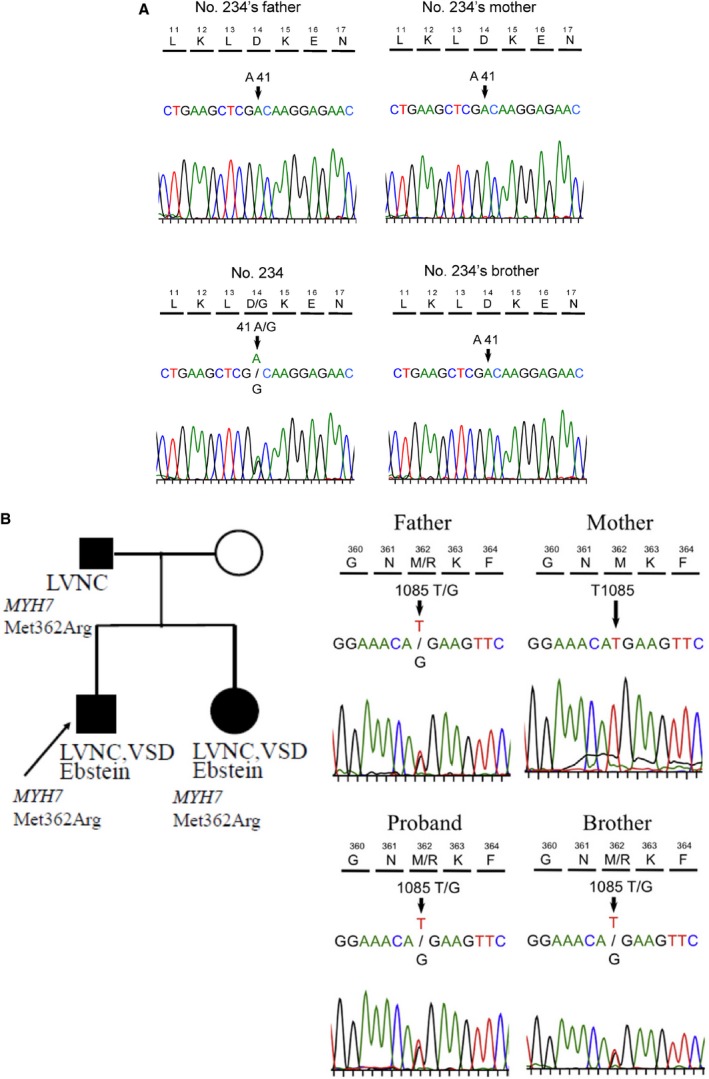
A, De novo variant of TPM1 c.41A>G (p. D14G) in an LVNC family. B, Familial LVNC and Ebstein anomaly associated with the MYH7 c.1085T>G p.Met362Arg. LVNC, left ventricular noncompaction.VSD, ventricular septal defect.
A variant in MYH7, c.1085T>G (p. Met362Arg), was identified in a family with LVNC and Ebstein anomaly (Figure 5B); we previously reported this variant14 using a candidate gene approach. However, no additional pathogenic variants, inherited from the unaffected mother, were identified in the offspring with Ebstein anomaly that could account for the phenotypic difference between the father and the children.
Discussion
In summary, use of a focused NGS strategy in a large cohort of 102 LVNC patients revealed a wide and specific spectrum of genetic variations and a high incidence of novel pathogenic variants in LVNC patients. In addition, we found poorer prognosis in the patients with pathogenic variants, and the detection of a pathogenic variant was an independent risk factor for death, HT, and ICD insertion.
There appears to be a distinct spectrum of gene variants in Japanese patients with LVNC. Variants in MYH7 appear to be a significant cause of LVNC, accounting for almost half of the pathogenic variants identified, whereas the prevalence of MYBPC3 variants were unexpectedly low. Furthermore, collapsing analysis confirmed that MYH7 variants increase the risk of developing LVNC, whereas MYBPC3 variants did not. This genetic spectrum is quite different from previous studies in patients with hypertrophic cardiomyopathy or dilated cardiomyopathy (Table S3). In patients with hypertrophic cardiomyopathy, mutations in MYBPC3 and MYH7 are most commonly detected.17, 18, 19, 20, 21 In contrast, in patients with dilated cardiomyopathy, variants in titin are most commonly detected, whereas variants in MYH7 and MYBPC3 account for <1%.22 Although the majority of the LVNC patients presented with the same phenotypic characteristics as patients with dilated cardiomyopathy, heart failure, dilated left ventricle, and decreased LVEF, they have a very different genetic etiology.
In the patients with MYH7 variants, we found that there was a broad spectrum in clinical manifestation, ranging from no symptoms to severe heart failure, as reported previously.9, 23 The mechanisms by which MYH7 variants induce cardiomyopathy are still unclear. Han et al identified abnormal long noncoding RNA transcripts from the MYH7 locus that may cause cardiomyopathy.24 Fang et al found that methylation levels in the promoters of MYH7 may play an important role in regulating embryonic cardiomyocyte gene expression, morphology, and function.25
Although previous studies have reported several MYBPC3 variants in LVNC patients,9 we identified only 1 pathogenic variant in MYBPC3, in a 3‐year‐old girl. She remained asymptomatic during the 5 years of follow‐up. Hypertrophic cardiomyopathy patients with MYBPC3 mutations also present with reduced or late penetrance, often during the fifth decade of life.26 Therefore, ongoing follow‐up is warranted, even in an asymptomatic patient with LVNC. Among the other sarcomere genes, ACTC1, TNNT2,27 and TPM1 mutations are less common in LVNC than other cardiomyopathies. ACTC1 was first reported to be associated with LVNC in 2008,8 and we reported 2 TPM1 mutations, as well as 2 ACTC1 mutations, in LVNC patients in 2011.28
TAZ variants may also increase the risk for LVNC, and survival analysis showed worse prognosis in patients with these variants. TAZ was identified in 1996 as the causative gene for Barth syndrome,29 and LVNC is frequently described in patients with Barth syndrome.30, 31, 32 However, half of the patients with TAZ variants identified in this study did not show any other manifestations of Barth syndrome. Consequently, male infants with severe heart failure should be considered for genetic analysis, including TAZ, even if they do not show any signs of Barth syndrome. In an animal model, tafazzin deficiency leads to ventricular noncompaction and early lethality.33 Wang et al used induced pluripotent stem cell–derived cardiomyocytes and elucidated that TAZ deficiency in Barth syndrome impairs sarcomere assembly and contractile stress generation. TAZ deficiency may increase reactive oxygen species production, which may cause features of Barth syndrome.34
Among channelopathy‐related genes, this is the first report of an ANK2 variant in LVNC. ANK2 variants have previously been associated with cardiac arrhythmia syndrome or long QT syndrome and were recently found in hypertrophic cardiomyopathy patients.35 Although none of our patients who carried variants in arrhythmia‐associated genes presented with severe arrhythmias, given the high risk of arrhythmia associated with these genes, close monitoring and consideration of ICD implantation to prevent sudden cardiac death is recommended.36
The variant in BMPR1A is also the first reported in a patient with LVNC. BMPs (Bone morphogenetic proteins) are members of the transforming growth factor family that play critical roles in cardiac development. BMP signaling is required in the myocardium of the atrioventricular canal for proper atrioventricular junction development, and an anomaly in BMPR1A‐mediated signaling may contribute to the development of cardiac hypertrophy and embryonic heart failure.37, 38, 39 In our study, the patient who carried both MYH7 and BMPR1A variants presented with bradycardia as a fetus and died of heart failure at 1 year of age. Although most patients with a single variant of MYH7 did not develop severe manifestations, the BMPR1A variant may act as genetic modifier and contribute to fetal heart failure. Functional studies of the BMPR1A variant are now under way in animal models.
The variant in TBX5 also represents the first in this gene in a patient with LVNC, as shown in the present study. Both TBX5 and TBX20 of the T‐box family are important for maintenance of mature cardiomyocyte function.40, 41 Kodo et al showed that proper activation of TGF‐β (transforming growth factor β) signaling in the embryonic heart is required to ensure compact layer remodeling. They used patient‐specific induced pluripotent stem cell–derived cardiomyocytes generated from an LVNC patient who carried a TBX20 mutation and found abnormal TGF‐β signaling.41 Functional studies of the TBX5 mutation are also under way in animal models.
The focused NGS strategy allows for rapid molecular diagnosis at a reasonable cost. In this study, we implemented strict pathogenic variant identification criteria that could prevent misinterpretation of the variants.42 We found that patients with pathogenic variants showed high morbidity and mortality. Furthermore, patients with double heterozygous variants presented with severe phenotypes during the fetal or neonatal periods and had very poor prognosis, as reported previously.43 The role of double variants in determining the severity of disease remains unknown and cannot be evaluated using in silico predictive algorithms at the present time. Our study suggests that comprehensive screening of multiple disease‐causing genes is necessary to identify high‐risk patients with LVNC, for whom earlier treatment strategies toward HT or ICD implantation should be considered.
Limitations
In this study, some parental samples were not available, limiting segregation analysis and the ability to determine whether variants were inherited or arose de novo; none of these patients reported family history, and the parents were healthy and without evidence of cardiomyopathy by ECG and echocardiography. In addition, we chose NGS panels of genes known to be associated with cardiac phenotypes or development; therefore, variants in novel genes would have been missed. Our sequencing approach lacked of ability to assess copy number and structural variants. Whole‐exome or ‐genome sequencing in this cohort might have uncovered additional variants, including copy number variations and structural variants, but at considerably higher cost. Genetic analysis using NGS is considered to have some limitations. Recent studies showed extended genetic noise (false positive), particularly within cardiac disease–associated genes, even if these variants were rare. Guidelines recommend that several in silico analyses be used to evaluate variants without familial and/or experimental evidence of pathogenicity because most algorithms used for missense variant prediction are only 65–80% accurate for known disease variants.12, 44 Further research will be focus on the mechanism presented in animal models and analysis of induced pluripotent stem cells developed from patients with known gene variants to identify the mechanisms that underlie the abnormal development of the failed compacted layer during the embryonic period.
Conclusion
A focused NGS approach revealed a wide and distinct spectrum of gene variants in a large cohort of patients with LVNC. Patients with pathogenic variants showed early age at onset and decreased LVEF. The identification of a pathogenic variant was an independent risk factor for death, HT, or ICD insertion. Survival analysis showed poorer prognosis in the patients with pathogenic variants, especially patients with multiple or TAZ variants. Our study suggests that comprehensive screening of multiple disease‐causing genes is necessary to identify high‐risk patients with LVNC, for whom earlier treatment strategies toward HT or ICD implantation should be considered.
Sources of Funding
This study was partially supported by the Ministry of Education, Culture, Sports, Science and Technology in Japan (Research Project Number: 15K09685, 24591571, and 17591072) and by a Japan Heart Foundation Research Grant on Dilated Cardiomyopathy awarded to Fukiko Ichida.
Disclosures
None.
Supporting information
Appendix S1. Left Ventricular Noncompaction Study Collaborators.
Table S1. List of 73 Analyzed Genes of Next‐Generation Sequencing
Table S2. Silico Predictive Algorithms Used in the Study
Table S3. Frequency of MYH7 and MYBPC3 in Patients With Left Ventricular Noncompaction, Hypertrophic Cardiomyopathy, or Dilated Cardiomyopathy
Table S4. Novel Mutations Absent from Exome Aggregation Consortium Database and Human Genetic Variation Database
Table S5. Gene Collapsing Test of Rare Variants
Table S6. Specific Variants Found in Participants With Systolic Dysfunction Versus Those Without Dysfunction
Table S7. Details for Each Participant
Acknowledgments
The authors are grateful to Professor Yuichi Adachi for the steadfast counsel and guidance. The authors gratefully acknowledge all left ventricular noncompaction study collaborators.
(J Am Heart Assoc. 2017;6:e006210 DOI: 10.1161/JAHA.117.006210.)28855170
Contributor Information
Xianyi Yu, Email: yuxy@sj-hospital.org.
Fukiko Ichida, Email: fukiko@med.u-toyama.ac.jp.
References
- 1. Chin TK, Perloff JK, Williams RG, Jue K, Mohrmann R. Isolated noncompaction of left ventricular myocardium. A study of eight cases. Circulation. 1990;82:507–513. [DOI] [PubMed] [Google Scholar]
- 2. Maron BJ, Towbin JA, Thiene G, Antzelevitch C, Corrado D, Arnett D, Moss AJ, Seidman CE, Young JB; American Heart Association; Council on Clinical Cardiology, Heart Failure and Transplantation Committee; Quality of Care and Outcomes Research and Functional Genomics and Translational Biology Interdisciplinary Working Groups; Council on Epidemiology and Prevention . Contemporary definitions and classification of the cardiomyopathies: an American Heart Association Scientific Statement from the Council on Clinical Cardiology, Heart Failure and Transplantation Committee; Quality of Care and Outcomes Research and Functional Genomics and Translational Biology Interdisciplinary Working Groups; and Council on Epidemiology and Prevention. Circulation. 2006;113:1807–1816. [DOI] [PubMed] [Google Scholar]
- 3. Ichida F. Left ventricular noncompaction. Circ J. 2009;73:19–26. [DOI] [PubMed] [Google Scholar]
- 4. Weiford BC, Subbarao VD, Mulhern KM. Noncompaction of the ventricular myocardium. Circulation. 2004;109:2965–2971. [DOI] [PubMed] [Google Scholar]
- 5. Kawel N, Nacif M, Arai AE, Gomes AS, Hundley WG, Johnson WC, Prince MR, Stacey RB, Lima JA, Bluemke DA. Trabeculated (noncompacted) and compact myocardium in adults: the multi‐ethnic study of atherosclerosis. Circ Cardiovasc Imaging. 2012;5:357–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Arbustini E, Weidemann F, Hall JL. Left ventricular noncompaction: a distinct cardiomyopathy or a trait shared by different cardiac diseases? J Am Coll Cardiol. 2014;64:1840–1850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Weir‐McCall JR, Yeap PM, Papagiorcopulo C, Fitzgerald K, Gandy SJ, Lambert M, Belch JJ, Cavin I, Littleford R, Macfarlane JA, Matthew SZ, Nicholas RS, Struthers AD, Sullivan F, Waugh SA, White RD, Houston JG. Left ventricular noncompaction: anatomical phenotype or distinct cardiomyopathy? J Am Coll Cardiol. 2016;68:2157–2165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Klaassen S, Probst S, Oechslin E, Gerull B, Krings G, Schuler P, Greutmann M, Hurlimann D, Yegitbasi M, Pons L, Gramlich M, Drenckhahn JD, Heuser A, Berger F, Jenni R, Thierfelder L. Mutations in sarcomere protein genes in left ventricular noncompaction. Circulation. 2008;117:2893–2901. [DOI] [PubMed] [Google Scholar]
- 9. Probst S, Oechslin E, Schuler P, Greutmann M, Boyé P, Knirsch W, Berger F, Thierfelder L, Jenni R, Klaassen S. Sarcomere gene mutations in isolated left ventricular noncompaction cardiomyopathy do not predict clinical phenotype. Circ Cardiovasc Genet. 2011;4:367–374. [DOI] [PubMed] [Google Scholar]
- 10. O'Connor TD, Kiezun A, Bamshad M, Rich SS, Smith JD, Turner E, NHLBIGO Exome Sequencing Project; ESP Population Genetics, Statistical Analysis Working Group , Leal SM, Akey JM. Fine‐scale patterns of population stratification confound rare variant association tests. PLoS One. 2013;8:e65834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Pearson RD. Bias due to selection of rare variants using frequency in controls. Nat Genet. 2011;43:392–393. [DOI] [PubMed] [Google Scholar]
- 12. Hata Y, Kinoshita K, Mizumaki K, Yamaguchi Y, Hirono K, Ichida F, Takasaki A, Mori H, Nishida N. Postmortem genetic analysis of sudden unexplained death syndrome under 50 years of age: a next‐generation sequencing study. Heart Rhythm. 2016;13:1544–1551. [DOI] [PubMed] [Google Scholar]
- 13. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier‐Foster J, Grody WW, Hegde M, Lyon E, Spector E, Voelkerding K, Rehm HL. Standards and Guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Cirulli ET, Lasseigne BN, Petrovski S, Sapp PC, Dion PA, Leblond CS, Couthouis J, Lu YF, Wang Q, Krueger BJ, Ren Z, Keebler J, Han Y, Levy SE, Boone BE, Wimbish JR, Waite LL, Jones AL, Carulli JP, Day‐Williams AG, Staropoli JF, Xin WW, Chesi A, Raphael AR, McKenna‐Yasek D, Cady J, Vianney de Jong JM, Kenna KP, Smith BN, Topp S, Miller J, Gkazi A; FALS Sequencing Consortium , Al‐Chalabi A, van den Berg LH, Veldink J, Silani V, Ticozzi N, Shaw CE, Baloh RH, Appel S, Simpson E, Lagier‐Tourenne C, Pulst SM, Gibson S, Trojanowski JQ, Elman L, McCluskey L, Grossman M, Shneider NA, Chung WK, Ravits JM, Glass JD, Sims KB, Van Deerlin VM, Maniatis T, Hayes SD, Ordureau A, Swarup S, Landers J, Baas F, Allen AS, Bedlack RS, Harper JW, Gitler AD, Rouleau GA, Brown R, Harms MB, Cooper GM, Harris T, Myers RM, Goldstein DB. Exome sequencing in amyotrophic lateral sclerosis identifies risk genes and pathways. Science. 2015;347:1436–1441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Bagnall RD, Crompton DE, Petrovski S, Lam L, Cutmore C, Garry SI, Sadleir LG, Dibbens LM, Cairns A, Kivity S, Afawi Z, Regan BM, Duflou J, Berkovic SF, Scheffer IE, Semsarian C. Exome‐based analysis of cardiac arrhythmia, respiratory control, and epilepsy genes in sudden unexpected death in epilepsy. Ann Neurol. 2016;79:522–534. [DOI] [PubMed] [Google Scholar]
- 16. Firth D. Bias reduction of maximum likelihood estimates. Biometrika. 1993;80:27–38. [Google Scholar]
- 17. Hirono K, Hata Y, Ibuki K, Yoshimura N. Familial Ebstein's anomaly, left ventricular noncompaction, and ventricular septal defect associated with an MYH7 mutation. J Thorac Cardiovasc Surg. 2014;148:e223–e226. [DOI] [PubMed] [Google Scholar]
- 18. Chida A, Inai K, Sato H, Shimada E, Nishizawa T, Shimada M, Furutani M, Furutani Y, Kawamura Y, Sugimoto M, Ishihara J, Fujiwara M, Soga T, Kawana M, Fuji S, Tateno S, Kuraishi K, Kogaki S, Nishimura M, Ayusawa M, Ichida F, Yamazawa H, Matsuoka R, Nonoyama S, Nakanishi T. Prognostic predictive value of gene mutations in Japanese patients with hypertrophic cardiomyopathy. Heart Vessels. 2017;32:700–707. [DOI] [PubMed] [Google Scholar]
- 19. Richard P, Charron P, Carrier L, Ledeuil C, Cheav T, Pichereau C, Benaiche A, Isnard R, Dubourg O, Burban M, Gueffet JP, Millaire A, Desnos M, Schwartz K, Hainque B, Komajda M; EUROGENE Heart Failure Project . Hypertrophic cardiomyopathy: distribution of disease genes, spectrum of mutations, and implications for a molecular diagnosis strategy. Circulation. 2003;107:2227–2232. [DOI] [PubMed] [Google Scholar]
- 20. Van Driest SL, Vasile VC, Ommen SR, Will ML, Tajik AJ, Gersh BJ, Ackerman MJ. Myosin binding protein C mutations and compound heterozygosity in hypertrophic cardiomyopathy. J Am Coll Cardiol. 2004;44:1903–1910. [DOI] [PubMed] [Google Scholar]
- 21. Burke MA, Cook SA, Seidman JG, Seidman CE. Clinical and mechanistic insights into the genetics of cardiomyopathy. J Am Coll Cardiol. 2016;68:2871–2886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Akinrinade O, Ollila L, Vattulainen S, Tallila J, Gentile M, Salmenperä P, Koillinen H, Kaartinen M, Nieminen MS, Myllykangas S, Alastalo TP, Koskenvuo JW, Heliö T. Genetics and genotype‐phenotype correlations in Finnish patients with dilated cardiomyopathy. Eur Heart J. 2015;36:2327–2337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Fiorillo C, Astrea G, Savarese M, Cassandrini D, Brisca G, Trucco F, Pedemonte M, Trovato R, Ruggiero L, Vercelli L, D'Amico A, Tasca G, Pane M, Fanin M, Bello L, Broda P, Musumeci O, Rodolico C, Messina S, Vita GL, Sframeli M, Gibertini S, Morandi L, Mora M, Maggi L, Petrucci A, Massa R, Grandis M, Toscano A, Pegoraro E, Mercuri E, Bertini E, Mongini T, Santoro L, Nigro V, Minetti C, Santorelli FM, Bruno C; Italian Network on Congenital Myopathies . MYH7‐related myopathies: clinical, histopathological and imaging findings in a cohort of Italian patients. Orphanet J Rare Dis. 2016;7:11–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Han P, Li W, Lin CH, Yang J, Shang C, Nurnberg ST, Jin KK, Xu W, Lin CY, Lin CJ, Xiong Y, Chien HC, Zhou B, Ashley E, Bernstein D, Chen PS, Chen HS, Quertermous T, Chang CP. A long noncoding RNA protects the heart from pathological hypertrophy. Nature. 2014;514:102–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Fang X, Poulsen RR, Wang‐Hu J, Shi O, Calvo NS, Simmons CS, Rivkees SA, Wendler CC. Knockdown of DNA methyltransferase 3a alters gene expression and inhibits function of embryonic cardiomyocytes. FASEB J. 2016;30:3238–3255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Niimura H, Bachinski LL, Sangwatanaroj S, Watkins H, Chudley AE, McKenna W, Kristinsson A, Roberts R, Sole M, Maron BJ, Seidman JG, Seidman CE. Mutations in the gene for cardiac myosin‐binding protein C and late‐onset familial hypertrophic cardiomyopathy. N Engl J Med. 1998;338:1248–1257. [DOI] [PubMed] [Google Scholar]
- 27. Luedde M, Ehlermann P, Weichenhan D, Will R, Zeller R, Rupp S, Müller A, Steen H, Ivandic BT, Ulmer HE, Kern M, Katus HA, Frey N. Severe familial left ventricular non‐compaction cardiomyopathy due to a novel troponin T (TNNT2) mutation. Cardiovasc Res. 2010;86:452–460. [DOI] [PubMed] [Google Scholar]
- 28. Chang B, Nishizawa T, Furutani M, Fujiki A, Tani M, Kawaguchi M, Ibuki K, Hirono K, Taneichi H, Uese K, Onuma Y, Bowles NE, Ichida F, Inoue H, Matsuoka R, Miyawaki T; Noncompaction study collaborators . Identification of a novel TPM1 mutation in a family with left ventricular noncompaction and sudden death. Mol Genet Metab. 2011;102:200–206. [DOI] [PubMed] [Google Scholar]
- 29. Bione S, D'Adamo P, Maestrini E, Gedeon AK, Bolhuis PA, Toniolo D. A novel X‐linked gene, G4.5 is responsible for Barth syndrome. Nat Genet. 1996;12:385–389. [DOI] [PubMed] [Google Scholar]
- 30. Ichida F, Tsubata S, Bowles KR, Haneda N, Uese K, Miyawaki T, Dreyer WJ, Messina J, Li H, Bowles NE, Towbin JA. Novel gene mutations in patients with left ventricular noncompaction or Barth syndrome. Circulation. 2001;103:1256–1263. [DOI] [PubMed] [Google Scholar]
- 31. Chen R, Tsuji T, Ichida F, Bowles KR, Yu X, Watanabe S, Hirono K, Tsubata S, Hamamichi Y, Ohta J, Imai Y, Bowles NE, Miyawaki T, Towbin JA; Noncompaction study collaborators . Mutation analysis of the G4.5 gene in patients with isolated left ventricular noncompaction. Mol Genet Metab. 2002;77:319–325. [DOI] [PubMed] [Google Scholar]
- 32. Brandner K, Mick DU, Frazier AE, Taylor RD, Meisinger C, Rehling P. Taz1, an outer mitochondrial membrane protein, affects stability and assembly of inner membrane protein complexes: implications for Barth Syndrome. Mol Biol Cell. 2005;16:5202–5214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Phoon CK, Acehan D, Schlame M, Stokes DL, Edelman‐Novemsky I, Yu D, Xu Y, Viswanathan N, Ren M. Tafazzin knockdown in mice leads to a developmental cardiomyopathy with early diastolic dysfunction preceding myocardial noncompaction. J Am Heart Assoc. 2012;2:e000455 DOI: 10.1161/JAHA.113.000455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Wang G, McCain ML, Yang L, He A, Pasqualini FS, Agarwal A, Yuan H, Jiang D, Zhang D, Zangi L, Geva J, Roberts AE, Ma Q, Ding J, Chen J, Wang DZ, Li K, Wang J, Wanders RJ, Kulik W, Vaz FM, Laflamme MA, Murry CE, Chien KR, Kelley RI, Church GM, Parker KK, Pu WT. Modeling the mitochondrial cardiomyopathy of Barth syndrome with induced pluripotent stem cell and heart‐on‐chip technologies. Nat Med. 2014;20:616–623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Lopes LR, Syrris P, Guttmann OP, O'Mahony C, Tang HC, Dalageorgou C, Jenkins S, Hubank M, Monserrat L, McKenna WJ, Plagnol V, Elliott PM. Novel genotype‐phenotype associations demonstrated by high‐throughput sequencing in patients with hypertrophic cardiomyopathy. Heart. 2015;101:294–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Priori SG, Blomström‐Lundqvist C, Mazzanti A, Blom N, Borggrefe M, Camm J, Elliott PM, Fitzsimons D, Hatala R, Hindricks G, Kirchhof P, Kjeldsen K, Kuck KH, Hernandez‐Madrid A, Nikolaou N, Norekvål TM, Spaulding C, Van Veldhuisen DJ. 2015 ESC guidelines for the management of patients with ventricular arrhythmias and the prevention of sudden cardiac death. Eur Heart J. 2015;36:2793–2867. [DOI] [PubMed] [Google Scholar]
- 37. Stroud DM, Gaussin V, Burch JB, Yu C, Mishina Y, Schneider MD, Fishman GI, Morley GE. Abnormal conduction and morphology in the atrioventricular node of mice with atrioventricular canal targeted deletion of Alk3/Bmpr1a receptor. Circulation. 2007;116:2535–2543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Shahid M, Spagnolli E, Ernande L, Thoonen R, Kolodziej SA, Leyton PA, Cheng J, Tainsh RE, Mayeur C, Rhee DK, Wu MX, Scherrer‐Crosbie M, Buys ES, Zapol WM, Bloch KD, Bloch DB. PABMP type I receptor ALK2 is required for angiotensin II‐induced cardiac hypertrophy. Am J Physiol Heart Circ Physiol. 2016;310:H984–H994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Nomura‐Kitabayashi A, Phoon CK, Kishigami S, Rosenthal J, Yamauchi Y, Abe K, Yamamura K, Samtani R, Lo CW, Mishina Y. Outflow tract cushions perform a critical valve‐like function in the early embryonic heart requiring BMPRIA‐mediated signaling in cardiac neural crest. Am J Physiol Heart Circ Physiol. 2009;297:H1617–H1628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Ang YS, Rivas RN, Ribeiro AJ, Srivas R, Rivera J, Stone NR, Pratt K, Mohamed TM, Fu JD, Spencer CI, Tippens ND, Li M, Narasimha A, Radzinsky E, Moon‐Grady AJ, Yu H, Pruitt BL, Snyder MP, Srivastava D. Disease model of GATA4 mutation reveals transcription factor cooperativity in human cardiogenesis. Cell. 2016;167:1734–1749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kodo K, Ong SG, Jahanbani F, Termglinchan V, Hirono K, InanlooRahatloo K, Ebert AD, Shukla P, Abilez OJ, Churko JM, Karakikes I, Jung G, Ichida F, Wu SM, Snyder MP, Bernstein D, Wu JC. iPSC‐derived cardiomyocytes reveal abnormal TGF‐β signalling in left ventricular non‐compaction cardiomyopathy. Nat Cell Biol. 2016;18:1031–1042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Manrai AK, Funke BH, Rehm HL, Olesen MS, Maron BA, Szolovits P, Margulies DM, Loscalzo J, Kohane IS. Genetic misdiagnoses and the potential for health disparities. N Engl J Med. 2016;375:655–665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Schaefer E, Helms P, Marcellin L, Desprez P, Billaud P, Chanavat V, Rousson R, Millat G. Next generation sequencing (NGS) as a fast molecular diagnosis tool for left ventricular noncompaction in an infant with compound mutations in the MYBPC3 gene. Eur J Med Genet. 2014;57:129–132. [DOI] [PubMed] [Google Scholar]
- 44. Ackerman MJ. Genetic purgatory and the cardiac channelopathies: exposing the variants of uncertain/unknown significance issue. Heart Rhythm. 2015;12:2325–2331. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1. Left Ventricular Noncompaction Study Collaborators.
Table S1. List of 73 Analyzed Genes of Next‐Generation Sequencing
Table S2. Silico Predictive Algorithms Used in the Study
Table S3. Frequency of MYH7 and MYBPC3 in Patients With Left Ventricular Noncompaction, Hypertrophic Cardiomyopathy, or Dilated Cardiomyopathy
Table S4. Novel Mutations Absent from Exome Aggregation Consortium Database and Human Genetic Variation Database
Table S5. Gene Collapsing Test of Rare Variants
Table S6. Specific Variants Found in Participants With Systolic Dysfunction Versus Those Without Dysfunction
Table S7. Details for Each Participant