

Abstract

While the use of triphenylphosphine as a reductant is common in organic synthesis, the resulting triphenylphosphine oxide (TPPO) waste can be difficult to separate from the reaction product. While a number of strategies to precipitate TPPO are available, none have been reported to work in more polar solvents. We report here that mixing ZnCl2 with TPPO precipitates a TPPO–Zn complex in high yield in several common polar organic solvents. The solvent compatibility of this procedure and the reliability of the precipitation in the presence of polar functional groups were examined to show the utility and limitations of this method.
Introduction
Triphenylphosphine is a versatile reagent in organic synthesis; reactions that use it to reduce functional groups, convert carbonyls to olefins, or effect substitution of an alcohol for another nucleophile are among the most common in organic chemistry.1 In transformations that employ triphenylphosphine (TPP) as a reductant, the triphenylphosphine is converted to triphenylphosphine oxide (TPPO). Although in some cases separation of TPPO from the reaction product is easily achieved, the difficulty of the separation is often cited as a barrier to using TPP as a reductant or reagent. This has inspired research on improved removal methods for TPPO, the development of alternative phosphines whose oxides are easier to remove, and reactions that are catalytic in phosphine. Although substantial progress continues to be made using insoluble phosphine reagents or catalytic strategies, triphenylphosphine is unlikely to be completely replaced in the near term. For this reason, additional methods for the removal of TPPO are still needed. We report here that TPPO is efficiently precipitated as ZnCl2(TPPO)2 in polar solvents and demonstrate how this discovery can be used to precipitate TPPO from reactions that would otherwise require purification by column chromatography.
Background
Reports on solutions to the problem of separating triphenylphosphine oxide (TPPO) byproducts in organic reactions fall into three broad categories: (1) improved methods for TPPO removal; (2) avoiding TPPO by using alternative phosphines2 with more easily separated phosphine oxides,3 and (3) avoiding stoichiometric phosphine oxide waste by developing reactions that are catalytic in phosphine4 or do not require phosphine at all.5 This report is focused on methods of TPPO removal, so strategies (2) and (3) will not be discussed further.
The separation of TPPO byproducts from reaction products has generally been accomplished by chromatography, but this can be tedious on a larger scale.1,6 Separation by distillation is useful for cases where the reaction product is sufficiently stable and low-boiling,7 but a liquid–liquid or liquid–solid phase separation would be more generally useful. TPPO can be precipitated in cases where the reaction product is soluble in very nonpolar solvents, such as cold hexanes and diethyl ether mixtures.8 Product precipitation or crystallization is also a common strategy, but success depends greatly upon the identity of the product.9
In cases where these strategies cannot be applied, several groups have described methods to convert TPPO into a more easily separated species. For example, Lipshutz demonstrated the removal of TPPO by alkylative trapping on Merrifield resin,10 and Gilheany described the use of oxalyl chloride to convert TPPO to triphenylphosphonium chloride, which is easily precipitated from cyclohexane.11
When direct precipitation cannot be used and reactive conversion is not possible, addition of a co-crystallization agent has been employed. TPPO has been demonstrated to co-crystallize with a wide variety of organic molecules with acidic protons in nonpolar solvents.12 For example, researchers at Shin-Etsu Chemical Co. removed TPPO by adding acetic acid to an n-hexane mixture of product and TPPO to form an immiscible TPPO–AcOH fluid phase that was separated from the product solution.13 As another example, chemists at Squibb found that, in toluene, they could remove TPPO and diisopropylurea as a 1:1 complex.14
Lewis acid TPPO adducts are also well-known and have been used in the extraction of metal salts but have been less frequently applied to the removal of TPPO. For example, crystals of ZnCl2(TPPO)2 have been known for over 100 years,15 yet we could only find one report where ZnCl2 was used to precipitate TPPO from ethereal reaction mixtures16 and one patent where precipitation was mentioned, without examples, alongside more detailed methods using MgCl2.17 Although the magnesium chloride method has been utilized on scale, it is notable that a switch to a less polar solvent was required for efficient separation.17b
All of these physical separation strategies were described using low-polarity solvents, such as hexanes, toluene, cyclohexane, and diethyl ether. To our knowledge, there are no methods for the precipitation of TPPO from more polar solvents (i.e., ethanol, ethyl acetate, tetrahydrofuran, etc.) that are commonly used in organic synthesis. Such a method would provide an additional tool in complex molecule synthesis.
Results and Discussion
In the course of large-scale (>50 g) preparation of 2,7-dibromocarbazole 2 from 4,4′-dibromo-2-nitrobiphenyl 1 by reductive cyclization with triphenylphosphine,18 one of us noted that triphenylphosphine could be conveniently separated from the carbazole product by precipitation from ethanolic zinc chloride (Scheme 1). This approach was inspired by the use of phosphine oxides to selectively extract metal ions from mixtures during the enrichment of uranium ores.19 In addition, the synthesis of pure crystals of ZnCl2(TPPO)2 from ethanol solutions has been reported previously but never applied to TPPO removal from reaction mixtures.15b,15d
Scheme 1. Removal of Triphenylphosphine Oxide by ZnCl2.

The reaction procedure was simple: a 1.8 M solution of ZnCl2 in warm ethanol was added to an ethanolic solution of the product/TPPO mixture at room temperature. After stirring and scraping to induce precipitation, the ZnCl2(TPPO)2 adduct precipitated from solution. The solution was filtered to remove the precipitate, and the filtrate concentrated to remove ethanol. Finally, the residue was slurried with acetone to separate the soluble product from any insoluble excess zinc chloride. The filtered solution was completely free of triphenylphosphine oxide by TLC analysis without the need for chromatography.
Given this promising initial result, we sought to examine the molar amount of zinc chloride added and find the optimal ratio of ZnCl2 to TPPO for complete precipitation of TPPO from solution (Table 1). This study was accomplished by dissolving a fixed quantity of commercial TPPO in ethanol and adding various equivalents of ZnCl2 to promote precipitation. After precipitation, the amount of TPPO remaining in solution was quantitated by GC analysis. We found that 90% of the TPPO was removed from solution with an equimolar ratio of ZnCl2 and TPPO. Increasing the ratio of ZnCl2 to TPPO increased TPPO precipitation; at a 3:1 ratio TPPO could no longer be detected in the filtered solution. Considering the diminishing returns on TPPO removal with higher equivalents of ZnCl2, a 2:1 ratio was selected as optimal for further studies. It should be noted that this precipitation is robust and the ratio of ZnCl2 to TPPO can be tuned to minimize TPPO or excess ZnCl2 depending on the requirements for reaction purification. Elemental analysis of several different precipitates suggested that the complex formed is ZnCl2(TPPO)215 but might contain a small amount of additional TPPO. The stoichiometry and literature precedent are consistent with ZnCl2(TPPO)2 as the precipitate.
Table 1. Effect of ZnCl2 Equivalents on Precipitation Efficiencya.

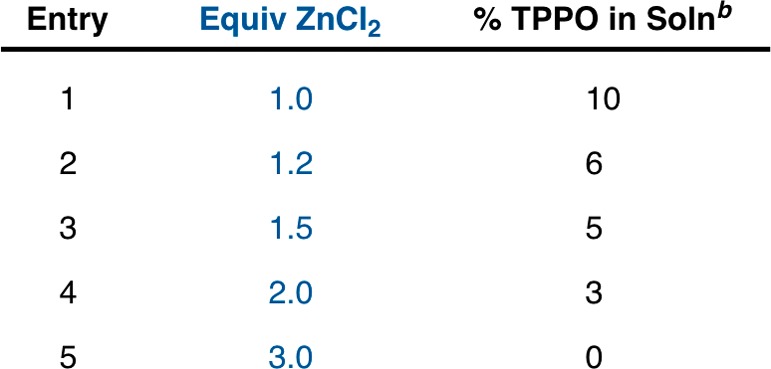
Experiments were performed with 1.0 g of TPPO (3.6 mmol) solvated in 10 mL of ethanol. To this solution was added the listed ratio of solid ZnCl2. A white precipitate formed immediately, and the reaction was allowed to stand for 18 h at 22 °C before the solid was separated by filtration and rinsed with 10 mL of ethanol. The filtrate was analyzed to determine the amount of TPPO in solution.
Percentage of TPPO remaining in solution was determined by corrected GC yield with dodecane as an internal standard.
Next, a systematic investigation of polar solvents frequently used in organic synthesis was undertaken to determine the general utility of this procedure (Table 2). TPPO was dissolved in each solvent and precipitated with a 2:1 ratio of ZnCl2 to TPPO. After filtration, GC analysis demonstrated that precipitation proceeds in solvents ranging from THF to iPrOH. EtOAc, iPrOAc, and iPrOH stood out as excellent solvents for this method (<5% TPPO), whereas THF, 2-MeTHF, and methyl ethyl ketone were tolerated (<15% TPPO). MeOH, MeCN, acetone, and DCM (no precipitate formed) did not allow for efficient precipitation (>15% TPPO) either due to increased solubility of the TPPO/ZnCl2 adducts or increased solvation of ZnCl2 reducing the equilibrium constant for adduct formation. Solvent mixtures were explored to determine if combinations of solvents could improve precipitation. Only the combination of DCM/iPrOH improved TPPO removal. Finally, the addition of excess EtOH can assist in precipitation of ZnCl2(TPPO)2 (vide infra).
Table 2. Solvent Effect on TPPO Precipitation with ZnCl2a.
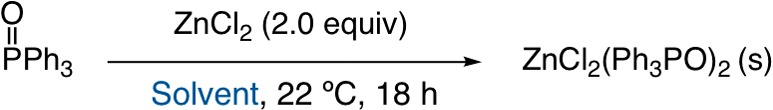
See Table 1 for experimental details. TPPO was precipitated with 0.98 g of ZnCl2 (7.2 mmol) in 10 mL of the listed solvent. An additional 10 mL of solvent was used to wash the solid precipitate.
Percentage of TPPO remaining in solution was determined by corrected GC yield with dodecane as an internal standard. EtOH = ethanol; iPrOH = 2-propanol; THF = tetrahydrofuran; 2-MeTHF = 2-methyltetrahydrofuran; MeCN = acetonitrile; EtOAc = ethyl acetate; iPrOAc = isopropyl acetate; MEK = methyl ethyl ketone; DCM = dichloromethane.
We next sought to assess the functional group compatibility of the method. While no brief survey could reveal all potential incompatibilities, we conducted several precipitations in the presence of small molecules bearing a representative array of functional groups (Table 3). We anticipated that some functional groups would prevent efficient separation by either solubilizing the ZnCl2 adduct or by co-precipitating with TPPO and ZnCl2.12 Removal of TPPO occurred efficiently (<5% TPPO) with alcohol, aldehyde, and amide functional groups in iPrOH20 with only a slight decrease in the recovery of benzaldehyde (78%) (entries 1, 3, and 4). A protected amino acid, Boc-Gly-OMe (entry 9), was also tolerated with minimal loss. Less positive results were obtained with substrates that appeared to co-precipitate with TPPO and ZnCl2 through basic nitrogen atoms: aniline, 4-methoxypyridine, and quinidine (entries 2, 7, and 10).21 A more hindered pyridine, 2,6-lutidine, worked well (entry 8), consistent with the coordination hypothesis. It may be possible to avoid coprecipitation by further modification of the conditions (solvent, amount of zinc used), but this was not investigated further.
Table 3. Functional Group Compatibility of TPPO Precipitationa.
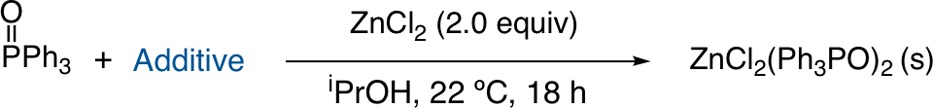
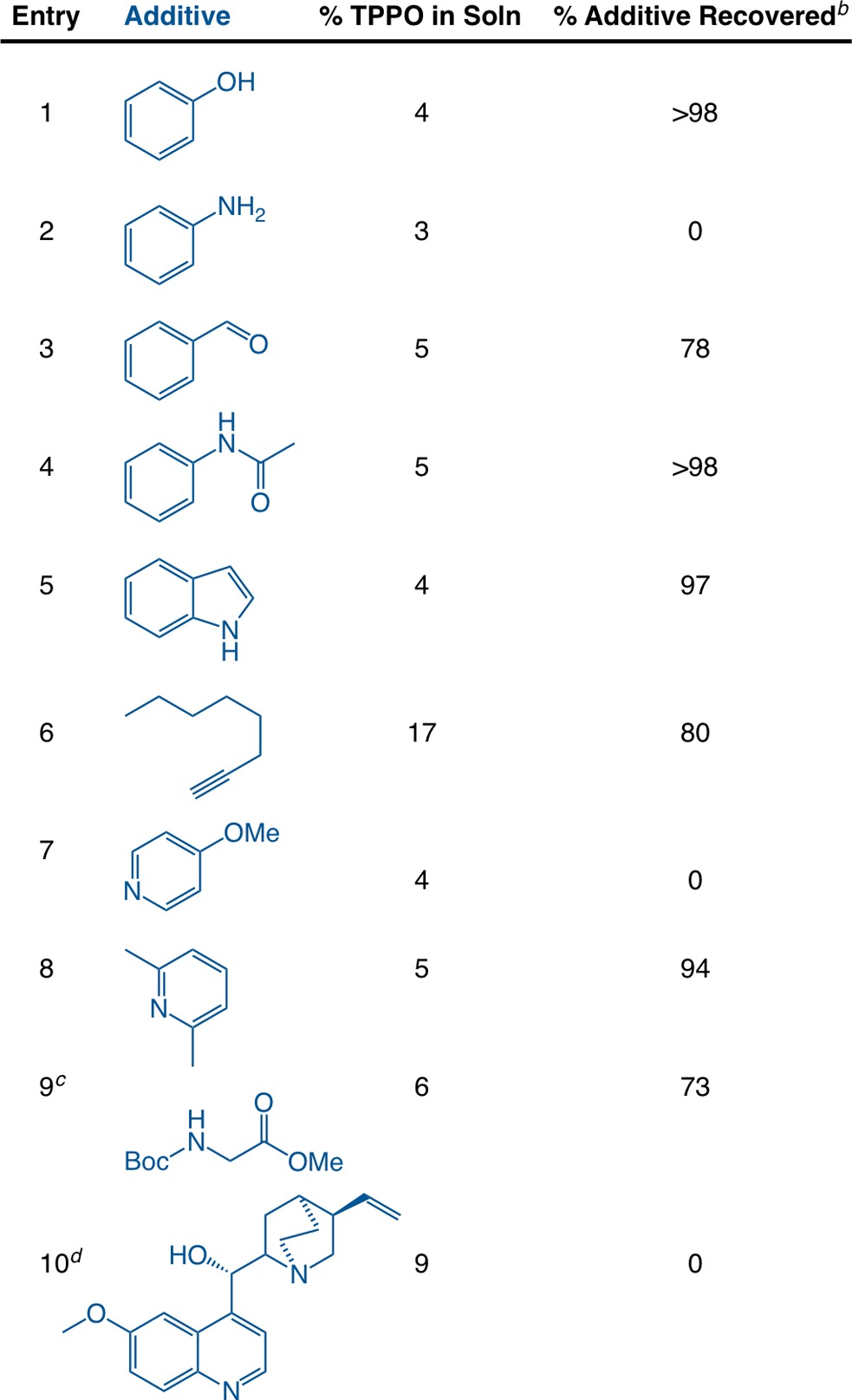
See Table 1 for experimental details. The listed additive (3.6 mmol) was added to the initial TPPO solution in iPrOH prior to the addition of 0.98 g of ZnCl2 (7.2 mmol). After precipitation, the reactions were filtered and washed, and the remaining solution was analyzed by GC.
The percentage of TPPO and additive remaining in solution was determined by GC comparison of the solution prior to ZnCl2 addition and after filtration using dodecane as an internal standard.
Reaction was run with double the solvent.
Control reactions showed that quinidine formed a precipitate with ZnCl2 in the absence of TPPO.
In addition to the initial carbazole-forming reaction (see Scheme 1), two classic organic reactions, the Corey–Fuchs22 and the Mitsunobu reaction,23 were explored to examine the new method in situations where TPPO separation often requires column chromatography.24 In the Corey–Fuchs reaction to synthesize 4 (Scheme 2), we found that precipitation of TPPO worked well. The crude reaction mixture was washed with H2O2 to convert any remaining phosphine and phosphonium species to TPPO, allowing efficient removal with a single precipitation with ZnCl2 (Scheme 2).25 Compound 4 could be obtained on multigram scale in 82% yield without the need for column chromatography.
Scheme 2. Corey–Fuchs Reaction with Removal of TPP and TPPO by Oxidation and Precipitation.

The Mitsunobu reaction to form 6 from l-menthol and 5 was similarly successful (Scheme 3).23 The crude reaction mixture was washed with sodium bicarbonate and peroxide to remove excess acid 5 and oxidize any remaining TPP to TPPO. Simple solvent evaporation followed by dilution with EtOH and precipitation with ZnCl2 (twice) removed the majority of TPPO and allowed for the crystallization of the product directly from the ethanol solution with no further manipulation; 6 was isolated in 68% yield without purification by column chromatography.
Scheme 3. Mitsunobu Reaction with Precipitation of TPPO by ZnCl2.

While the procedure worked well for the synthesis of 2, 4, and 6, we did find several limitations to the method. Besides the potential for co-precipitation (vide supra), we also found, in one case, that an acid-sensitive β-lactone26 reacted with solvent under these Lewis acidic conditions. In addition, the use of an excess of ZnCl2 means that, in cases where zinc contamination would be a concern, additional washes or manipulations could be required (i.e., distillation, extraction with water).
In conclusion, we have discovered that triphenylphosphine oxide can be conveniently removed from solution by precipitation with ZnCl2 in ethanol. This precipitation functions in several polar organic solvents, such as EtOAc, iPrOAc, and iPrOH, and is tolerant of many common functional groups including alcohols, aldehydes, amides, and some nitrogen heterocycles. The method was showcased in the large-scale, chromatography-free isolation of products from three reactions that generate TPPO byproducts. We anticipate that this method will provide an additional purification tool for many reactions that use triphenylphosphine as a reductant.
Experimental Section
Zinc chloride was ACS grade. All other solvents and reagents used were commercial, ACS grade, or better.
Trial Precipitation of the Zinc Chloride Complex of Triphenylphosphine Oxide
In a blank experiment, 20.0 g (76.3 mmol) of triphenylphosphine was suspended in 200 mL of ethanol with gentle refluxing. Air was bubbled through the solution overnight, yielding a clear, colorless solution of triphenylphosphine oxide. To this mixture was added a solution of 10.4 g (76.3 mmol) of zinc chloride in 100 mL of warm ethanol. The mixture was stirred and allowed to cool, whereupon a heavy, white precipitate separated. After the mixture had cooled to rt, the solid zinc chloride complex was collected by vacuum filtration on a Buchner funnel. The solid was then washed with water (100 mL), ethanol (100 mL), and ether (100 mL) before it was air-dried at room temperature, yielding 23.8 g. This corresponds to a 90% yield for Zn(O=PPh3)2Cl2.
Trial Reaction of Zinc Chloride with Triphenylphosphine
A second experiment on the same scale was run to see if alcohol solutions of triphenylphosphine and zinc chloride would also form a complex at reflux. The starting triphenylphosphine was recovered, and no complex was detected.
Trial Reaction of Triphenylphosphine Oxide with Magnesium Chloride in Ethanol
A final experiment was run to see if, under the same conditions, triphenylphosphine oxide would form an alcohol insoluble complex with magnesium chloride.17 No precipitation occurred and only triphenylphosphine oxide was recovered.
General Procedure for Table 1: Optimizing Equivalents of ZnCl2
A 20 mL scintillation vial was charged with a stirbar, triphenylphosphine oxide (1.0 g, 3.6 mmol, 1 equiv), and EtOH (10 mL). To this solution was added the listed equivalents of solid ZnCl2 accompanied by the immediate formation of a white precipitate. The mixture was allowed to stir for 18 h at 22 °C before the solid was separated by filtration and rinsed with EtOH (10 mL). The filtrate was collected, and dodecane (100 uL, 0.44 mmol) was added as an internal standard. The percentage of triphenylphosphine oxide remaining in the solution was determined by GC analysis, corrected.
General Procedure for Table 2: Solvent Effect on TPPO Precipitation
A 20 mL scintillation vial was charged with a stirbar, triphenylphosphine oxide (1.0 g, 3.6 mmol, 1 equiv), and solvent(s) (10 mL total). To this solution was added ZnCl2 (0.98 g, 7.2 mmol, 2 equiv). The mixture was allowed to stir for 18 h at 22 °C before any precipitate was separated by filtration and rinsed with the listed solvent (10 mL). The filtrate was collected, and dodecane (100 μL, 0.44 mmol) was added as an internal standard. The percentage of triphenylphosphine oxide remaining in the solution was determined by GC analysis, corrected.
General Procedure for Table 3: Functional Group Compatibility
A 20 mL scintillation vial was charged with a stirbar, triphenylphosphine oxide (1.0 g, 3.6 mmol, 1 equiv), and the listed additive (1.0 equiv). The solids were dissolved in iPrOH (10 mL), which was used because phase separation occurred when EtOH was used to solvate 1-octyne. Following complete dissolution, dodecane (100 μL, 0.44 mmol) was added to the homogeneous solution as an internal standard. The solution was analyzed by GC to determine baseline ratios of triphenylphosphine oxide and additive to dodecane. ZnCl2 (0.98 g, 7.2 mmol, 2 equiv) was then added to the solution, initiating the formation of a white precipitate. The mixture was allowed to stir for 18 h at 22 °C before the precipitate was separated by filtration and rinsed with iPrOH (10 mL). The filtrate was collected and analyzed by GC to determine the ratios of triphenylphosphine oxide and additive to dodecane. The percentage of triphenylphosphine oxide and additive remaining in the solution was determined by comparing the GC ratios before and after zinc addition.
2,7-Dibromocarbazole (2).18
A 5-L, three-neck flask fitted with an air-powered mechanical stirrer, reflux condenser, and thermometer was set in an electric heating mantle. Into this flask was added 214 g (0.600 mol) of 4,4′-dibromo-2-nitrobiphenyl,18 1.18 L of 1,2-dichlorobenzene, and 388 g (1.48 mol) of triphenylphosphine. The stirred mixture was heated to reflux (190 °C) under a blanket of argon and held at this temperature for 6 h. The dark-colored mixture was allowed to cool slowly to rt with stirring overnight. The mixture was concentrated at reduced pressure on a rotary evaporator to remove 1,2-dichlorobenzene. The dark, viscous residue was poured into a Pyrex glass tray, the residue remaining in the evaporating flask was rinsed with 200–300 mL of petroleum ether, and this was added to the main body of crude product in the Pyrex tray. The tray was placed in a vacuum oven and warmed to remove the petroleum ether and any remaining 1,2-dichlorobenzene. The tray was allowed to cool to room temperature, whereupon the black material solidified. The crude solid, containing the product and triphenylphosphine oxide, was powdered using a mortar and pestle or a Waring blender. The triphenylphosphine oxide content was assumed to be equivalent to the molar quantity of triphenylphosphine used.
The solid was dissolved in 1.8–2.0 L of ethanol with warming (about 35–40 °C) in a 5-L, three-neck flask equipped with a stirrer, thermometer, and reflux condenser and set in an electric heating mantle. A solution of 202 g (1.48 mol) of zinc chloride in 1.0 L of ethanol was made by gently warming the mixture. The resulting solution was poured rapidly into the stirred solution of the mixture of product and triphenylphosphine oxide in the 5-L flask, forming a clear dark solution. Within minutes, a heavy white precipitate separated from the dark solution. After filtration of the solids (presumed to be Zn(O=PPh3)2Cl2) and rinsing of the solids with fresh ethanol, the filtrate was shown by TLC analysis to contain only the product and no detectible triphenylphosphine or its oxide.
Following evaporation of the ethanol from the 2,7-dibromocarbazole, the pure product was recrystallized from methylcyclohexane/toluene to yield 111 g of product (75% yield). NMR (1H) and mass spectrometry of this material were consistent with the structure and the 1H NMR in DMSO matched that obtained from a commercial sample (Sigma-Aldrich, copies of both in the SI).181H NMR (400 MHz; DMSO-d6): δ 11.52 (s, 1H), 8.10 (d, J = 8.3 Hz, 2H), 7.71 (d, J = 1.4 Hz, 2H), 7.33 (dd, J = 8.3, 1.5 Hz, 2H).
(4,4-Dibromobut-3-enyl)benzene (4)22
A 2-L, three-neck flask was fitted with a stirbar, addition funnel, and septum before being flame-dried under vacuum and backfilled with N2. To the dry flask were added carbon tetrabromide (41.3 g, 0.249 mol, 2 equiv) and dry dichloromethane (200 mL). The flask was cooled in an ice water bath to 0 °C, and a solution of triphenylphosphine (65.2 g, 0.497 mol, 4 equiv) in anhydrous dichloromethane (200 mL) was added under N2 at 0 °C over 30 min using the addition funnel. The mixture was stirred at 0 °C for 10 min before a solution of 3-phenylpropionaldehyde (8.2 mL, 0.124 mol, 1 equiv) in anhydrous dichloromethane (133 mL) was added via the addition funnel over 10 min while holding the reaction mixture at 0 °C. The mixture was further stirred at 0 °C for 1 h before 250 mL of water was added and the mixture was transferred to a 2-L separatory funnel. The aqueous layer was separated and extracted with dichloromethane (3 × 100 mL). The combined dichloromethane extracts were washed with a 10% solution of hydrogen peroxide (2 × 100 mL) to oxidize any remaining phosphorus species to TPPO. The organic layer was then washed with a saturated solution of Na2SO3 (2 × 100 mL), brine (2 × 100 mL), and dried over sodium sulfate. The resulting solution was filtered and concentrated on a rotary evaporator to an orange residue.
The residue was dissolved in 400 mL of absolute ethanol in a 2-L Erlenmeyer flask, and a homogeneous solution of ZnCl2 (37.5 g, 0.275 mol, 2.2 equiv) dissolved in EtOH (150 mL) was prepared with stirring and gentle warming. Both solutions were allowed to cool to room temperature before the zinc chloride solution was poured into the stirred solution of the product over a 1 min period. Stirring was continued, and formation of a white precipitate was induced by scratching the interior wall of the Erlenmeyer flask with a glass rod. After being stirred for 30 min, the zinc complex was collected by filtration and rinsed with additional EtOH (50 mL). Analysis of the filtrate by GC with dodecane as an internal standard showed that <1% triphenylphosphine oxide remained in solution. The filtrate was concentrated under reduced pressure on a rotary evaporator to yield an orange oil. The crude material was further purified by distillation to yield 29.5 g (82% yield) of (4,4-dibromobut-3-enyl)benzene as a colorless oil boiling at 90–92 °C at 0.3 mmHg. Analysis by 1H NMR and TLC (10:1 hexanes/ethyl acetate) and comparison with authentic samples of triphenylphosphine and triphenylphosphine oxide showed the product to be pure and that no phosphine impurities were present.22
(1R,2S,5R)-2-Isopropyl-5-methylcyclohexyl 4-Nitrobenzoate (6)23
A 1-L, round-bottom, three-neck flask, equipped with a thermometer, dropping funnel, argon inlet, and magnetic stirrer bar, was charged with l-menthol (6.00 g, 38.4 mmol), TPP (40.1 g, 152.4 mmol), 4-nitrobenzoic acid (25.8 g, 154.4 mmol), and THF (300 mL). While the mixture was stirred under an argon atmosphere, the solution was cooled in an ice bath to 0 °C and diethyl azodicarboxylate (24.2 mL, 154 mmol) was added slowly by addition funnel while maintaining the temperature below 10 °C. Upon completion of the addition, the cooling bath was removed, and the yellow solution was allowed to warm to rt and stir for 18 h. The reaction mixture was warmed to 35–40 °C for 3 h before being transferred to a 2-L separatory funnel and diluted with EtOAc (400 mL). The solution was washed with saturated sodium bicarbonate (2 × 200 mL), 6% hydrogen peroxide (2 × 250 mL), and water (400 mL). The organic layer was isolated and dried over anhydrous sodium sulfate before being filtered and concentrated to dryness by rotary evaporation. The resulting slightly yellow solid was dissolved in 600 mL of ethanol in a 1-L Erlenmeyer flask with gentle warming and stirring. To the stirred solution was added, in a single portion, a warm solution of anhydrous zinc chloride (42.0 g, 310 mmol) in ethanol (200 mL). Stirring was continued while the inner wall of the flask was scratched with a glass rod to initiate the precipitation of the zinc chloride complex of triphenylphosphine oxide. The precipitation was complete after stirring for 1 h at rt, and the mixture was filtered to remove the ZnCl2(TPPO)2. The white solid was rinsed with 50 mL of warm ethanol, and the filtrate was treated with decolorizing carbon (500 mg) before being filtered through a Whatman glass microfiber filter. Analysis (GC with dodecane as an internal standard) showed that 30% of the TPPO had been removed. A second precipitation with ZnCl2 resulted in 85% of the TPPO being removed. The product was recrystallized directly from the second ethanol filtrate as a thick mat of white crystalline needles. The product was isolated by filtration to give 7.9 g (68% yield) of the pure compound (1R,2S,5R)-2-isopropyl-5-methylcyclohexyl 4-nitrobenzoate. Additional yield could be obtained from concentration of the mother liquor and recrystallization but was not combined with the initial recrystallization due to a slight triphenylphosphine oxide impurity. 1H and 13C NMR matched literature data.23
Acknowledgments
Research reported in this publication was supported in part by the National Institute of General Medical Sciences of the National Institutes of Health under Award No. R01GM097243. We thank Jalil Shojae (University of Rochester) and Astrid Olivares (University of Rochester) for assistance with compound characterization. We also thank Stella Wu for assistance with video editing.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.joc.7b00459.
The authors declare no competing financial interest.
This paper was published ASAP on September 28, 2017. Table 3 was replaced. The revised paper was reposted on October 6, 2017.
Supplementary Material
References
- Cobb J. E.; Cribbs C. M.; Henke B. R.; Uehling D. E.; Hernan A. G.; Martin C.; Rayner C. M., Triphenylphosphine. In Encyclopedia of Reagents for Organic Synthesis; John Wiley & Sons, Ltd.: 2001. [Google Scholar]
- For reviews, see:; a Fletcher S. Org. Chem. Front. 2015, 2, 739–752. 10.1039/C5QO00016E. [DOI] [Google Scholar]; b Dembinski R. Eur. J. Org. Chem. 2004, 2004, 2763–2772. 10.1002/ejoc.200400003. [DOI] [Google Scholar]; c Swamy K.; Kumar N.; Balaraman E.; Kumar K. Chem. Rev. 2009, 109, 2551–2651. 10.1021/cr800278z. [DOI] [PubMed] [Google Scholar]
- a Grynkiewicz G.; Jurczak J.; Zamojski A. Tetrahedron 1975, 31, 1411–1414. 10.1016/0040-4020(75)87072-4. [DOI] [Google Scholar]; b Tsunoda T.; Ozaki F.; Itô S. Tetrahedron Lett. 1994, 35, 5081–5082. 10.1016/S0040-4039(00)73326-0. [DOI] [Google Scholar]; c O’Neil I. A.; Thompson S.; Murray C. L.; Kalindjian S. B. Tetrahedron Lett. 1998, 39, 7787–7790. 10.1016/S0040-4039(98)01702-X. [DOI] [Google Scholar]; d Camp D.; Jenkins I. Aust. J. Chem. 1988, 41, 1835–1839. 10.1071/CH9881835. [DOI] [Google Scholar]; e Dandapani S.; Curran D. P. J. Org. Chem. 2004, 69, 8751–8757. 10.1021/jo0488098. [DOI] [PubMed] [Google Scholar]; f Muramoto N.; Yoshino K.; Misaki T.; Sugimura T. Synthesis 2013, 45, 931–935. 10.1055/s-0032-1318327. [DOI] [Google Scholar]
- a Voituriez A.; Saleh N. Tetrahedron Lett. 2016, 57, 4443–4451. 10.1016/j.tetlet.2016.08.036. [DOI] [Google Scholar]; b O’Brien C. J.; Tellez J. L.; Nixon Z. S.; Kang L. J.; Carter A. L.; Kunkel S. R.; Przeworski K. C.; Chass G. A. Angew. Chem., Int. Ed. 2009, 48, 6836–6839. 10.1002/anie.200902525. [DOI] [PubMed] [Google Scholar]; c Harris J. R.; Haynes M. T.; Thomas A. M.; Woerpel K. A. J. Org. Chem. 2010, 75, 5083–5091. 10.1021/jo1008367. [DOI] [PubMed] [Google Scholar]; d van Kalkeren H. A.; Leenders S. H. A. M.; Hommersom C. R. A.; Rutjes F. P. J. T.; van Delft F. L. Chem. - Eur. J. 2011, 17, 11290–11295. 10.1002/chem.201101563. [DOI] [PubMed] [Google Scholar]; e Kosal A. D.; Wilson E. E.; Ashfeld B. L. Angew. Chem., Int. Ed. 2012, 51, 12036–12040. 10.1002/anie.201206533. [DOI] [PubMed] [Google Scholar]; f Werner T.; Hoffmann M.; Deshmukh S. Eur. J. Org. Chem. 2014, 2014, 6630–6633. 10.1002/ejoc.201402941. [DOI] [Google Scholar]; g Buonomo J. A.; Aldrich C. C. Angew. Chem., Int. Ed. 2015, 54, 13041–13044. 10.1002/anie.201506263. [DOI] [PMC free article] [PubMed] [Google Scholar]; h Zhao W.; Yan P. K.; Radosevich A. T. J. Am. Chem. Soc. 2015, 137, 616–619. 10.1021/ja511889y. [DOI] [PubMed] [Google Scholar]
- a Kuhn F.; Santos A. Mini-Rev. Org. Chem. 2004, 1, 55–64. 10.2174/1570193043488971. [DOI] [Google Scholar]; b An J.; Denton R. M.; Lambert T. H.; Nacsa E. D. Org. Biomol. Chem. 2014, 12, 2993–3003. 10.1039/c4ob00032c. [DOI] [PubMed] [Google Scholar]; c Shi L.; Wang W.; Wang Y.; Huang Y. J. Org. Chem. 1989, 54, 2027–2028. 10.1021/jo00270a001. [DOI] [Google Scholar]; d Huang Y.-Z.; Shi L.-L.; Li S.-W.; Wen X.-Q. J. Chem. Soc., Perkin Trans. 1 1989, 2397–2399. 10.1039/p19890002397. [DOI] [Google Scholar]
- Anderson N. G.Practical Process Research and Development – A Guide for Organic Chemists; Academic Press: Burlington, VT, 2012. [Google Scholar]
- Matsui M.; Yabuta G. Agric. Biol. Chem. 1968, 32, 1044–1045. 10.1271/bbb1961.32.1044. [DOI] [Google Scholar]
- Dodge J. A.; Nissen J. S.; Presnell M.; Okabe M.; Sun R. C.; Coffen D. Org. Synth. 1996, 73, 110. 10.15227/orgsyn.073.0110. [DOI] [Google Scholar]
- For example, see:Yee N. K.; Farina V.; Houpis I. N.; Haddad N.; Frutos R. P.; Gallou F.; Wang X.-j.; Wei X.; Simpson R. D.; Feng X.; Fuchs V.; Xu Y.; Tan J.; Zhang L.; Xu J.; Smith-Keenan L. L.; Vitous J.; Ridges M. D.; Spinelli E. M.; Johnson M.; Donsbach K.; Nicola T.; Brenner M.; Winter E.; Kreye P.; Samstag W. J. Org. Chem. 2006, 71, 7133–7145. 10.1021/jo060285j. [DOI] [PubMed] [Google Scholar]
- Lipshutz B. H.; Blomgren P. A. Org. Lett. 2001, 3, 1869–1871. 10.1021/ol0159219. [DOI] [PubMed] [Google Scholar]
- Byrne P. A.; Rajendran K. V.; Muldoon J.; Gilheany D. G. Org. Biomol. Chem. 2012, 10, 3531–3537. 10.1039/c2ob07074j. [DOI] [PubMed] [Google Scholar]
- Etter M. C.; Baures P. W. J. Am. Chem. Soc. 1988, 110, 639–640. 10.1021/ja00210a076. [DOI] [Google Scholar]
- Fukumoto T.; Yamamoto A. (Shin-Etsu Chemical Co., Ltd., Tokyo, Japan). Phosphine oxide removal from compounds formed by a Wittig reaction. US Patent 5,292,973, Mar 8, 1994.
- Anderson N. G.; Lust D. A.; Colapret K. A.; Simpson J. H.; Malley M. F.; Gougoutas J. Z. J. Org. Chem. 1996, 61, 7955–7958. 10.1021/jo9609539. [DOI] [PubMed] [Google Scholar]
- a Pickard R. H.; Kenyon J. J. Chem. Soc., Trans. 1906, 89, 262–273. 10.1039/CT9068900262. [DOI] [Google Scholar]; b Rose J. P.; Lalancette R. A. Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem. 1980, 36, 2409–2411. 10.1107/S0567740880008874. [DOI] [Google Scholar]; c Kosky C. A.; Gayda J. P.; Gibson J. F.; Jones S. F.; Williams D. J. Inorg. Chem. 1982, 21, 3173–3179. 10.1021/ic00138a051. [DOI] [Google Scholar]; d Liu X.; Wang G.; Dang Y.; Zhang S.; Tian H.; Ren Y.; Tao X. CrystEngComm 2016, 18, 1818–1824. 10.1039/C5CE02002F. [DOI] [Google Scholar]
- Weygand F.; Bestmann H. J.. Syntheses Using Diazoketones. In Newer Methods of Preparative Organic Chemistry; Foerst W., Ed.; Academic Press: New York, 1964; Vol. 3, p 470; translated by H. Birnbaum. [Google Scholar]
- a Isola A. M.; Holman N. J.; Tometzki G. B.; Watts J. P.; Koser S.; Klintz R.; Münster P.. Triphenylphosphine oxide complex process. US Patent 6011181A, 2000.; b Lukin K.; Kishore V.; Gordon T. Org. Process Res. Dev. 2013, 17, 666–671. 10.1021/op300345v. [DOI] [Google Scholar]
- Freeman A. W.; Urvoy M.; Criswell M. E. J. Org. Chem. 2005, 70, 5014–5019. 10.1021/jo0503299. [DOI] [PubMed] [Google Scholar]
- a Burger L. L. J. Phys. Chem. 1958, 62, 590–593. 10.1021/j150563a017. [DOI] [Google Scholar]; b Burger L. L. Nucl. Sci. Eng. 1963, 16, 428–439. 10.13182/NSE63-A26555. [DOI] [Google Scholar]
- Reactions were conducted in iPrOH because some of the less polar molecules were not sufficiently soluble in ethanol.
- We found that quinidine combined with ZnCl2 resulted in precipitation of quinidine from solution, even in the absense of TPPO, presumably as a zinc chloride complex.
- Mori M.; Tonogaki K.; Kinoshita A.; Denmark S. E.; Kobayashi T. Org. Synth. 2005, 81, 1–13. 10.15227/orgsyn.081.0001. [DOI] [Google Scholar]; Mori M.; Tonogaki K.; Kinoshita A.. Organic Syntheses; Wiley, 2009; Collect. Vol. 11, pp 561–569. [Google Scholar]
- a Dodge J. A.; Trujillo J. I.; Presnell M. J. Org. Chem. 1994, 59, 234–236. 10.1021/jo00080a039. [DOI] [Google Scholar]; b Dodge J. A.; Nissen J. S.; Presnell M.; Okabe M.; Sun R. C.; Coffen D. Org. Synth. 1996, 73, 110. 10.1002/0471264180.os073.11. [DOI] [Google Scholar]; Dodge J. A.; Nissen J. S.; Presnell M.; Okabe M.; Sun R. C.; Coffen D.. Organic Syntheses; Wiley, 1998; Collect. Vol. 9, p 607. [Google Scholar]
- We examined procedures in Organic Syntheses where triphenylphosphine was used as a reagent and the resulting triphenylphosphine oxide was removed by chromatography.
- Proctor A. J.; Beautement K.; Clough J. M.; Knight D. W.; Li Y. Tetrahedron Lett. 2006, 47, 5151–5154. 10.1016/j.tetlet.2006.05.070. [DOI] [Google Scholar]
- Pansare S. V.; Arnold L. D.; Vederas J. C.; Manchand P. S.; Mastrodonato-DeLora P.; Coffen D. L. Org. Synth. 1992, 70, 10. 10.15227/orgsyn.070.0010. [DOI] [Google Scholar]; Pansare S. V.; Arnold L. D.; Vederas J. C.; Manchand P. S.; Mastrodonato-DeLora P.; Coffen D. L.. Organic Syntheses; Wiley, 1998; Collect. Vol. 9, p 24. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.