

Abstract
Background
With chronic ischemia after myocardial infarction, the resulting scar tissue result in electrical and structural remodeling vulnerable to an arrhythmogenic substrate. The cholinergic anti‐inflammatory pathway elicited by vagal nerve via α7 nicotinic acetylcholine receptors (α7‐nAChR) can modulate local and systemic inflammatory responses. Here, we aimed to clarify a novel mechanism for the antiarrhythmogenic properties of vagal nerve during the ischemic cardiomyopathy (ICM).
Methods and Results
Left anterior descending artery of adult male Sprague‐Dawley rats was ligated for 4 weeks to develop ICM. Western blot revealed that eliciting the cholinergic anti‐inflammatory pathway by nicotine treatment showed a significant reduction in the amounts of collagens, cytokines, and other inflammatory mediators in the left ventricular infarcted border zone via inhibited NF‐κB activation, whereas it increased the phosphorylated connexin 43. Vagotomy inhibited the anti‐inflammatory, anti‐fibrosis, and anti‐arrhythmogenic effect of nicotine administration. And immunohistochemistry confirmed that the nicotine administration‐induced increase of connexin 43 was located in intercellular junctions. Furthermore nicotine treatment suppressed NF‐κB activation in lipopolysaccharide‐stimulated RAW264.7 cells, and α‐bungarotoxin (an α7‐nAChR selective antagonist) partly inhibited the nicotine‐treatment effect. In addition, 4‐week nicotine administration slightly improved the cardiac function, increased cardiac parasympathetic tone, decreased the prolonged QTc, and decreased the arrhythmia score of programmed electric stimulation‐induced ventricular arrhythmia.
Conclusions
Eliciting the cholinergic anti‐inflammatory pathway exerts anti‐arrhythmogenic effects against ICM‐induced ventricular arrhythmia accompanied by downregulation of cytokines, downgenerating of collagens, decrease in sympathetic/parasympathetic ratio, and prevention of the loss of phosphorylated connexin 43 during ICM. Our findings may suggest a promising therapy for the generation of ICM‐induced ventricular arrhythmia by eliciting the cholinergic anti‐inflammatory pathway.
Keywords: acetylcholine, cholinergic anti‐inflammatory pathway, ischemic cardiomyopathy, vagus nerve, ventricular arrhythmia
Subject Categories: Arrhythmias, Electrophysiology, Ventricular Fibrillation
Clinical Perspective
What Is New?
We found that regional anti‐inflammatory and antifibrotic properties, along with improving cardiac autonomic function, are responsible for the antiarrhythmogenic effects of the cholinergic anti‐inflammatory pathway in ischemic cardiomyopathy.
What Are the Clinical Implications?
Based on our results, preventing the progression of regional inflammation and fibrosis accompanied by decreases in the sympathetic‐to‐parasympathetic ratio may protect against heart failure and ventricular arrhythmias in patients with ischemic cardiomyopathy.
Introduction
Ventricular tachyarrhythmia is a common complication among patients surviving myocardial infarction (MI)1 and is the major cause of death in the United States, contributing 15% to 20% of deaths worldwide.2
The scar tissue generation and long‐term existing cytokine result in structural and electrical remodeling during ischemic cardiomyopathy (ICM) and causing an arrhythmogenic substrate eventually. In particular, the intercellular electrical uncoupling of ventricular cardiomyocytes also represents a major role in arrhythmogenesis during chronic ischemic heart disease.3 The gap‐junctional protein connexin 43 (Cx43) coordinates with cardiomyocyte activation and allows intercellular communication,4 whereas its downregulation and nonuniformity increases the risk of ventricular arrhythmia directly.5 Inhibiting interleukin‐1β (IL‐1β) following MI increases the expression of Cx43, thereby representing a novel antiarrhythmic property.6 Polymorphonuclear neutrophils accumulate in ischemic myocardium, releasing myeloperoxidase, promoting inflammation, increasing myocardial collagen, and remodeling myocardium ultimately. Consequently, the degradation of the Cx43 exhibits ventricular arrhythmogenesis.7 Moreover, the nerve regeneration that increases the sympathetic nerve density and increases nerve sprouting abnormally after MI has a correlation with the occurrence of ventricular arrhythmia and sudden cardiac death.8
Activating the efferent vagal nerve fibers can modulate local and systemic inflammatory responses via the cholinergic anti‐inflammatory pathway (CAP).9 Eliciting the CAP by vagal electrical stimulation has been reported to have a protective role against fatal ventricular arrhythmia in myocardial ischemia/reperfusion animal models,10, 11 and vagal electrical stimulation also improves chronic heart failure in rats by increasing survival rate in long‐term myocardial infarction.12 It is well established that the beneficial effects of the CAP are mediated by acetylcholine receptors.13, 14 In addition, the α7‐nicotinic acetylcholine receptor (α7‐nAChR) subunit, which plays a crucial role in CAP, is expressed widely on the surface of inflammatory cells, and it significantly influences the anti‐inflammatory effect of the vagus nerve.15
In this study, we hypothesized and examined potential cardioprotective effects of CAP during ICM and a potential novel therapeutic effect that could modify cardiac autonomic neural remodeling and exert an antiarrhythmic effect. Therefore, we wanted to address (1) the antifibrosis and anti‐inflammatory effects of CAP in ICM rat models; (2) the results of using nicotine administration to stimulate CAP and improve cardiac autonomic nerve function and heart function; (3) the anti‐arrhythmic effect of CAP; and (4) the underlying mechanisms behind CAP.
Methods
Animal Preparation
All animal experiments were performed according to the regulations of, and approved by, the Animal Ethics Committee of Wenzhou Medical University (number wydw2014‐0058) and conformed to the Guide for the Care and Use of Laboratory Animals by the National Institutes of Health. Male Sprague‐Dawley rats (SPF class, 220–250 g, 6 to 8 weeks old) were purchased from Shanghai Laboratory Animal Center of China and were maintained in the Wenzhou Medical University animal facilities under specific pathogen‐free conditions (24±1°C, 45±10% humidity) with a 12‐hour light/dark cycle daily with food and water available ad libitum.
Myocardial Infarction
After induction of anesthesia (urethane, 1.5 g/kg intraperitoneally), the rat was ventilated artificially by rodent respirator (Inspira, Harvard Apparatus, Holliston, MA) in volume‐controlled mode at 80 strokes per minute. After a left parasternotomy, the heart and pericardium were exposed fully. The left anterior descending coronary artery was ligated around 2 mm from its origin (near the main pulmonary artery) using a 6‐0 Prolene suture. Four weeks after the surgery, left ventricular ejection fraction (LVEF) of all surviving rats was measured by echocardiography. Rats with LVEF <50% and/or development of a large akinetic aneurysm with some hypokinesis at nonischemic sites within the left ventricle (LV) were selected (Figure 1). Vagotomy of the afferent nerve was performed 4 weeks after left anterior descending coronary artery ligation.16 In vagotomized animals, after a ventral cervical midline incision, right‐side vagus trunks were exposed, ligated with a 4‐0 silk suture, and divided. All rats were assigned to 6 groups receiving the following treatments: sham‐operated rats (Control, n=8), sham‐operated rats treated with unilateral (right‐sided) cervical vagotomy (Vag, n=8), ischemic cardiomyopathy rats (ICM, n=8), ischemic cardiomyopathy rats with vagotomy (ICM+Vag, n=8), ischemic cardiomyopathy rats with nicotine administration (ICM+Nic, n=8), and ischemic cardiomyopathy rats with vagotomy and nicotine administration (ICM+N+V, n=8). Rats received intraperitoneal injection of nicotine (0.4 mg/kg in saline) the day after vagotomy.17
Figure 1.
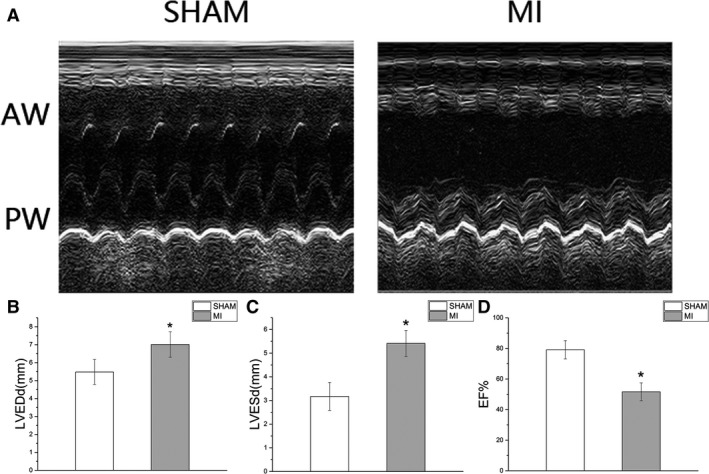
A, The M‐mode echocardiograms of the short‐axis midventricular view from a sham‐operated rat (SHAM) and a rat with 4‐week LAD ligation (MI). B through D, The changes of LVEDd, LVESd and LVEF between SHAM group and MI group. (SHAM, sham‐operated rats, n=16; MI, rats with 4‐week LAD ligation, n=32. The ICM rats showed evident thinning and akinesis of anterior wall, dilated left ventricular cavity, and hypokinesia posterior wall. Data are expressed as means±SD. AW indicates anterior wall; ICM, ischemic cardiomyopathy; LAD, left anterior descending coronary artery; LVEDd, left ventricular end‐diastolic dimension; LVEF, left ventricular ejection fraction; LVESd, left ventricular end‐systolic dimension; MI, myocardial infarction; PW, posterior wall. *P<0.05 vs SHAM group.
Doppler Echocardiography Study
Transthoracic echocardiography was performed at 4 and 8 weeks (4 weeks of nicotine administration) after ligation with M‐mode transducer (12‐MHz phased‐array transducer; Sonos 5500, Philips USA, Bothell, WA).18 At the papillary muscle level, short‐axis views of M‐mode tracings were recorded through the anterior and posterior LV walls to measure LV end‐diastolic dimension (LVEDd), LV end‐systolic dimension (LVESd), and fraction shortening (FS). LVEF was obtained using the Simpson approach. All measurements were performed by an experienced technician who was blinded to study groups.
ECG and Heart Rate Variability Measurements
Rats were anesthetized with pentobarbital sodium (30 mg/kg intraperitoneally). ECG was performed with a computer‐based electrical physiology system (PowerLab 8/36; AD Instruments, Colorado Springs, CO), and 2 biopotential leads were anchored subcutaneously over the right pectoral muscle and on the lower left side of the chest wall, respectively. The RR, PR, QRS, and QT durations were recorded using the PowerLab data acquisition system via LabChart 8 software (AD Instruments). The QTc was corrected for heart rate using a modification of the Bazett formula. Heart rate variability (HRV) was calculated from 5‐minute segments of ECG data. The HRV parameters were analyzed by the LabChart Pro HRV Analysis Module (AD Instruments). Ectopic beats and artifacts were manually excluded. The standard deviation of the averages of normal‐to‐normal intervals (SDNN) as well as frequency‐domain parameters such as low‐frequency (LF) power (0.06 to 0.6 Hz), high‐frequency (HF) power (0.6 to 3 Hz),19 and LF‐HF ratio were measured.
Ventricular Programmed Electric Stimulation
A ventral midline incision was performed on the thorax by sternotomy at the end of the ECG and HRV measurements. Two Teflon‐coated silver wire stimulating‐electrodes were inserted into thorax and contacted with the cardiac apex.20 Pacing was performed by means of programmable stimulator (PowerLab 8/36; AD Instruments) and the programmed electric stimulation (PES) protocol used was similar to that described by Bélichard et al.21 To determine the effective refractory period, a premature stimulus was applied after 8 paced beats at a basic cycle length of 100 ms. To induce the ventricular arrhythmias, simulation at a basic cycle (S1) with single (S2), double (S3), or triple (S4) extra stimuli was performed (Figure 2A). The PES was stopped when it had induced a ventricular tachyarrhythmia consisting of at least 6 consecutive nondriven ventricular extra stimulus beats. Either no ventricular premature beats or only self‐terminating salvos of <6 beats were considered “noninducible.” A ventricular tachyarrhythmia was considered nonsustained when it lasted ≤15 beats and sustained when it lasted >15 beats before terminating spontaneously or by overdrive pacing. There was no difference between sustained ventricular tachycardia and ventricular fibrillation.22
Figure 2.
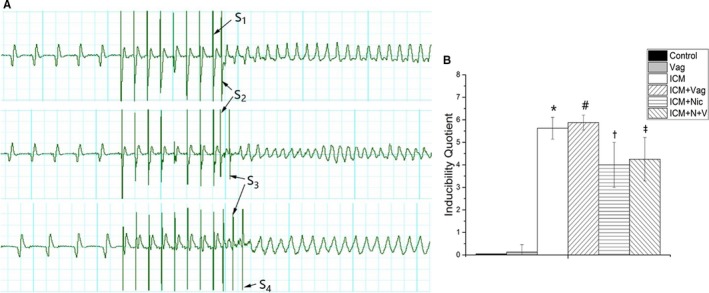
A, Three typical examples of sustained ventricular arrhythmias induced by PES. S1=8 paced beats at a basic cycle length of 100 ms, and S2, S3, and S4 represent extra stimuli. B, Inducibility quotient of ventricular arrhythmias obtained from 6 groups. The scoring system was listed in methods section. (Control group, n=8; Vag group, n=8; ICM group, n=8; ICM+Vag group, n=8; ICM+Nic group, n=8; ICM+N+V group, n=8; S1, basic cycle stimuli; S2, S3, S4, extra stimuli.) Data are expressed as means±SD. ICM indicates ischemic cardiomyopathy; Nic, nicotine; PES, programmed electric stimulation; Vag, vagotomy. *P<0.05 vs Control group. # P<0.05 vs ICM group. † P<0.05 vs ICM group. ‡ P<0.05 vs ICM+Nic group.
An arrhythmia scoring system was used: 0, noninducible preparations; 1, nonsustained tachyarrhythmias induced with 3 extra stimuli; 2, sustained tachyarrhythmias induced with 3 extra stimuli; 3, nonsustained tachyarrhythmias induced with 2 extra stimuli; 4, sustained tachyarrhythmias induced with 2 extra stimuli; 5, nonsustained tachyarrhythmias induced with 1 extrastimulus; 6, sustained tachyarrhythmias induced with 1 extrastimulus; 7, tachyarrhythmias induced during the 20 paced beats at a basic cycle length of 100 ms; and 8, heart stopped before the PES or during the preparation.23
Cell Culture
RAW264.7 cells were purchased from the American Type Culture Collection. All cells were maintained in Dulbecco modified Eagle medium (Thermo Fisher, Waltham, MA); 10% fetal bovine serum (Gibco, Gaithersburg, MD) and penicillin/streptomycin (1:100; Sigma Chemical Co, St. Louis, MO) were supplied in cell culture. The constant temperature incubator was maintained (37°C) with a 95% humidified atmosphere and 5% CO2. Cells pretreated with nicotine (1 μmol/L) for 1 hour were stimulated with lipopolysaccharide (100 ng/mL).15 Furthermore, to examine the effect of vagotomy, cells were treated with α‐bungarotoxin (100 mmol/L, Sigma)24 for 30 minutes before being subjected to nicotine.
Western Blotting
The protein of each sample was extracted from the border zone (2 mm away from the infarct edge).25 The tissues were lysed in 1 mL RIPA lysis (ThermoFisher, 89900) with protease inhibitors (Beyotime, Jiangsu, China, ST506‐2) and phosphatase inhibitors (Applygen Technologies, Beijing, China, P1260) for 30 minutes under rotating agitation, and the protein concentration was determined with a BCA protein assay kit (ThermoFisher, 23235). Then 20 μg protein was taken and mixed with one fifth volume of 5× loading buffer. The mixture was denatured by boiling at 100°C for 5 minutes. Then each sample was separated by 10% SDS‐PAGE and transferred to a polyvinylidene difluoride membrane. The membrane was blocked in 5% nonfat dry milk. After being incubated with specific antibody (overnight, 4°C) and secondary antibody, the membrane was detected with the enhanced chemiluminescence detection system (Millipore, Billerica, MA). The antibodies against phospho‐Akt (Ser473), phospho‐IκBα (Ser32), NF‐κB p65, phospho‐NF‐κB p65 (Ser536), and Cx43 were purchased from Cell Signaling Technology (Danvers, MA). The antibodies against phospho‐IKKα/β (Ser180/181), β‐actin, and GAPDH were purchased from Bioworld Technology (Nanjing, China). The antibodies against IL‐1β and IL‐6 were purchased from R&D System Technology (Minneapolis, MN). The antibodies against interferon (IFN)‐γ, tumor necrosis factor (TNF)‐α, transforming growth factor‐β, collagen I, and collagen III were purchased from Abcam (Shanghai, China).
Fibrosis Size Measurement
LV middle ring (the middle 1/3 of the left ventricle) was embedded in paraffin. Then samples were sectioned into 5‐μm‐thick slices and applied Mallory trichrome staining. The fibrosis size was expressed as the mean percentage of the total border zone.
Immunocytochemistry
Samples were embedded in paraffin and sectioned at a thickness of 4 μm. After being deparaffinized, sections were boiled in citrate buffer in a microwave oven for 5 minutes to enhance specific immunostaining. Then sections were incubated overnight with anti‐Cx43 (1:100, CST) antibody and incubated for 2 hours in FITC‐conjugated goat anti‐rabbit IgG (1:200, Bioworld, Nanjing, China). Fluorescence of FITC was observed with a fluorescence microscope system (DP72, Olympus, Waltham, MA).
The NF‐κB p65 nuclei translocation was assessed by fluorescence microscope system (DP72, Olympus). RAW264.7 cells, growing on glass coverslips, were fixed with 4% paraformaldehyde. The anti‐NF‐κB p65 antibody (diluted 1:100) was used for staining at 4°C incubated overnight. And then cells were incubated with FITC‐conjugated goat anti‐rabbit second antibody (1:250, Bioworld) for 1 hour at room temperature. The fluorescent dye 4′,6‐diamidino‐2‐phenylindole dihydrochloride (DAPI) was used to stain nuclei (3 minutes) after PBS washing.
Statistical Methods
Data are expressed as means±SD. All outcomes are compared among groups using 1‐way ANOVA followed by the Dunnett multiple‐comparison test. Chi‐squared test was used on inducibility of ventricular arrhythmias. Statistical analyses were performed using SPSS 14 software (Unicom, Mission Hills, CA). A value of P<0.05 was considered significant.
Results
Effect of CAP on the Cardiac Function
After 4‐week ligation, all rats were measured by echocardiography, and 32 rats were included in this experiment. LVEDd and LVESd were significantly increased at 4 weeks after left anterior descending coronary artery ligation in the MI group compared with the sham group (MI versus sham; LVEDd, LVESd; 7.0±0.7 versus 5.5±0.7, P<0.05; 5.4±0.5 versus 3.2±0.6, P<0.05). LVEF was markedly reduced in the MI group compared with the sham group (MI versus sham; 50.6±2.1 versus 82.3±2.6, P<0.05). The M‐mode image showed a large akinetic anterior wall. Mallory trichrome staining showed the collagens staining the border zone (stained in blue) in the MI group. As shown in Figure 3, LVEDd, LVESd, LVEF, and FS were similar between the Control group and Vag group (Control versus Vag; LVEDd, LVESd, LVEF, FS; 5.5±0.7 versus 5.6±0.6, P>0.05; 3.0±0.5 versus 3.1±0.3, P>0.05; 82.3±2.6 versus 82.7±2.3, P>0.05; 44.9±3.8 versus 44.0±2.4, P>0.05). Four‐week nicotine administration slightly decreased LVESd and LVEDd and improved LVEF and FS in the ICM+Nic group compared with the ICM group (ICM+Nic versus ICM; LVEDd, LVESd, LVEF, FS; 7.3±0.2 versus 7.8±0.3, P<0.05; 5.3±0.1 versus 6.0±0.2, P<0.05; 55.3±1.6 versus 50.6±2.1, P<0.05; 27.0±1.2 versus 23.0±1.1, P<0.05). These effects of nicotine in the ICM+Nic group were attenuated when vagotomy was performed in the ICM+Nic group (ICM+Nic versus ICM+N+V; LVEDd, LVESd, LVEF, FS; 7.3±0.2 versus 7.4±0.2, P>0.05; 5.3±0.1 versus 5.6±0.2, P>0.05; 55.3±1.6 versus 53.2±1.3, P<0.05; 27.0±1.2 versus 24.6±0.8, P<0.05).
Figure 3.
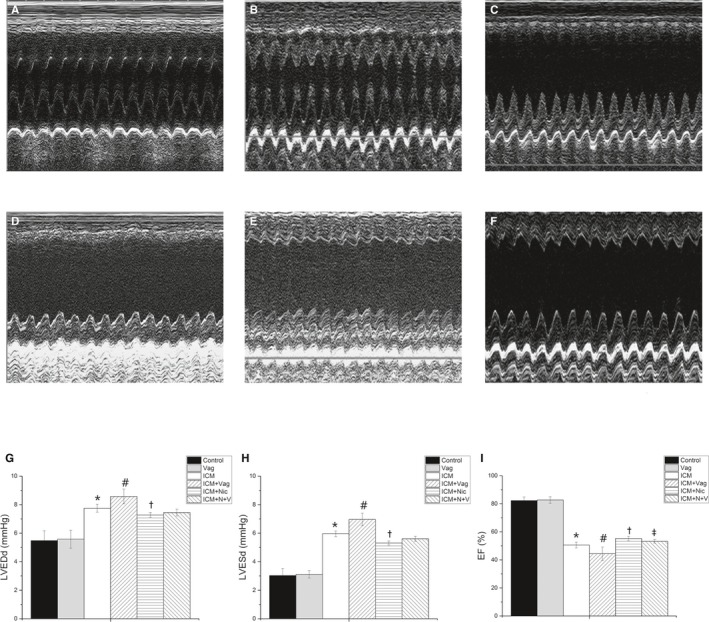
A through F, The typical M‐mode echocardiograms of the short‐axis midventricular view from 6 groups. A, Control group, n=8; B, Vag group, n=8; C, ICM group, n=8; D, ICM+Vag group, n=8; E, ICM+Nic group, n=8; F, ICM+N+V group, n=8. G through I, Nicotine administration slightly decreased LVESd and LVEDd and improved LVEF and FS in ICM+Nic group compared with ICM group. Vagotomy aggravated the dilation of left ventricular cavity and decreased LVEF and FS of ICM rats. And vagotomy operation attenuated the effect of nicotine on ICM rats. LVEDd, LVESd, LVEF and FS were similar among Control group and Vag group (P>0.05). Data are expressed as means±SD; FS, fractional shortening. ICM indicates ischemic cardiomyopathy; LVEDd, left ventricular end‐diastolic dimension; LVEF, left ventricular ejection fraction; LVESd, left ventricular end‐systolic dimension; Nic, nicotine; Vag, vagotomy. *P<0.05 vs Control group. # P<0.05 vs ICM group. † P<0.05 vs ICM group. ‡ P<0.05 vs ICM+Nic group.
Effect of CAP on Cardiac Autonomic Nerve Function
The time domain and frequency domain of HRV were performed to assess cardiac autonomic function (Figures 4 and 5). The Control group and Vag group had similar SDNN and LF/HF ratios (Control versus Vag; SDNN, LF/HF; 12.5±3.8 versus 13.9±1.4, P>0.05; 0.3±0.1 versus 0.3±0.1, P>0.05). The ICM group showed a significant decrease in SDNN and increase in LF/HF ratio compared with the Control group (ICM versus Control; SDNN, LF/HF; 4.3±1.8 versus 12.5±3.8, P<0.05; 2.3±1.3 versus 0.3±0.1, P<0.05). However 4‐week nicotine administration increased SDNN and decreased the LF/HF ratio in the ICM group (ICM+Nic versus ICM; SDNN, LF/HF; 9.9±3.2 versus 4.3±1.8, P<0.05; 0.2±0.1 versus 2.3±1.3, P<0.05). Moreover, vagotomy attenuated the effect of nicotine administration in the ICM+Nic group (ICM+N+V versus ICM+Nic; SDNN, LF/HF; 7.3±1.7 versus 9.9±3.2, P<0.05; 0.4±0.1 versus 0.2±0.1, P<0.05).
Figure 4.
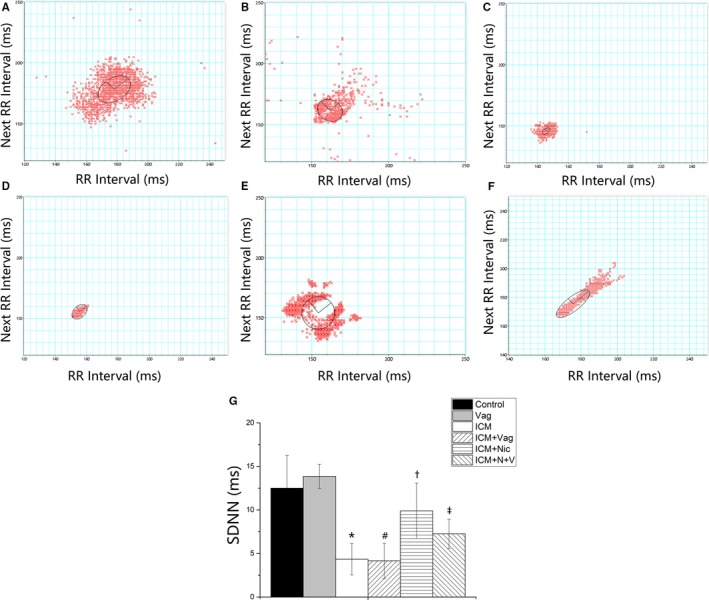
A through F, Time domain parameter of heart rate variability (HRV) obtained from 6 groups (A, Control group, n=8; B, Vag group, n=8; C, ICM group, n=8; D, ICM+Vag group, n=8; E, ICM+Nic group, n=8; F, ICM+N+V group, n=8). SDNN indicates standard deviation of the averages of normal‐to‐normal intervals. G, ICM group showed a significant decrease in SDNN compared with the Control group. Nicotine administration increased the SDNN of ICM rats. Vagotomy operation attenuated the effect of nicotine on ICM rats. Data are expressed as means±SD. ICM indicates ischemic cardiomyopathy; Nic, nicotine; Vag, vagotomy. *P<0.05 vs Control group. # P<0.05 vs ICM group. † P<0.05 vs ICM group. ‡ P>0.05 vs ICM+Nic group.
Figure 5.
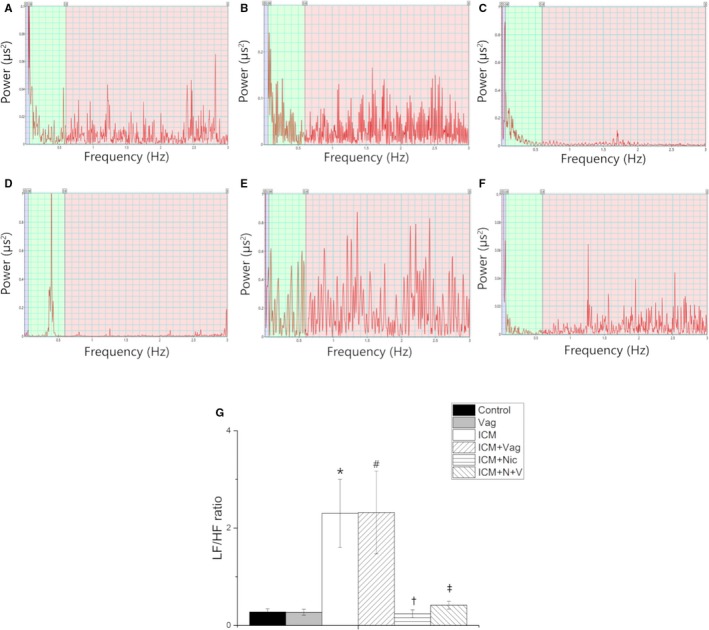
A through F, LF/HF ratio of HRV obtained from 6 groups (A: Control group, n=8; B: Vag group, n=8; C: ICM group, n=8; D: ICM+Vag group, n=8; E: ICM+Nic group, n=8; F: ICM+N+V group, n=8); G, ICM group showed a significant increase in LF/HF ratio compared with the Control group. Nicotine administration decreased the SDNN of ICM rats. Vagotomy operation attenuated the effect of nicotine on ICM rats. Data are expressed as means±SD. HF indicates high‐frequency power; HRV, heart rate variability; ICM, ischemic cardiomyopathy; LF, low‐frequency power; Nic, nicotine; SDNN, standard deviation of the averages of normal‐to‐normal intervals; Vag, vagotomy. *P<0.05 vs Control group. # P>0.05 vs ICM group. † P<0.05 vs ICM group. ‡ P<0.05 vs ICM+Nic group.
Effect of CAP on PES‐Induced Ventricular Tachyarrhythmia
ECG parameters were shown in Table (means±SD). RR interval, PR interval, duration of P‐wave, and duration of QRS‐wave were similar among 6 groups (P>0.05). Four‐week nicotine administration attenuated the prolonging of QTc in ICM group (ICM+Nic versus ICM; QTc; 145.9±8.5 versus 170.4±4.7, P<0.05). This effect of nicotine in ICM+Nic group was attenuated slightly when vagotomy was performed in ICM+Nic group (ICM+Nic group versus ICM group; QTc 145.9±8.5 versus 147.5±4.9, P>0.05).
Table 1.
Results of ECG Parameters Among 6 Groups
| RR (ms) | P (ms) | PR (ms) | QRS (ms) | QTc (ms) | |
|---|---|---|---|---|---|
| Control | 197.0±33.2 | 18.3±0.5 | 52.3±1.0 | 24.2±1.5 | 133.1±2.1 |
| Vag | 191.4±27.6 | 19.0±1.2 | 55.0±4.5 | 23.1±1.0 | 130.7±5.6 |
| ICM | 186.5±27.6 | 17.8±0.8 | 50.5±2.2 | 24.6±0.8 | 169.4±7.7a |
| ICM+Vag | 204.9±36.8 | 18.6±0.7 | 55.4±5.0 | 25.5±1.5 | 175.8±10.2b |
| ICM+Nic | 217.1±33.2 | 18.1±1.4 | 54.5±3.1 | 24.2±1.8 | 145.9±8.5c |
| ICM+N+V | 224.5±54.9 | 18.5±1.0 | 52.2±1.1 | 24.2±1.0 | 147.5±4.9d |
Data are expressed as means±SD. Control group, n=8; Vag group, n=8; ICM group, n=8; ICM+Vag group, n=8; ICM+Nic group, n=8; ICM+N+V group, n=8; RR, R‐R interval; P, P‐wave duration; PR, P‐R duration; QRS, QRS‐wave duration; QTc, heart rate‐corrected QT interval. ICM indicates ischemic cardiomyopathy; Nic, nicotine; Vag, vagotomy.
P<0.05 vs control group.
P>0.05 vs ICM group.
P<0.05 vs ICM group.
P>0.05 vs ICM+Nic group.
As shown in Figure 2B, rats with vagotomy only did not have ventricular arrhythmias induced by PES. Considering the severity of the arrhythmia induced by calculating the inducibility quotient, nicotine‐treated rats had a lower quotient than other 2 groups regardless of vagotomy operation (ICM+Nic versus ICM, P<0.05; ICM+N+V versus ICM+Vag, P<0.05).
Effect of CAP on Fibrosis of Border Zone and Cx43 Distribution
The fibrosis was examined by Mallory trichrome staining and Western blotting (Figures 6 and 7A). The fibrosis of the border zone was significantly increased in the ICM group (ICM versus Control, 72.5±5.0 versus 5.1±0.6, P<0.05). Additionally, the collagen synthesis/release was significantly reduced in the ICM+Nic group compared with the ICM group (ICM+Nic versus ICM; 52.4±4.6 versus 72.5±5.0, P<0.05). Vagotomy decreased the CAP antifibrosis effect (ICM+N+V versus ICM+Nic; 61.3±6.0 versus 52.4±4.6, P<0.05).
Figure 6.
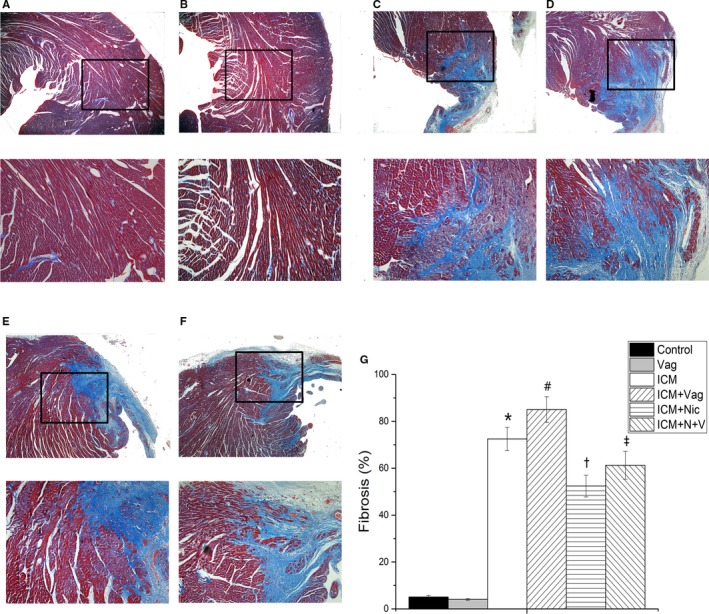
A through F, Comparison of fibrosis area (%) of border zone among the 6 groups (A, Control group, n=8; B, Vag group, n=8; C, ICM group, n=8; D, ICM+Vag group, n=8; E, ICM+Nic group, n=8; F, ICM+N+V group, n=8; upper magnification ×200; lower magnification ×400). G, The ICM group showed an evident increase of fibrosis in border zone compared with the Control group. Vagotomy operation aggravated the abnormality of ICM rats. The collagen synthesis/release was significantly reduced in ICM rats with nicotine treatment. Vagotomy also weakened the effect of nicotine on ICM rats. Data are expressed as means±SD. ICM indicates ischemic cardiomyopathy; Nic, nicotine; Vag, vagotomy. *P<0.05 vs Control group. # P<0.05 vs ICM group. † P<0.05 vs ICM group. ‡ P<0.05 vs ICM+Nic group.
Figure 7.
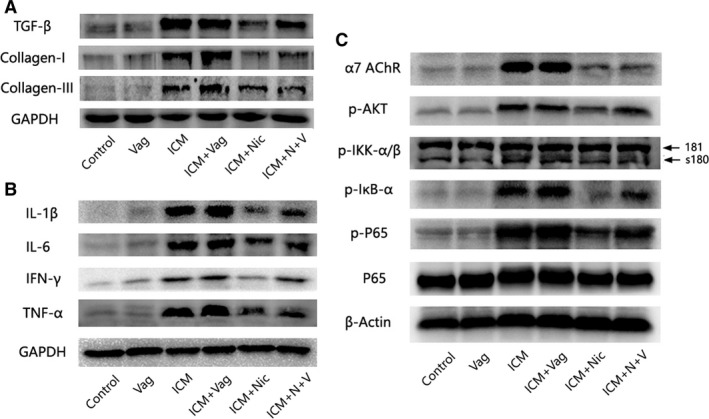
Western blot analysis demonstrating protein expression of fibrosis, inflammation, and the NF‐κB signaling pathway of 6 groups (n=6 for each group). A, ICM rats with 4‐week nicotine administration had a lower TGF‐β, collagen I, and collagen III expression, whereas vagotomy aggregated the abnormality of ICM rats and partly blocked the effect of nicotine. B, Eliciting CAP by nicotine treatment decreased the expression of proinflammatory cytokines (IL‐1β, IL‐6, IFN‐γ, TNF‐α) in the border zone. Vagotomy operation increased the expression of proinflammatory cytokines in either ICM rats or ICM rats with nicotine treatment. C, Effect of nicotine on α7‐nAChR, Akt activation, and NF‐κB. Nicotine substantially inhibited the enhanced phosphorylation of NF‐κB p65, IκB, IKKα/β, and Akt in the ICM and ICM+Vag groups. Vagotomy operation attenuated the effect of nicotine on ICM rats. The expression of α7‐nACnR was significantly increased in the ICM and ICM+Vag groups, but nicotine administration blocked this effect regardless of vagotomy operation. CAP indicates cholinergic anti‐inflammatory pathway; ICM, ischemic cardiomyopathy; TGF=β, transforming growth factor‐β; Vag, vagotomy.
As shown in Figure 8, the ICM group had a significant decrease of Cx43 compared with the Control group, and distribution of Cx43 in the ICM+Nic group tended to be denser than that in the ICM group. Additionally, surgical vagotomy attenuated this effect slightly (ICM+Nic versus ICM+N+V). Same changes had been shown in the Western blotting result.
Figure 8.
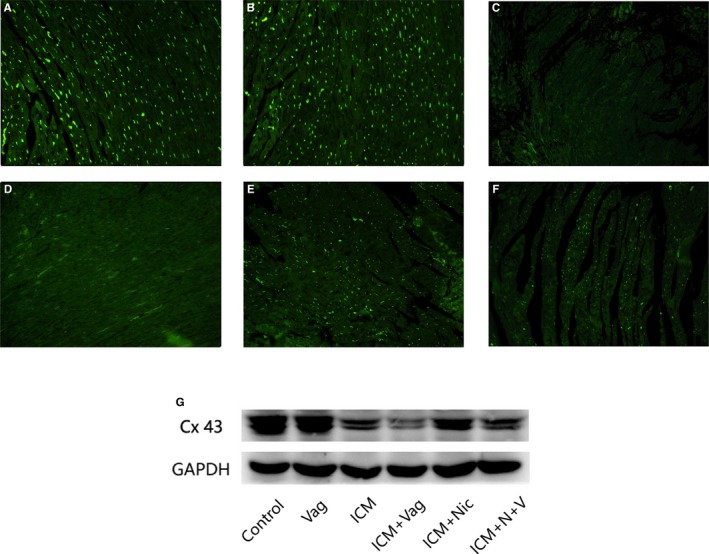
A through F, Representative immunofluorescence images of Cx43 from rat left ventricles (A, Control group, n=8; B, Vag group, n=8; C, ICM group, n=8; D, ICM+Vag group, n=8; E, ICM+Nic group, n=8; F, ICM+N+V group, n=8; magnification ×200). The positive immunoreactive signals (green) were accumulated in intercellular positions. The ICM group had a significant decrease of Cx43 when compared with the Control group, and the Cx43 distribution tended to be higher in the ICM+Nic group than in the ICM group. Vagotomy treatment attenuated the effect of nicotine on the ICM group slightly. G, Same changes as shown in Western‐blotting result (n=6 for each group). Cx43 indicates connexion 43; ICM, ischemic cardiomyopathy; Nic, nicotine; Vag, vagotomy.
Effect of CAP on Proinflammatory Cytokine Production in Border Zone
To determine the inhibitory action of CAP on the proinflammatory cytokines in the border zone, IL‐1β, IL‐6, IFN‐γ, and TNF‐α were analyzed by Western blotting. As shown in Figure 7B, the production of IL‐1β, IL‐6, IFN‐γ, and TNF‐α was significantly increased in the ICM group and the ICM+Vag group. Nicotine administration reduced the production of proinflammatory cytokines in the ICM group (ICM+Nic versus ICM); additionally vagotomy attenuated this effect of nicotine on proinflammatory cytokine release (ICM+Nic versus ICM+N+V).
CAP Suppressed NF‐κB Activation in ICM Rat Model
NF‐κB is a transcriptional regulator that has marked effects on inflammatory response. As shown in Figure 7C, nicotine substantially inhibited the enhanced phosphorylation of Ser536 of NF‐κB p65 that was stimulated in the ICM and ICM+Vag groups. The IκB protein, as an inactive NF‐κB–IκB complex, inhibits NF‐κB activity in the cytoplasm, and IKK is an important upstream kinase for the phosphorylation of IκB. As expected, nicotine administration blocked the phosphorylation of IKKα/β and IκBα. The PI3K/Akt pathway is involved in the regulation of NF‐κB activation, and in our study, p‐Akt was significantly increased in the ICM group, and nicotine dramatically blocked this effect. Furthermore, vagotomy attenuated the effect of nicotine inhibiting NF‐κB activation (ICM+Nic versus ICM+N+V).
Nicotine Treatment Suppressed NF‐κB Activation in Lipopolysaccharide‐Stimulated RAW264.7 Cells
NF‐κB, a pivotal transcriptional regulator, evidently affects inflammatory response. As shown in Figure 9, nicotine treatment significantly inhibited the nuclear translocation of LPS‐induced NF‐κB p65, and α‐bungarotoxin attenuated the effect of nicotine on inhibiting NF‐κB activation.
Figure 9.
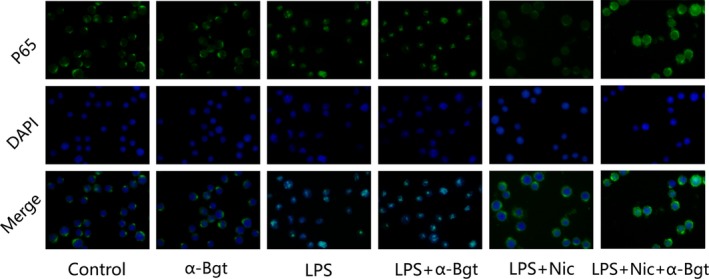
Nicotine treatment suppressed NF‐κB activation in LPS‐stimulated RAW264.7 cells (n=8 for each group). α‐Bungarotoxin (α‐Bgt) partly inhibited the effect of nicotine on nuclear translocation of LPS‐induced NF‐κB p65. NF‐κB p65 antibody labeled in green with FITC. The nuclei were stained in blue with 40,6‐diamidino‐2‐phenylindole. LPS indicates lipopolysaccharide; Nic, nicotine.
Effect of Nicotine on α7‐NAChR
As shown in Figure 7C, 4 weeks after operation, α7‐nAChR had a significantly increasing in ICM and ICM+Vag group. Nicotine administration blocked this effect regardless of vagotomy operation (ICM+Nic versus ICM; ICM+N+V versus ICM+Vag).
Discussion
In the present study, we investigated whether eliciting CAP could attenuate ICM‐induced arrhythmogenic properties via modulation of the inflammation and fibrosis in infarcted border zone. Additionally, we found that eliciting CAP by 4‐week nicotine administration improved the heart rate variability, increased the parasympathetic/sympathetic ratio, and consequently improved the cardiac autonomic function. Furthermore, our study suggested that the principal cardiac gap‐junction protein Cx43 was increased by eliciting CAP. Although the precise mechanism by which CAP has its significant effect on cardiac autonomic function and Cx43 remains unknown, it is likely that CAP exerts its antiarrhythmogenic effects not only through its anti‐inflammation and antifibrosis effects but by modulating cardiac autonomic function and Cx43 as well.
Limiting the inflammatory response after myocardial infarction ameliorates cardiac dysfunction.26 It has already been reported that CAP interacting with α7‐nAChR expressed by macrophages and other cytokine‐producing cells15, 27, 28, 29 has markedly influenced the anti‐inflammatory response. Utilizing the major vagal neurotransmitter ACh and nicotine as AChR agonists that interact with α7‐nAChR on macrophages to reduce the release of TNF‐α and other proinflammatory cytokines confirmed that this modulation is mediated through the vagus nerve.15 Our present study demonstrated for the first time in an ICM rat model that eliciting the CAP by nicotine not only reduced the cytokines (IL‐1β, IL‐6, IFN‐γ, TNF‐α, transforming growth factor‐β) and collagens (collagen I, collagen III) of the infarction border zone but also improved the cardiac autonomic function, increased ejection fraction, and attenuated arrhythmogenic properties. Rats with ICM showed uncontrollable inflammation and fibrosis in the border zone and had dysfunctional autonomic function, lower ejection fraction, and higher arrhythmogenic properties compared with those subjected to a sham operation. These abnormalities were ameliorated by a 4‐week nicotine treatment. In addition, 4 weeks of nicotine decreased the expression of a7‐nAChR receptors in border zone and inhibited NF‐ĸB activation. Intriguingly, all abnormalities became more severe or the effects of nicotine were attenuated with a vagotomy operation when the ICM group was compared with the ICM+Vag group or the ICM+Nic group with the ICM+N+V group. However, because vagotomy evokes a tachycardic effect, the question remains whether the transient heart rate acceleration by vagotomy has an effect on antiarrhythmic properties during ICM. In a preliminary study, we confirmed that vagotomy performed in normal rats does not have any effects on inflammation, fibrosis, cardiac function, or arrhythmogenic properties (comparing the results between the Control group and the Vag group).
To explain the above findings, we hypothesize that regional myocardial infarction induces an accumulation of monocytes/macrophages accompanied by an increase of cytokines that, in turn, can support angiogenesis and extracellular matrix synthesis by providing vascular endothelial growth factor, transforming growth factor‐β, and profibrotic transforming growth factor‐α after acute wound.30 Takamura et al reported that regulating the activation of proinflammatory macrophages post–myocardial infarction improves cardiac remodeling.31 Indeed, Ly‐6Clow macrophage has been shown to play a crucial role in the pathogenesis and progression of the healing myocardium.30 In addition, macrophage‐induced fibrosis and several cytokines could aggravate arrhythmogenic properties.32, 33 We found that levels of fibrosis and inflammation were significantly elevated after myocardial infarction. In addition, there were significant inverse correlations between inflammatory cytokine release and the CAP elicitation. In our study of RAW264.7 cells, we suggested that the nuclear translocation of LPS‐induced NF‐κB p65 was significantly inhibited by nicotine treatment, which may indicate the modulation of nicotine treatment on macrophages in ICM rat. Consistent with the present result, a study has documented a link between α7‐nAChR and Ly‐6C macrophages.34
The clinical syndrome of ICM is a challenging problem because few treatment options are available to prevent the development and progression of heart failure and infarction‐induced fatal ventricular arrhythmia. Increased sympathetic and/or decreased parasympathetic activity has been closely related to risks of cardiac outcomes such as heart failure and ventricular arrhythmias.35 Subsequent research has identified neural remodeling after myocardial infarction that may cause a fatal ventricular arrhythmia. Cao et al suggested increased density of postinjury sympathetic nerves is responsible for the occurrence of ventricular arrhythmia.8 Improving the cardiac autonomic function markedly attenuated ICM‐induced arrhythmogenic properties.22 To find evidence for the identification of a protective, melanocortin‐activated efferent vagal cholinergic pathway, Mioni et al stimulated efferent vagal fibers electrically after bilateral cervical vagotomy, suggesting that an efferent vagal cholinergic pathway served a protective role against myocardial ischemia/reperfusion‐induced ventricular arrhythmia.10 Zuanetti et al reported that the protective effect of vagal stimulation against reperfusion ventricular fibrillation is mediated by the decreased heart rate; however, they also found no difference in occurrence of complex reperfusion arrhythmias between neutrally intact and postvagotomy animals.11 Additionally, vagal nerve stimulation prevented pumping failure and cardiac remodeling in chronic heart failure rats with ligation of the left coronary artery and improved their long‐term survival.12 Our present study also showed that eliciting CAP during ICM modulates the cardiac parasympathetic/sympathetic ratio, and that, in turn, decreased the arrhythmia score of PES‐induced ventricular arrhythmia. In addition, Cx43, which contributes to intercellular communication and electrical coupling, is the principal component of ventricular gap‐junction proteins. Genetically engineered Cx43‐deficient (Cx43+/− or Cx43−/−) mice have been reported to be markedly susceptible to ischemia‐induced ventricular tachycardia.36, 37, 38 Ando et al39 reported that increased vagal tone could prevent the loss of Cx43 during acute MI and thus exert antiarrhythmogenic effects. In the present study, we found that eliciting CAP decreased the arrhythmia score of PES‐induced ventricular arrhythmia partly by preventing the loss of Cx43 during ICM. Additionally, nicotine administration improved cardiac autonomic function and enhanced vagal tone. The decreased releasing of IL‐1β also inhibited the loss of Cx43. The functional preservation of Cx43 by eliciting CAP would play an irreplaceable role in antiarrhythmogenic properties during ICM. Interestingly, Shirai et al40 reported that bilateral vagotomy attenuated the cardiac sympathetic nerve activity response to acute MI that, in turn, reduced the incidence of arrhythmic episodes. In contrast, our study suggested that unilateral vagotomy not only aggravated the abnormality of ICM rats and increased the inducibility quotient of ventricular arrhythmias but also attenuated the effects of eliciting CAP. It was also reported that vagotomy attenuated the anti‐inflammatory effect of CAP and thereby enhanced cytokine release in response to LPS treatment.41, 42 Thus, vagotomy, performed in an ICM rat, increased the release of cytokines and collagens and altered the sympathetic/parasympathetic ratio, which served as a harmful role of ventricular arrhythmia.
The anti‐inflammatory activity of CAP involves the Jak2/STAT3 signaling pathway.43 Another critical component of the nicotine anti‐inflammatory pathway appears to be mediated by the inhibition of NF‐ĸB.44 Our data suggest that inhibition of NF‐ĸB activation is associated with a better outcome after eliciting CAP. However, these effects are attenuated by a vagotomy operation. In addition, as our data showed, a vagotomy operation aggravates the abnormalities of ICM.
In summary, we demonstrated that eliciting CAP could exert protective effects against ICM‐induced ventricular arrhythmia. The underlying mechanism might include (1) a direct effect of CAP on regional anti‐inflammation and antifibrosis; (2) the effect of CAP to improve cardiac autonomic function; and (3) the effect of CAP to prevent the loss of Cx43 during ICM. There, CAP could be an alternative therapeutic strategy for ICM‐induced ventricular arrhythmia.
Sources of Funding
These studies were supported by grant 81570342 and grant 81570299 from the National Natural Science Foundation of China.
Disclosures
None.
Acknowledgments
We appreciate the helpful comments on this paper received from our reviewers.
(J Am Heart Assoc. 2017;6:e006510 DOI: 10.1161/JAHA.117.006510.)28928157
References
- 1. Huikuri HV, Castellanos A, Myerburg RJ. Sudden death due to cardiac arrhythmias. N Engl J Med. 2001;345:1473–1482. [DOI] [PubMed] [Google Scholar]
- 2. Hayashi M, Shimizu W, Albert CM. The spectrum of epidemiology underlying sudden cardiac death. Circ Res. 2015;116:1887–1906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Severs NJ. Gap junction remodeling in heart failure. J Card Fail. 2002;8:S293–S299. [DOI] [PubMed] [Google Scholar]
- 4. Wijesurendra RS, Liu A, Eichhorn C, Ariga R, Levelt E, Clarke WT, Rodgers CT, Karamitsos TD, Bashir Y, Ginks M, Rajappan K, Betts T, Ferreira VM, Neubauer S, Casadei B. Lone atrial fibrillation is associated with impaired left ventricular energetics that persists despite successful catheter ablation. Circulation. 2016;134:1068–1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. van Rijen HV, Eckardt D, Degen J, Theis M, Ott T, Willecke K, Jongsma HJ, Opthof T, de Bakker JM. Slow conduction and enhanced anisotropy increase the propensity for ventricular tachyarrhythmias in adult mice with induced deletion of connexin43. Circulation. 2004;109:1048–1055. [DOI] [PubMed] [Google Scholar]
- 6. De Jesus NM, Wang L, Lai J, Rigor RR, Francis Stuart SD, Bers DM, Lindsey ML, Ripplinger CM. Antiarrhythmic effects of interleukin 1 inhibition after myocardial infarction. Heart Rhythm. 2017;14:727–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Mollenhauer M, Friedrichs K, Lange M, Gesenberg J, Remane L, Kerkenpass C, Krause J, Schneider J, Ravekes T, Maass M, Halbach M, Peinkofer G, Saric T, Mehrkens D, Adam M, Deuschl FG, Lau D, Geertz B, Manchanda K, Eschenhagen T, Kubala L, Rudolph TK, Wu Y, Tang WW, Hazen SL, Baldus S, Klinke A, Rudolph V. Myeloperoxidase mediates postischemic arrhythmogenic ventricular remodeling. Circ Res. 2017;121:56–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Cao JM, Fishbein MC, Han JB, Lai WW, Lai AC, Wu TJ, Czer L, Wolf PL, Denton TA, Shintaku IP, Chen PS, Chen LS. Relationship between regional cardiac hyperinnervation and ventricular arrhythmia. Circulation. 2000;101:1960–1969. [DOI] [PubMed] [Google Scholar]
- 9. Tracey KJ. The inflammatory reflex. Nature. 2002;420:853–859. [DOI] [PubMed] [Google Scholar]
- 10. Mioni C, Bazzani C, Giuliani D, Altavilla D, Leone S, Ferrari A, Minutoli L, Bitto A, Marini H, Zaffe D, Botticelli AR, Iannone A, Tomasi A, Bigiani A, Bertolini A, Squadrito F, Guarini S. Activation of an efferent cholinergic pathway produces strong protection against myocardial ischemia/reperfusion injury in rats. Crit Care Med. 2005;33:2621–2628. [DOI] [PubMed] [Google Scholar]
- 11. Zuanetti G, De Ferrari GM, Priori SG, Schwartz PJ. Protective effect of vagal stimulation on reperfusion arrhythmias in cats. Circ Res. 1987;61:429–435. [DOI] [PubMed] [Google Scholar]
- 12. Li M, Zheng C, Sato T, Kawada T, Sugimachi M, Sunagawa K. Vagal nerve stimulation markedly improves long‐term survival after chronic heart failure in rats. Circulation. 2004;109:120–124. [DOI] [PubMed] [Google Scholar]
- 13. Li DL, Liu JJ, Liu BH, Hu H, Sun L, Miao Y, Xu HF, Yu XJ, Ma X, Ren J, Zang WJ. Acetylcholine inhibits hypoxia‐induced tumor necrosis factor‐α production via regulation of MAPKs phosphorylation in cardiomyocytes. J Cell Physiol. 2011;226:1052–1059. [DOI] [PubMed] [Google Scholar]
- 14. Tracy KJ. Physiology and immunology of the cholinergic antiinflammatory pathway. J Clin Invest. 2007;117:289–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Wang H, Yu M, Ochani M, Amella CA, Tanovic M, Susarla S, Li JH, Wang H, Yang H, Ulloa L, Al‐Abed Y, Czura CJ, Tracey KJ. Nicotinic acetylcholine receptor alpha7 subunit is an essential regulator of inflammation. Nature. 2003;421:384–388. [DOI] [PubMed] [Google Scholar]
- 16. Nishina T, Nishimura K, Yuasa S, Miwa S, Nomoto T, Sakakibara Y, Handa N, Hamanaka I, Saito Y, Komeda M. Initial effects of the left ventricular repair by plication may not last long in a rat ischemic cardiomyopathy model. Circulation. 2001;104:I‐241–I‐245. [DOI] [PubMed] [Google Scholar]
- 17. Yeboah MM, Xue X, Duan B, Ochani M, Tracey KJ, Susin M, Metz CN. Cholinergic agonists attenuate renal ischemia‐reperfusion injury in rats. Kidney Int. 2008;74:62–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Cury AF, Bonilha A, Saraiva R, Campos O, Carvalho AC, De Paola AA, Fischer C, Tucci PF, Moises VA. Myocardial performance index in female rats with myocardial infarction: relationship with ventricular function parameters by Doppler echocardiography. J Am Soc Echocardiogr. 2005;18:454–460. [DOI] [PubMed] [Google Scholar]
- 19. Henze M, Tiniakov R, Samarel A, Holmes E, Scrogin K. Chronic fluoxetine reduces autonomic control of cardiac rhythms in rats with congestive heart failure. Am J Physiol Heart Circ Physiol. 2013;304:h444–h454. [DOI] [PubMed] [Google Scholar]
- 20. Shiba Y, Fernandes S, Zhu WZ, Filice D, Muskheli V, Kim J, Palpant NJ, Gantz J, Moyes KW, Reinecke H, Van Biber B, Dardas T, Mignone JL, Izawa A, Hanna R, Viswanathan M, Gold JD, Kotlikoff MI, Sarvazyan N, Kay MW, Murry CE, Laflamme MA. Human ES‐cell‐derived cardiomyocytes electrically couple and suppress arrhythmias in injured hearts. Nature. 2012;489:322–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Be′lichard P, Savard P, Cardinal R, Nadeau R, Gosselin H, Paradis P, Rouleau JL. Markedly different effects on ventricular remodeling result in a decrease in inducibility of ventricular arrhythmias. J Am Coll Cardiol. 1994;23:505–513. [DOI] [PubMed] [Google Scholar]
- 22. Zhang H, Yuan X, Jin PF, Hou JF, Wang W, Wei YJ, Hu S. Alteration of parasympathetic/sympathetic ratio in the infarcted myocardium after Schwann cell transplantation modified electrophysiological function of heart: a novel antiarrhythmic therapy. Circulation. 2010;122:S193–S200. [DOI] [PubMed] [Google Scholar]
- 23. Nguyen T, El Salibi E, Rouleau JL. Postinfarction survival and inducibility of ventricular arrhythmias in the spontaneously hypertensive rat effects of ramipril and hydralazine. Circulation. 1998;98:2074–2080. [DOI] [PubMed] [Google Scholar]
- 24. Yue Y, Liu R, Cheng W, Hu Y, Li J, Pan X, Peng J, Zhang P. GTS‐21 attenuates lipopolysaccharide‐induced inflammatory cytokine production in vitro by modulating the Akt and NF‐κB signaling pathway through the α7 nicotinic acetylcholine receptor. Int Immunopharmacol. 2015;29:504–512. [DOI] [PubMed] [Google Scholar]
- 25. Kohno T, Anzai T, Naito K, Sugano Y, Maekawa Y, Takahashi T, Yoshikawa T, Ogawa S. Angiotensin‐receptor blockade reduces border zone myocardial monocyte chemoattractant protein‐1 expression and macrophage infiltration in post‐infarction ventricular remodeling. Circ J. 2008;72:1685–1692. [DOI] [PubMed] [Google Scholar]
- 26. Liu ML, Nagai T, Tokunaga M, Iwanaga K, Matsuura K, Takahashi T, Kanda M, Kondo N, Naito AT, Komuro I, Kobayashi Y. Anti‐inflammatory peptides from cardiac progenitors ameliorate dysfunction after myocardial infarction. J Am Heart Assoc. 2014;3:e001101 DOI: 10.1161/JAHA.114.001101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. van Maanen MA, Stoof SP, van der Zanden EP, de Jonge WJ, Janssen RA, Fischer DF, Vandeghinste N, Brys R, Vervoordeldonk MJ, Tak PP. The α7 nicotinic acetylcholine receptor on fibroblast‐like synoviocytes and in synovial tissue from rheumatoid arthritis patients: a possible role for a key neurotransmitter in synovial inflammation. Arthritis Rheum. 2009;60:1272–1281. [DOI] [PubMed] [Google Scholar]
- 28. Waldburger JM, Boyle DL, Pavlov VA, Tracey KJ, Firestein GS. Acetylcholine regulation of synoviocyte cytokine expression by the α7 nicotinic receptor. Arthritis Rheum. 2008;58:3439–3449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Sato KZ, Fujii T, Watanabe Y, Yamada S, Ando T, Kazuko F, Kawashima K. Diversity of mRNA expression for muscarinic acetylcholine receptor subtypes and neuronal nicotinic acetylcholine receptor subunits in human mononuclear leukocytes and leukemic cell lines. Neurosci Lett. 1999;266:17–20. [DOI] [PubMed] [Google Scholar]
- 30. Nahrendorf M, Swirski FK, Aikawa E, Stangenberg L, Wurdinger T, Figueiredo JL, Libby P, Weissleder R, Pittet MJ. The healing myocardium sequentially mobilizes two monocyte subsets with divergent and complementary functions. J Exp Med. 2007;204:3037–3047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Takamura M, Kurokawa K, Ootsuji H, Inoue O, Okada H, Nomura A, Kaneko S, Usui S. Long‐term administration of eicosapentaenoic acid improves post‐myocardial infarction cardiac remodeling in mice by regulating macrophage polarization. J Am Heart Assoc. 2017;6:e004560 DOI: 10.1161/JAHA.116.004560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Nguyen TP, Qu Z, Weiss JN. Cardiac fibrosis and arrhythmogenesis: the road to repair is paved with perils. J Mol Cell Cardiol. 2014;70:83–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Perrier E, Kerfant BG, Lalevee N, Bideaux P, Rossier MF, Richard S, Gomez AM, Benitah JP. Mineralocorticoid receptor antagonism prevents the electrical remodeling that precedes cellular hypertrophy after myocardial infarction. Circulation. 2004;110:776–783. [DOI] [PubMed] [Google Scholar]
- 34. Jiang W, St‐Pierre S, Roy P, Morley BJ, Hao J, Simard AR. Infiltration of CCR2+Ly6Chigh proinflammatory monocytes and neutrophils into the central nervous system is modulated by nicotinic acetylcholine receptors in a model of multiple sclerosis. J Immunol. 2016;196:2095–2108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Jandackova VK, Scholes S, Britton A, Steptoe A. Are changes in heart rate variability in middle‐aged and older people normative or caused by pathological conditions? Findings from a large population‐based longitudinal cohort study. J Am Heart Assoc. 2016;5:e002365 DOI: 10.1161/JAHA.115.002365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Lerner DL, Yamada KA, Schuessler RB, Saffitz JE. Accelerated onset and increased incidence of ventricular arrhythmias induced by ischemia in Cx43‐deficient mice. Circ J. 2000;101:547–552. [DOI] [PubMed] [Google Scholar]
- 37. Gutstein DE, Morley GE, Tamaddon H, Vaidya D, Schneider MD, Chen J, Chien KR, Stuhlmann H, Fishman GI. Conduction slowing and sudden arrhythmic death in mice with cardiac‐restricted inactivation of connexin43. Circ Res. 2001;88:333–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Yao JA, Gustein D, Liu F, Fishman GI, Wit AL. Cell coupling between ventricular myocyte pairs from connexin43‐deficient murine hearts. Circ Res. 2003;93:736–743. [DOI] [PubMed] [Google Scholar]
- 39. Ando M, Katare RG, Kakinuma Y, Zhang D, Yamasaki F, Muramoto K, Sato T. Efferent vagal nerve stimulation protects heart against ischemia‐induced arrhythmias by preserving connexin43 protein. Circulation. 2005;112:164–170. [DOI] [PubMed] [Google Scholar]
- 40. Shirai M, Joe N, Tsuchimochi H, Sonobe T, Schwenke DO. Ghrelin suppresses sympathetic hyperexcitation in acute heart failure in male rats: assessing centrally and peripherally mediated pathways. Endocrinology. 2015;156:3309–3316. [DOI] [PubMed] [Google Scholar]
- 41. Borovikova LV, Ivanova S, Zhang M, Yang H, Botchkina GI, Watkins LR, Wang H, Abumrad N, Eaton JW, Tracey KJ. Vagus nerve stimulation attenuates the systemic inflammatory response to endotoxin. Nature. 2000;405:458–461. [DOI] [PubMed] [Google Scholar]
- 42. van Westerloo DJ, Giebelen IA, Florquin S, Daalhuisen J, Bruno MJ, de Vos AF, Tracey KJ, van der Poll T. The cholinergic anti‐inflammatory pathway regulates the host response during septic peritonitis. J Infect Dis. 2005;191:2138–2148. [DOI] [PubMed] [Google Scholar]
- 43. de Jonge WJ, van der Zanden EP, The FO, Bijlsma MF, van Westerloo DJ, Bennink RJ, Berthoud HR, Uematsu S, Akira S, van den Wijngaard RM, Boeckxstaens GE. Stimulation of the vagus nerve attenuates macrophage activation by activating the Jak2‐STAT3 signaling pathway. Nat Immunol. 2005;6:844–851. [DOI] [PubMed] [Google Scholar]
- 44. de Jonge WJ, Ulloa L. The alpha7 nicotinic acetylcholine receptor as a pharmacological target for inflammation. Br J Pharmacol. 2007;151:915–929. [DOI] [PMC free article] [PubMed] [Google Scholar]