

Abstract
Background
Trimethylamine‐N‐oxide (TMAO) has recently been identified as a novel and independent risk factor for promoting atherosclerosis through inducing vascular inflammation. However, the exact mechanism is currently unclear. Studies have established a central role of nucleotide‐binding oligomerization domain–like receptor family pyrin domain–containing 3 (NLRP3) inflammasome in the pathogenesis of vascular inflammation. Here, we examined the potential role of the NLRP3 inflammasome in TMAO‐induced vascular inflammation in vitro and in vivo and the underlying mechanisms.
Methods and Results
Experiments using liquid chromatography‐tandem mass spectrometry, Western blot, and fluorescent probes showed that TMAO‐induced inflammation in human umbilical vein endothelial cells (HUVECs) and aortas from ApoE−/− mice. Moreover, TMAO promoted NLRP3 and activated caspase‐1 p20 expression and caspase‐1 activity in vitro and in vivo. Notably, a caspase‐1 inhibitor (YVAD), an NLRP3 inhibitor (MCC950), as well as NLRP3 short interfering RNA attenuated TMAO‐induced activation of the NLRP3 inflammasome, subsequently leading to suppression of inflammation in HUVECs. TMAO additionally stimulated reactive oxygen species (ROS) generation, in particular, mitochondrial ROS, while inhibiting manganese superoxide dismutase 2 (SOD2) activation and sirtuin 3 (SIRT3) expression in HUVECs and aortas from ApoE−/− mice. TMAO‐induced endothelial NLRP3 inflammasome activation was ameliorated by the mitochondrial ROS scavenger Mito‐TEMPO, or SIRT3 overexpression in HUVECs. Conversely, TMAO failed to further inhibit magnesium SOD2 and activate the NLRP3 inflammasome or induce inflammation in SIRT3 short interfering RNA–treated HUVECs and aortas from SIRT3−/− mice.
Conclusions
TMAO promoted vascular inflammation by activating the NLRP3 inflammasome, and the NLRP3 inflammasome activation in part was mediated through inhibition of the SIRT3‐SOD2–mitochondrial ROS signaling pathway.
Keywords: atherosclerosis, NOD‐like receptor family pyrin domain containing 3 inflammasome, sirtuin 3, trimethylamine‐N‐oxide, vascular inflammation
Subject Categories: Angiogenesis, Inflammation, Endothelium/Vascular Type/Nitric Oxide, Mechanisms
Clinical Perspective
What Is New?
Trimethylamine‐N‐oxide has recently been identified as a novel and independent risk factor for promoting atherosclerosis through inducing vascular inflammation; however, the exact mechanism is currently unclear.
Our results demonstrated for the first time that trimethylamine‐N‐oxide promoted vascular inflammation by activating the nucleotide‐binding oligomerization domain–like receptor family pyrin domain–containing 3 inflammasome, and that inflammasome activation was mediated in part through inhibition of the sirtuin 3–superoxide dismutase 2–mitochondrial reactive oxygen species signaling pathway.
What Are the Clinical Implications?
Our findings provide new insights to interpret the potential mechanism of the promoting effects of trimethylamine‐N‐oxide on atherosclerosis, in which the nucleotide‐binding oligomerization domain–like receptor family pyrin domain–containing 3 inflammasome may play a critical role.
These results indicate that the nucleotide‐binding oligomerization domain–like receptor family pyrin domain–containing 3 inflammasome may become an interesting target for pharmacological or dietary interventions to decrease the risk of developing cardiovascular diseases.
Introduction
The incidence of cardiovascular diseases such as atherosclerosis (AS) is increasing globally, creating a serious and expensive public health issue.1 Accumulating evidence indicates that AS is the result of a prolonged and excessive inflammatory processes in the vascular wall, which often begins with inflammatory changes in the endothelium and is characterized by the expression of adhesion molecules.2, 3 A recent metabolomics approach identified plasma trimethylamine‐N‐oxide (TMAO), a choline metabolite, as a novel and independent risk factor for promoting AS.4, 5 Previous studies suggest that TMAO partially contributes to the development of AS by regulating the major pathway of cholesterol metabolism and the bile acid synthetic pathway.4, 5, 6 More recently, TMAO has been shown to promote vascular inflammation, leading to AS.7 However, the precise mechanism by which circulating TMAO promotes vascular inflammation remains to be established.
Nucleotide‐binding oligomerization domain–like receptor family pyrin domain containing 3 (NLRP3) inflammasome is an interleukin (IL)‐1β family cytokine‐activating protein complex involved in regulation of innate immune and inflammatory responses.8 The NLRP3 inflammasome plays a key role in the pathogenesis of diverse inflammatory diseases, including AS, type 2 diabetes mellitus, and gout.9 Oxidized low‐density lipoprotein and cholesterol crystals activate the inflammasome, thereby leading to the processing and secretion of the proinflammatory cytokines IL‐1β and IL‐18 and ultimately triggering vascular inflammation via upregulation of adhesion molecules.9, 10 Xiao et al11 additionally reported that atheroprone flow induces the NLRP3 inflammasome in endothelium through serol regulatory element binding protein 2 activation. This increased innate immunity in endothelium synergizes with hyperlipidemia to cause topographical distribution of atherosclerotic lesions.11 Several studies have suggested that ablation of NLRP3 protects endothelial cells from inflammatory damage, leading to attenuation of AS.12, 13 Thus, the NLRP3 inflammasome is proposed to be a key player in the pathogenesis of vascular inflammation. In 2016, Seldin and co‐workers7 showed that TMAO promotes vascular inflammation through signaling of mitogen‐activated protein kinase and nuclear factor‐κB (NF‐κB). However, limited information is available regarding the potential contribution of the NLRP3 inflammasome to TMAO‐induced vascular inflammation and the underlying mechanisms.
Following decades of research, the most well‐characterized mechanism underlying activation of the NLRP3 inflammasome is reactive oxygen species (ROS), particularly mitochondrial ROS (mtROS), generation. The majority of NLRP3 activators increase mtROS levels, and chemical scavengers of mtROS effectively inhibit NLRP3 activation.14, 15 Sirtuin 3 (SIRT3), an important member of the Sir2 family, has been identified as the primary mitochondrial acetylysine deacetylase that modulates various proteins to control mitochondrial function and mtROS homeostasis.16 SIRT3 primarily regulates mtROS levels by altering acetylation of the major mitochondrial antioxidant enzymes, including manganese superoxide dismutase 2 (SOD2), isocitrate dehydrogenase 2 and glutathione peroxidase.17 More importantly, SIRT3 can directly bind and deacetylate SOD2, leading to increased SOD2 activity and subsequently exerting significant effects on mtROS homeostasis and NLRP3 inflammasome activation.18, 19, 20 Thus, in the current study we intended to investigate the role of the NLRP3 inflammasome in TMAO‐induced vascular inflammation as well as the potential involvement of the SIRT3‐SOD2‐mtROS signaling pathway in vitro in cultured human umbilical vein endothelial cells (HUVECs) and in vivo in aortas from 129/SvJ, ApoE−/− and SIRT3−/− mice.
Our results demonstrated for the first time that TMAO promoted vascular inflammation by activating the NLRP3 inflammasome, and the NLRP3 inflammasome activation was mediated in part through inhibition of the SIRT3‐SOD2‐mtROS signaling pathway.
Materials and Methods
Reagents and Antibodies
Cell culture medium HyQ M199/EBSS (M199; SH30351.01) and fetal bovine serum (SH30370.03) were purchased from Hyclone Laboratories (Erie, UK); choline (C7017), TMAO (317594), and mito‐TEMPO (TEMPO, SML0737) from Sigma‐Aldrich (St. Louis, MO); zYVAD‐fmk (YVAD, sc‐3071) from Santa Cruz Biotechnology (Dallas, TX); MCC950 (HY‐12815) from MedChem Express (Monmouth Junction, NJ); and the Cell Counting Kit (CCK‐8; CK04) from Dojindo Laboratories (Rockville, MD). DCFH‐DA, caspase‐1 activity kit (C1101), and SOD activity kit (S0103) were obtained from the Beyotime Institute of Biotechnology (Jiangsu, China). MitoSOX™ Red (M36008) and Lipofectamine™ 2000 transfection reagent (11668‐019) were acquired from Invitrogen (Carlsbad, CA). The SIRT3 overexpression plasmid (13814) was purchased from Addgene (Cambridge, MA). Caspase‐1 antibody (NBP1‐45433) was obtained from NOVUS Biologicals (Littleton, CO), whereas antibodies against NLRP3 (15101), IL‐1β (12703), and SIRT3 (5490) were from Cell Signaling Technology, Inc (Danvers, MA). The antibody against SOD2 (sc‐33254) was from Santa Cruz Biotechnology (Dallas, TX), and those against Ac‐SOD2 (Ab137037) were from Abcam (Cambridge, UK). Antibodies against ACTB/‐actin (TA‐09), intercellular adhesion molecule 1 (ICAM‐1, ZS‐8439), and matrix metallopeptidase 9 (MMP‐9, zs‐6840) were obtained from Zhongshan Jinqiao Biotechnology Co (Beijing, China).
Cell Culture and Treatment
HUVECs were isolated from umbilical cord vein provided by XinQiao Hospital (Chongqing, China) as described previously21 and cultured on gelatin‐coated plastic dishes (Dibco Biocult, Uxbridge Waltham, MA, 1‐50350) in a humidified atmosphere in a 5% CO2 incubator at 37°C with M199 medium supplemented with 10% fetal bovine serum.
All experiments were performed on cells following 3 to 6 passages at 80% to 90% confluence. Cells were treated with different concentrations of TMAO (0, 150, 300, 600, and 900 μmol/L) for 24 hours or a fixed concentration of TMAO (600 μmol/L) for different time periods (0, 4, 12, and 24 hours). Where indicated, cells were treated with YVAD (10 μmol/L), MCC950 (10 μmol/L), or TEMPO (50 μmol/L) for 2 hours following the addition of TMAO (600 μmol/L) for another 24 hours. The ethics review board at the Third Military Medical University approved this study, and informed consent was obtained from all patients before participation.
Animals and Treatment
Eight‐week‐old female 129/SvJ (wild‐type, WT), SIRT3−/− and ApoE−/− mice were purchased from Jackson Laboratory (Bar Harbor, ME). Animals were maintained on a standard chow diet (NIH31 modified mouse/rat diet; Harlan Teklad/Envigo, Hyderabad, India) or a chow diet supplemented with 1% choline for 4 months. Mice were kept under controlled temperature conditions (22±2°C) under a 12‐hour light/dark period with ad libitum access to water. Pentobarbital sodium anesthesia (50 mg/kg) was administered before surgical procedures, with maximal efforts to minimize suffering of animals. At the end of the treatment period, mice were fasted for 4 hours before collection of blood and tissues for analysis. Animals were euthanized and killed by cervical dislocation followed by decapitation. Blood and aorta samples were collected immediately, snap‐frozen in liquid nitrogen, and stored at −80°C until use. Animal experiments were carried out in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals by the National Institutes of Health and approved by the Animal Care and Use Committee of the Third Military Medical University (Chongqing, China; Approval SYXC‐2014‐00112).
Cell Viability Measurements
The CCK‐8 detection kit was used to measure cell viability as described previously.21 Briefly, HUVECs were seeded in a 96‐well microplate (Corning Life Sciences, Corning, NY; 3650) at a density of 8000 cells/well and treated with a series of concentrations of TMAO for 24 hours (0, 50, 100, 200, 300, 400, 600, 800, 1000, and 2000 μmol/L) or a fixed concentration (600 μmol/L) for the indicated time periods (0, 4, 8, 12, 24, 36, and 72 hours). Subsequently, CCK‐8 solution (20 μL/well) was added to the wells, and the plate was incubated at 37°C for 2 hours. Viable cells were counted by absorbance measurements with a monochromator microplate reader (Safire II; Tecan Group Ltd, Männedorf, Switzerland) at a wavelength of 450 nm. The optical density value at 450 nm was assessed as the percentage of cell viability in relation to the control group (set as 100%).
Monocyte Adhesion and Transmigration Assays
Monocyte adhesion to endothelial cells was measured using fluorescently labeled monocytic THP‐1 cells as described before.22 Briefly, the THP‐1 cells were labeled with Cell Tracker CM‐Dil (ThermoFisher Scientific, Waltham, MA) according to the manufacturer's protocol; the cells were washed 3 times with phosphate‐buffered saline and resuspended in RPMI medium at a concentration of 2×106 cells/mL. HUVECs (3×105 per 100‐mm dish) were seeded into 24‐well plates. After 24 hours, fluorescently labeled THP‐1 cells were added to wells containing a confluent endothelial monolayer and incubated at 37°C for 30 minutes; then the cocultured wells were washed twice with phosphate‐buffered saline in order to eliminate the nonadherent cells. Adherent THP‐1 cells were visualized via fluorescence microscopy and were counted in the high power field (×40 phase‐contrast objective) in 3 separate fields in each well. The average number of adherent monocytes was calculated in the 3 fields for each set of 3 wells, and the data were represented as percentage of control.
Quantitation of Plasma TMAO Levels
Based on our published results,23 75 μL of 80% acetonitrile was added to 25 μL serum for precipitation of proteins. As the internal standard, d9‐(trimethyl) TMAO was added to plasma samples for its respective native compounds. After 30 minutes, samples were centrifuged (14 000g, 4°C, 30 minutes) and analyzed via liquid chromatography‐tandem mass spectrometry using an Agilent 6410 Series Triple Quadrupole mass spectrometer (Agilent Technologies, Wilmington, DE) equipped with an electrospray ionization source. The capillary voltage was set at +4000 V, and it was heated to 350°C. TMAO was monitored in the multiple‐reaction–monitoring mode using characteristic precursor‐product ion transition m/z 76→58. An Agilent Technologies (Santa Clara, CA) 1260 high performance liquid chromatography system was equipped with a G1322A vacuum degasser, a G1312B binary pump, a G1316B column oven, and a G1367D autosampler. Chromatographic separation was performed on an XBridge™ HILIC column (150×2.1 mm, internal diameter of 3.5 μm; Waters, Milford, MA) protected by a flex capillary XBridge™ HILIC guard column (10×2.1 mm, internal diameter of 3.5 μm; Waters). A mobile phase containing methanol with 0.1% formic acid (phase A) and water with 0.1% formic acid (phase B) was used at a ratio of 32:68 (phase A: phase B) with a flow rate of 0.25 mL/min. Various concentrations of TMAO standards were added to control plasma to generate a calibration curve that allowed quantification of plasma TMAO levels.
Western Blot Analysis
Cells and aorta tissues were collected, lysed, and subjected to Western blot as described previously.21 Briefly, proteins were resolved via 10% to 15% sodium dodecyl sulfate polyacrylamide gel electrophoresis and transferred to polyvinylidene difluoride membranes. Following incubation with Tris‐buffered saline containing 0.1% Tween‐20 with 5% skimmed milk, blots were probed with primary antibodies at dilutions of 1:200 and 1:1000 overnight at 4°C, followed by horseradish peroxidase–conjugated secondary antibodies (Thermo Scientific Lab Vision, Waltham, MA; 31340 and 31455) for 24 hours. Protein bands were visualized using the enhanced chemiluminescence system, and densitometric analysis was performed using Scion Image‐Release Beta 4.02 software (Scion Software, Frederick, MD) (http://scion-corporation.software.informer.com/).
Measurement of Cellular Caspase‐1 Activity
Cells in the logarithmic growth phase were seeded in a 6‐well microplate with 3 replicate wells for each condition. After various treatments, cells were collected, and cellular caspase‐1 activity assayed using the caspase‐1 assay kit according to the manufacturer's instructions. Results were calculated as the enzyme activity unit of caspase‐1 contained per unit weight protein.
siRNA Assay
siRNAs for NLRP3 (human, sc‐45469) and SIRT3 (human, sc‐61555) were purchased from Santa Cruz Biotechnology (Dallas, TX), along with control siRNA (sc‐44230) and siRNA Transfection Reagent (sc‐29528). Endothelial cells were transfected with 100 nmol/L siRNA for 5 to 7 hours, in keeping with the manufacturer's protocol. Then, the cells were switched into M199 medium and incubated for an additional 24 hours. Where indicated, cells were treated with TMAO (600 μmol/L) for 24 hours and thereafter harvested and subjected to Western blot and other analyses.
Plasmids and Transfection
Endothelial cells were transfected with plasmids expressing SIRT3 or control plasmids using Lipofectamine 2000 according to the manufacturer's instructions (Invitrogen, Carlsbad, CA; 11668‐019). After 24 hours, cells were washed and processed for immunoblotting and other indicated treatments.
ROS Assessment
Total ROS and mtROS levels were determined using DCFH‐DA (Beyotime Institute of Biotechnology, Jiangsu, China) or MitoSOX™ Red (Invitrogen) according to the manufacturer's instructions. HUVECs were added to a 96‐well microplate at a concentration of 8000 cells/well and treated as indicated. Next, cells were loaded with DCFH‐DA (10 μmol/L) or MitoSOX reagents (5 μmol/L) at 37°C for 30 minutes in the dark and washed gently 3 times with warm phosphate buffer saline. Fluorescence intensities of total ROS and mtROS were determined using an Infinite™ M200 Microplate Reader (Tecan Group Ltd, Männedorf, Switzerland).
Measurement of SOD2 Enzyme Activity
SOD2 enzymatic activity was assayed using a SOD1 and SOD2 Assay Kit with WST‐8 (Beyotime Institute of Biotechnology, S0103) following the manufacturer's instructions. One unit of SOD was defined as the amount of enzyme that inhibits the rate of nitro blue tetrazolium reduction observed in a blank sample by 50%. SOD isoforms were determined by adding SOD1 inhibitors A and B (to detect SOD2). Absorption at 450 nm was measured using an Infinite™ M200 Microplate Reader (Tecan Group).
Ultrasound Detection of Aortic Root Lesions
A high‐frequency ultrasound system (Vevo 2100, Visualsonics, Toronto, Canada) equipped with a linear array transducer (MS 550D, 22‐55 MHz) was used to detect atherosclerotic lesions at the aortic sinus as described previously.24 Briefly, ApoE−/− mice were anesthetized with an intraperitoneal injection of 50 mg/kg pentobarbital sodium (1% in normal saline). Mice were placed on a heated procedural board, and their limbs were taped to ECG electrodes coated with electrode cream. A rectal thermometer was inserted to assist with maintaining normothermia (37°C internal temperature). Fur at the imaging location was shaved and warm ultrasound gel liberally applied to ensure optimal image quality. The aortic sinus was imaged and visualized in a long‐axis view. A cine loop of 100 frames was stored for later off‐line analysis. The time‐gain compensation curve was adjusted to produce a uniform intensity of echoes. The gain was set to 30 dB, and the dynamic range to 65 dB. To reduce variability, image parameters remained constant throughout the experiment. All examinations were performed by an experienced operator, and all measurements were repeated 3 times at the same site. A total of 60 aortic sinus regions of interest from 20 ApoE−/− mice (n=10 per group) were analyzed.
Quantitation of AS
Atherosclerotic lesions were quantified by en face analysis of the aorta (including aortic arch, thoracic, and abdominal regions) and cross‐sectional analysis of the aortic root as reported previously.25 For en face preparations, the aorta was opened longitudinally and stained with Oil‐red O (Sigma‐Aldrich) to detect lipids and determine the lesion area. En face images of the aorta were obtained with a Canon EOS 7D digital camera (Canon, Tokyo, Japan) and analyzed using ImagePro Plus (Media Cybernetics, Rockville, MD). Atherosclerotic lesions of the aorta were expressed as a percentage of total surface area. To measure atherosclerotic lesions at the aortic sinus, the upper sections of hearts were embedded in OCT compound (Sigma‐Aldrich) and frozen at −20°C. Sections (10 μm thickness) were collected, beginning at the aortic root and extending for 400 μm as described earlier.26 Lesions from 10 alternating sections were stained with Oil‐Red‐O and hematoxylin and quantified using Optimas Image Analysis software (Bioscan Inc, Edmonds, WA).
Statistical Analysis
Quantitative data are presented as means±SE of 3 experiments. Statistical analysis was conducted with the t test and 1‐way analysis of variance using SPSS 13.0 statistical software (SPSS Inc, Chicago, IL). Data were considered significant at a P<0.05, and the Tukey‐Kramer post hoc test was applied at this value.
Results
TMAO Induced Inflammation in Endothelial Cells
To investigate the cytotoxicity of TMAO to endothelial cells, HUVECs were treated with various concentrations of TMAO (50, 100, 200, 300, 400, 600, 800, 1000, and 2000 μmol/L) or a fixed dose of TMAO (600 μmol/L) at different time points (4, 8, 12, 24, 36, 48, and 72 hours), following which cell viability was determined. As shown in Figure 1A and 1B, TMAO was tolerated by endothelial cells and exerted no significant effects on cell viability but induced a significant increase in IL‐1β, ICAM‐1, and MMP‐9 expression in endothelial cells (Figure 1C through 1F). Moreover, treatment with TMAO (600 μmol/L) for 24 hours also caused a significant increase in the adhesion of monocytes to endothelial cells (Figure S1A and S1B). These results indicated that TMAO triggered inflammation with no significant effects on cell viability in endothelial cells.
Figure 1.
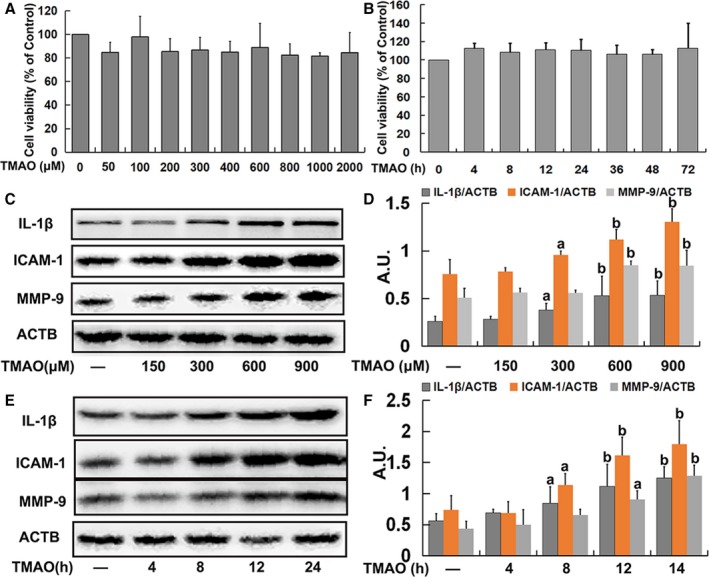
Trimethylamine‐N‐oxide (TMAO)‐induced inflammation in endothelial cells. A, Human umbilical vein endothelial cells (HUVECs) were incubated with different concentrations of TMAO (50, 100, 200, 300, 400, 600, 800, 1000, and 2000 μmol/L) for 24 hours. Thereafter, cell viability was determined. B, Cells were treated with TMAO (600 μmol/L) for different time‐periods (4, 8, 12, 24, 36, 48, and 72 hours), and cell viability was detected. C, Cells were treated as described in A, and the expression of IL‐1β, ICAM‐1, and MMP‐9 was detected via Western blot. D, Bar charts show the quantification of the indicated proteins. E, Cells were treated as described in B, and the expression of IL‐1β, ICAM‐1, and MMP‐9 was analyzed via Western blot. F, Bar graphs show the quantification of the indicated proteins. Values are presented as means±SE (n=3); a P<0.05, b P<0.01 vs the vehicle‐treated control group; AU indicates arbitrary units; ICAM, intercellular adhesion molecule; IL, interleukin; MMP, matrix metallopeptidase.
TMAO Induced Inflammation via NLRP3 Inflammasome Activation in Endothelial Cells
To determine whether the NLRP3 inflammasome was activated by TMAO in HUVECs, we investigated the expression of NLRP3 and activated caspase‐1 (p20), which are considered accurate indicators of inflammasome activation.27 TMAO promoted NLRP3 and caspase‐1 p20 expression in a concentration‐ and time‐dependent manner (Figure 2A through 2D). As shown in Figure 2E and 2F, TMAO additionally induced a marked increase in caspase‐1 activity, indicating activation of the NLRP3 inflammasome. The collective results suggested that TMAO treatment promoted cellular NLRP3 inflammasome activation in HUVECs.
Figure 2.
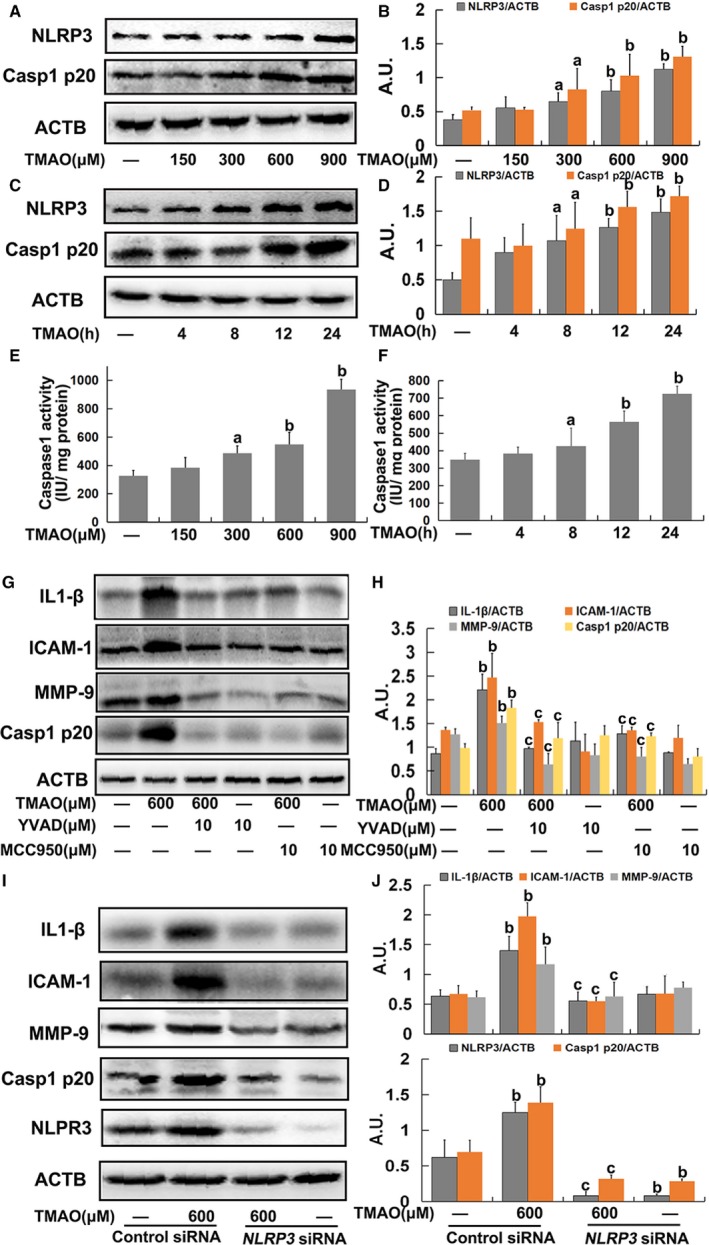
Trimethylamine‐N‐oxide (TMAO)‐induced inflammation via nucleotide‐binding oligomerization domain–like receptor family pyrin domain–containing 3 (NLRP3) inflammasome activation in endothelial cells. A, Cells were treated with TMAO at a series of concentrations (150, 300, 600, and 900 μmol/L) for 24 hours, and the expression of NLRP3 and caspase‐1 p20 was detected via Western blot. B, Bar charts showing quantification of endogenous NLRP3 and caspase‐1 p20. C, Cells were incubated with 600 μmol/L TMAO for different time intervals (4, 8, 12, and 24 hours), and the expression of NLRP3 and caspase‐1 p20 was detected via Western blot. D, Bar charts showing quantification of endogenous NLRP3 and caspase‐1 p20. Cells were treated as described for A and B. Thereafter, caspase‐1 activity was measured using caspase‐1 activity kits (E and F). G, Cells were pretreated with YVAD (10 μmol/L) or MCC950 (10 μmol/L) for 2 hours, and then exposed to TMAO (600 μmol/L) for a further 24 hours. Expression of IL‐1β, ICAM‐1, MMP‐9, and caspase‐1 p20 was detected via Western blot. H, Bar charts showing quantification of the indicated proteins. I, Human umbilical vein endothelial cells (HUVECs) were transfected with NLRP3 siRNA as described in Materials and Methods. After 24 hours, cells were incubated with TMAO (600 μmol/L) for 24 hours, and the expression of IL‐1β, ICAM‐1, MMP‐9 and Casp1 p20 was detected via Western blot. J, Bar charts showing quantification of indicated proteins. Values are expressed as means±SE (n=3). a P<0.05, b P<0.01 vs vehicle‐treated control group; c P<0.01 vs TMAO‐treated group; AU indicates arbitrary units; ICAM, intercellular adhesion molecule; IL, interleukin; MMP, matrix metallopeptidase.
To further confirm the role of the NLRP3 inflammasome in TMAO‐induced inflammation in endothelial cells, an NLRP3 inhibitor (MCC950) and caspase‐1 inhibitor (YVAD) were used. Pretreatment with MCC950 (10 μmol/L) or YVAD (10 μmol/L) markedly inhibited TMAO‐induced NLRP3 and caspase‐1 p20 expression, followed by decreased expression of IL‐1β, ICAM‐1, and MMP9 in endothelial cells (Figure 2G and 2H). Furthermore, the TMAO‐induced increases in NLRP3, caspase‐1 p20, IL‐1β, ICAM‐1, and MMP9 expression were blocked on suppression of NLRP3 via transfection with specific siRNA (Figure 2I and 2J). Additionally, pretreatment with MCC950 (10 μmol/L) or YVAD (10 μmol/L) also attenuated the increased adhesion of monocytes to endothelial cells induced by TMAO (Supplementary Figure S1A and S1B). Based on these findings, we postulated that the NLRP3 inflammasome was required for TMAO‐induced inflammation in endothelial cells.
mtROS Played a Key Role in TMAO‐Induced Activation of the NLRP3 Inflammasome in Endothelial Cells
We further explored the mechanism underlying TMAO‐mediated NLRP3 inflammasome activation in endothelial cells. ROS generation is one of the known mechanisms through which the NLRP3 inflammasome is activated, of which mtROS formation is particularly critical for NLRP3 activation.14 Accordingly, the relationship between NLRP3 inflammasome induction and ROS generation, especially mtROS, in endothelial cells was investigated. Total ROS levels measured based on DCFH‐DA and mtROS levels were evaluated using MitoSOX™ Red, a highly selective fluorescent probe for detection of generated within mitochondria.28 Our results showed an increase in both ROS and mtROS levels in response to TMAO in a dose‐ and time‐dependent manner. Time kinetic analysis revealed that ROS and mtROS reached maximum levels at 24 hours after TMAO treatment (Figure 3A through 3D). The specific mtROS scavenger, TEMPO, was further employed to confirm the contributory role of mtROS. We found that pretreatment with TEMPO (50 μmol/L) markedly inhibited TMAO‐induced activation of the NLRP3 inflammasome followed by decreased expression of IL‐1β, ICAM‐1, and MMP‐9 (Figure 3E and 3F) as well as the adhesion of monocytes to endothelial cells (Supplementary Figure S1A and S1B), validating the requirement of mtROS for this process.
Figure 3.
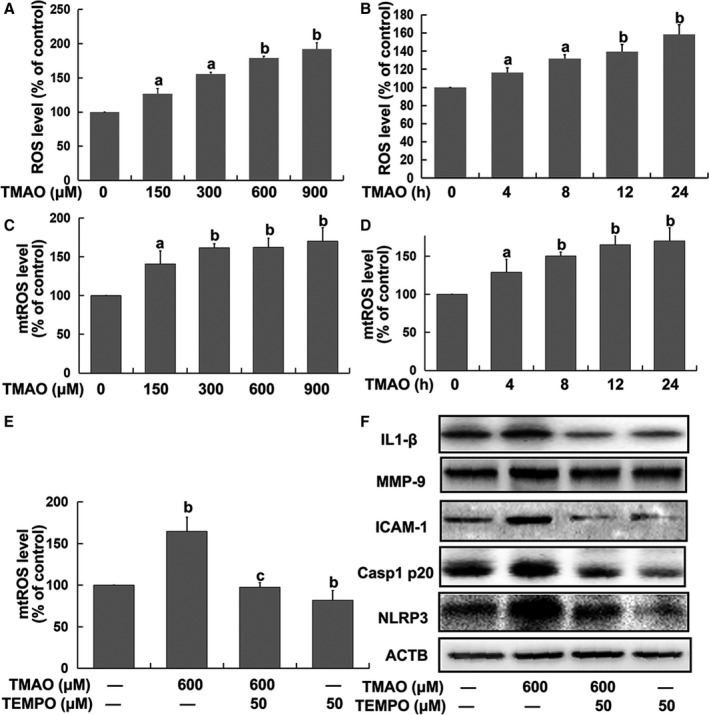
Mitochondrial reactive oxygen species (mtROS) played a key role in trimethylamine‐N‐oxide (TMAO)‐induced activation of the nucleotide‐binding oligomerization domain–like receptor family pyrin domain–containing 3 (NLRP3) inflammasome in endothelial cells. Cells were treated with TMAO at a series of concentrations (150, 300, 600, and 900 μmol/L) for 24 hours or 600 μmol/L TMAO for the indicated time intervals (4, 8, 12, and 24 hours). A and B, Total ROS levels were detected via DCFH‐DA. C and D, mtROS levels were detected with MitoSOX ™ Red. Human umbilical vein endothelial cells (HUVECs) were pretreated with TEMPO (50 μmol/L) for 2 hours followed by the addition of TMAO (600 μmol/L) for a further 24 hours. E, mtROS and (F) expression of the indicated protein were detected. Values are expressed as means±SE (n=3). a P<0.05, b P<0.01 vs the vehicle‐treated control group; c P<0.01 vs TMAO‐treated group; AU, arbitrary units.
TMAO Induced mtROS Accumulation via the SIRT3‐SOD2 Pathway in Endothelial Cells
In view of the finding that SOD2, the primary mitochondrial oxidative scavenger, plays a crucial role in regulation of mtROS,29 the effects of TMAO on SOD2 expression and activity were investigated. TMAO induced significant suppression of SOD2 activity in a dose‐ and time‐dependent manner (Figure 4A and 4B). SOD2 activity is known to be tightly regulated via acetylation at its lysine residues.20 Accordingly, we measured SOD2 acetylation levels using Western blot with an anti‐acetyl‐lysine antibody. As shown in Figure 4C through 4F, acetylation levels of SOD2 were markedly increased by TMAO in a dose‐ and time‐dependent manner. Furthermore, because SOD2 is mainly regulated by the deacetylation of specific conserved lysines in a reaction catalyzed by the mitochondrial sirtuin SIRT3,20 we investigated the effects of TMAO on SIRT3 expression. As expected, TMAO treatment led to a significant decrease in SIRT3 expression (Figure 4G through 4J), revealing a suppressive effect on both expression of SIRT3 and activity of SOD2.
Figure 4.
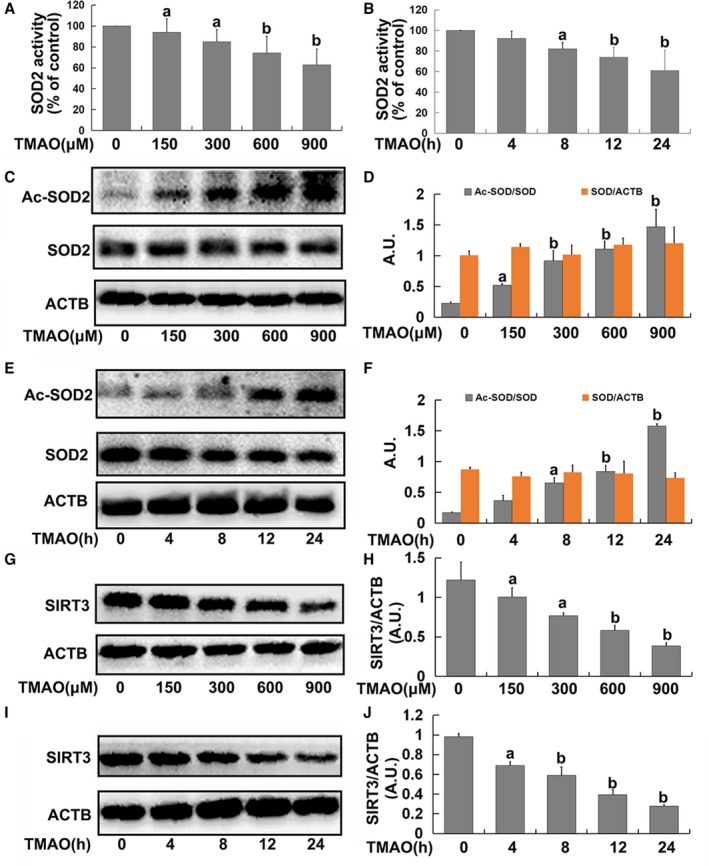
Trimethylamine‐N‐oxide (TMAO) exposure decreased superoxide dismutase (SOD)2 activity and sirtuin‐3 (SIRT3) expression. Cells were treated with TMAO at a series of concentrations (150, 300, 600, and 900 μmol/L) for 24 hours or incubated with 600 μmol/L TMAO at indicated time intervals (4, 8, 12, and 24 hours). A and B, SOD2 enzymatic activity was assayed using SOD1 and SOD2 Assay Kits with WST‐8 following the manufacturer's instructions. C and E, Western blot analysis of Ac‐SOD2 and SOD2 expression. D and F, The bar graph shows quantification of endogenous Ac‐SOD2 and SOD2. G and I, SIRT3 expression was detected via Western blot. H and J, The bar graph shows quantification of endogenous SIRT3. Values are expressed as means±SE (n=3); a P<0.05, b P<0.01 vs the vehicle‐treated control group; AU, arbitrary units.
To further determine the role of the SIRT3‐SOD2 pathway in TMAO‐induced mtROS accumulation, HUVECs were transfected with SIRT3 siRNA or overexpression plasmid as described in Materials and Methods. Transfection with SIRT3 siRNA alone led to decreased SOD2 activity and increased total ROS and mtROS levels followed by an increase in caspase‐1 activity and NLRP3 and caspase‐1 p20 expression, similar to the results obtained with TMAO and SIRT3 siRNA cotreatment (Figure 5A through 5F). Conversely, transfection with the SIRT3 overexpression plasmid significantly diminished the effects of TMAO on SOD2 activity, mtROS/ROS generation, and NLRP3 inflammasome activation (Figure 5A through 5F). Additionally, SIRT3 siRNA had similar effects on IL‐1β, ICAM‐1, and MMP‐9 expression as TMAO. Treatment with TMAO failed to further increase IL‐1β, ICAM‐1, and MMP‐9 expression on siRNA‐induced suppression of SIRT3. However, the TMAO‐induced increase in IL‐1β, ICAM‐1, and MMP‐9 was markedly attenuated on transfection with SIRT3 overexpression plasmid (Figure 5G and 5H). Our data clearly indicated that the SIRT3‐SOD2 pathway was required for mtROS accumulation and subsequent activation of the NLRP3 inflammasome induced by TMAO in endothelial cells.
Figure 5.
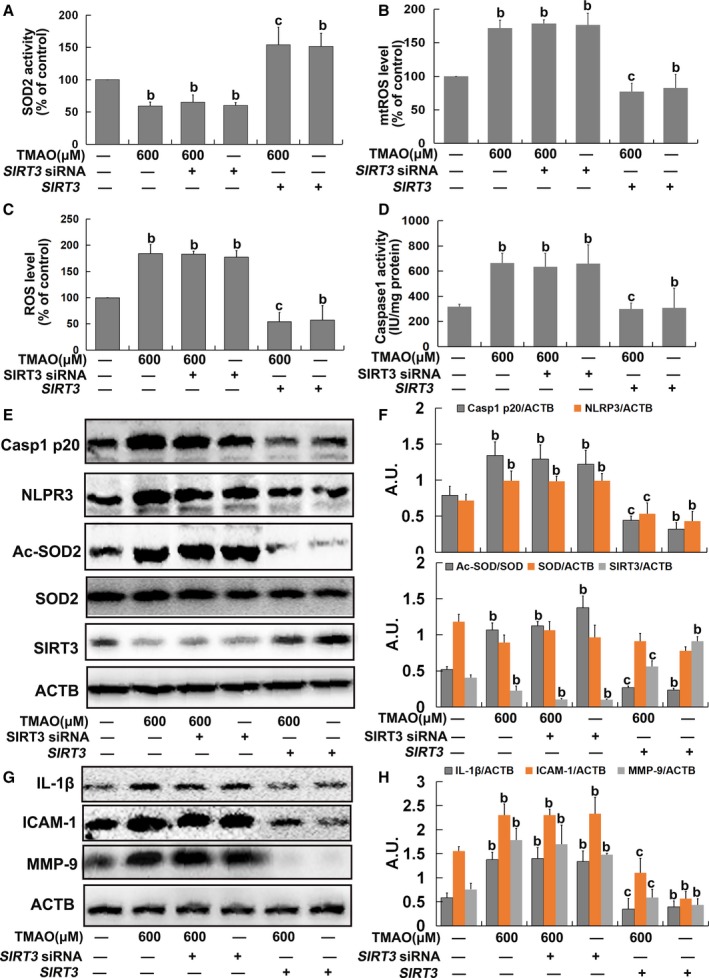
Trimethylamine‐N‐oxide (TMAO) induced mitochondrial reactive oxygen species (mtROS) accumulation via the sirtuin‐3–superoxide dismutase‐2 (SIRT3‐SOD2) pathway in endothelial cells. Human umbilical vein endothelial cells (HUVECs) were transfected with SIRT3 siRNA or a plasmid overexpressing SIRT3 as described in Materials and Methods. After 24 hours, cells were incubated with 600 μmol/L TMAO for 24 hours. A, Assay of SOD2 enzymatic activity using SOD1 and SOD2 Assay Kits with WST‐8 following the manufacturer's instructions. B, Detection of mtROS levels with MitoSOX ™ Red. C, Detection of total ROS levels using DCFH‐DA. D, Measurement of caspase‐1 activity with a caspase‐1 activity assay kit. E, Western blot analysis of caspase‐1 p20, nucleotide‐binding oligomerization domain–like receptor family pyrin domain–containing 3 (NLRP3), Ac‐SOD2, SOD2, and SIRT3 contents. F, Bar graphs showing quantification of the indicated proteins. G, Western blot analysis of IL‐1β, ICAM‐1, and MMP‐9 expression. H, Bar graphs showing quantification of the indicated proteins. Values are expressed as means±SE (n=3); b P<0.01 vs the vehicle‐treated control group; c P<0.01 vs TMAO‐treated group; AU, arbitrary units; ICAM, intercellular adhesion molecule; IL, interleukin; MMP, matrix metallopeptidase.
TMAO Induced Vascular Inflammation via NLRP3 Inflammasome Activation In Vivo
We additionally investigated the effect of TMAO on vascular inflammation in aorta in vivo. Compared with chow‐fed ApoE−/− mice, the atherosclerotic lesion areas in whole aorta and aortic root, as indices of AS severity, were significantly higher in mice fed choline (Figure 6A through 6C). Choline induced a significant increase in NLRP3 and caspase‐1 p20 expression and caspase‐1 activity, followed by upregulation of IL‐1β, ICAM‐1, and MMP‐9 expression in aorta (Figure 6D through 6F). Additionally, the addition of 1% choline led to a marked increase in the levels of plasma TMAO in ApoE−/− mice (Figure 6G). The results suggested that TMAO induced vascular inflammation via NLRP3 inflammasome activation, ultimately promoting AS in vivo.
Figure 6.
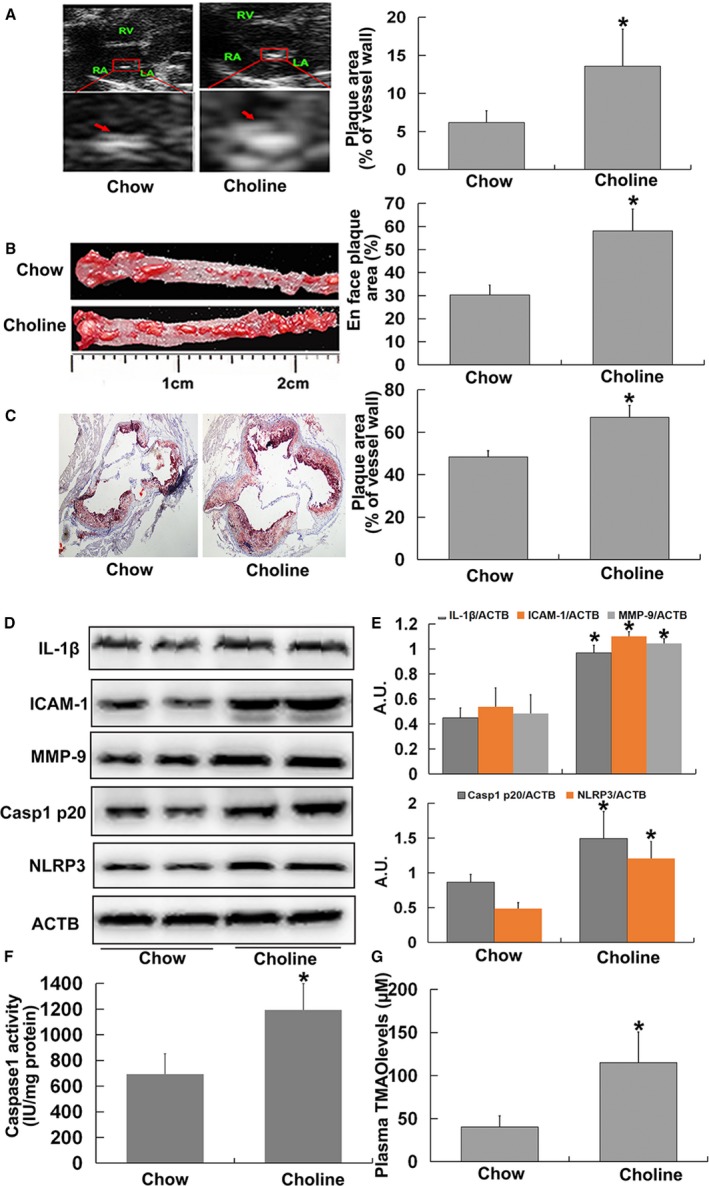
Trimethylamine‐N‐oxide (TMAO) induced vascular inflammation via nucleotide‐binding oligomerization domain–like receptor family pyrin domain–containing 3 (NLRP3) inflammasome activation in vivo. Eight‐week‐old female ApoE−/− mice (n=10 per group) were fed chow diet or chow diet combined with choline (1%) for 4 months. Mice were killed, and their blood and aorta samples were collected immediately, snap‐frozen in liquid nitrogen, and stored at −80°C until required. A, Ultrasound B‐mode images of aortic sinus and quantification. Arrows indicate the regions of interest. B, Oil‐red O staining of whole aortas, including aortic arch and thoracic and abdominal regions, and their quantitation. C, Oil‐red O–stained aortic root (counterstained with hematoxylin) and quantification. D, Western blot analysis of caspase‐1 p20, NLRP3, IL‐1β, ICAM‐1, and MMP‐9 contents in aortas. E, Bar graphs showing quantification of the indicated proteins. F, Caspase‐1 activity in aortas. G, Measurement of plasma TMAO levels using liquid chromatography–tandem mass spectrometry. Values are expressed as means±SE (n=10). *P<0.01 vs vehicle‐treated control group; AU, arbitrary units; ICAM, intercellular adhesion molecule; IL, interleukin; MMP, matrix metallopeptidase.
TMAO Induced Vascular NLRP3 Inflammasome Activation in a SIRT3‐Dependent Manner In Vivo
To further confirm the involvement of a SIRT3‐dependent mechanism in TMAO‐mediated NLRP3 inflammasome activation and vascular inflammation in vivo, ApoE−/−, WT, and SIRT3−/− mice were fed a diet with or without 1% choline for 4 months. As depicted in Figure 7A and 7B, choline promoted SIRT3 expression and SOD2 acetylation as well as inhibition of SOD2 activity in ApoE−/− mice. Similar results were observed with choline‐fed WT mice (Figure 7C through 7E). Compared with chow‐fed WT mice, expression of Ac‐SOD2, NLRP3, caspase‐1 p20, IL‐1β, ICAM‐1, MMP‐9, and caspase‐1 activity were higher and SOD2 activity was lower in the aortas of chow‐fed SIRT3−/− mice, similar to results that were observed in aortas from choline‐fed WT and SIRT3−/− mice (Figure 7C through 7H). Accordingly, we concluded that SIRT3 was necessary for TMAO‐induced SOD2 inhibition and NLRP3 inflammasome activation and subsequent vascular inflammation in vivo.
Figure 7.
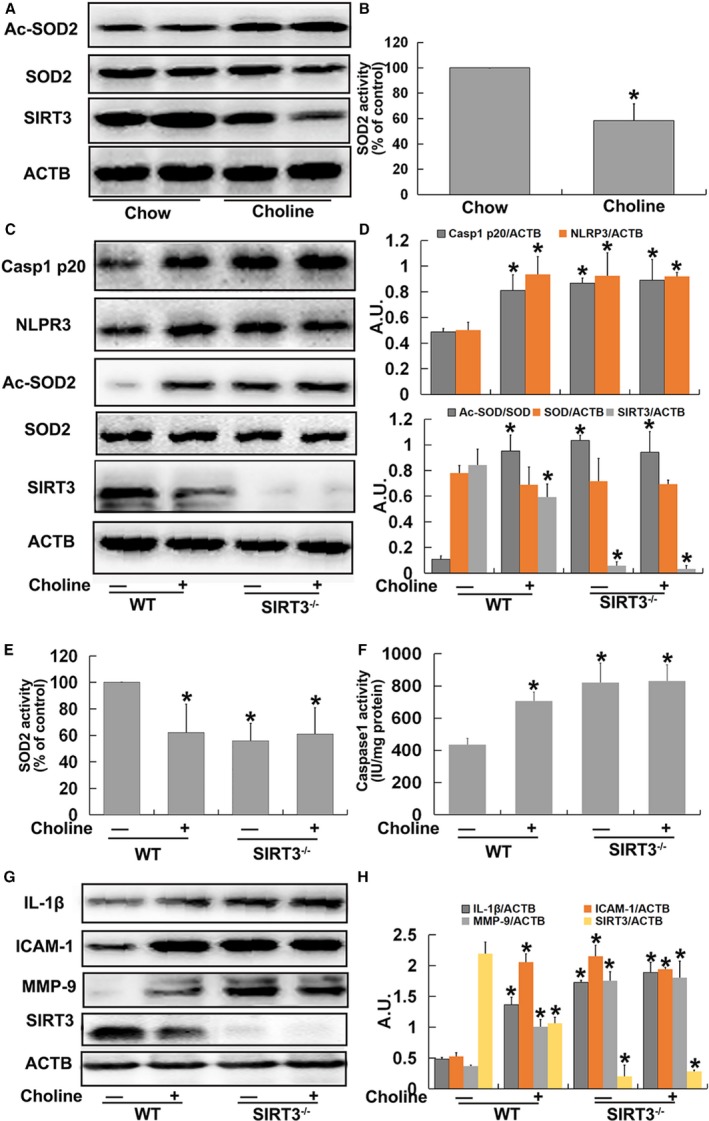
Trimethylamine‐N‐oxide (TMAO) induced vascular nucleotide‐binding oligomerization domain–like receptor family pyrin domain–containing 3 (NLRP3) inflammasome activation in a sirtuin‐3 (SIRT3)‐dependent manner in vivo. Eight‐week‐old female ApoE−/− mice were treated with or without 1% choline for 4 months. Mice were killed, and their aorta samples were collected immediately, snap‐frozen in liquid nitrogen, and stored at −80°C until required. A, Western blot detection of Ac‐SOD2, SOD2, and SIRT3 expression in aortas. B, Bar graphs showing quantification of the indicated proteins. Eight‐week‐old female wild type (WT) and SIRT3−/− mice were fed with or without 1% choline for 4 months. Mice were killed, and their aorta samples were collected immediately, snap‐frozen in liquid nitrogen, and stored at −80°C until required. C, Western blot analysis of caspase‐1 p20, NLRP3, Ac‐SOD2, SOD2, and SIRT3 contents in aortas. D, Bar graphs showing quantification of the indicated proteins. E, SOD2 enzymatic activity in aortas was assayed using a SOD1 and SOD2 Assay Kit with WST‐8 following the manufacturer's instructions. F, Measurement of caspase‐1 activity in aortas. G, Western blot analysis of IL‐1β, ICAM‐1, MMP‐9 and SIRT3 expression. H, Bar graphs showing quantification of the indicated proteins. Values are expressed as means±SE (n=10); *P<0.01 vs the vehicle‐treated control group; AU, arbitrary units; ICAM, intercellular adhesion molecule; IL, interleukin; MMP, matrix metallopeptidase; SOD, superoxide dismutase.
Discussion
Recently, plasma TMAO was identified as a novel and independent risk factor for promoting AS. In a large independent clinical cohort (n=4007), elevated TMAO levels were associated with significantly increased risk for major adverse cardiovascular events (2.54 for highest versus lowest TMAO quartile; P<0.001).5 Within the cohort examined, the hazard ratio for TMAO was significantly higher than that for traditional risk factors such as LDL cholesterol. Furthermore, elevated TMAO levels retained a strong prognostic value for predicting the incidence of adverse cardiovascular events, even after adjustment for traditional risk factors and renal function.5 Given that blood levels of TMAO are causatively linked to AS risk, the next obvious question is the mechanism by which circulating TMAO promotes AS, which is under active investigation. Researchers have confirmed that TMAO contributes to the development of AS, in part, by affecting cholesterol metabolism through regulating the major pathway for cholesterol elimination from the body and the bile acid synthetic pathway at multiple levels.4, 6 Recent studies have shown that physiological levels of TMAO can induce expression of cytokines and adhesion molecules, at least in part via the NF‐κB signaling pathway in vascular endothelial and smooth muscle cells, both in vivo and in vitro.7 These results support a role for TMAO in the activation of inflammatory pathways in cells of the vasculature, leading to endothelial cell leukocyte recruitment and AS. However, the exact mechanisms remain to be elucidated.
In the present study we showed for the first time that TMAO triggered vascular inflammation via an NLRP3 inflammasome‐dependent mechanism. The NLRP3 inflammasome is an IL‐1β family cytokine‐activating protein complex consisting of the pattern recognition receptor NLRP3, adaptor protein apoptotic speck‐like protein, and inactive pro‐caspase‐1, and involved in the regulation of innate immunity and inflammatory response.30 Several endogenous metabolic stress molecules, such as oligomers of islet amyloid polypeptide, glucose, ceramid, oxidized LDL, and cholesterol crystals, are sensed by NLRP3, stimulating NLRP3 inflammasome complex assembly and, in turn, leading to activation of caspase‐1 and ultimately to the processing and secretion of the proinflammatory cytokines IL‐1β and IL‐18.9 IL‐1β is a major atheroprone factor that plays a significant role in promoting the development of lipid plaques and destabilizing plaques by upregulating adhesion molecules, including vascular cell adhesion molecule‐1 and ICAM‐1.31, 32, 33 Primed macrophages secrete large amounts of IL‐1β in response to cholesterol crystals in an NLRP3 inflammasome‐dependent manner in mouse and human cells.34, 35 AS is decreased in several rodent models lacking NLRP3 or IL‐1 molecules, whereas in contrast, mice deficient in the IL‐1 receptor antagonist show increased AS.10 Moreover, recent studies have demonstrated that sterol regulatory element binding protein 2 activation of NLRP3 inflammasome in endothelium mediates hemodynamic‐induced AS susceptibility,11 whereas ablation of NLRP3 protects endothelial cells from inflammatory damage, subsequently attenuating AS.12, 13 These results clearly indicate that the NLRP3 inflammasome plays a key role in the pathogenesis of vascular inflammation, thereby contributing to AS development. Inflammation participates centrally in all stages of AS from the initial lesion to end‐stage thrombotic complications. The process often begins with inflammatory changes in the endothelium, characterized by expression of adhesion molecules.2, 3 AS is a chronic disease that develops over many years, and one can assume that lower concentrations of TMAO would have some impact, although it may be hard to detect experimentally. Therefore, according to the results of the concentration‐course experiments, 600 μmol/L TMAO was used in our time‐course and mechanism studies in vitro, although this concentration of TMAO was higher than that in humans. Data from the present study showed that TMAO activated the NLRP3 inflammasome and IL‐1β release, subsequently causing inflammation, both in vitro in cultured HUVECs and in vivo in aortas from ApoE−/− mice, and ultimately promoting AS. Our findings provide new insights into the potential mechanism underlying the promoting effects of TMAO on AS, in which the NLRP3 inflammasome may play a critical role.
The potential mechanisms of TMAO‐induced activation of the NLRP3 inflammasome were further investigated. Previous studies have ascertained that ROS generation is the most common pathway in inflammasome assembly. Inflammasome activators, such as uric acid crystals, enhance ROS production, thereby promoting association of thioredoxin‐interacting protein with NLRP3 and activation of the inflammasome.36 Conversely, treatment with ROS inhibitors has been shown to suppress cadmium‐, silica‐, and asbestos‐induced NLRP3 inflammasome activation.37, 38 Importantly, mitochondria are the major source of cellular ROS. Several studies have shown that inhibition of mitochondrial complex I by rotenone or of complex III by antimycin A induces robust ROS production by mitochondria, which is sufficient to drive NLRP3 inflammasome activation.39, 40 These results suggest that mtROS serve as direct activators of the NLRP3 inflammasome. In the present investigation, MitoSOX™ Red (a highly selective fluorescent probe for the detection of generated within mitochondria) was employed to determine the levels of mtROS, which could differentiate ROS generation in mitochondria from cytoplasmic and exogenous sources. As expected, TMAO treatment induced mtROS generation to a significant extent. On the other hand, TEMPO, a specific mtROS scavenger, mitigated mtROS levels, thereby inhibiting TMAO‐induced NLRP3 inflammasome activation and ultimately leading to attenuation of inflammation in endothelial cells. These findings indicate that TMAO‐triggered NLRP3 inflammasome is mainly preceded by the generation of mtROS in endothelial cells. It has also been found that the mtROS‐dependent activation of the endothelial NLRP3 inflammasome by hyperglycemia may be an important initiating mechanism for endothelial dysfunction,13 and endothelial senescence could be mediated through mtROS and NLRP3 inflammasome signaling pathways.41 Data from our current investigation complement those from previous studies on the role of mtROS and NLRP3 inflammasome in endothelial dysfunction, highlighting a potentially effective target for the prevention and treatment of endothelial dysfunction‐related cardiovascular disease.
Finally, our results supported a key role of the SIRT3‐SOD2 linked pathway in TMAO‐mediated mtROS production and NLRP3 inflammasome activation in HUVECs in vitro and aortas in vivo. Sirtuins are NAD+‐dependent enzymes implicated in a range of physiological and pathophysiological conditions, including AS, diabetes mellitus, cancer, longevity, and neurodegeneration, through deacetylation of numerous substrates.42 SIRT3, a mitochondrial sirtuin, serves as a primary regulator of mitochondrial protein acetylation, which is involved in the regulation of energy production and mtROS homeostasis.16 SIRT3 deacetylates a number of mitochondrial proteins including SOD2, which is responsible for limiting the accumulation of mtROS.20 Specifically, SIRT3 directly binds and deacetylates SOD2, thereby increasing SOD2 activity and subsequently leading to a significant effect on mtROS homeostasis, which plays a key protective role in high glucose–,43 angiotensin II–,44 and hypoxia45‐induced endothelial dysfunction. Recently, Zhao and colleagues18 demonstrated a protective role of SIRT3 against mitochondrial damage in the kidney through activating SOD2 and decreasing ROS production, subsequently inhibiting the NRLP3 inflammasome and downregulating IL‐1β and IL‐18. Traba et al19 additionally confirmed that a 24‐hour fast in human subjects activates SIRT3 biology, thereby increasing SOD2 activity, in turn conferring resistance to NLRP3 inflammasome activation via blunting of the mtROS level.19 Data from the present study showed that TMAO markedly suppressed SIRT3 expression and SOD2 activity in vitro and in vivo. Moreover, TMAO failed to further inhibit SOD2 and activate NLRP3 inflammasome and vascular inflammation in SIRT3 siRNA‐transfected HUVECs and SIRT3−/− mice. Conversely, SIRT3 overexpression significantly abrogated TMAO‐induced SOD2 inhibition and NLRP3 inflammasome activation, thereby downregulating inflammation caused by TMAO in endothelial cells. Our findings provided preliminary evidence that SIRT3‐SOD2 was a major signaling pathway mediating TMAO‐induced NLRP3 inflammasome activation and vascular inflammation, both in vitro and in vivo. These results, together with previously published data, have identified a potential SIRT3‐dependent program to attenuate the NLRP3 inflammasome and support the modulation of this pathway as a strategy to alleviate NLRP3‐linked inflammation. Additionally, it has been demonstrated that NF‐κB–mediated signaling is also involved in the NLRP3 inflammasome activation by regulating the transcription of inflammasome‐related components, including inactive NLRP3, pro‐IL‐1β, and pro‐IL‐18.46 More recently, TMAO has been found to induce phosphorylation of the NF‐κB pathway, thereby enhancing p65 NF‐κB nuclear localization and ultimately causing inflammatory transcripts (cyclooxygenase 2, IL‐6, E‐selectin, and ICAM‐1) in endothelial cells.7 Accordingly, TMAO might activate NF‐κB–mediated signaling, thereby upregulating transcription of inflammasome‐related components in endothelial cells. However, the exact role of NF‐κB in TMAO‐induced NLRP3 inflammasome activation needs further studies.
Overall, our findings indicated a novel mechanism of TMAO‐induced vascular inflammation through activation of the NLRP3 inflammasome. Moreover, the SIRT3‐SOD2‐mtROS signaling pathway played a critical role in modulating TMAO‐mediated NLRP3 inflammasome activation (Figure 8). Hence, these results open a new avenue of research regarding the potential mechanism by which TMAO promotes AS and are important complements to the role of the NLRP3 inflammasome in vascular inflammation.
Figure 8.
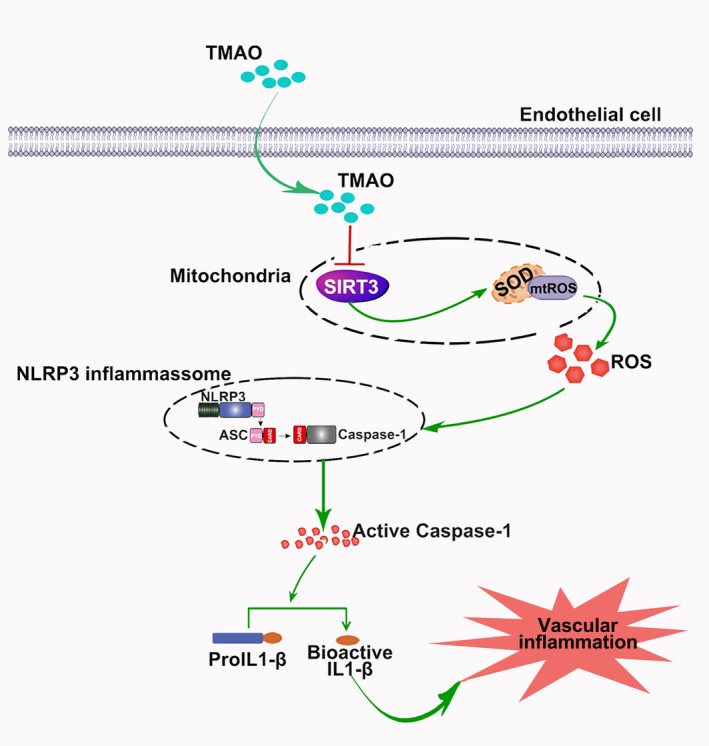
Trimethylamine‐N‐oxide (TMAO) mediated vascular inflammation by activating the nucleotide‐binding oligomerization domain–like receptor family pyrin domain–containing 3 (NLRP3) inflammasome via the sirtuin‐3 (SIRT3)–supeoxide dismutase (SOD)–mitochondrial reactive oxygen species (mtROS) signaling pathway.
Sources of Funding
This work was supported by the National Natural Science Foundation of China (81470562).
Disclosures
None.
Supporting information
Figure S1. The effect of TMAO on the adhesion of monocytes to endothelial cells. A, HUVECs were pretreated with YVAD (10 μmol/L), MCC950 (10 μmol/L), or TEMPO (50 μmol/L) for 2 hours followed by the addition of TMAO (600 μmol/L) for a further 24 hours. Monocyte adhesion to endothelial cells was measured using fluorescently labeled monocytic THP‐1 cells as described in Materials and Methods section. B, Quantification of adhered monocytes of A. Values are expressed as means±SE (n=3); b P<0.01 vs the vehicle‐treated control group; c P<0.01 vs TMAO‐treated group. HUVECs indicates human umbilical vein endothelial cells; TMAO, trimethylamine N‐oxide.
(J Am Heart Assoc. 2017;6:e006347 DOI: 10.1161/JAHA.117.006347.)28871042
References
- 1. Murray CJ, Lopez AD. Global mortality, disability, and the contribution of risk factors: Global Burden of Disease Study. Lancet. 1997;349:1436–1442. [DOI] [PubMed] [Google Scholar]
- 2. Maskrey BH, Megson IL, Whitfield PD, Rossi AG. Mechanisms of resolution of inflammation: a focus on cardiovascular disease. Arterioscler Thromb Vasc Biol. 2011;31:1001–1006. [DOI] [PubMed] [Google Scholar]
- 3. Wong BW, Meredith A, Lin D, McManus BM. The biological role of inflammation in atherosclerosis. Can J Cardiol. 2012;28:631–641. [DOI] [PubMed] [Google Scholar]
- 4. Wang Z, Klipfell E, Bennett BJ, Koeth R, Levison BS, Dugar B, Feldstein AE, Britt EB, Fu X, Chung YM, Wu Y, Schauer P, Smith JD, Allayee H, Tang WH, DiDonato JA, Lusis AJ, Hazen SL. Gut flora metabolism of phosphatidylcholine promotes cardiovascular disease. Nature. 2011;472:57–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Tang WH, Wang Z, Levison BS, Koeth RA, Britt EB, Fu X, Wu Y, Hazen SL. Intestinal microbial metabolism of phosphatidylcholine and cardiovascular risk. N Engl J Med. 2013;368:1575–1584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Koeth RA, Wang Z, Levison BS, Buffa JA, Org E, Sheehy BT, Britt EB, Fu X, Wu Y, Li L, Smith JD, DiDonato JA, Chen J, Li H, Wu GD, Lewis JD, Warrier M, Brown JM, Krauss RM, Tang WH, Bushman FD, Lusis AJ, Hazen SL. Intestinal microbiota metabolism of l‐carnitine, a nutrient in red meat, promotes atherosclerosis. Nat Med. 2013;19:576–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Seldin MM, Meng YH, Qi HX, Zhu WF, Wang ZE, Hazen SL, Lusis AJ, Shih DM. Trimethylamine N‐oxide promotes vascular inflammation through signaling of mitogen‐activated protein kinase and nuclear factor‐κB. J Am Heart Assoc. 2016;5:e002767. DOI: 10.1161/JAHA.115.002767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Lamkanfi M, Dixit VM. Inflammasomes and their roles in health and disease. Annu Rev Cell Dev Biol. 2012;28:137–161. [DOI] [PubMed] [Google Scholar]
- 9. Yin Y, Pastrana JL, Li XY, Huang X, Mallilankaraman K, Choi ET, Madesh M, Wang H, Yang XF. Inflammasomes: sensors of metabolic stresses for vascular inflammation. Front Biosci (Landmark Ed). 2013;18:638–649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Duewell P, Kono H, Rayner KJ, Sirois CM, Vladimer G, Bauernfeind FG, Abela GS, Franchi L, Nunez G, Schnurr M, Espevik T, Lien E, Fitzgerald KA, Rock KL, Moore KJ, Wright SD, Hornung V, Latz E. NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature. 2010;464:1357–1361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Xiao H, Lu M, Lin TY, Chen Z, Chen G, Wang WC, Marin T, Shentu TP, Wen L, Gongol B, Sun W, Liang X, Chen J, Huang HD, Pedra JHF, Johnson DA, Shyy JYJ. Sterol regulatory element binding protein 2 activation of NLRP3 inflammasome in endothelium mediates hemodynamic‐induced atherosclerosis susceptibility. Circulation. 2013;128:632–642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Wu JJ, Xu XS, Li Y, Kou JP, Huang F, Liu BL, Liu K. Quercetin, luteolin and epigallocatechin gallate alleviate TXNIP and NLRP3‐mediated inflammation and apoptosis with regulation of AMPK in endothelial cells. Eur J Pharmacol. 2014;745:59–68. [DOI] [PubMed] [Google Scholar]
- 13. Chen Y, Wang L, Pitzer AL, Li X, Li PL, Zhang Y. Contribution of redox‐dependent activation of endothelial NLRP3 inflammasomes to hyperglycemia‐induced endothelial dysfunction. J Mol Med. 2016;94:1335–1347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Zhou RB, Yazdi AS, Menu P, Tschopp J. A role for mitochondria in NLRP3 inflammasome activation. Nature. 2011;469:221–225. [DOI] [PubMed] [Google Scholar]
- 15. Alfonso‐Loeches S, Urena‐Peralta JR, Morillo‐Bargues MJ, Oliver‐De La Cruz J, Guerri C. Role of mitochondria ROS generation in ethanol‐induced NLRP3 inflammasome activation and cell death in astroglial cells. Front Cell Neurosci. 2014;8:216–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Bause AS, Haigis MC. SIRT3 regulation of mitochondrial oxidative stress. Exp Gerontol. 2013;48:634–639. [DOI] [PubMed] [Google Scholar]
- 17. Qiu XL, Brown K, Hirschey MD, Verdin E, Chen D. Calorie restriction reduces oxidative stress by SIRT3‐mediated SOD2 activation. Cell Metab. 2010;12:662–667. [DOI] [PubMed] [Google Scholar]
- 18. Zhao WY, Zhang L, Sui MX, Zhu YH, Zeng L. Protective effects of sirtuin 3 in a murine model of sepsis‐induced acute kidney injury. Sci Rep. 2016;6:33201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Traba J, Kwarteng‐Siaw M, Okoli TC, Li J, Huffstutler RD, Bray A, Waclawiw MA, Han K, Pelletier M, Sauve AA, Siegel RM, Sack MN. Fasting and refeeding differentially regulate NLRP3 inflammasome activation in human subjects. J Clin Invest. 2015;125:4592–4600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Tao R, Coleman MC, Pennington JD, Ozden O, Park SH, Jiang H, Kim HS, Flynn CR, Hill S, Hayes McDonald W, Olivier AK, Spitz DR, Gius D. Sirt3‐mediated deacetylation of evolutionarily conserved lysine 122 regulates MnSOD activity in response to stress. Mol Cell. 2010;40:893–904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Chen ML, Yi L, Jin X, Liang XY, Zhou Y, Zhang T, Xie Q, Zhou X, Chang H, Fu YJ, Zhu JD, Zhang QY, Mi MT. Resveratrol attenuates vascular endothelial inflammation by inducing autophagy through the cAMP signaling pathway. Autophagy. 2013;9:2033–2045. [DOI] [PubMed] [Google Scholar]
- 22. Chen CY, Yi L, Jin X, Zhang T, Fu YJ, Zhu JD, Mi MT, Zhang QY, Ling WH, Yu B. Inhibitory effect of delphinidin on monocyte‐endothelial cell adhesion induced by oxidized low‐density lipoprotein via ROS/p38MAPK/NF‐κB pathway. Cell Biochem Biophys. 2011;61:337–348. [DOI] [PubMed] [Google Scholar]
- 23. Chen ML, Yi L, Zhang Y, Zhou X, Ran L, Yang JN, Zhu JD, Zhang QY, Mi MT. Resveratrol attenuates trimethylamine‐N‐oxide (TMAO)‐induced atherosclerosis by regulating TMAO synthesis and bile acid metabolism via remodeling of the gut microbiota. MBio. 2016;7:e02210–e02215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Niu LL, Qian M, Yang W, Meng L, Xiao Y, Wong KKL, Abbott D, Liu X, Zheng HR. Surface roughness detection of arteries via texture analysis of ultrasound images for early diagnosis of atherosclerosis. PLoS One. 2013;8:e76880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Wang DL, Xia M, Yan X, Li D, Wang L, Xu YX, Jin TR, Ling WH. Gut microbiota metabolism of anthocyanin promotes reverse cholesterol transport in mice via repressing miRNA‐10b. Circ Res. 2012;111:967–981. [DOI] [PubMed] [Google Scholar]
- 26. Paigen B, Morrow A, Holmes PA, Mitchell D, Williams RA. Quantitative assessment of atherosclerotic lesions in mice. Atherosclerosis. 1987;68:231–240. [DOI] [PubMed] [Google Scholar]
- 27. Vanaja SK, Rathinam VA, Fitzgerald KA. Mechanisms of inflammasome activation: recent advances and novel insights. Trends Cell Biol. 2015;25:308–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Chowdhury SK, Dobrowsky RT, Fernyhough P. Nutrient excess and altered mitochondrial proteome and function contribute to neurodegeneration in diabetes. Mitochondrion. 2011;11:845–854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Tao R, Vassilopoulos A, Parisiadou L, Yan Y, Gius D. Regulation of MnSOD enzymatic activity by SIRT3 connects the mitochondrial acetylome signaling networks to aging and carcinogenesis. Antioxid Redox Signal. 2014;20:1646–1654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Gross O, Thomas CJ, Guarda G, Tschopp J. The inflammasome: an integrated view. Immunol Rev. 2011;243:136–151. [DOI] [PubMed] [Google Scholar]
- 31. De Nardo D, Latz E. NLRP3 inflammasomes link inflammation and metabolic disease. Trends Immunol. 2011;32:373–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Connat JL. [Inflammasome and cardiovascular diseases]. Ann Cardiol Angeiol (Paris). 2011;60:48–54. [DOI] [PubMed] [Google Scholar]
- 33. Alcocer‐Gomez E, Cordero MD. NLRP3 inflammasome: common nexus between depression and cardiovascular diseases. Nat Rev Cardiol. 2017;14:124. [DOI] [PubMed] [Google Scholar]
- 34. Rajamaki K, Lappalainen J, Oorni K, Valimaki E, Matikainen S, Kovanen PT, Eklund KK. Cholesterol crystals activate the NLRP3 inflammasome in human macrophages: a novel link between cholesterol metabolism and inflammation. PLoS One. 2010;5:e11765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Abdul‐Muneer PM, Alikunju S, Mishra V, Schuetz H, Szlachetka AM, Burnham EL, Haorah J. Activation of NLRP3 inflammasome by cholesterol crystals in alcohol consumption induces atherosclerotic lesions. Brain Behav Immun. 2017;62:291–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Zhou R, Tardivel A, Thorens B, Choi I, Tschopp J. Thioredoxin‐interacting protein links oxidative stress to inflammasome activation. Nat Immunol. 2010;11:136–140. [DOI] [PubMed] [Google Scholar]
- 37. Dostert C, Petrilli V, Van Bruggen R, Steele C, Mossman BT, Tschopp J. Innate immune activation through Nalp3 inflammasome sensing of asbestos and silica. Science. 2008;320:674–677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Chen H, Lu Y, Cao Z, Ma Q, Pi H, Fang Y, Yu Z, Hu H, Zhou Z. Cadmium induces NLRP3 inflammasome‐dependent pyroptosis in vascular endothelial cells. Toxicol Lett. 2016;246:7–16. [DOI] [PubMed] [Google Scholar]
- 39. Huang LS, Cobessi D, Tung EY, Berry EA. Binding of the respiratory chain inhibitor antimycin to the mitochondrial bc1 complex: a new crystal structure reveals an altered intramolecular hydrogen‐bonding pattern. J Mol Biol. 2005;351:573–597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Won JH, Park S, Hong S, Son S, Yu JW. Rotenone‐induced impairment of mitochondrial electron transport chain confers a selective priming signal for NLRP3 inflammasome activation. J Biol Chem. 2015;290:27425–27437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Sun C, Fan S, Wang X, Lu J, Zhang Z, Wu D, Shan Q, Zheng Y. Purple sweet potato color inhibits endothelial premature senescence by blocking the NLRP3 inflammasome. J Nutr Biochem. 2015;26:1029–1040. [DOI] [PubMed] [Google Scholar]
- 42. Haigis MC, Sinclair DA. Mammalian sirtuins: biological insights and disease relevance. Annu Rev Pathol. 2010;5:253–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Liu G, Cao M, Xu Y, Li Y. SIRT3 protects endothelial cells from high glucose‐induced cytotoxicity. Int J Clin Exp Pathol. 2015;8:353–360. [PMC free article] [PubMed] [Google Scholar]
- 44. Liu H, Chen T, Li N, Wang S, Bu P. Role of SIRT3 in angiotensin II‐induced human umbilical vein endothelial cells dysfunction. BMC Cardiovasc Disord. 2015;15:81–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Tseng AH, Wu LH, Shieh SS, Wang DL. SIRT3 interactions with FOXO3 acetylation, phosphorylation and ubiquitinylation mediate endothelial cell responses to hypoxia. Biochem J. 2014;464:157–168. [DOI] [PubMed] [Google Scholar]
- 46. Shao BZ, Xu ZQ, Han BZ, Su DF, Liu C. NLRP3 inflammasome and its inhibitors: a review. Front Pharmacol. 2015;6:262–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. The effect of TMAO on the adhesion of monocytes to endothelial cells. A, HUVECs were pretreated with YVAD (10 μmol/L), MCC950 (10 μmol/L), or TEMPO (50 μmol/L) for 2 hours followed by the addition of TMAO (600 μmol/L) for a further 24 hours. Monocyte adhesion to endothelial cells was measured using fluorescently labeled monocytic THP‐1 cells as described in Materials and Methods section. B, Quantification of adhered monocytes of A. Values are expressed as means±SE (n=3); b P<0.01 vs the vehicle‐treated control group; c P<0.01 vs TMAO‐treated group. HUVECs indicates human umbilical vein endothelial cells; TMAO, trimethylamine N‐oxide.