

Abstract
Background
Heterozygous loss of function mutations in the KCNK3 gene cause hereditary pulmonary arterial hypertension (PAH). KCNK3 encodes an acid‐sensitive potassium channel, which contributes to the resting potential of human pulmonary artery smooth muscle cells. KCNK3 is widely expressed in the body, and dimerizes with other KCNK3 subunits, or the closely related, acid‐sensitive KCNK9 channel.
Methods and Results
We engineered homomeric and heterodimeric mutant and nonmutant KCNK3 channels associated with PAH. Using whole‐cell patch‐clamp electrophysiology in human pulmonary artery smooth muscle and COS7 cell lines, we determined that homomeric and heterodimeric mutant channels in heterozygous KCNK3 conditions lead to mutation‐specific severity of channel dysfunction. Both wildtype and mutant KCNK3 channels were activated by ONO‐RS‐082 (10 μmol/L), causing cell hyperpolarization. We observed robust gene expression of KCNK3 in healthy and familial PAH patient lungs, but no quantifiable expression of KCNK9, and demonstrated in functional studies that KCNK9 minimizes the impact of select KCNK3 mutations when the 2 channel subunits co‐assemble.
Conclusions
Heterozygous KCNK3 mutations in PAH lead to variable loss of channel function via distinct mechanisms. Homomeric and heterodimeric mutant KCNK3 channels represent novel therapeutic substrates in PAH. Pharmacological and pH‐dependent activation of wildtype and mutant KCNK3 channels in pulmonary artery smooth muscle cells leads to membrane hyperpolarization. Co‐assembly of KCNK3 with KCNK9 subunits may provide protection against KCNK3 loss of function in tissues where both KCNK9 and KCNK3 are expressed, contributing to the lung‐specific phenotype observed clinically in patients with PAH because of KCNK3 mutations.
Keywords: ion channel, pathophysiology, pharmacology, potassium channels, pulmonary hypertension
Subject Categories: Electrophysiology, Pulmonary Hypertension, Ion Channels/Membrane Transport, Pathophysiology, Genetics
Clinical Perspective
What Is New?
Heterozygous KCNK3 mutations associated with pulmonary arterial hypertension result in loss of potassium channel function by differing mechanisms, and with varying severity.
Activation of mutant and nonmutant KCNK3 channels in a pulmonary artery smooth muscle cell line by the phospholipase A2 inhibitor, ONO‐RS‐082, or by extracellular alkalosis, leads to graded cell hyperpolarization.
KCNK9 heterodimerizes with mutant and nonmutant KCNK3 channel subunits, and minimizes the impact of KCNK3 loss‐of‐function mutations when KCNK9 and KCNK3 co‐assembled.
What Are the Clinical Implications?
Homodimeric and heterodimeric mutant and nonmutant KCNK3 potassium channels represent potential therapeutic targets in pulmonary arterial hypertension.
The absence of KCNK9 expression in the lung may underlie the lung‐specific phenotype observed in pulmonary arterial hypertension patients with heterozygous KCNK3 mutations.
Developing pharmacologic agents that specifically regulate KCNK3 activity, and an animal model to study the physiological consequences of such agents, warrant further investigation in the context of pulmonary hypertension treatment.
Introduction
Pulmonary arterial hypertension (PAH) is a progressive primary illness of the lung, characterized by increased pulmonary artery pressure ≥25 mm Hg, increased pulmonary vascular resistance, right‐sided heart failure, and high mortality rates.1 Pulmonary arterial endothelial and smooth muscle cell proliferation and excessive vasoconstriction are pathogenetic mechanisms in PAH. Mutations in the BMPR2 gene, a component of the transforming growth factor‐β signaling pathway, account for most genetic cases of PAH, while less frequently, mutations in other genes underlie the disease.2
We recently identified KCNK3 as the first ion channelopathy associated with PAH.3 Using exome sequencing, 6 distinct mutations in the KCNK3 potassium channel were identified in patients with hereditary PAH. All patients were heterozygous at the KCNK3 gene locus, possessing 1 mutant and 1 wildtype (WT) channel. Each of the 6 KCNK3 mutations led to loss of channel function, and some mutant channels were functionally rescued by ONO‐RS‐082 (ONO), a phospholipase A2 inhibitor and KCNK3 activator.3
KCNK3 encodes an acid‐sensitive 2‐pore domain potassium channel (also referred to as TASK‐1), extremely sensitive to extracellular pH, especially within the range of physiological pH 7.4.4 Strongly inhibited by extracellular acidosis and hypoxia, KCNK3 regulates cellular excitability, and is thought to contribute to hypoxic pulmonary vasoconstriction.5 The voltage‐insensitivity of KCNK3 renders the channel open across all voltages, leading to potassium efflux from cells expressing the channel, which contributes to the negative resting potential. KCNK3 is widely expressed in the human body, including in the central nervous system, heart, and pulmonary artery smooth muscle cells (PASMCs).6, 7
KCNK3 currents contribute to the PASMC resting potential and impact pulmonary arterial tone and smooth muscle cell growth; KCNK3 downregulation or inhibition leads to PASMC depolarization, proliferation, and pulmonary arterial constriction.5, 7, 8, 9 Notably, KCNK3 downregulation was recently identified as a pathogenic hallmark of PAH in humans and in a monocrotaline‐induced pulmonary hypertension rat model, and administration of ONO alleviated signs of pulmonary hypertension in the animal model, further implicating KCNK3 as a therapeutic target in PAH.3, 8
KCNK3 dimerizes in vivo, forming functional channels from 2 channel subunits linked together.10 Adding to the complexity of KCNK3 regulation, the closely related acid‐sensitive KCNK9 channel dimerizes with KCNK3, forming KCNK9‐KCNK3 heterodimeric channels in tissues where both channels are expressed.11, 12, 13, 14, 15 KCNK9 is more maximally activated at pH 7.4 than KCNK3.16, 17 The channels are co‐expressed in a variety of tissues, promoting tissue‐specific diversity of channel function.
In this study, we investigate (1) mechanisms of heterozygous KCNK3 loss of function mutations in PAH by studying channel function over a broad pH range; (2) the capacity of mutant and WT KCNK3 channels to serve as therapeutic targets in PAH; (3) the impact of selected KCNK3 mutations on channel function and pharmacology in physiologically relevant heterozygous conditions; and (4) the potential role for KCNK9 in underlying the lung‐specific disease phenotype conferred upon patients with heterozygous KCNK3 mutations.
Methods
Data S1 include further details of materials and methods.
Study Patients
Human lung parenchymal samples were obtained from 10 control patients (failed donor lungs); 5 patients with familial PAH; and 5 patients with congenital heart disease–associated PAH. cDNA samples were provided by the Pulmonary Hypertension Breakthrough Initiative.18 We were not required to obtain informed consent. The protocol, “Studying Gene Expression in Pulmonary Arterial and Lung Tissue in Healthy and Diseased Samples,” (#AAAQ2454) was approved by the Institutional Review Board at Columbia University Medical Center. KCNK3 mutations in PAH patients were identified as previously reported.3
Quantitative Real‐Time Polymerase Chain Reaction
The TaqMan gene expression system was used to quantify mRNA expression (Applied Biosystems). No‐template controls lacking cDNA were included. Experiments were performed in duplicate for each sample. Data are expressed as means of the average cycle threshold (Ct) value of duplicates, and as fold changes in expression (2▵▵Ct method).
Molecular Biology
Mutations were engineered into human KCNK3 cDNA in a pcDNA3.1+ expression vector by site‐directed mutagenesis using QuickChange (Stratagene).3 Human KCNK9 cDNA in a pIRES‐GFP and pcDNA3.1+ vector was used. Where noted, KCNK3 constructs were tagged with a C‐terminal green fluorescent protein (GFP). Tandem‐linked KCNK3‐KCNK3 and KCNK9‐KCNK3 dimer constructs were engineered by joining 2 KCNK subunits with a glycine‐rich linker, subcloned into a pcDNA3.1+ vector.
Materials
ONO‐RS‐082 (Enzo), ML365 (MedChem Express), ruthenium red, and dimethylsulfoxide (Life Technologies) were purchased commercially.
Cell Culture and Heterologous Channel Expression
KCNK3 and KCNK9 channel constructs were expressed in cultured human PASMC and COS7 cell lines. GFP was co‐expressed or tagged to the C‐terminus of the channel as a marker of transfection. A previously established transfection protocol using Lipofectamine reagents was used in COS7 cells,3 with modifications to optimize efficiency in human PASMCs (hPASMCs).
Electrophysiology
KCNK3 channel current and membrane potential changes were recorded by whole‐cell patch clamp in hPASMCs and COS7 cells. An Axopatch 200B amplifier (Axon Instruments), Digidata 1440A model, and pClamp 10 software were used for recording and analysis (Molecular Devices, CA). For voltage clamp experiments, cells were held at −80 mV and a 500‐ms voltage ramp was applied once every 3 s, with voltage increasing linearly from −120 mV to +60 mV before returning to holding. Expressed KCNK3, KCNK9, and tandem dimer constructs were recorded. For current clamp experiments, changes in membrane potential were recorded over time after first applying the voltage ramp to verify stability of the patch and expression of KCNK3 channels.
For experiments in COS7 cells, solutions were prepared as previously reported3: pipette (internal) solution (in mmol/L) contained: 150 KCl, 3 MgCl2, 5 EGTA, 10 HEPES, adjusted to pH 7.2 with KOH. Bath (extracellular) solution (in mmol/L) contained: 150 NaCl, 5 KCl, 1 MgCl2, 1.8 CaCl2, 10 HEPES adjusted to pH 6.4, 7.4, or 8.4 with NaOH. In hPASMCs, solutions were adapted from a prior study.5 Pipette (internal) solution (in mmol/L) contained: 135 K‐methanesulphonate, 20 KCl, 2 Na2ATP, 1 MgCl2, 1 EGTA, 20 HEPES, adjusted to pH 7.2 with KOH. Bath (extracellular) solution (in mmol/L) contained: 140.5 NaCl, 5.5 KCl, 1.5 CaCl2, 1 MgCl2, 10 glucose, 0.5 Na2HPO4, 0.5 KH2PO4, 10 HEPES, adjusted to pH 6.4, 7.4, or 8.4 with NaOH. In COS7 cells and hPASMCs, solutions at pH 5.0 contained 10 mmol/L 2‐(N‐morpholino)ethane sulfonic acid instead of HEPES. Solutions at pH 10.4 contained 10 mmol/L Tris‐Base instead of HEPES.
Statistical Analyses
Graphic analysis was performed with Origin 7.0 and 9.0 (Microcal Software, Northampton, MA). pClamp 10 software was used for electrophysiological analysis. Data are reported as means±SEM, based on n observations. Student t tests and 1‐way ANOVA with post hoc Tukey tests were applied as indicated, and significant differences were determined based on P<0.05. Statistical tests were performed using Origin and Excel software (Microsoft, Bellevue, WA).
Results
Functional Characterization of KCNK3 Variants Identifies a Unique Acid‐Sensing Phenotype
We previously identified 6 loss of function mutations in KCNK3 causing PAH.3 Given the extreme sensitivity of KCNK3 channels to extracellular pH changes within the physiological pH range, here we investigated the mechanistic basis of loss of channel function focusing on possible mutation‐induced changes in the pH dependence of the expressed channels. WT or mutant KCNK3 channels were expressed in COS7 cells, and a whole‐cell voltage ramp was applied across a physiological voltage range (Figure 1A, top). As expected for WT KCNK3 channels, we observed appreciable K+ current at physiological extracellular pH 7.4; inhibition of current by acidosis, pH 6.4; and activation of current by alkalosis, pH 10.4 (Figure 1A and 1B).4
Figure 1.
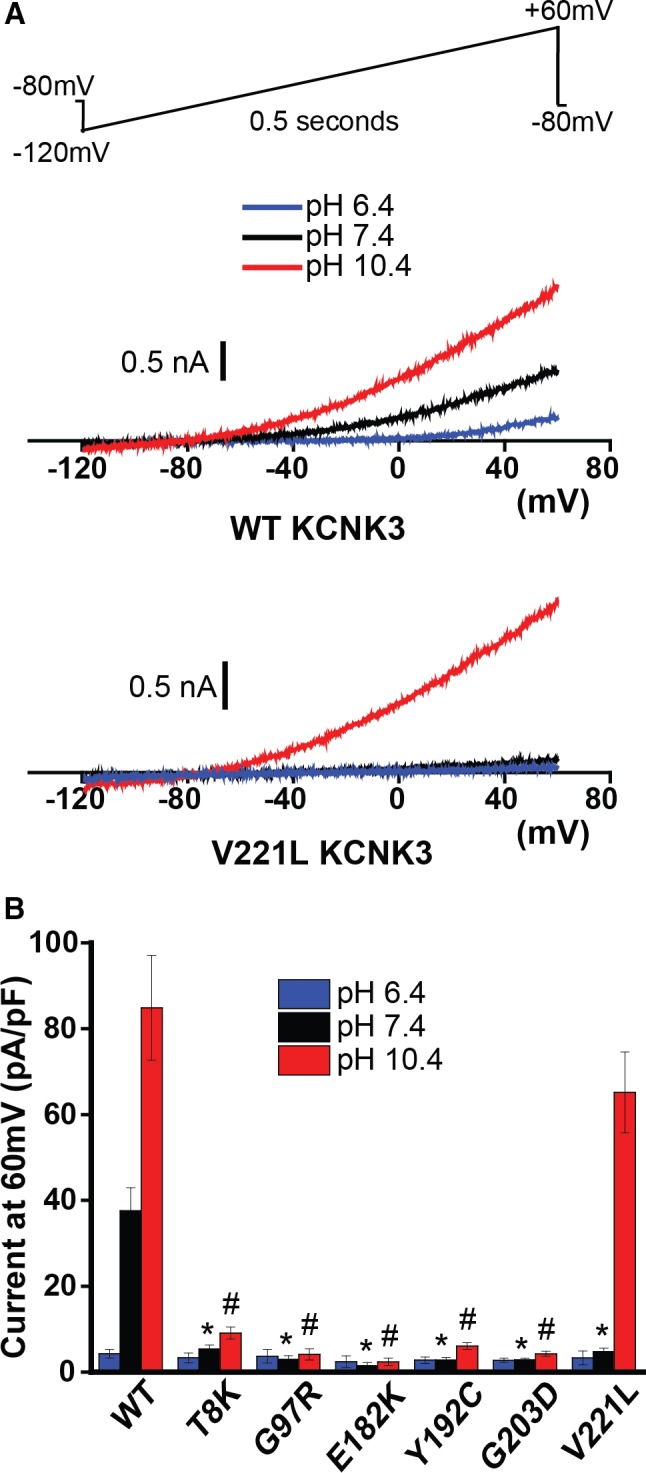
Pulmonary arterial hypertension (PAH)‐associated mutant KCNK3 channels demonstrate mutation‐specific severity of loss of function, across a broad pH range in COS7 cells. A, Typical voltage clamp recordings of wildtype (WT, top) and V221L (bottom) KCNK3. Sample current traces at pH 6.4 (blue), 7.4 (black), and 10.4 (red) are shown. A voltage ramp (top) was applied, −120 mV to +60 mV over 0.5 s, every 3 s, from a holding potential of −80 mV for all voltage clamp recordings in this study. B, Summary of current density (pA/pF at 60 mV) of cells expressing WT or one of the PAH‐associated mutant KCNK3 channels (n=3–9 cells at pH 6.4; n=6–33 cells at pH 7.4; n=6–32 cells at pH 10.4). Bars show mean±SEM. *P<0.05 at pH 7.4; # P<0.05 at pH 10.4, for the comparison of WT and each KCNK3 mutant channel by 1‐way ANOVA (P<0.05) and post hoc Tukey test.
Compared with WT KCNK3 channels, all 6 PAH‐associated mutant KCNK3 channels exhibited loss of function at pH 7.4 and pH 10.4 (Figure 1B), with 1 notable exception: V221L KCNK3 conferred loss of function at physiological pH 7.4, with recovery of function observed at alkalotic pH 10.4 (Figure 1A and 1B). Given its unique alteration in pH‐dependent regulation and readily discernible robust current activity in alkalotic conditions, V221L KCNK3 served as a model KCNK3 mutant to study in more physiologic conditions, using cultured hPASMCs.
Cultured hPASMCs Provide a Physiological Platform for KCNK3 Expression
KCNK3 currents contribute to the resting potential of native PASMCs, and the downregulation or inhibition of KCNK3 causes PASMC depolarization, excessive proliferation, and pulmonary arterial constriction.5, 7, 8, 9 Cultured PASMCs have been used previously to study overexpressed potassium channel activity, regulation, and pharmacology in the context of PAH.19, 20 We adapted this approach to test the hypothesis that both WT and mutant (eg, V221L) KCNK3 channels represent therapeutic targets in PAH (Figure 2).
Figure 2.
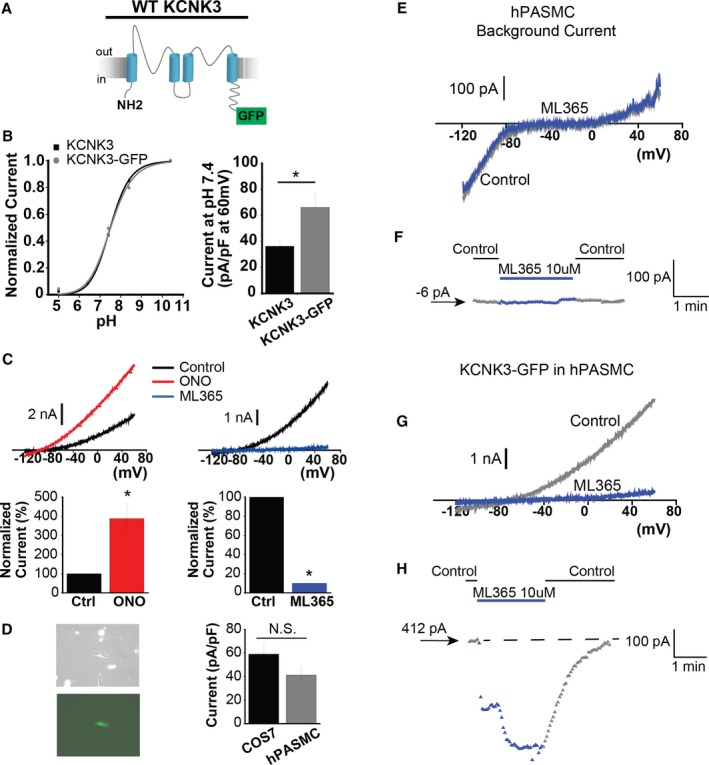
KCNK3 expression platform in human pulmonary artery smooth muscle cells (hPASMCs). A, KCNK3, tagged with GFP at the C‐terminus, was engineered (KCNK3‐GFP). B, KCNK3‐GFP (gray curve) vs KCNK3 (black curve) expression in COS7 cells across a broad pH range, with current normalized to max current at pH 10.4 (n=9–25 cells per pH data point for KCNK3; n=9–12 cells for KCNK3‐GFP; fitted by the Hill equation. C, KCNK3‐GFP is activated by ONO‐RS‐082 (ONO) 10 μmol/L (red trace, left) and inhibited by ML365 10 μmol/L (blue trace, right), under voltage‐clamp shown in COS7 cells. Predrug (control, black traces) and drug conditions are at pH 7.4. Bar graphs (bottom) show percent change in current at −50 mV after ONO (n=8 cells) or ML365 (n=12 cells) application, compared with control. D, Cultured hPASMCs (left, top) expressing KCNK3‐GFP fluoresce green (left, bottom). Bar graph (right) shows KCNK3‐GFP current activity at pH 7.4 (pA/pF at 60 mV) in hPASMCs (gray, n=20 cells) vs COS7 cells (black, n=26 cells). E, Background hPASMC current at pH 7.4 (control, gray trace), and after application of ML365 10 μmol/L (blue trace). F, Sample ML365 time course of action in control and drug conditions, measured at −50 mV, from a starting current amplitude of −6 pA indicated by the arrow. G, Current from hPASMC expressing KCNK3‐GFP is shown at pH 7.4 (control, gray trace), and in ML365 10 μmol/L (blue trace). H, Sample ML365 time course of action in control and drug conditions, measured at −50 mV, from a starting current amplitude of 412 pA indicated by the arrow. Horizontal dashed line is drawn at the starting level of current in control solution. Bar graphs show mean±SEM. *P<0.05 by the paired (B) and unpaired (C) Student t test. N.S. indicates no significant difference.
As loss of function could result from lack of KCNK3 expression, we first engineered a C‐terminal GFP‐tagged KCNK3 channel (KCNK3‐GFP, Figure 2A) and screened its function in COS7 cells. We observed a similar pH dependence for KCNK3‐GFP versus KCNK3, as well as a significant increase in current density (pA/pF, measured at 60 mV) for KCNK3‐GFP (Figure 2B). In voltage‐clamp experiments, ONO 10 μmol/L activated KCNK3‐GFP with similar efficacy to KCNK3 channels (Figure 2C; and compare with KCNK3 activation previously reported3). The recently developed KCNK3 inhibitor, ML365 10 μmol/L,21, 22 produced robust inhibition of KCNK3‐GFP currents (Figure 2C). Mean current densities (pA/pF) in response to the drugs are summarized in Figure 2C, measured at −50 mV to minimize interference of background ionic currents, including minimizing nonselective effects of ONO observed at more depolarized potentials.
The use of KCNK3‐GFP thus did not appreciably change KCNK3 function or pharmacology, while providing the advantage of increased current density. Next, KCNK3‐GFP was expressed in cultured hPASMCs, identified by green fluorescence, and produced similar current density (pA/pF at 60 mV) compared with KCNK3‐GFP expression in COS7 cells (Figure 2D).
Loss of native KCNK3 expression was previously demonstrated in cultured PASMCs,23 and confirmed functionally in our system of cultured hPASMCs by applying ML365 10 μmol/L in nontransfected hPASMCs, and observing no significant effect on cell currents (Figure 2E, 2F, and S1B). The lack of effect of ML365 in nontransfected cells highlights its strong selectivity for KCNK3 inhibition when KCNK3 is expressed. In comparison, KCNK3‐GFP expression in hPASMCs produced robust channel activity, inhibited by ML365 10 μmol/L (Figure 2G, 2H, S1B, and S1C). We thus developed a platform to compare the relative impact of expressed mutant versus WT KCNK3 channels on hPASMC membrane potential, and the effect of KCNK3 pharmacological agents in a more physiological environment (Figure 3).
Figure 3.
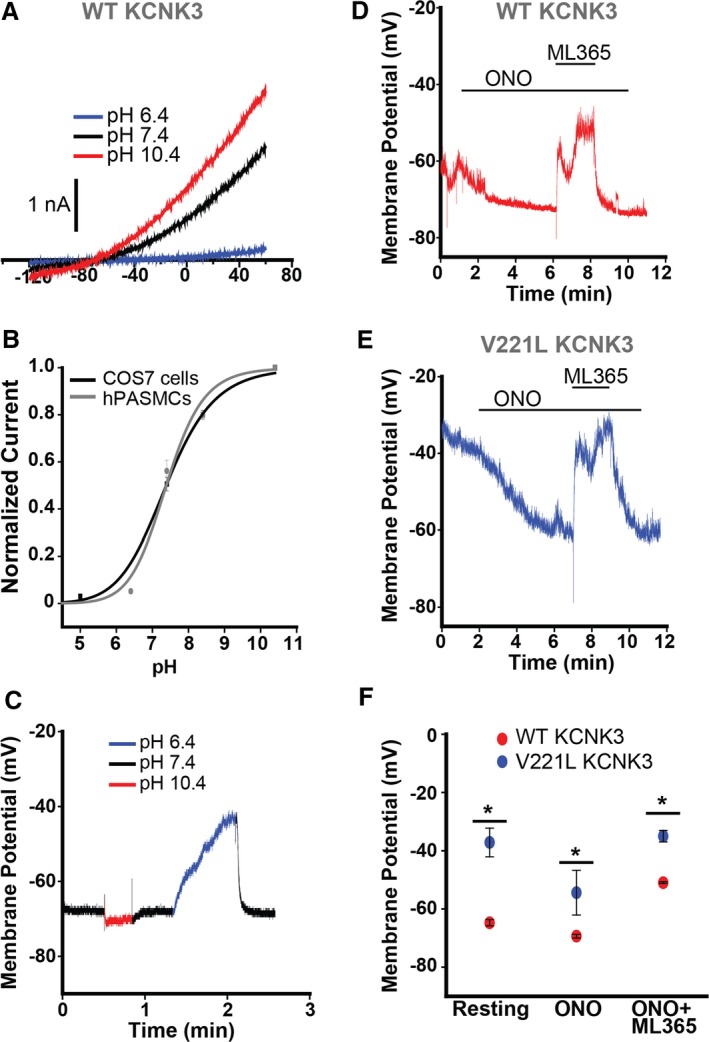
Robust response of KCNK3 channels to pH changes and pharmacological modulators in human pulmonary artery smooth muscle cells (hPASMCs). A, Voltage clamp recording of KCNK3‐GFP expressed in hPASMCs at pH 6.4 (blue), pH 7.4 (black), and pH 10.4 (red). B, KCNK3‐GFP expression in hPASMCs (gray curve) vs COS7 cells (black curve) across a broad pH range, with current normalized to max current at pH 10.4 (n=9–12 cells per pH data point in COS7; n=2–6 cells per pH data point in hPASMCs; fitted by the Hill equation). C, Current clamp recording of wildtype (WT) KCNK3‐GFP, with changes in membrane potential (mV) measured at pH 6.4 (blue), pH 7.4 (black), and pH 10.4 (red). D, Current clamp recording of WT KCNK3‐GFP (red trace), showing changes in membrane potential (mV) upon application of ONO‐RS‐082 (ONO), ONO+ML365, or pH 8.4. E, Current clamp recording of V221L KCNK3‐GFP (blue trace), showing changes in membrane potential (mV) upon application of ONO, or ONO+ML365. F, Current clamp summary of WT (red) and V221L (blue) KCNK3‐GFP for resting potential, ONO, and ONO+ML365 conditions (n=3–11 cells per condition). Drugs applied at a concentration of 10 μmol/L in all experiments. Data plots represent means±SEM. *P<0.05 by the unpaired Student t test.
Activation of PAH‐Associated Mutant KCNK3 Channels in hPASMCs
KCNK3‐GFP expression in hPASMCs produced a consistent pH dependence of expressed channel activity compared with expression in COS7 cells (Figure 3A and 3B). We recorded membrane potentials in cells expressing KCNK3‐GFP at physiological pH 7.4, upon channel activation by pH 10.4, and upon inhibition by pH 6.4. A sample current clamp trace (Figure 3C) reveals a resting potential of −68 mV at pH 7.4, hyperpolarization to −70 mV at pH 10.4, and depolarization to −44 mV at pH 6.4 (summarized in Figure S1A).
Next, we determined the relative impact of pharmacological activation and inhibition of KCNK3 in hPASMCs expressing WT or V221L KCNK3‐GFP. A recording of a cell expressing WT KCNK3‐GFP (Figure 3D) demonstrates a resting potential of −62 mV at physiological pH 7.4; hyperpolarization by ONO 10 μmol/L to −73 mV; and depolarization to −51 mV upon co‐application of ML365 10 μmol/L. By comparison, a recording of a cell expressing V221L KCNK3‐GFP (Figure 3E) reveals a resting potential of −40 mV at physiological pH 7.4; hyperpolarization by ONO 10 μmol/L to −61 mV; and depolarization to −33 mV upon co‐application of ML365 10 μmol/L. A summary of resting potentials, and responses to ONO 10 μmol/L, and to ML365 10 μmol/L, is shown in Figure 3F, for WT and V221L KCNK3‐GFP. In each condition, V221L KCNK3‐GFP responses are significantly different from WT.
KCNK3 Heterodimeric Channels Are Functional Reporters of KCNK3 Heterozygosity
After demonstrating that PAH‐associated mutant KCNK3 channels are potential therapeutic targets in hPASMCs, we investigated the consequence of heterozygosity on channel function more closely. All patients harboring PAH‐associated KCNK3 mutations in our study are heterozygous at the KCNK3 locus, possessing 1 mutant and 1 WT KCNK3 channel subunit.3 KCNK3 is known to dimerize, forming functional channels from 2 subunits joined together.10 For heterozygous expression, net current is therefore a result of 3 populations of channels: homodimers of WT or mutant KCNK3, and heterodimers with 1 of each subunit. We engineered tandem‐linked KCNK3 dimers by joining 2 KCNK3 subunits with a glycine‐rich linker; by forcing assembly of mutant and/or WT KCNK3 subunits to one another, we assessed function of discrete proportions of channels (ie, heterodimers) that form in a heterozygous patient (Figure 4).
Figure 4.
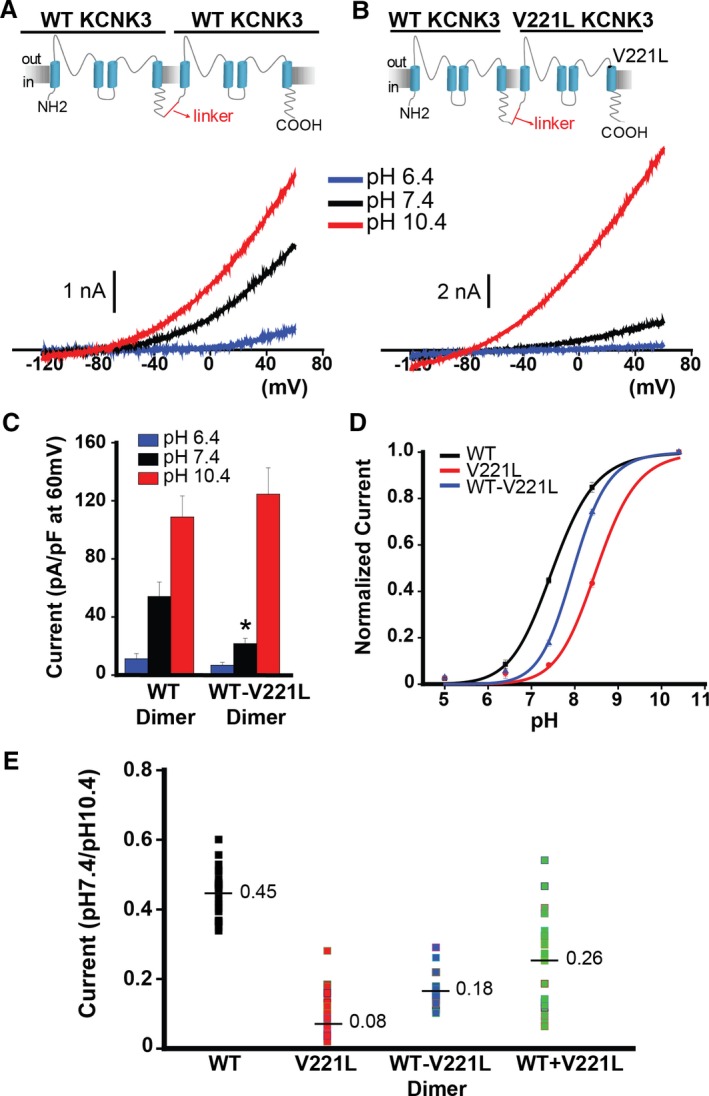
Tandem‐linked KCNK3 heterodimeric channels are functional reporters of KCNK3 heterozygosity. A and B, KCNK3 dimers were engineered by interconnecting 2 KCNK3 subunits with a glycine‐rich linker. The wildtype (WT) KCNK3 homodimer (A) and the WT−V221L KCNK3 heterodimer (B) are depicted, with sample voltage clamp recordings for each condition. Current traces at pH 6.4 (blue), 7.4 (black), and 10.4 (red) are shown. C, Summary of current densities (pA/pF at 60 mV) at pH 6.4, 7.4, and 10.4 (n=4–16 cells per pH bar). D, KCNK3 current activity is depicted for WT (black curve), V221L (red curve), and WT−V221L heterodimer (blue curve), at extracellular pH 5.0 through 10.4, with current normalized to max current at pH 10.4 (n=5–33 cells per pH value plotted; fitted by the Hill equation). E, Scatterplot of current at pH 7.4 normalized to current at pH 10.4, for WT, V221L, WT−V221L heterodimer, and WT+V221L co‐expression. Each dot represents an independent cell recording. Mean current in each condition is displayed at the horizontal line (n=16–33 cells per condition, measured at 60 mV). Bar graphs and pH curve values show means±SEM. *P<0.05 for the comparison of WT vs WT−V221L KCNK3 dimer conditions in panel C by the unpaired Student t test; (E) P<0.05 for the comparison of all KCNK3 conditions, calculated by 1‐way ANOVA (P<0.05) and post hoc Tukey test.
We tested function of mutant heterodimeric channels containing the V221L mutation. Figure 4A and 4B depict the engineered WT KCNK3 homodimer and WT−V221L mutant KCNK3 heterodimer, respectively, with sample current traces shown at extracellular pH 6.4, 7.4, and 10.4 from voltage clamp experiments. Figure 4C summarizes current density measurements (pA/pF at 60 mV) for each dimer. Significant loss of function was observed at pH 7.4 for WT−V221L heterodimeric channels. We recorded V221L‐containing KCNK3 function over a broad pH range, 5.0 through 10.4, and notably, WT−V221L KCNK3 produced an intermediate rightward shift in pH dependence (Figure 4D, blue curve), with intermediate channel activity at physiological pH 7.4, compared with WT KCNK3 (Figure 4D, black curve), and V221L KCNK3 (Figure 4D, red curve), for which we observed an extreme rightward shift in pH dependence.
The dominant‐negative impact of KCNK3 heterozygosity on channel function was quantified further. Figure 4E shows a scatterplot of individual voltage clamp recordings (n=16–33 cells per condition: WT homomeric, V221L homomeric, WT−V221L heterodimeric, and WT+V221L monomeric subunit co‐expression), with each data point representing the current recorded at physiological pH 7.4, normalized to maximal current at pH 10.4, measured at 60 mV. Loss of function was observed for all conditions containing V221L mutant channels (homomeric V221L, WT−V221L heterodimer, and co‐expression of WT+V221L). Importantly, we observed greater variation in current when co‐expressing WT+V221L channels compared with WT−V221L heterodimer expression. The co‐expression population, an equal 1:1 stoichiometric ratio of WT to V221L KCNK3 channel expressed, includes randomly assembled homodimers of WT KCNK3, V221L KCNK3, and WT−V221L heterodimers and therefore fully recapitulates heterozygous conditions. The minimal variation in current in the WT homomer, V221L homomer, and WT−V221L heterodimer lanes in Figure 4E supports the notion that we dissected current activity of discrete proportions of formed channels that would assemble in a heterozygous patient.
Pharmacological Activation of Heterodimeric Mutant KCNK3 Channels
In voltage clamp recordings, WT KCNK3 homodimers (Figure 5A) and WT−V221L mutant heterodimers (Figure 5B) were activated by ONO 10 μmol/L (shown in red), and inhibited by ML365 10 μmol/L (shown in blue), in voltage‐clamp recordings. PAH‐associated KCNK3 heterodimeric channels thus represent therapeutic targets in PAH, susceptible to pharmacological recovery. Patients with PAH caused by heterozygous KCNK3 mutation therefore possess a significant proportion of assembled KCNK3 channels (WT and mutant) that, dependent on the mutation, represent viable targets for treatment.
Figure 5.
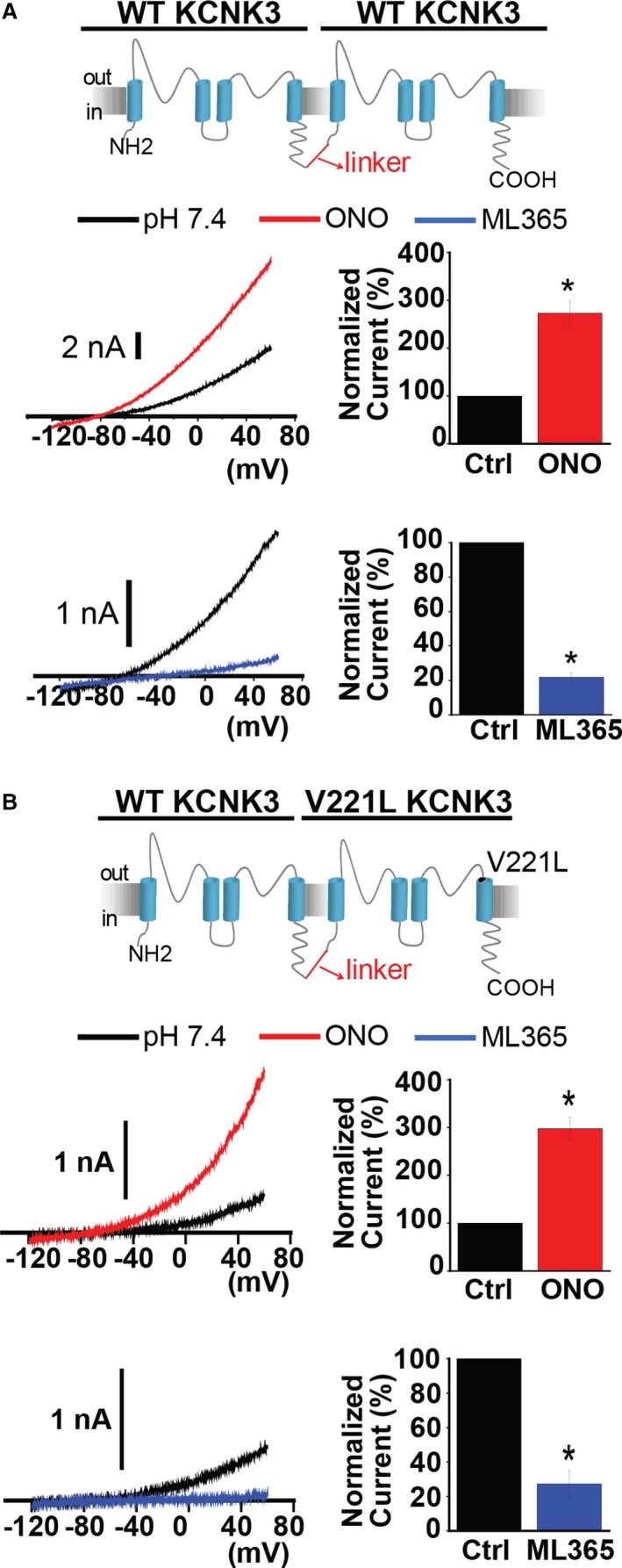
Wildtype (WT) and mutant KCNK3 dimers respond to pharmacological modulation. A, WT KCNK3 dimer (top) is activated by ONO‐RS‐082 (ONO) 10 μmol/L (red trace) and inhibited by ML365 10 μmol/L (blue trace), in current recordings from voltage clamp experiments. Control (predrug, pH 7.4) traces are shown in black. Bar graphs show fold change in current at −50 mV for the WT KCNK3 dimer, after ONO (red, n=4 cells) or ML365 (blue, n=7 cells) application. B, WT−V221L KCNK3 heterodimer (top) is activated by ONO 10 μmol/L (red trace), and inhibited by ML365 10 μmol/L (blue trace), and heterodimer channel activity was confirmed by channel activation at extracellular pH 10.4 (gray dotted traces). Control (predrug, pH 7.4) traces are shown in black. Bar graphs show fold change in current at −50 mV for the WT−V221L heterodimer, after ONO (red, n=5 cells) or ML365 (blue, n=3 cells) application. Bar graphs display mean±SEM. *P<0.05 by the paired Student t test.
As shown previously,3 cells expressing T8K or E182K KCNK3 channels are modulated by ONO 10 μmol/L. Importantly, ONO is not a KCNK3‐specific activator and produces variable responses in the presence and absence of KCNK3. As such, it is difficult to quantify the drug's effect, especially in the setting of loss‐of‐function KCNK3 mutant channels that produce small currents at baseline (Figure S2A through S2C), yet we observed variable activation of current of T8K‐, E182K‐, and V221L‐based KCNK3 constructs in the presence of ONO.
In addition to the WT−V221L KCNK3 heterodimer, the WT‐E182K KCNK3 heterodimer responds robustly to current activation by ONO 10 μmol/L, suggesting that a significant population of channels (WT and mutant heterodimeric) in patients with heterozygous KCNK3 mutations respond to pharmacological activation (Figure S2D).
KCNK3 and KCNK9 Form Functional Heterodimeric Channels
To elucidate the broader impact of KCNK3 heterozygosity on channel function, we investigated the interactions of KCNK3 with a closely related acid‐sensitive, 2‐pore domain potassium channel, KCNK9 (Figure 6A, top). Previous reports have shown that KCNK3 heterodimerization with KCNK9 results in functional channels consisting of 1 KCNK3 subunit and 1 KCNK9 subunit.11, 12, 13, 14, 15 We engineered the first known tandem‐linked human KCNK9‐KCNK3 heterodimer (Figure 6B, top).
Figure 6.
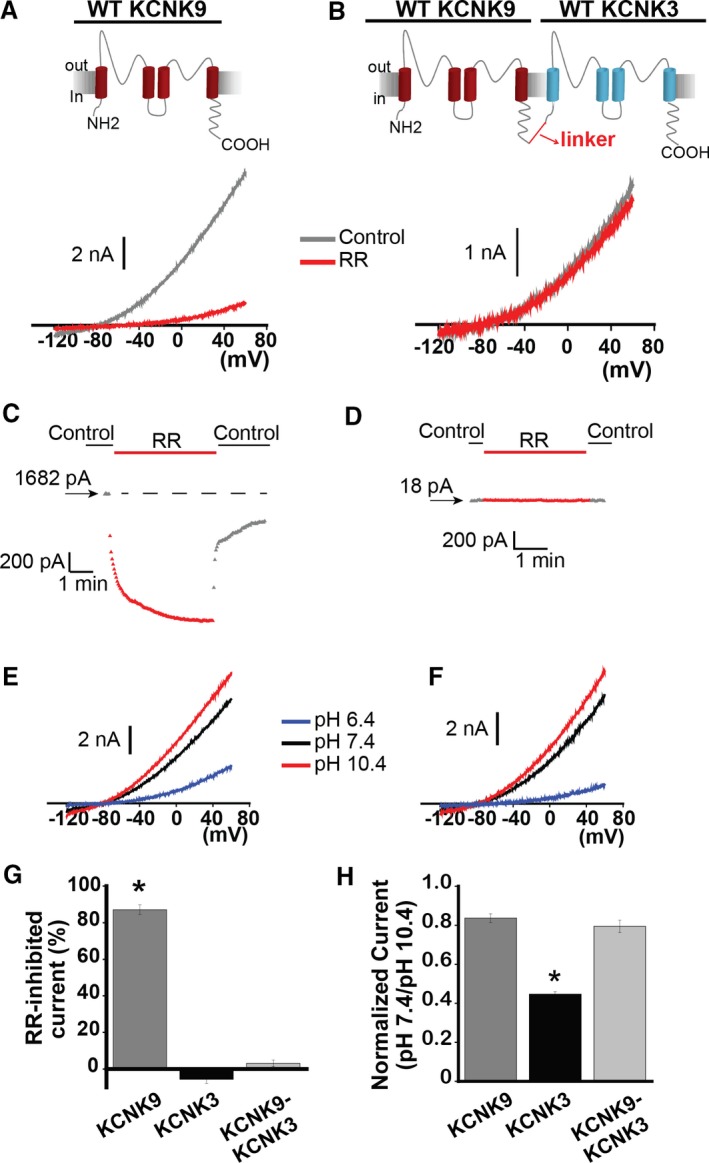
KCNK9 forms functional heterodimers with KCNK3. A, The effect of ruthenium red (RR) 10 μmol/L (red trace) on KCNK9 channels. Control trace (predrug, pH 7.4) shown in gray. B, The effect of RR 10 μmol/L (red trace) on KCNK9‐KCNK3 heterodimeric channels. Control trace (predrug, pH 7.4) shown in gray. C, Sample RR time course of action on KCNK9 in control and drug conditions, measured at −50 mV, from a starting current amplitude of 1682 pA indicated by the arrow. Horizontal dashed line is drawn at the starting level of current in control solution. D, Sample RR time course of action on KCNK9‐KCNK3 heterodimers in control and drug conditions, measured at −50 mV, from a starting current amplitude of 18 pA indicated by the arrow. E, Voltage clamp recording of KCNK9. F, Voltage clamp recording of KCNK9‐KCNK3. E and F, sample current traces at pH 6.4 (blue), 7.4 (black), and 10.4 (red) are shown. G, Summary of RR's effect on KCNK9, KCNK3, and KCNK9‐KCNK3, measured by percent‐inhibited current at −50 mV (n=5–8 cells per condition). H, Summary of mean current at pH 7.4 normalized to current at pH 10.4, measured at 60 mV, for KCNK9, KCNK3, and KCNK9‐KCNK3 (n=10–25 cells per condition). Bar graphs show mean±SEM. *P<0.05 by the paired Student t test for the comparison of control vs RR (G), and * indicates significance by 1‐way ANOVA (P<0.05) and post hoc Tukey test for the comparison of KCNK9, KCNK3, and KCNK9‐KCNK3 (H).
The true dimeric assembly of KCNK9 with KCNK3 to form KCNK9‐KCNK3 heterodimers was confirmed by ruthenium red (RR) sensitivity analysis: KCNK9 was previously shown to be inhibited by RR (10 μmol/L), while KCNK9‐KCNK3 heterodimers and KCNK3 channels were not inhibited by RR 10 μmol/L as KCNK3‐containing channels lack a necessary glutamic acid residue (E70) for RR binding and channel pore block.11 Figure 6A (bottom) shows inhibition of KCNK9 currents by RR 10 μmol/L at pH 7.4, and Figure 6B (bottom) shows no effect by RR 10 μmol/L on KCNK9‐KCNK3 heterodimer currents. Sample drug time courses are shown in Figure 6C and 6D, for KCNK9 and KCNK9‐KCNK3, respectively; RR sensitivity data are summarized in Figure 6G, for KCNK9, KCNK3, and KCNK9‐KCNK3 (and see Figure S3A through S3C). The lack of RR sensitivity of the KCNK9‐KCNK3 heterodimer confirms true assembly of KCNK9 with KCNK3, while ensuring that the tandem dimers do not combine with other tandem dimers to form functional channels; if this were the case, there would have been a RR‐sensitive component to the current produced by KCNK9‐KCNK3 expression, based on assembly of KCNK9 homomeric channels.
After verifying the formation and expression of the KCNK9‐KCNK3 tandem dimer, we evaluated the pH dependence of KCNK9‐containing channels. KCNK9 has a left‐shifted pH dependence profile compared with KCNK3, rendering it more maximally activated at physiological pH 7.4 (Figure 6E, and compare with Figure 1A). We observed increased K+ current activity at pH 7.4 for KCNK9‐KCNK3 compared with KCNK3 homomeric channels, as expected (Figure 6F, and summarized in Figure 6H).
KCNK3, But Not KCNK9, Is Expressed in Healthy and PAH Patient Lungs
KCNK9 is thought to be co‐expressed with KCNK3 in various tissues outside of the lung, including in the central nervous system, heart, and adrenal glands, and functional heterodimerization of KCNK3 with KCNK9 in multiple tissues has already been studied.6, 11, 12, 13, 17 KCNK3 is expressed in hPASMCs, while KCNK9 has been reported absent from hPASMCs.5 We performed gene expression analysis on whole human lung tissue samples from healthy (control) subjects, and from patients with familial PAH. KCNK3 expression was observed in lungs of control and familial PAH patients, while no quantifiable expression of KCNK9 was observed in control or familial PAH patient lungs (Figure 7A, top; and see Figure S3D and S3E). Therefore, of all possible KCNK3/KCNK9 channel combinations, only KCNK3 homomeric channels would be expected to form in human lung tissue (Figure 7A, bottom). KCNK9‐KCNK3 heterodimerization adds to the complexity of KCNK3 activity, particularly in PAH patients heterozygous at the KCNK3 gene locus.
Figure 7.
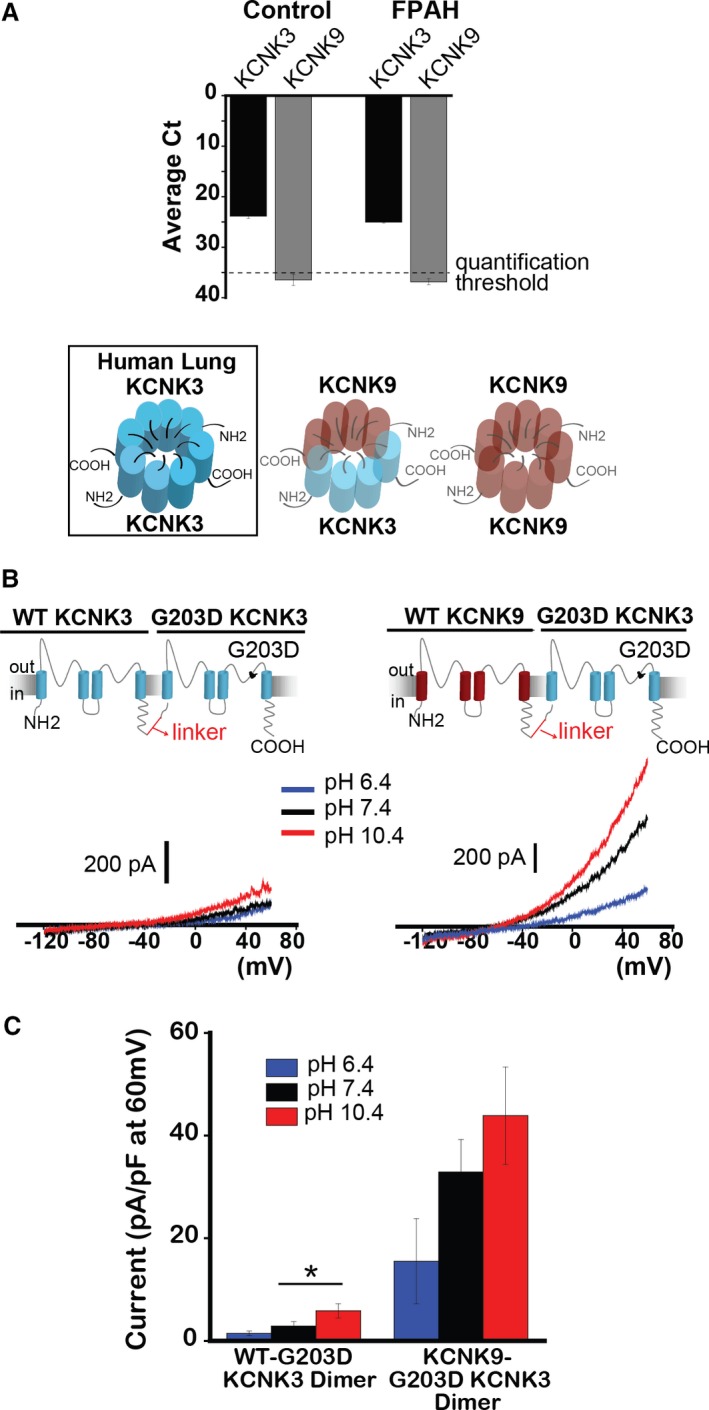
KCNK9 protects against KCNK3 dysfunction. A, Quantitative real‐time PCR analysis of human lung samples from healthy (Control) and familial PAH (FPAH) patient lungs. Expression of KCNK3 (black bars), and KCNK9 (gray bars) are compared, based on mean cycle threshold (Ct) values observed for each gene; Ct>35 indicates no quantifiable gene expression (n=5 patient lungs for each lane). Of the possible KCNK3+KCNK9 channel combinations, only KCNK3 homomeric channels (boxed, left) are predicted to form in human lungs. B, Voltage clamp recordings of the WT‐G203D KCNK3 heterodimer (left), and the KCNK9‐G203D KCNK3 heterodimer (right), showing current traces at pH 6.4 (blue), 7.4 (black), and 10.4 (red). C, Summary of current densities (pA/pF at 60 mV) at pH 6.4, 7.4, and 10.4 (n=4–14 cells per pH bar). Bar graphs show mean±SEM. *P<0.05 by the unpaired Student t test. PAH indicates pulmonary arterial hypertension; PCR, polymerase chain reaction; WT, wildtype.
KCNK9 Protects Against KCNK3 Dysfunction
The lung‐specific disease phenotype in patients with heterozygous KCNK3 mutation, despite widespread tissue expression of KCNK3, remains a puzzling phenomenon: Are lungs particularly susceptible to KCNK3 loss of function due, in part, to the absence of KCNK9? We sought to determine whether KCNK9 can recover function of even the severe mutant G203D KCNK3 channel, in order to test the hypothesis that co‐assembly of KCNK9 with KCNK3 provides protection against heterozygous KCNK3 mutation when the channels are co‐expressed, while the absence of KCNK9 in the lungs underlies the lung‐specific phenotype in the setting of heterozygous KCNK3 loss of function.
G203D KCNK3 disrupts the conserved “GxG” potassium selectivity filter amino acid sequence in 1 of the channel's 2 pore‐loop domains, a region sensitive to dominant‐negative mutations.24, 25, 26 We first engineered and studied the WT‐G203D KCNK3 heterodimer (Figure 7B, left), and indeed observed severe dominant‐negative dysfunction, as the mutant channels produced small currents across pH 6.4 through 10.4 (Figure 7B and 7C; also see Figure S4).
Next, we engineered a KCNK9‐G203D KCNK3 mutant heterodimer to determine the impact of KCNK9 assembly with G203D KCNK3 (Figure 7B, right). We observed appreciable pH‐sensitive currents, with significantly increased current density at physiological pH 7.4 and pH 10.4, compared with WT‐G203D KCNK3 heterodimeric channels (Figure 7B and 7C). KCNK9 thus assembles with mutant KCNK3 channels to produce functional recovery and provide protection against KCNK3 channel dysfunction (see also Figure S3F through S3H for modeling heterozygous co‐expression of mutant KCNK3 [eg, V221L] with KCNK9 channels).
Discussion
Our initial characterization of KCNK3 mutations associated with PAH showed loss of function for all disease‐associated mutant channels.3 Here, we have reported 5 major findings, summarized schematically in Figure 8. First, KCNK3 mutations associated with PAH harbor mutation‐specific severity of loss of function that can occur by distinct underlying mechanisms. Second, mutant and WT KCNK3 channels can be pharmacologically activated in hPASMCs to cause membrane hyperpolarization, and thus represent potential targets for PAH treatment. Third, mutant heterodimeric tandem‐linked KCNK3 channels provide a valuable tool for evaluating the functional impact of clinically relevant heterozygous KCNK3 conditions, as they report function of a substantial, discrete proportion of formed KCNK3 channels in heterozygous patients. Fourth, mutant heterodimeric KCNK3 channels can be pharmacologically targeted for activation, dependent on the mutation. Fifth, KCNK9 co‐expression and assembly with KCNK3 channels protect against KCNK3 dysfunction, which we hypothesize contributes to the lung‐specific phenotype observed clinically in patients with PAH‐associated heterozygous KCNK3 mutation.
Figure 8.
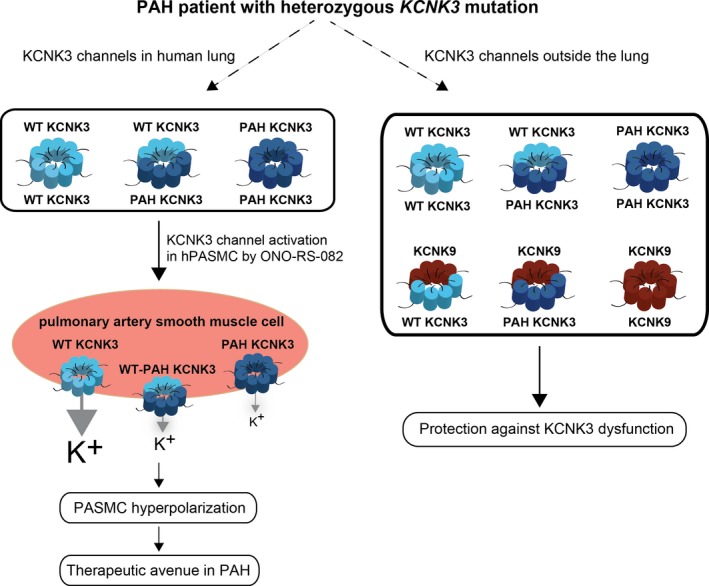
Schematic of the proposed impact of heterozygous potassium channel subfamily K member 3 (KCNK3) mutation in pulmonary arterial hypertension (PAH). Wildtype KCNK3 (light blue) and mutant (“PAH”) KCNK3 (dark blue) homomeric, and heterodimeric channels are expressed in human lung. Additional interactions of KCNK3 with KCNK9 (brown) channel subunits occur outside of the lung, protecting against KCNK3 loss of function. However, in human pulmonary artery smooth muscle cells (hPASMCs), only KCNK3 (and not KCNK9) is expressed, and the greater proportion of mutant KCNK3 channels in hPASMCs promotes membrane depolarization. ONO‐RS‐082, a KCNK3 activator, recovers function of some mutant and wildtype KCNK3 channels leading to PASMC hyperpolarization, which may represent a therapeutic avenue in PAH. PAH indicates pulmonary arterial hypertension; PASMC, pulmonary artery smooth muscle cell; WT, wildtype.
A Unique pH‐Dependent Mechanism of Mutant KCNK3 Channel Dysfunction in PAH
Potassium channel mutations confer loss of function by discrete mechanisms of varying severity. In well‐studied channelopathies, mechanisms of dysfunction include defects in trafficking, channel assembly, and electrophysiological function, dependent on the location of the mutation.27, 28
In our study, V221L KCNK3 confers a pH‐dependent mechanism of loss of function. Extracellular histidine residues lining the KCNK3 channel pore account for nearly all of the pH sensitivity of KCNK3 directly, as protonation of these residues leads to inhibition of potassium conductance.10, 29 However, other regions, including the extracellular portion of the fourth transmembrane segment of KCNK3—the location of the V221 residue—may contribute to pH sensing and channel inhibition indirectly by stabilizing the channel pore and the selectivity filter within it.29, 30 The unique properties of V221L KCNK3 can be exploited for KCNK3 structure–function studies in relation to the channel's crucial regulation by pH.
Mutant and WT KCNK3 Channels Are Pharmacological Targets in hPASMCs
Potassium channels regulate the resting membrane potential of pulmonary artery smooth muscle cells.31, 32, 33 Voltage‐gated potassium channels are involved in the hypoxic pulmonary vasoconstrictive response,34 while downregulation of Kv channel expression in PASMCs of PAH patients promotes PASMC excitability and proliferation, and excessive pulmonary arterial vasoconstriction.20, 35, 36 Potassium channel openers may provide therapeutic benefit by opposing such deleterious pulmonary arterial remodeling37; recently, pharmacological KCNK3 activation by ONO‐RS‐082 (ONO) attenuated development of pulmonary hypertension in a monocrotaline‐induced rat model.8
We therefore studied mutant and WT KCNK3 activation in human PASMCs. KCNK3 expression is nearly completely lost in cultured PASMCs, as a proliferative versus contractile smooth muscle cell phenotype predominates concomitant with loss of potassium channel expression and cell depolarization.23 Previous studies have used anandamide and A293, inhibitors of KCNK3 channel activity, to analyze the functional contributions of KCNK3 on hPASMC excitability.8, 9, 38, 39 We confirmed loss of native KCNK3 channel activity in cultured hPASMCs using a recently developed, more selective KCNK3 inhibitor, ML365 (Figure 2E through 2H and S1B).21
We exploited the properties of cultured hPASMCs to compare the relative impact of expressed mutant versus WT KCNK3 channels in a more physiological environment. The current clamp results in Figure 3 demonstrate that (1) a gradient exists for the relative contribution of mutant and WT KCNK3 to the resting potential of hPASMCs, with cells expressing mutant KCNK3 more depolarized at rest; and (2) activation of mutant or WT KCNK3 by ONO hyperpolarizes hPASMCs, highlighting PAH‐associated mutant and WT KCNK3 channels as viable pharmacological targets. Given the overexpression of KCNK3 in our system, the results are not meant to suggest that the magnitude of membrane voltage responses is physiological, but rather provide proof of principle for the graded responses of mutant and WT KCNK3 channels to pharmacologic activation in hPASMCs.
Based on the variable time course of activation of KCNK3 by ONO (seconds to minutes, not instantaneous), we hypothesize that ONO does not bind directly to the channel. More likely, ONO acts on membrane phospholipase pathways to alter KCNK3 phosphorylation. It has been shown that phospholipase C activation leads to KCNK3 inhibition,39 and endothelin‐1, a vasoconstrictor and mediator of PAH pathogenesis, inhibits KCNK3 in hPASMCs leading to depolarization, the sensitivity of which requires endothelin‐A receptors, phospholipase C, phosphatidylinositol 4,5‐bisphosphate, diacylglycerol, and protein kinase C.40 Diacylglycerol was recently identified as a direct regulator of KCNK3 downstream of activated G protein–coupled receptors.41 Ultimately, elucidating the specific pathways involved in KCNK3 activation by ONO will further cultivate KCNK3 activation as a PAH treatment paradigm.42
Mutant KCNK3 Heterodimeric Channel Function and Recovery
Mature KCNK3 channels dimerize, forming functional channels from 2 individual KCNK3 subunits that co‐assemble.10 In this study, tandem‐linked KCNK3 heterodimers served as a functional reporter of a discrete and substantial population of formed channels in heterozygous patients. The dominant‐negative phenotype conferred by the V221L KCNK3 mutation did not prevent pharmacological recovery of function: heterodimeric WT−V221L KCNK3 channels responded to pharmacological activation by ONO. As KCNK3 downregulation occurs in PAH irrespective of a patient's KCNK3 genetic status,8 KCNK3 activation indeed represents a more generalizable therapeutic approach in pulmonary hypertension from any cause.
While appreciable activation of homomeric mutant channels with marked loss of function at baseline was difficult to discern, we have demonstrated robust current activation of mutant heterodimeric KCNK3 channels by ONO (Figure 5; and see Figure S2). Thus, in PAH patients heterozygous at the KCNK3 gene locus, the majority of formed channels—WT homodimeric and mutant heterodimeric KCNK3 channels—may respond markedly to pharmacological activation and represent targetable therapeutic substrates.
KCNK9 Minimizes the Impact of KCNK3 Loss of Function
Interestingly, none of the PAH patients in our cohort with heterozygous KCNK3 mutations possessed a primary clinical phenotype outside of the lung, despite widespread KCNK3 expression in the body.4 KCNK9, which has been shown to heterodimerize with KCNK3,11 is absent from human lungs of healthy controls and PAH patients (Figure 7A; and see Figure S3D and S3E), but assembles with KCNK3 in tissues where both KCNK9 and KCNK3 are expressed, leading to diversity of channel currents.12, 13, 14 We observed that KCNK9 heterodimerization with WT and G203D KCNK3 increased current activity at physiological pH 7.4 compared with equivalent KCNK3‐only based channels, and suggest that KCNK9 may protect against KCNK3 dysfunction.
We hypothesize that the co‐expression of KCNK9 with KCNK3 in tissues outside the lung provides redundancy of channel function and protection against KCNK3 mutations of varying severity, sparing tissues outside the lung of disease. Meanwhile, the absence of KCNK9 in the lung would leave PASMCs susceptible to excessive depolarization, proliferation, and vasoconstriction in the setting of KCNK3 dysfunction.
Incomplete penetrance was observed in individuals with PAH‐associated KCNK3 mutations.3 Conceivably, KCNK9 provides greater protection in some individuals than others. Some patients with heterozygous KCNK3 mutation may develop PAH at a later age of onset, or not develop PAH at all, dependent, in part, on KCNK9 channel activity.
Limitations
The impact of KCNK3 heterozygosity at the pulmonary arterial level remains undetermined. Because of its altered pH dependence easily discerned in electrophysiological studies, the V221L KCNK3 mutation represents a model mutation to incorporate into a rat model (eg, using CRISPR knock‐in technology) to recapitulate KCNK3 heterozygosity and evaluate the pathophysiological consequences in the lungs and outside of them.8, 43
Uncovering the cellular pathways involved in KCNK3 activation by ONO has broader implications than for PAH pathogenesis and treatment alone. For instance, enhanced KCNK3 activity was recently demonstrated in atrial myocytes of patients with chronic atrial fibrillation, leading to shortening of the action potential duration.44 Mechanistic understanding of ONO's regulation of KCNK3 would aid the development of more selective pharmacological modulators of channel activity.
Conclusions
We have demonstrated, for the first time, that KCNK3 mutations associated with PAH harbor differing underlying mechanisms of loss of function of varying severity; that mutant KCNK3 channels represent viable therapeutic targets in PAH via activation by ONO‐RS‐082 in pulmonary artery smooth muscle cells; that engineered mutant heterodimeric KCNK3 channels report function of a substantial proportion of formed KCNK3 channels in heterozygous patients, and that heterodimeric KCNK3 channels can be pharmacologically activated; and lastly, that heterodimerization of KCNK9 with KCNK3 can serve a protective role to minimize the impact of heterozygous KCNK3 loss of function, which may underlie the PAH‐specific phenotype observed clinically in patients with heterozygous KCNK3 mutation.
Sources of Funding
Funding support was provided by the National Heart, Lung, and Blood Institute (NHLBI) F30 HL129656. Funding for the PHBI is provided under an NHLBI R24 grant, #R24HL123767, and by the Cardiovascular Medical Research and Education Fund (CMREF).
Disclosures
None.
Supporting information
Data S1. Supplemental Methods.
Figure S1. KCNK3‐GFP expression and activity in hPASMCs. A, Summary of current clamp results of KCNK3‐GFP expressed in hPASMCs, with mean membrane potentials (mV) measured at pH 6.4 (blue), pH 7.4 (black), and pH 10.4 (red) shown (n=2 to 4 cells per pH value). B, Comparison of the ML365‐sensitive current (pA/pF at −50 mV, pH 7.4), in hPASMCs expressing KCNK3‐GFP (n=3 cells) vs no transfection (n=6 cells). C, Gradient of current expression (pA/pF at 60 mV, pH 7.4) in hPASMCs expressing KCNK3‐GFP (n=20 cells), V221L KCNK3‐GFP (n=7 cells), and GFP only (n=3 cells). D, Sample current trace of V221L KCNK3‐GFP in hPASMC at pH 7.4 (black) and pH 8.4 (red). E, Summary of V221L KCNK3 expression in COS7 cells vs V221L KCNK3‐GFP expression in hPASMCs (n=4 to 7 cells per lane), showing greater current activity of expressed channels in hPASMCs (pA/pF at 60 mV, pH 7.4 in black, pH 8.4 in red). Data are represented as means±SEM. *P<0.05 by the unpaired Student t test.
Figure S2. The effect of ONO‐RS‐082 on homomeric and heterodimeric mutant KCNK3 channels associated with PAH. Voltage clamp recordings in COS7 cells are depicted. A through D, The effect of ONO‐RS‐082 10 μmol/L (red traces) at extracellular pH 7.4 on currents from cells expressing T8K KCNK3 (A); V221L KCNK3 (B); E182K KCNK3 (C); and heterodimeric WT‐E182K KCNK3 (D). Control (predrug) conditions at extracellular pH 7.4 (black traces) are shown for each recording.
Figure S3. The impact of KCNK9 expression and current activity on KCNK3 function. A, The effect of ruthenium red (RR) 10 μmol/L (red trace) on KCNK3 channels in COS7 cells is shown. Control trace (predrug, pH 7.4) is depicted in gray. B, Sample RR time course of action on KCNK3 in control and drug conditions, measured at −50 mV, from a starting current amplitude of 196 pA indicated by the arrow. C, The effect of RR on KCNK9, KCNK3, and KCNK9‐KCNK3 channels is summarized, showing fold change in current at −50 mV (n=5–8 cells per condition). D, Quantitative real‐time PCR analysis of human lung samples from healthy (Control) and congenital cardiac defect–associated PAH (APAH) patient lungs. Expression of KCNK3 (black bars) and KCNK9 (gray bars) are compared, based on mean cycle threshold (Ct) values observed for each gene; Ct>35 indicates no quantifiable gene expression (n=5 patient lungs for each lane). Bars show mean±SEM. E, Fold difference in KCNK3 gene expression, calculated by the 2−▵▵Ct method, in FPAH and APAH vs Control patient lungs. No significant (N.S.) fold changes were observed compared with control. F and G, Co‐expression of KCNK9 with WT KCNK3 channels (F), and KCNK9 with V221L KCNK3 channels (G) with sample voltage clamp recordings shown for each condition at pH 6.4 (blue), pH 7.4 (black), and pH 10.4 (red). H, Summary of current activity for co‐expression of KCNK9 with WT KCNK3 (left) vs V221L KCNK3 (right). Current at pH 6.4 (blue) and pH 7.4 (black) is normalized to maximum current at pH 10.4 (n=5–8 cells per lane). Experiments were performed in COS7 cells. Bar graphs show mean±SEM. *P<0.05 by the paired Student t test for the comparison of control vs ruthenium red conditions in C; *P<0.05 by the unpaired Student t test in (H).
Figure S4. KCNK3 heterodimeric GFP fusion dimer confirms the more severe loss of function in G203D vs V221L‐containing KCNK3 channels. A, A WT‐G203D KCNK3‐GFP fusion heterodimer was engineered by interconnecting 2 KCNK3 subunits with a glycine‐rich linker, and inserting a C‐terminal GFP tag. Given the relative severity of G203D KCNK3 dysfunction, the GFP fusion construct ensured channel expression in fluorescent cells studied. Sample traces from a voltage clamp recording are shown at extracellular pH 5 (blue), pH 7.4 (black), and pH 10.4 (red), revealing small currents across the pH range. B, Summary of current densities (pA/pF at 60 mV) at pH 5 (blue), pH 7.4 (black), and pH 10.4 (red) for the WT−V221L KCNK3 heterodimer vs the WT‐G203D KCNK3‐GFP heterodimer (n=6 to 16 cells per pH bar). Bars show mean±SEM. *P<0.05 at pH 7.4 and pH 10.4, by the unpaired Student t test.
Acknowledgments
We thank the PAH patients and their families for their generous contribution. We thank Mark Geraci for data/tissue samples provided through the Pulmonary Hypertension Breakthrough Initiative (PHBI). We thank Monica Goldklang and the D'armiento laboratory at Columbia University for assisting with gene expression studies. Thanks to Jenny Rao for laboratory technical support.
(J Am Heart Assoc. 2017;6:e006465 DOI: 10.1161/JAHA.117.006465.)28889099
References
- 1. Galie N, Humbert M, Vachiery JL, Gibbs S, Lang I, Torbicki A, Simonneau G, Peacock A, Vonk Noordegraaf A, Beghetti M, Ghofrani A, Gomez Sanchez MA, Hansmann G, Klepetko W, Lancellotti P, Matucci M, McDonagh T, Pierard LA, Trindade PT, Zompatori M, Hoeper M, Aboyans V, Vaz Carneiro A, Achenbach S, Agewall S, Allanore Y, Asteggiano R, Paolo Badano L, Albert Barbera J, Bouvaist H, Bueno H, Byrne RA, Carerj S, Castro G, Erol C, Falk V, Funck‐Brentano C, Gorenflo M, Granton J, Iung B, Kiely DG, Kirchhof P, Kjellstrom B, Landmesser U, Lekakis J, Lionis C, Lip GY, Orfanos SE, Park MH, Piepoli MF, Ponikowski P, Revel MP, Rigau D, Rosenkranz S, Voller H, Luis Zamorano J. 2015 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension: the Joint Task Force for the Diagnosis and Treatment of Pulmonary Hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS): endorsed by: association for European Paediatric and Congenital Cardiology (AEPC), International Society for Heart and Lung Transplantation (ISHLT). Eur Heart J. 2016;37:67–119. [DOI] [PubMed] [Google Scholar]
- 2. Ma L, Chung WK. The role of genetics in pulmonary arterial hypertension. J Pathol. 2017;241:273–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Ma L, Roman‐Campos D, Austin ED, Eyries M, Sampson KS, Soubrier F, Germain M, Tregouet DA, Borczuk A, Rosenzweig EB, Girerd B, Montani D, Humbert M, Loyd JE, Kass RS, Chung WK. A novel channelopathy in pulmonary arterial hypertension. N Engl J Med. 2013;369:351–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Duprat F, Lesage F, Fink M, Reyes R, Heurteaux C, Lazdunski M. TASK, a human background K+ channel to sense external pH variations near physiological pH. EMBO J. 1997;16:5464–5471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Olschewski A, Li Y, Tang B, Hanze J, Eul B, Bohle RM, Wilhelm J, Morty RE, Brau ME, Weir EK, Kwapiszewska G, Klepetko W, Seeger W, Olschewski H. Impact of TASK‐1 in human pulmonary artery smooth muscle cells. Circ Res. 2006;98:1072–1080. [DOI] [PubMed] [Google Scholar]
- 6. Enyedi P, Czirjak G. Molecular background of leak K+ currents: two‐pore domain potassium channels. Physiol Rev. 2010;90:559–605. [DOI] [PubMed] [Google Scholar]
- 7. Gurney AM, Osipenko ON, MacMillan D, McFarlane KM, Tate RJ, Kempsill FE. Two‐pore domain K channel, TASK‐1, in pulmonary artery smooth muscle cells. Circ Res. 2003;93:957–964. [DOI] [PubMed] [Google Scholar]
- 8. Antigny F, Hautefort A, Meloche J, Belacel‐Ouari M, Manoury B, Rucker‐Martin C, Pechoux C, Potus F, Nadeau V, Tremblay E, Ruffenach G, Bourgeois A, Dorfmuller P, Breuils‐Bonnet S, Fadel E, Ranchoux B, Jourdon P, Girerd B, Montani D, Provencher S, Bonnet S, Simonneau G, Humbert M, Perros F. Potassium channel subfamily K member 3 (KCNK3) contributes to the development of pulmonary arterial hypertension. Circulation. 2016;133:1371–1385. [DOI] [PubMed] [Google Scholar]
- 9. Gardener MJ, Johnson IT, Burnham MP, Edwards G, Heagerty AM, Weston AH. Functional evidence of a role for two‐pore domain potassium channels in rat mesenteric and pulmonary arteries. Br J Pharmacol. 2004;142:192–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Lopes CM, Zilberberg N, Goldstein SA. Block of Kcnk3 by protons. Evidence that 2‐P‐domain potassium channel subunits function as homodimers. J Biol Chem. 2001;276:24449–24452. [DOI] [PubMed] [Google Scholar]
- 11. Czirjak G, Enyedi P. Formation of functional heterodimers between the TASK‐1 and TASK‐3 two‐pore domain potassium channel subunits. J Biol Chem. 2002;277:5426–5432. [DOI] [PubMed] [Google Scholar]
- 12. Berg AP, Talley EM, Manger JP, Bayliss DA. Motoneurons express heteromeric TWIK‐related acid‐sensitive K+ (TASK) channels containing TASK‐1 (KCNK3) and TASK‐3 (KCNK9) subunits. J Neurosci. 2004;24:6693–6702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Rinne S, Kiper AK, Schlichthorl G, Dittmann S, Netter MF, Limberg SH, Silbernagel N, Zuzarte M, Moosdorf R, Wulf H, Schulze‐Bahr E, Rolfes C, Decher N. TASK‐1 and TASK‐3 may form heterodimers in human atrial cardiomyocytes. J Mol Cell Cardiol. 2015;81:71–80. [DOI] [PubMed] [Google Scholar]
- 14. Kang D, Han J, Talley EM, Bayliss DA, Kim D. Functional expression of TASK‐1/TASK‐3 heteromers in cerebellar granule cells. J Physiol. 2004;554:64–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kim D, Cavanaugh EJ, Kim I, Carroll JL. Heteromeric TASK‐1/TASK‐3 is the major oxygen‐sensitive background K+ channel in rat carotid body glomus cells. J Physiol. 2009;587:2963–2975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Rajan S, Wischmeyer E, Xin Liu G, Preisig‐Muller R, Daut J, Karschin A, Derst C. TASK‐3, a novel tandem pore domain acid‐sensitive K+ channel. An extracellular histiding as pH sensor. J Biol Chem. 2000;275:16650–16657. [DOI] [PubMed] [Google Scholar]
- 17. Kim Y, Bang H, Kim D. TASK‐3, a new member of the tandem pore K(+) channel family. J Biol Chem. 2000;275:9340–9347. [DOI] [PubMed] [Google Scholar]
- 18. Stearman RS, Cornelius AR, Lu X, Conklin DS, Del Rosario MJ, Lowe AM, Elos MT, Fettig LM, Wong RE, Hara N, Cogan JD, Phillips JA III, Taylor MR, Graham BB, Tuder RM, Loyd JE, Geraci MW. Functional prostacyclin synthase promoter polymorphisms. Impact in pulmonary arterial hypertension. Am J Respir Crit Care Med. 2014;189:1110–1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Brevnova EE, Platoshyn O, Zhang S, Yuan JX. Overexpression of human KCNA5 increases IK V and enhances apoptosis. Am J Physiol Cell Physiol. 2004;287:C715–C722. [DOI] [PubMed] [Google Scholar]
- 20. Remillard CV, Tigno DD, Platoshyn O, Burg ED, Brevnova EE, Conger D, Nicholson A, Rana BK, Channick RN, Rubin LJ, O'Connor DT, Yuan JX. Function of Kv1.5 channels and genetic variations of KCNA5 in patients with idiopathic pulmonary arterial hypertension. Am J Physiol Cell Physiol. 2007;292:C1837–C1853. [DOI] [PubMed] [Google Scholar]
- 21. Zou B, Flaherty DP, Simpson DS, Maki BE, Miller MR, Shi J, Wu M, McManus OB, Golden JE, Aube J, Li M. ML365: Development of Bis‐Amides as Selective Inhibitors of the KCNK3/TASK1 Two Pore Potassium Channel Probe Reports from the NIH Molecular Libraries Program Bethesda (MD); 2010. [PubMed]
- 22. Skarsfeldt MA, Jepps TA, Bomholtz SH, Abildgaard L, Sorensen US, Gregers E, Svendsen JH, Diness JG, Grunnet M, Schmitt N, Olesen SP, Bentzen BH. pH‐dependent inhibition of K(2)P3.1 prolongs atrial refractoriness in whole hearts. Pflugers Arch. 2016;468:643–654. [DOI] [PubMed] [Google Scholar]
- 23. Manoury B, Etheridge SL, Reid J, Gurney AM. Organ culture mimics the effects of hypoxia on membrane potential, K(+) channels and vessel tone in pulmonary artery. Br J Pharmacol. 2009;158:848–861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Yuill KH, Stansfeld PJ, Ashmole I, Sutcliffe MJ, Stanfield PR. The selectivity, voltage‐dependence and acid sensitivity of the tandem pore potassium channel TASK‐1: contributions of the pore domains. Pflugers Arch. 2007;455:333–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Doyle DA, Morais Cabral J, Pfuetzner RA, Kuo A, Gulbis JM, Cohen SL, Chait BT, MacKinnon R. The structure of the potassium channel: molecular basis of K+ conduction and selectivity. Science. 1998;280:69–77. [DOI] [PubMed] [Google Scholar]
- 26. Pei L, Wiser O, Slavin A, Mu D, Powers S, Jan LY, Hoey T. Oncogenic potential of TASK3 (Kcnk9) depends on K+ channel function. Proc Natl Acad Sci USA. 2003;100:7803–7807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Vandenberg JI, Perry MD, Perrin MJ, Mann SA, Ke Y, Hill AP. hERG K(+) channels: structure, function, and clinical significance. Physiol Rev. 2012;92:1393–1478. [DOI] [PubMed] [Google Scholar]
- 28. Nerbonne JM, Kass RS. Molecular physiology of cardiac repolarization. Physiol Rev. 2005;85:1205–1253. [DOI] [PubMed] [Google Scholar]
- 29. Yuill K, Ashmole I, Stanfield PR. The selectivity filter of the tandem pore potassium channel TASK‐1 and its pH‐sensitivity and ionic selectivity. Pflugers Arch. 2004;448:63–69. [DOI] [PubMed] [Google Scholar]
- 30. Morton MJ, O'Connell AD, Sivaprasadarao A, Hunter M. Determinants of pH sensing in the two‐pore domain K(+) channels TASK‐1 and ‐2. Pflugers Arch. 2003;445:577–583. [DOI] [PubMed] [Google Scholar]
- 31. Platoshyn O, Remillard CV, Fantozzi I, Mandegar M, Sison TT, Zhang S, Burg E, Yuan JX. Diversity of voltage‐dependent K+ channels in human pulmonary artery smooth muscle cells. Am J Physiol Lung Cell Mol Physiol. 2004;287:L226–L238. [DOI] [PubMed] [Google Scholar]
- 32. Tennant BP, Cui Y, Tinker A, Clapp LH. Functional expression of inward rectifier potassium channels in cultured human pulmonary smooth muscle cells: evidence for a major role of Kir2.4 subunits. J Membr Biol. 2006;213:19–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Cui Y, Tran S, Tinker A, Clapp LH. The molecular composition of K(ATP) channels in human pulmonary artery smooth muscle cells and their modulation by growth. Am J Respir Cell Mol Biol. 2002;26:135–143. [DOI] [PubMed] [Google Scholar]
- 34. Archer SL, Souil E, Dinh‐Xuan AT, Schremmer B, Mercier JC, El Yaagoubi A, Nguyen‐Huu L, Reeve HL, Hampl V. Molecular identification of the role of voltage‐gated K+ channels, Kv1.5 and Kv2.1, in hypoxic pulmonary vasoconstriction and control of resting membrane potential in rat pulmonary artery myocytes. J Clin Invest. 1998;101:2319–2330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Yuan JX, Aldinger AM, Juhaszova M, Wang J, Conte JV Jr, Gaine SP, Orens JB, Rubin LJ. Dysfunctional voltage‐gated K+ channels in pulmonary artery smooth muscle cells of patients with primary pulmonary hypertension. Circulation. 1998;98:1400–1406. [DOI] [PubMed] [Google Scholar]
- 36. Yuan XJ, Wang J, Juhaszova M, Gaine SP, Rubin LJ. Attenuated K+ channel gene transcription in primary pulmonary hypertension. Lancet. 1998;351:726–727. [DOI] [PubMed] [Google Scholar]
- 37. Morecroft I, Murray A, Nilsen M, Gurney AM, MacLean MR. Treatment with the Kv7 potassium channel activator flupirtine is beneficial in two independent mouse models of pulmonary hypertension. Br J Pharmacol. 2009;157:1241–1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Maingret F, Patel AJ, Lazdunski M, Honore E. The endocannabinoid anandamide is a direct and selective blocker of the background K(+) channel TASK‐1. EMBO J. 2001;20:47–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Schiekel J, Lindner M, Hetzel A, Wemhoner K, Renigunta V, Schlichthorl G, Decher N, Oliver D, Daut J. The inhibition of the potassium channel TASK‐1 in rat cardiac muscle by endothelin‐1 is mediated by phospholipase C. Cardiovasc Res. 2013;97:97–105. [DOI] [PubMed] [Google Scholar]
- 40. Tang B, Li Y, Nagaraj C, Morty RE, Gabor S, Stacher E, Voswinckel R, Weissmann N, Leithner K, Olschewski H, Olschewski A. Endothelin‐1 inhibits background two‐pore domain channel TASK‐1 in primary human pulmonary artery smooth muscle cells. Am J Respir Cell Mol Biol. 2009;41:476–483. [DOI] [PubMed] [Google Scholar]
- 41. Wilke BU, Lindner M, Greifenberg L, Albus A, Kronimus Y, Bunemann M, Leitner MG, Oliver D. Diacylglycerol mediates regulation of TASK potassium channels by Gq‐coupled receptors. Nat Commun. 2014;5:5540. [DOI] [PubMed] [Google Scholar]
- 42. Olschewski A. Targeting TASK‐1 channels as a therapeutic approach. Adv Exp Med Biol. 2010;661:459–473. [DOI] [PubMed] [Google Scholar]
- 43. Manoury B, Lamalle C, Oliveira R, Reid J, Gurney AM. Contractile and electrophysiological properties of pulmonary artery smooth muscle are not altered in TASK‐1 knockout mice. J Physiol. 2011;589:3231–3246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Schmidt C, Wiedmann F, Voigt N, Zhou XB, Heijman J, Lang S, Albert V, Kallenberger S, Ruhparwar A, Szabo G, Kallenbach K, Karck M, Borggrefe M, Biliczki P, Ehrlich JR, Baczko I, Lugenbiel P, Schweizer PA, Donner BC, Katus HA, Dobrev D, Thomas D. Upregulation of K(2P)3.1 K+ current causes action potential shortening in patients with chronic atrial fibrillation. Circulation. 2015;132:82–92. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1. Supplemental Methods.
Figure S1. KCNK3‐GFP expression and activity in hPASMCs. A, Summary of current clamp results of KCNK3‐GFP expressed in hPASMCs, with mean membrane potentials (mV) measured at pH 6.4 (blue), pH 7.4 (black), and pH 10.4 (red) shown (n=2 to 4 cells per pH value). B, Comparison of the ML365‐sensitive current (pA/pF at −50 mV, pH 7.4), in hPASMCs expressing KCNK3‐GFP (n=3 cells) vs no transfection (n=6 cells). C, Gradient of current expression (pA/pF at 60 mV, pH 7.4) in hPASMCs expressing KCNK3‐GFP (n=20 cells), V221L KCNK3‐GFP (n=7 cells), and GFP only (n=3 cells). D, Sample current trace of V221L KCNK3‐GFP in hPASMC at pH 7.4 (black) and pH 8.4 (red). E, Summary of V221L KCNK3 expression in COS7 cells vs V221L KCNK3‐GFP expression in hPASMCs (n=4 to 7 cells per lane), showing greater current activity of expressed channels in hPASMCs (pA/pF at 60 mV, pH 7.4 in black, pH 8.4 in red). Data are represented as means±SEM. *P<0.05 by the unpaired Student t test.
Figure S2. The effect of ONO‐RS‐082 on homomeric and heterodimeric mutant KCNK3 channels associated with PAH. Voltage clamp recordings in COS7 cells are depicted. A through D, The effect of ONO‐RS‐082 10 μmol/L (red traces) at extracellular pH 7.4 on currents from cells expressing T8K KCNK3 (A); V221L KCNK3 (B); E182K KCNK3 (C); and heterodimeric WT‐E182K KCNK3 (D). Control (predrug) conditions at extracellular pH 7.4 (black traces) are shown for each recording.
Figure S3. The impact of KCNK9 expression and current activity on KCNK3 function. A, The effect of ruthenium red (RR) 10 μmol/L (red trace) on KCNK3 channels in COS7 cells is shown. Control trace (predrug, pH 7.4) is depicted in gray. B, Sample RR time course of action on KCNK3 in control and drug conditions, measured at −50 mV, from a starting current amplitude of 196 pA indicated by the arrow. C, The effect of RR on KCNK9, KCNK3, and KCNK9‐KCNK3 channels is summarized, showing fold change in current at −50 mV (n=5–8 cells per condition). D, Quantitative real‐time PCR analysis of human lung samples from healthy (Control) and congenital cardiac defect–associated PAH (APAH) patient lungs. Expression of KCNK3 (black bars) and KCNK9 (gray bars) are compared, based on mean cycle threshold (Ct) values observed for each gene; Ct>35 indicates no quantifiable gene expression (n=5 patient lungs for each lane). Bars show mean±SEM. E, Fold difference in KCNK3 gene expression, calculated by the 2−▵▵Ct method, in FPAH and APAH vs Control patient lungs. No significant (N.S.) fold changes were observed compared with control. F and G, Co‐expression of KCNK9 with WT KCNK3 channels (F), and KCNK9 with V221L KCNK3 channels (G) with sample voltage clamp recordings shown for each condition at pH 6.4 (blue), pH 7.4 (black), and pH 10.4 (red). H, Summary of current activity for co‐expression of KCNK9 with WT KCNK3 (left) vs V221L KCNK3 (right). Current at pH 6.4 (blue) and pH 7.4 (black) is normalized to maximum current at pH 10.4 (n=5–8 cells per lane). Experiments were performed in COS7 cells. Bar graphs show mean±SEM. *P<0.05 by the paired Student t test for the comparison of control vs ruthenium red conditions in C; *P<0.05 by the unpaired Student t test in (H).
Figure S4. KCNK3 heterodimeric GFP fusion dimer confirms the more severe loss of function in G203D vs V221L‐containing KCNK3 channels. A, A WT‐G203D KCNK3‐GFP fusion heterodimer was engineered by interconnecting 2 KCNK3 subunits with a glycine‐rich linker, and inserting a C‐terminal GFP tag. Given the relative severity of G203D KCNK3 dysfunction, the GFP fusion construct ensured channel expression in fluorescent cells studied. Sample traces from a voltage clamp recording are shown at extracellular pH 5 (blue), pH 7.4 (black), and pH 10.4 (red), revealing small currents across the pH range. B, Summary of current densities (pA/pF at 60 mV) at pH 5 (blue), pH 7.4 (black), and pH 10.4 (red) for the WT−V221L KCNK3 heterodimer vs the WT‐G203D KCNK3‐GFP heterodimer (n=6 to 16 cells per pH bar). Bars show mean±SEM. *P<0.05 at pH 7.4 and pH 10.4, by the unpaired Student t test.