

Abstract
Genetic syndromes are well characterized by the phenotypic point of view, but little is known about their progression and patients’ quality of life. We report a 10-year neuropsychiatric follow-up of a boy with duplication of chromosome 19. Cytogenetic investigation was requested at the age of 5 years for psychomotor and speech delay. The genomic study identified an 8.17 Mb duplication on chromosome 19q12q13.2. We propose that the long-term follow-up of our patient would help to delineate the neuropsychiatric phenotype associated with 19q duplication. This study could be a model for further long-term research in the neuropsychiatric follow-up of patients with 19q duplication syndrome.
Keywords: 19q duplication, neuropsychiatric follow-up, array-CGH
Introduction
Genetic syndromes are mainly characterized by developmental delay, psychomotor retardation and language difficulties. In literature, the phenotype of these cases has been described in detail because facial dysmorphisms and structural anomalies are important for a correct diagnostic classification, but little is known about disease progression and patients’ quality of life. Most congenital defects may be dealt with surgery, secondary symptoms may be treated with drugs, but basic information about socialization and daily living skills are often absent.
Here, we report a 10-year neuropsychiatric follow-up of a boy with duplication of chromosome 19. Partial trisomy of 19q has been reported in literature in different cases, but the origin can be very different due to the presence of a small supernumerary marker, a duplication or derivative from translocation in combination with the relative loss of translocation partner. These distinct mechanisms influence gene expression and its regulation, complicating the identification of a unique clinical noxa.
We propose that the long-term follow-up of our patient would help to delineate the neuropsychiatric phenotype associated with 19q duplication. This study could be a model for further long-term research in the neuropsychiatric follow-up of patients with 19q duplication syndrome.
Case presentation
Clinical report
The patient (A.S.) is the second child of a nonconsanguineous couple. Pregnancy was uneventful; at birth, APGAR scores were 6, 6 and 10 after 1, 5 and 10 minutes, respectively. No major medical problems were observed during the neonatal period except for transitory tachypnea. Growth has always been normal in height and weight, while head circumference showed macrocephaly.
The results of various clinical and instrumental examinations (electrocardiography, audiometry, routine laboratory tests) were normal. The boy was referred to the genetic department of our hospital for psychomotor and speech delay at the age of 5 years and a cytogenetic investigation was requested.
Cytogenetic conventional analysis showed an abnormal chromosome 19q, defined and confirmed as direct duplication (19q12q13.2) after fluorescence in situ hybridization investigation with bacterial artificial chromosome probes mapped in the region involved in duplication (data not shown). Parental karyotypes were normal, indicating a de novo origin of the abnormality.
Array-comparative genomic hybridization (CGH) analysis, performed using CGH 105K microarray kit (Agilent Technologies, Santa Clara, CA, USA), identified an 8.17 Mb duplication on chromosome 19q arm from nt 28,740,636 to nt 36,906,767. The karyotype, defined following International System of Chromosome Nomenclature 2016, was: 46,XY,dup (19) (q12q13.2).arr [GRCh37] 19q12q13.2 (28,740,636_36,906,767) ×3 (Figure 1).
Figure 1.
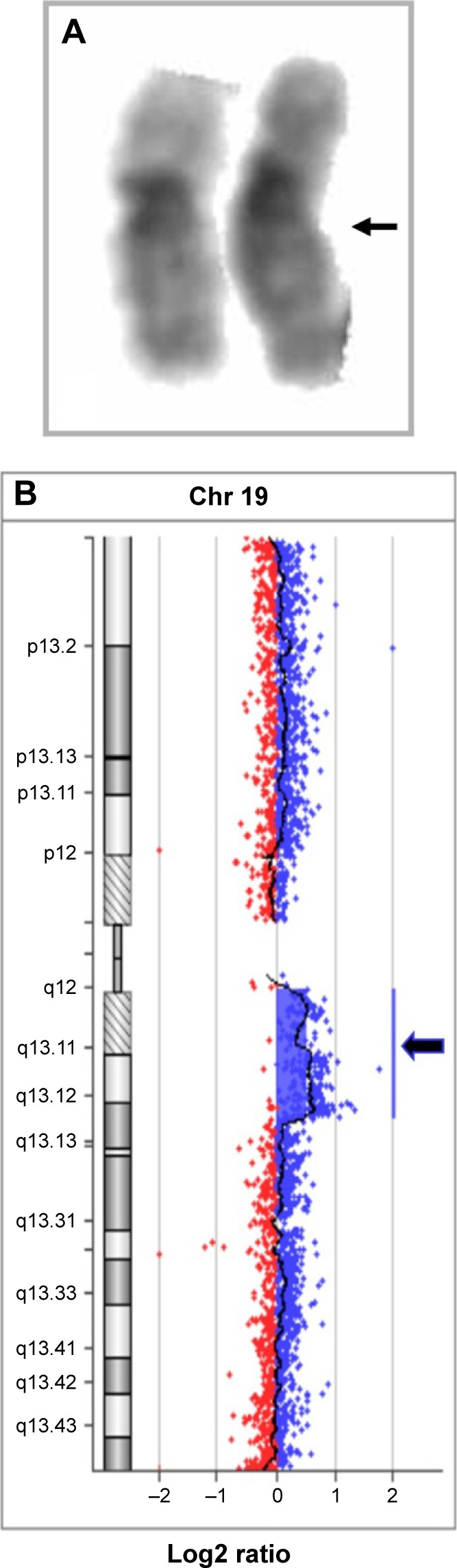
Cytogenetic and array-CGH characterization.
Notes: (A) Proband’s GTG-banded chromosomes 19; the arrow shows the duplication. (B) Chromosome 19 view showing the duplication in array-CGH (blue arrow).
Abbreviations: CGH, comparative genomic hybridization; GTG, Giemsa Trypsin-Giemsa.
The duplicated region contained 105 genes, 17 associated with disease (Online Mendelian Inheritance in Man, https://www.ncbi.nlm.nih.gov/omim/).
Neuropsychiatric report
When the child was 3 years old, the parents requested service for a first evaluation because of speech difficulties. Speech development began on time, but showed slow evolution and poor communicative intent: the first words “mamma, papà” (“mummy, daddy”) appeared when he was 1 year old. Between 12 and 36 months, the proband produced only a few mono - or two-syllable words and some onomatopoeic sounds, and he used gestures to indicate or respond to requests; there was a discrepancy between the deficient expressive skills and the verbal comprehension, which was discrete.
The psychomotor evaluation conducted at 41 months, confirmed a disharmonious developmental delay of about 1 year, with a significant gap in speech (21 month), understanding (24 month), space–time organization (24 month) and perception (26 month). This delay tended to worsen over time: after 6 months, the gaps in space–time organization (27 month) and speech (28 month) were more evident, associated with passiveness, tendency to feel frustrated, and repetitive play. At the age of 3 years, the proband was also clumsy and uncoordinated, without major neurological dysfunctions and he had a bilateral pronator syndrome with valgus knee. For this reason, for a few months, he underwent physiotherapy sessions to reinforce the arch of the foot and improve his motor skills.
The speech evaluation at 42 months of age showed: a preferential use of onomatopoeic sounds and a few words combined in simple sentences of some terms, without verbs; deficit in articulation movements and coordination; use of gestures to denominate; discrete verbal comprehension; malocclusion with overbite. Later, a phonological and morphosyntactic deficit was observed.
Moreover, in the same period, the child showed relationship difficulties: he was passive and clingy with peers and adults and interested only in playing with his brother; over time, withdrawal behavior was detected in group settings.
The multidisciplinary evaluation led to the diagnosis of expressive language disorder with childhood dyspraxia of speech and mixed disorder of development.
The therapy project included counseling to parents and educators; and psychomotor and speech therapy carried out in a rehabilitation center for 3 and 4 years, respectively, once a week.
The cognitive development at 4 years 11 months, measured by the Wechsler Preschool and Primary Scale of Intelligence (WPPSI) scale, was in a borderline range (full scale IQ =79), with a significant discrepancy between verbal and performance scores, due to lower scores in verbal subtest, especially in lexical and semantic expressiveness and in the logical and abstraction area (VIQ =67). Performance score was in the normal range (PIQ =96).
The WPPSI Scale results at 5 and 11 years were similar to the first evaluation (VIQ =66, PIQ =95, total IQ =78), but differences between verbal subtests were observed: “vocabulary” increased and “comprehension” worsened.
In fact, after 2 years of therapy, the proband had achieved a good phonetic repertoire, a better expressive vocabulary than in the past, with an emerging awareness of his language difficulties and communicative intention, despite a significant delay in morphosyntactic production and comprehension and the persistence of clumsiness.
At the beginning of primary school, the proband needed a support teacher because of the clinical complexity of language disorder and the praxis and relational difficulties. He achieved good writing and reading abilities and, thanks to this, he improved his language skills the phonological way, so logopedic rehabilitation was stopped.
Neuropsychiatric follow-up, together with counseling to parents and teachers continued during the school period.
At the age of 10, the cognitive development of the proband, measured by the Wechsler Intelligence Scale for Children-III (WISC-III) Scale, was in the lower limits of the normal range (Full Scale IQ =80), with a significant discrepancy between verbal (VIQ =74) and performance (PIQ =90) scores.
The worst subtests were the following: “information” (weighted score =3), corresponding to a poor wealth of knowledge acquired; “arithmetic” (=5), difficulties in the logical area and calculation; “digit span” (=6), low auditory verbal short-term memory; “block design” (=6), expression of poor ability to analyze and synthesize an abstract design and reproduce that design from colored plastic blocks. Less obvious deficiencies were found in “vocabulary” (=7) that measures verbal fluency and concept formation, word knowledge, and word use and in “coding” (=7) that investigates visual-motor dexterity, associative nonverbal learning, and nonverbal short-term memory.
The other subtests scores were within normal ranges: the critical judgment (“comprehension” =8), verbal abstraction (“similarities” =8), recognition of causal and temporal connections in illustrated sequences (“picture arrangement” =9). Finally, the ability of perceptive analysis of visual stimuli seemed to be normal (“picture completion =10). Also, the proband showed non-specific difficulties in learning, when logic skills and problem-solving abilities were required.
Moreover, toward the end of the school cycle, behavioral problems began, such as oppositional traits, impulsiveness, together with integration and relationship problems among peers.
At the same time, the cognitive development worsened: after 1 year, the global level, measured by the re-test with the WISC-III, was at the borderline range of normal (Full Scale IQ =76), with a more marked discrepancy between verbal (VIQ =62) and performance (PIQ =96) scores. For these issues, psychological support and neuropsychological therapy were introduced, as well as educational and social help.
During adolescence, his capability to interact with peers and parents worsened, due to atypical alteration of his psychological development, characterized by emotional and behavioral disorder (EBD). The main features of his disease were an increased level of impulsivity, agitation crisis with aggressive reactions, kleptomania and compulsive behaviors.
From a psycho-dynamic perspective, these events fit into the context of a very weak “Self”, with an affective immaturity and lack of emotional auto-regulation; as a consequence of that, fluctuations between maniacal excitement and anguish states with pseudo-depressive symptoms were observed.
Moreover, regression in communication and conversational skills was observed: stammer, reduced phono-articulatory control, and a general decrease in his interest in communication.
The proband had a brief hospitalization and later started psychotherapy supported by pharmacological treatment. He initially interrupted the pharmacological therapy of benzodiazepine and risperidone as it was not showing significant benefits. However, without any pharmacologic treatment, he quickly relapsed into aggressive behavioral symptoms. Following an urgent first aid visit, he was treated with a first-generation anti-psychotic (promazina) at low dosage. After a short period, the treatment was suspended as the proband showed collateral effects: sedation, apathy, lack of appetite with loss of weight.
Finally, his condition significantly improved by therapy based on a second-generation anti-psychotic at low dosage (olanzapine, 5 mg), which he still continues to take. His parents are currently being supported by counseling.
Thanks to the measures applied, after the most complicated phase observed between the ages of 13 and 15 years, the proband is now experiencing increased wellness. Although some relational difficulties in the acquisition of social autonomy are still present, he is now able to study in a professional school with the help of a learning support teacher.
Discussion
Partial duplication of the long arm of chromosome 19 is a rare chromosomal anomaly that can originate from different mechanisms explaining the phenotypic, intellectual and behavioral differences of the affected subjects.
Here, we report a duplication of chromosome 19q12q13.2 that can be compared, for the onset mechanism and the size, with only another two cases reported in the literature (Table 1, Figure 2).1,2 Moreover, focusing only on the neuropsychiatric aspects, the comparison appears very difficult due to the short term and poor follow-up description.
Table 1.
Comparison with literature data
| Present case | Bralo et al1 | Lugli et al2 | Qorri et al3 | Wilson et al4 | Quack et al5 | Zung et al6 | Hall et al7 | |
|---|---|---|---|---|---|---|---|---|
| Cytogenetic abnormality | dirdup q12q13.2 | dirdup q12q13.2 | dirdup q12q13.2 | dirdup q13.1q13.3 | dup at mos q12q13.2 | ring q11.05q13.2 | SMC q12q13.2 | SMC at mos q13.11q13.2 |
| Sex | Male | Female | Male | Female | Male | Male | Male | Male |
| Psychomotor or mental retardation | + | + | + | + | + | + | + | + |
| Growth retardation | − | − | − | − | − | − | − | − |
| Obesity | − | − | − | − | + | + | + | + |
| Tall stature | − | − | − | − | + | + | + | − |
| Follow-up | 16 years | 15 months | 3 years | 2 years, 3 months | 3 years, 2 months | 3 years | 14 years* | 5 years, 4 months |
Note:
Not neuropsychiatric follow-up.
Abbreviations: dirdup, direct duplication; Mos, mosaic; SMC, supernumerary marker chromosome; dup, duplication.
Figure 2.

Facial features of the patient.
Note: Our patient at the age of 13 months.
The other two cases reported for a pure 19q duplication, have an incomplete cytogenetics definition or a mosaic duplication, making the correlation with our case difficult.3,4 Finally, another three patients showed 19q duplication due to the presence of ring or small supernumerary chromosomes, but no long-term follow-up was reported.5–7
Comparing chromosomal duplication observed in our patient with those reported in genomic variants databases, we found only two comparable cases for size and position: one in Decipher database (patient 254971, https://decipher.sanger.ac.uk/) and one in the Italian Database of Troina (patient 11629, http://dbcnv.oasi.en.it/gvarianti/index.php), but unfortunately, these cases lack in clinical description.
In the last few years, the use of comparative genomic hybridization array refined the molecular bases of developmental disabilities and psychiatric disorders associated with chromosome deletion/duplication.8,9
Different from the other studies, obesity and seizures were not present in our proband (Table 1). He did not show motor milestones delay but had mild psychomotor difficulties, being clumsy and uncoordinated for his age. Most importantly, a neuropsychiatric follow-up, which continues still today, has enabled us to detect the onset of a psychiatric disorder not described previously. Our unique opportunity to follow the proband through adolescence (he is now 16 years old) fills a gap of knowledge about the relationship between chromosome alteration and EBDs over time.
Psychiatric comorbidity is common in children with neurodevelopmental and communication disorders. Our proband had disorders affecting multiple domains, including motor skills, language and cognition, as well as impulsiveness and relationship problems among peers. A growing number of reports underscore the genetic overlap between previously distinct clinical disorders. Behavioral and psychiatric features are increasingly identified in association with intellectual developmental disorders.10
To conclude, we wish to stress the importance, on the proband’s side, of a neuropsychiatric long-term follow-up and, on the parents’ side, of support and counseling to face the different implications of their son’s disease. We strongly recommend providing integrative support also to teachers at school. It is crucial that not only the educational aspects, but also the emotional and relational sides of the proband’s life outside the domestic environment are taken into account.
Acknowledgments
Written informed consent was obtained from the patient’s parents for publication of this case report and any accompanying images.
Footnotes
Disclosure
The authors report no conflicts of interest in this work.
References
- 1.Palomares Bralo M, Delicado A, Lapunzina P, et al. Direct tandem duplication in chromosome 19q characterized by array CGH. Eur J Med Genet. 2008;51(3):257–263. doi: 10.1016/j.ejmg.2008.01.003. [DOI] [PubMed] [Google Scholar]
- 2.Lugli L, Malacarne M, Cavani S, Pierluigi M, Ferrari F, Percesepe A. A 12.4 Mb direct duplication in 19q12-q13 in a boy with cardiac and CNS malformations and developmental delay. J Appl Genet. 2011;52(3):335–339. doi: 10.1007/s13353-011-0033-5. [DOI] [PubMed] [Google Scholar]
- 3.Qorri M, Oei P, Dockery H, McGaughran J. A rare case of a de novo dup(19q) associated with a mild phenotype. J Med Genet. 2002;39(10):E61. doi: 10.1136/jmg.39.10.e61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wilson BT, Newby R, Watts K, Hellens SW, Zwolinski SA, Splitt MP. A case of mosaic trisomy 19q12-q13.2 with high BMI, macrocephaly, and speech delay: does USF2 determine size in the 19q phenotypes? Clin Dysmorphol. 2012;21(1):33–36. doi: 10.1097/MCD.0b013e32834e7f9f. [DOI] [PubMed] [Google Scholar]
- 5.Quack B, Van Roy N, Verschraegen-Spae MR, Klein F. Interstitial deletion and ring chromosome derived from 19q. Proximal 19q trisomy phenotype. Ann Genet. 1992;35(4):241–244. [PubMed] [Google Scholar]
- 6.Zung A, Rienstein S, Rosensaft J, Aviram-Goldring A, Zadik Z. Proximal 19q trisomy: a new syndrome of morbid obesity and mental retardation. Horm Res. 2007;67(3):105–110. doi: 10.1159/000096419. [DOI] [PubMed] [Google Scholar]
- 7.Hall CE, Cunningham JJ, Hislop RG, Berg JN. A boy with supernumerary mosaic trisomy 19q, involving 19q13.11–19q13.2, with macrocephaly, obesity and mild facial dysmorphism. Clin Dysmorphol. 2010;19(4):218–221. doi: 10.1097/MCD.0b013e32833bff06. [DOI] [PubMed] [Google Scholar]
- 8.Barone R, Fichera M, De Grandi M, et al. Familial 18q12.2 deletion supports the role of RNA-binding protein CELF4 in autism spectrum disorders. Am J Med Genet A. 2017;173(6):1649–1655. doi: 10.1002/ajmg.a.38205. [DOI] [PubMed] [Google Scholar]
- 9.Polimanti R, Squitti R, Pantaleo M, Giglio S, Zito G. Duplication of FOXP2 binding sites within CNTNAP2 gene in a girl with neurodevelopmental delay. Minerva Pediatr. 2017;69(2):162–164. doi: 10.23736/S0026-4946.16.04326-7. [DOI] [PubMed] [Google Scholar]
- 10.King BH. Psychiatric comorbidities in neurodevelopmental disorders. Curr Opin Neurol. 2016;29(2):113–117. doi: 10.1097/WCO.0000000000000299. [DOI] [PubMed] [Google Scholar]