

Although typically considered a pediatric disease, Ewing sarcoma can occur adults. Current standard treatment for localized Ewing sarcoma is a multimodality approach, combining chemotherapy and local therapy consisting of surgery and/or radiation therapy. This article evaluates outcomes for adults with localized Ewing sarcoma treated exclusively with cyclophosphamide, doxorubicin, and vincristine followed by ifosfamide and etoposide in combination with local therapy.
Keywords: Ewing sarcoma, Localized, Adult, Radiotherapy, Surgery, Pelvis
Abstract
Background.
In children with localized Ewing sarcoma (ES), addition of ifosfamide and etoposide to cyclophosphamide, doxorubicin, and vincristine (VDC/IE) improved 5‐year overall survival (OS) to 70%–80%. Prior to delivery of VDC/IE in adults, 5‐year OS was <50%. We reviewed our institutional outcomes for adults with ES who received VDC/IE‐based treatment.
Materials and Methods.
Between 1997–2013, 67 adults with localized ES were treated with curative intent. Local recurrence‐free survival (LRFS), progression‐free survival (PFS), and OS were determined using Kaplan‐Meier method; comparisons were assessed with log‐rank. Proportional hazard models were used to determine predictive factors.
Results.
All patients received VDC/IE (median 14 cycles.) Local therapy was surgery for 33, radiation therapy for 17, or both for 17. Median follow‐up for living patients was 5.2 years. Six patients had disease progression on therapy. Site of first failure was local for three, local and distant for two, and distant for ten. Five‐year LRFS was 91%; 5‐year LRFS was 96% for nonpelvic disease and 64% for pelvic disease (p = .003). Five‐year PFS was 66%, and 5‐year OS was 79%. On multivariate analysis, pelvic site had a 3.3 times increased risk of progression (p = .01).
Conclusion.
Survival for adults with localized ES treated with VDC/IE‐based multimodality therapy appears to be better than historical data and similar to excellent outcomes in children. Pelvic site of disease remains a predictor of worse outcome. Given the paucity of literature for adult ES, these data help validate VDC/IE‐based therapy as an appropriate treatment approach for this rare disease in adults.
Implications for Practice.
Ewing sarcoma (ES) is rare in adults. Treatment approaches for adults have been extrapolated from the pediatric experience, and there is a sense that adults fare less well than children. We reviewed treatment outcomes in adults with localized ES treated with cyclophosphamide, doxorubicin, and vincristine in alternation with ifosfamide and etoposide (VDC/IE) as part of multimodality therapy. Survival outcomes appear to be better than historical data for adults and similar to the excellent outcomes for children. These data help validate VDC/IE‐based therapy as an appropriate treatment approach for this rare disease in adults.
Introduction
Although typically considered a pediatric disease, about 30% of Ewing sarcoma (ES) cases arise in adults [1]. Current standard treatment for localized ES is a multimodality approach, combining chemotherapy and local therapy consisting of surgery and/or radiation therapy (RT) [2], [3], [4], [5], [6], [7], [8].
Advances in chemotherapy regimens for ES have improved survival dramatically over the past 40 years. In the 1970s, addition of doxorubicin to vincristine, actinomycin D, and cyclophosphamide improved 5‐year overall survival (OS) from 28% to 65% for localized disease [2]. In 2003, Grier showed the addition of ifosfamide and etoposide, alternating with cyclophosphamide, doxorubicin, and vincristine (VDC/IE), further improved survival [4]. Most recently, VDC/IE given every 2 weeks as opposed to every 3 weeks has increased 5‐year OS to 83% [6]. These landmark trials were conducted primarily in pediatric populations, and although patients of any age [2], up to age 30 [4], or up to age 50 [6], were eligible for participation, numbers of patients included over age 18 were small, with median age enrolled being 12–13.
Despite substantial high‐quality data on localized ES, the majority of it is derived from the pediatric age group; data evaluating outcomes in adults are scarce. There are no randomized trials that specifically address treatment in adults; thus, standard treatment is extrapolated from literature derived from a younger patient population. It is not clear whether outcomes are different in adults than children, as nearly all reviews pertaining to adults have small patient populations or also included patients less than 18 [9–16]. Furthermore, the majority of reviews were performed prior to the VDC/IE era. Accordingly, we evaluated outcomes for adults with localized ES treated exclusively with VDC/IE in combination with local therapy at our institutions.
Materials and Methods
Patients
We conducted an Institutional Review Board‐approved review of all adult patients (≥18 years old) with newly diagnosed localized ES treated at Dana‐Farber Cancer Institute/Brigham and Women's Hospital and Massachusetts General Hospital in Boston, Massachusetts, between 1997 and 2013. Inclusion criteria were treatment with VDC‐IE‐based multimodality therapy, delivery of at least one treatment modality at our institutions, and availability of complete treatment information. For patients diagnosed at outside hospitals, pathology review was performed to confirm a diagnosis of ES. Patients with a prior diagnosis of cancer were excluded. The resulting cohort was composed of 67 patients. We collected patient, disease, treatment, recurrence, and survival characteristics through medical record review.
Treatment
Patients were treated with alternating VDC‐IE chemotherapy. Chemotherapy doses were administered as follows: vincristine 2 mg/m2 (capped at 2 mg) on day 1, doxorubicin 37.5 mg/m2 on days 1 and 2, cyclophosamide 600 mg/m2 on days 1 and 2, ifosfamide 1,800 mg/m2 on days 1–5, and etoposide 100 mg/m2 on days 1–5. Each cycle of chemotherapy was supported by filgrastim. Doxorubicin was omitted after achieving a cumulative dose of 350 mg/m2. At the beginning of the study period, patients were treated in 3‐week intervals for 14–17 cycles [4]. Following publication of the randomized trial showing superiority of dose dense treatment with 2‐week intervals [6], patients were considered for 14 cycles using 2‐week intervals with a schedule reduction to 3‐week intervals based on tolerance.
Patients were evaluated for local therapy at about week 12. Surgery was the preferred local modality unless it was judged to be associated with significant morbidity, in which case RT was recommended. RT doses were 55.8 Gy for gross disease and 50.4 Gy for resected disease with positive margins. RT technique consisted of three‐dimensional conformal therapy or intensity modulated RT.
Outcomes
Local recurrence‐free survival (LRFS) was defined from date of diagnosis to localized recurrence as a first site of recurrence or death. Patients were censored at the time of regional or distant recurrence (DR), and those without a recurrence were censored at date of last assessment. Progression‐free survival (PFS) was defined from date of diagnosis to first reported event (recurrence, progression, or death). Patients without an event were censored at date of last assessment. OS was defined from date of diagnosis to death or date last known alive.
Statistical Analysis
Descriptive statistics were used to report patient characteristics. Radiographic response to chemotherapy prior to local therapy was defined for extra‐osseous tumors following Response Evaluation Criteria In Solid Tumors 1.1 guidelines as complete response, partial response, or stable disease [17]. LRFS, PFS, and OS were calculated using the Kaplan‐Meier method. A log‐rank test was used to identify disease characteristics associated with time to event distributions. Step‐down proportional hazard models were constructed to identify factors associated with PFS and OS. STATA v. 13 (StataCorp, College Station, Texas, http://www.stata.com/company/) was used for all analyses.
Results
Patient Characteristics
Our cohort included 67 patients with localized ES. Patient characteristics are shown in Table 1. Median age at diagnosis was 28 years (range, 19–69); 13 (19%) patients were ≥40 years. Sixty percent of patients were male. Median follow‐up for patients still alive was 5.2 years (range, 0.8–15.6). Primary tumor locations included extremity (29), pelvis (12), and other (26). Other sites included superficial and deep trunk (14), spine (5), central nervous system (4), and head and neck (3). Primary tumor tissue type was extraosseous for 31 and osseous for 36. Median age for osseous ES was 25 years (range, 19–51), and median age for extraosseous was 33 (range, 21–69). Median tumor size was 7.7 cm (range, 1.3–28.3).
Table 1. Patient characteristics for localized Ewing sarcoma patients treated with curative intent vincristine, doxorubicin, cyclophosphamide, and ifosfamide, etoposide chemotherapy (n = 67).
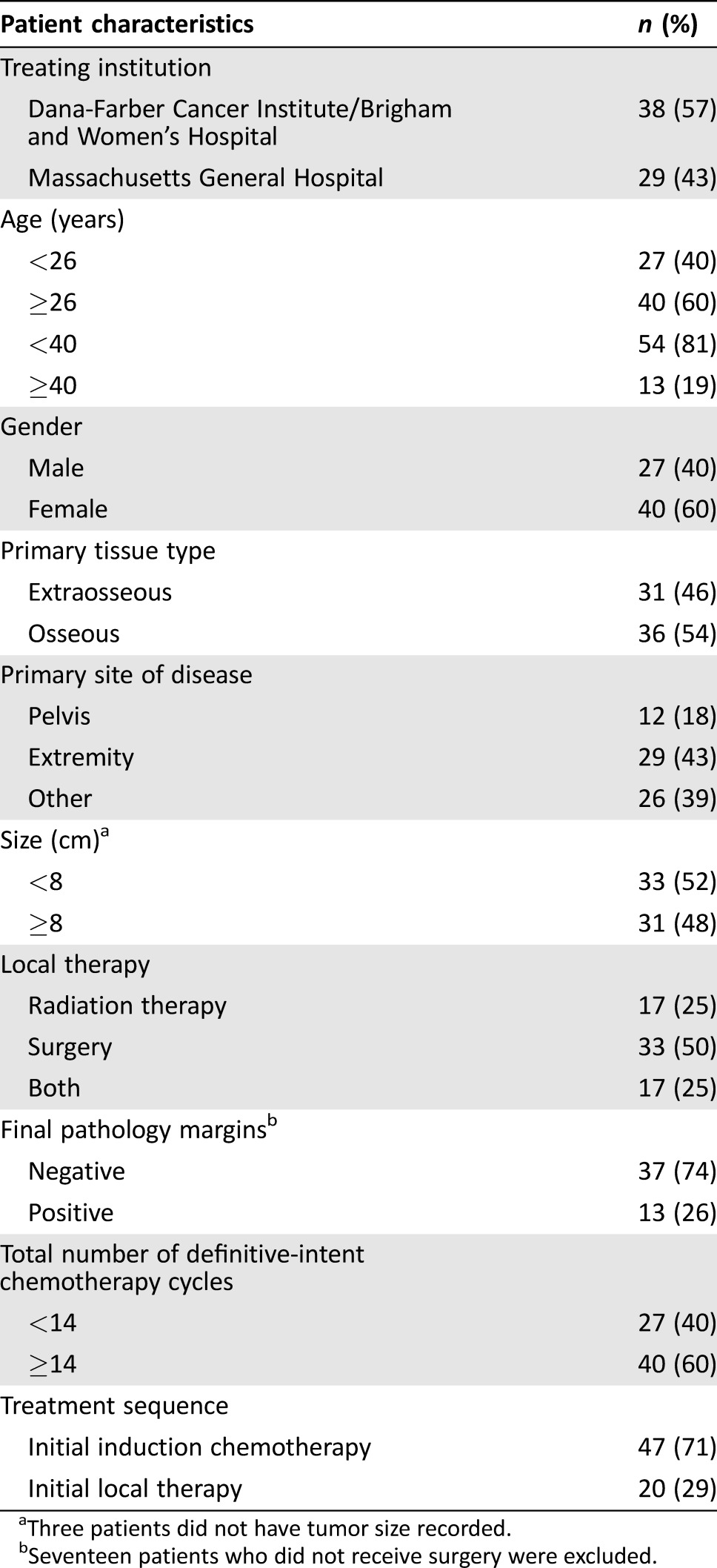
Three patients did not have tumor size recorded.
Seventeen patients who did not receive surgery were excluded.
Treatment
All patients received VDC/IE. The median number of definitive‐intent chemotherapy cycles delivered was 14 (range, 1–17); the number of patients who received <12 cycles was 11 (16%), 12–13 cycles was 16 (24%), 14 cycles was 28 (42%), and 15–17 cycles was 12 (18%). The median number of cycles delivered for those <40 years old was 14 (range, 2–17) and for those ≥40 was 13 (range, 1–15). Thirty‐one (46%) patients were treated with intended 2‐week interval cycles. Forty‐seven patients (71%) received initial induction chemotherapy. Twenty patients (29%) did not receive chemotherapy prior to local therapy. Of these 20 patients, 19 initiated treatment with local therapy (18 surgery and 1 concurrent chemotherapy and RT) because a diagnosis of ES was not anticipated prior to treatment initiation, and one received partial resection and spinal decompression as first therapy because the patient required immediate symptomatic relief.
Local therapy consisted of surgery (33), RT (17), or surgery and RT (17). Median tumor size among patients treated with surgery alone was 8.0 cm (range, 1.3–18 cm), with RT alone being 5.8 cm (range, 3.7–28.3), and with both surgery and RT being 8.4 cm (range, 3.8–28). Differences in sizes between local treatment groups were not statistically significant. Among pelvic tumors, 83% (10/12) received RT as a component of local therapy and only 17% (2/12) received surgery alone; among extremity tumors, 79% (23/29) were treated with surgery alone. Among patients who received RT only as local therapy, the median dose was 57.6 Gy (range, 45–68.4). Among patients who received RT and surgery, the median dose was 54 Gy (range, 45–60). Among patients who underwent surgery, 74% had negative margins. Eleven of 13 patients with positive margins received RT.
Four patients underwent RT without concurrent chemotherapy; their tumors were located in the central nervous system (two), pelvis (one), and deep trunk (one), respectively. The two patients with tumors in brain parenchyma received initial surgery followed by cranial‐spinal RT; chemotherapy was held during RT due to toxicity concerns and was delivered following local therapy. The patient with a pelvic tumor received chemotherapy prior to and following RT. The patient with a retroperitoneal tumor received two cycles of induction chemotherapy but experienced severe toxicity such that systemic therapy was discontinued.
Response and Patterns of Failure
Thirty‐five of 42 (83%) patients with an extraosseous component of disease who were treated with initial induction chemotherapy were evaluable for radiographic response to chemotherapy prior to local therapy. Among these 35 evaluable patients, 2 (6%) had complete response, 28 (80%) had partial response, and 5 (14%) had stable disease.
Six patients experienced disease progression during treatment (four during adjuvant chemotherapy, one after surgery, one during RT); tumor sites were pelvis (two), spine (two), deep trunk (one), and extremity (one). Fifteen patients experienced recurrence, and 15 died. Among the 15 patients who had recurrence, the site of first recurrence was local for 3, local and distant for 2, and distant for 10.
LRFS
Among the 61 patients who did not experience disease progression during treatment, there were 5 local recurrences (LR) as first site of recurrence and 2 LRs following DR. The median time to LR was 1.2 years (range, 0.9–2.3). As shown in Figure 1A, 5‐year LRFS for all patients was 91% (95% confidence interval [CI]: 80%–96%). Five‐year LRFS for patients with primary pelvic disease (n = 10) was 64% (95% CI: 23%–87%) and for nonpelvic primary disease (n = 51) was 96% (95% CI: 84%–99%, p = .003). Among patients with nonpelvic disease, 5‐year LRFS for extremity (n = 28) was 96% (95% CI: 76%–99%) and for other (nonpelvis, nonextremity, n = 23) was 95% (95% CI: 72%–99%; p = .01). There were too few LR events to perform a multivariate analysis.
Figure 1.
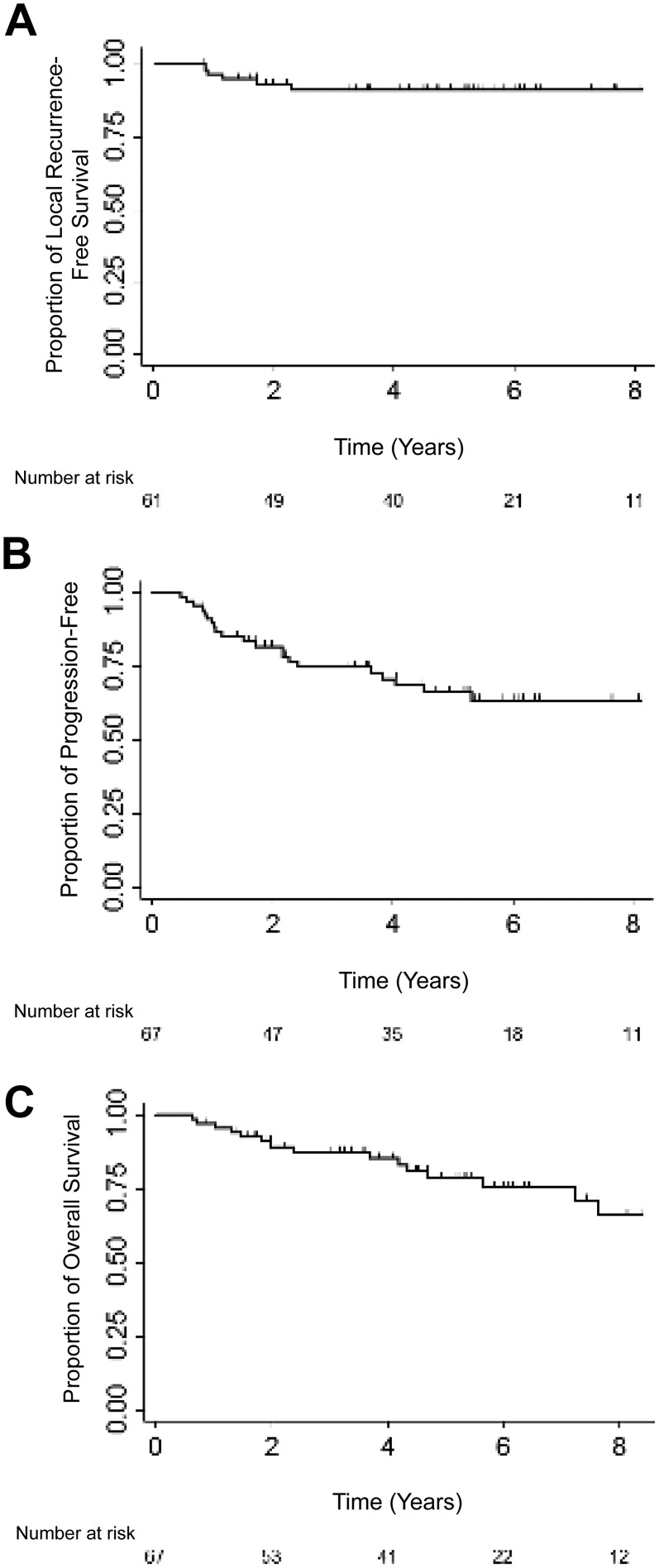
Kaplan‐Meier plots for local recurrence‐free survival (A), progression‐free survival (B), and overall survival (C).
Survival (PFS and OS)
Among our 67 patients, there were 21 events. Median time to progression was 1.5 years (range: 0.5–5.3 years), Figure 1B. Five‐year PFS was 66% (95% CI: 52%–77%). Five‐year PFS for pelvic site was 27%, for extremity was 78%, and for other sites was 69%, (p = .02). PFS according to local treatment modality showed borderline statistical significance (p = .05); 5‐year PFS for patients treated with RT was 44%, with surgery was 64%, and with both was 88%. On univariate analysis, there was no difference in PFS according to age, gender, tumor tissue type (osseous vs. extraosseous), tumor size, margin status, or treatment sequence (Table 2).
Table 2. Univariate predictors for PFS and OS.
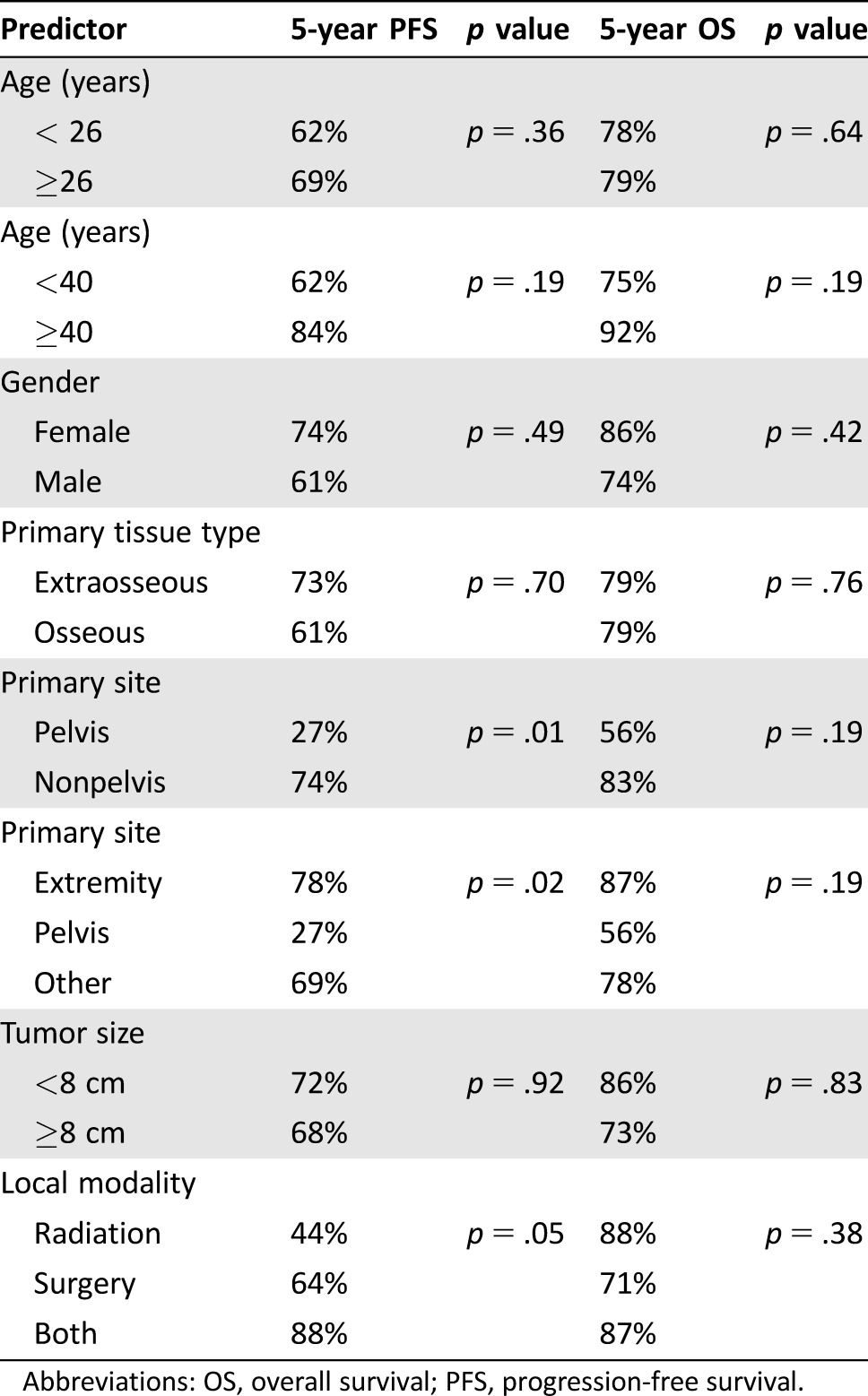
Abbreviations: OS, overall survival; PFS, progression‐free survival.
Multivariate analysis showed patients with pelvic primary had a 3.3 times increased risk of progression/recurrence compared with nonpelvic primary sites (hazard ratio [HR]: 3.3, 95% CI: 1.3–8.3, p = .01). In a second model, pelvic primary had a 4.4 times increased risk of progression/recurrence compared with extremity primary (HR: 4.4, 95% CI: 1.5–13.4, p = .008).
Among the 67 patients, 15 died. Median time to death was 2.4 years (range, 0.6–7.6). As shown in Figure 1C, 5‐year OS rate was 79% (95% CI: 65%–87%). No patient characteristics were prognostic for OS on univariate analysis (Table 2). Although only associated with borderline statistical significance, multivariate analysis showed pelvic primary had a 3.3 times increased risk of death compared with extremity primary (HR: 3.3, 95% CI: 0.8–13.4, p = .09).
Discussion
To our knowledge, this is the largest series of adults with localized ES treated with VDC/IE chemotherapy. We report 5‐year OS of 79%. This compares favorably with 5‐year OS rates reported in historical series, which range from 20%–60% [9], [10], [11], [12], [13], [14]. The previously reported 5‐year OS for adults with localized disease from our institution was 49% [13]. This dramatic improvement is largely attributed to the use of VDC/IE as part of treatment. All the patients in our prior series received either no or alternative chemotherapy. A recent series by Ahmed et al. [15] demonstrated that treatment era was an independent prognostic factor for survival. They compared outcomes of adults with localized ES treated between 1977–1992 and 1993–2007. The modern‐era cohort was comprised of 52 patients, of whom 83% received VDC/IE, a population quite similar to the group reported herein. Five‐year OS was 49% for patients treated between 1977–1992 and 73% for those treated between 1993–2007.
Usage of VDC/IE in adults began as an extrapolation from the data‐driven pediatric experience [2], [3], [4], [5], [6]. Although these trials allowed enrollment of adults, they had relatively small numbers ≥18 years old, ranging from 9%–15% of study populations. It has been hypothesized by some investigators that adults with ES do worse than their pediatric counterparts and perhaps have tumors with inherently different biologies [9], [18]. Subgroup analyses within some prospective trials have demonstrated worse outcomes for older patients [2], [4], [5], [6], with 5‐year event‐free survival reported as 44%–47% for patients ≥18 [4, 6]. However, the trials that established VDC/IE as the standard regimen report 5‐year OS ranging from 72%–83% across the study population [4], [6], and with such small numbers of adults included, power to make conclusions for this subgroup is limited. The 5‐year OS of 79% reported herein and the recent 5‐year OS of 73% reported by Ahmed [15] are in pure adult populations and are consistent with survival rates reported for recent (predominantly pediatric) trials. Furthermore (in this issue of The Oncologist), Wagner [19] reports favorable 68% 5‐year OS for 42 adults with localized ES treated with a vincristine, ifosfamide, and doxorubicin chemotherapy backbone. These similar survival rates suggest, with modern‐era systemic therapy, ES in adults may have comparable outcome to children. It is also worth noting that results from retrospective single institutions trials are often favorable; accordingly, it will be important to confirm these outcomes in adults with ES in a prospective multicenter setting.
Age was not a significant prognostic factor in the present study. Although some prior reports have also reported no survival differences between older and younger adults [11], [14], [20], others have suggested a significant adverse outcome for older patients [2], [4], [6], [10], [21], [22].
Both size and location of primary tumor have been proposed as predictors of survival for ES. In this study, only pelvic site of disease had worse outcomes. Many prior reports have shown similar adverse prognosis for pelvic sites [2], [6], [21], [22], [23], although others have failed to show this difference [13], [15], [24], [25]. In our analysis, there was no difference in outcomes for tumor size ≥8 cm compared with <8 cm. Although there generally is more agreement in the literature with respect to large tumor size portending a poorer outcome [8], [10], [11], [23], the definition of tumor bulk is variable, and size may be correlated with tumor location.
The 5‐year LR rate in this series was 9%; this is comparable to the LR rates reported in other studies, which range from 7%–15% [2], [3], [4], [5], [6], [15]. The most recent randomized study by Womer reported LR rates of 7%–8% [6], while the adult retrospective study published by Ahmed reported 14% [15]. No prospective trial has directly compared surgery to radiation, thus choice of local therapy is appropriately based on patient and tumor characteristics in an effort to minimize morbidity. Some series suggest better local control with surgery [5], [14], [15], [20], [26], [27], but others suggest no difference between the two modalities [21]. These series may be prone to inherent biases in patient selection in that larger, less resectable tumors are more likely treated with RT. However, because of risk of second malignancy and other late effects following RT, surgery is often the preferred modality if feasible with acceptable morbidity, particularly for children and young adults [8]. Consistent with this thinking, Ahmed et al. report an increased utilization of surgery over time [15].
Conclusion
We report outcomes and prognostic factors for 67 adults with localized ES treated with VDC/IE‐based multimodality therapy. Although this is retrospective, it is the largest series of adults treated exclusively with chemotherapy considered standard of care for pediatric patients. PFS and OS rates are excellent, are better than historical outcomes for adults, and are comparable to current pediatric outcomes. We also report very good local control following treatment with a mix of surgery and RT; we do not support one local modality as superior to the other and encourage continued case‐specific choices in this regard. Consistent with pediatric literature, pelvic location is an adverse predictor for PFS and OS, such that these patients could benefit from alternative treatment strategies or treatment intensification.
Footnotes
Editor's Note: See the companion paper, “Vincristine, Ifosfamide, and Doxorubicin for Initial Treatment of Ewing Sarcoma in Adults,” by Michael J. Wagner, Vanceswaran Gopalakrishnan, Vinod Ravi et al., on page 1271 of this issue.
Author Contributions
Conception/design: Elizabeth H. Baldini, Constance M. Barysauskas, Suzanne George, Jason L. Hornick, Chandrajit P. Raut, Edwin Choy, Francis Hornicek, Thomas F. DeLaney
Provision of study material or patients: Elizabeth H. Baldini, Constance M. Barysauskas, Suzanne George, Jason L. Hornick, Chandrajit P. Raut, Edwin Choy, Francis Hornicek, Thomas F. DeLaney
Collection and/or assembly of data: Elizabeth H. Baldini, Constance M. Barysauskas, Suzanne George, Jason L. Hornick, Karen C. Marcus, John E. Ready, Chandrajit P. Raut, Edwin Choy, Francis Hornicek, Thomas F. DeLaney
Data analysis and interpretation Elizabeth H. Baldini, Constance M. Barysauskas, Suzanne George, Jason L. Hornick, Chandrajit P. Raut, Edwin Choy, Francis Hornicek, Thomas F. DeLaney
Manuscript writing: Elizabeth H. Baldini, Constance M. Barysauskas, Suzanne George, Jason L. Hornick, Chandrajit P. Raut, Edwin Choy, Francis Hornicek, Thomas F. DeLaney
Final approval of manuscript: Elizabeth H. Baldini, Constance M. Barysauskas, Suzanne George, Jason L. Hornick, Chandrajit P. Raut, Edwin Choy, Francis Hornicek, Thomas F. DeLaney, Karen C. Marcus, John E. Ready
Disclosures
Suzanne George: AstraZeneca, Deciphera (C/A), Bayer, Pfizer, Blueprint Medicines, Deciphera (RF); Thomas F. DeLaney: RBC Consulting (C/A), UpToDate, Oakstone Medical Publishing (H). The other authors indicate no financial relationships.
(C/A) Consulting/advisory relationship; (RF) Research funding; (E) Employment; (ET) Expert testimony; (H) Honoraria received; (OI) Ownership interests; (IP) Intellectual property rights/inventor/patent holder; (SAB) Scientific advisory board
References
- 1. Glass AG, Fraumeni JF Jr. Epidemiology of bone cancer in children. J Natl Cancer Inst 1970;44:187–199. [PubMed] [Google Scholar]
- 2. Nesbit ME Jr, Gehan EA, Burgert EO Jr et al. Multimodal therapy for the management of primary, nonmetastatic Ewing's sarcoma of bone: A long‐term follow‐up of the First Intergroup study. J Clin Oncol 1990;8:1664–1674. [DOI] [PubMed] [Google Scholar]
- 3. Burgert EO Jr, Nesbit ME, Garnsey LA et al. Multimodal therapy for the management of nonpelvic, localized Ewing's sarcoma of bone: Intergroup study IESS‐II. J Clin Oncol 1990;8:1514–1524. [DOI] [PubMed] [Google Scholar]
- 4. Grier HE, Krailo MD, Tarbell NJ et al. Addition of ifosfamide and etoposide to standard chemotherapy for Ewing's sarcoma and primitive neuroectodermal tumor of bone. N Engl J Med 2003;348:694–701. [DOI] [PubMed] [Google Scholar]
- 5. Granowetter L, Womer R, Devidas M et al. Dose‐intensified compared with standard chemotherapy for nonmetastatic Ewing sarcoma family of tumors: A Children's Oncology Group Study. J Clin Oncol 2009;27:2536–2541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Womer RB, West DC, Krailo MD et al. Randomized controlled trial of interval‐compressed chemotherapy for the treatment of localized Ewing sarcoma: A report from the Children's Oncology Group. J Clin Oncol 2012;30:4148–4154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Lin PP, Jaffe N, Herzog CE et al. Chemotherapy response is an important predictor of local recurrence in Ewing sarcoma. Cancer 2007;109:603–611. [DOI] [PubMed] [Google Scholar]
- 8. Gaspar N, Hawkins DS, Dirksen U et al. Ewing sarcoma: Current management and future approaches through collaboration. J Clin Oncol 2015;33:3036–3046. [DOI] [PubMed] [Google Scholar]
- 9. Sinkovics JG, Plager C, Ayala AG et al. Ewing sarcoma: its course and treatment in 50 adult patients. Oncology 1980;37:114–119. [DOI] [PubMed] [Google Scholar]
- 10. Picci P, Bohling T, Bacci G et al. Chemotherapy‐induced tumor necrosis as a prognostic factor in localized Ewing's sarcoma of the extremities. J Clin Oncol 1997;15:1553–1559. [DOI] [PubMed] [Google Scholar]
- 11. Verrill MW, Judson IR, Wiltshaw E et al. The use of paediatric chemotherapy protocols at full dose is both a rational and feasible treatment strategy in adults with Ewing's family tumours. Ann Oncol 1997;8:1099–1105. [DOI] [PubMed] [Google Scholar]
- 12. Fizazi K, Dohollou N, Blay JY et al. Ewing's family of tumors in adults: Multivariate analysis of survival and long‐term results of multimodality therapy in 182 patients. J Clin Oncol 1998;16:3736–3743. [DOI] [PubMed] [Google Scholar]
- 13. Baldini EH, Demetri GD, Fletcher CD et al. Adults with Ewing's sarcoma/primitive neuroectodermal tumor: Adverse effect of older age and primary extraosseous disease on outcome. Ann Surg 1999;230:79–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Bacci G, Balladelli A, Forni C et al. Adjuvant and neoadjuvant chemotherapy for Ewing sarcoma family tumors in patients aged between 40 and 60: Report of 35 cases and comparison of results with 586 younger patients treated with the same protocols in the same years. Cancer 2007;109:780–786. [DOI] [PubMed] [Google Scholar]
- 15. Ahmed SK, Robinson SI, Okuno SH et al. Adult ewing sarcoma: Survival and local control outcomes in 102 patients with localized disease. Sarcoma 2013;2013:681425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Oksüz DC, Tural D, Dincbas FÖ et al. Non‐metastatic Ewing's sarcoma family of tumors of bone in adolescents and adults: Prognostic factors and clinical outcome‐single institution results. Tumori 2014;100:452–458. [DOI] [PubMed] [Google Scholar]
- 17. Eisenhauer EA, Therasse P, Bogaerts J et al. New response evaluation criteria in solid tumours: Revised RECIST guideline (version 1.1). Eur J Cancer 2009;45:228–247. [DOI] [PubMed] [Google Scholar]
- 18. Karski EE, Matthay KK, Neuhaus JM et al. Characteristics and outcomes of patients with Ewing sarcoma over 40 years of age at diagnosis. Cancer Epidemiol 2013;37:29–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Wagner MJ, Ravi V, Gopalakrishan V et al, manuscript submitted for publication. Vincristine, ifosfamide, and oxorubicin (VID) for initial treatment of Ewing sarcoma in adults. [DOI] [PMC free article] [PubMed]
- 20. Wilkins RM, Pritchard DJ, Burgert EO Jr et al. Ewing's sarcoma of bone. Experience with 140 patients. Cancer 1986;58:2551–2555. [DOI] [PubMed] [Google Scholar]
- 21. Kinsella TJ, Miser JS, Waller B et al. Long‐term follow‐up of Ewing's sarcoma of bone treated with combined modality therapy. Int J Radiat Oncol Biol Phys 1991;20:389–395. [DOI] [PubMed] [Google Scholar]
- 22. Bacci G, Forni C, Longhi A et al. Long‐term outcome for patients with non‐metastatic Ewing's sarcoma treated with adjuvant and neoadjuvant chemotherapies. 402 patients treated at Rizzoli between 1972 and 1992. Eur J Cancer 2004;40:73–83. [DOI] [PubMed] [Google Scholar]
- 23. Bacci G, Toni A, Avella M et al. Long‐term results in 144 localized Ewing's sarcoma patients treated with combined therapy. Cancer 1989;63:1477–1486. [DOI] [PubMed] [Google Scholar]
- 24. Evans RG, Nesbit ME, Gehan EA et al. Multimodal therapy for the management of localized Ewing's sarcoma of pelvic and sacral bones: A report from the second intergroup study. J Clin Oncol 1991;9:1173–1180. [DOI] [PubMed] [Google Scholar]
- 25. Sauer R, Jurgens H, Burgers JM et al. Prognostic factors in the treatment of Ewing's sarcoma. The Ewing's Sarcoma Study Group of the German Society of Paediatric Oncology CESS 81. Radiother Oncol 1987;10:101–110. [DOI] [PubMed] [Google Scholar]
- 26. Dunst J, Sauer R, Burgers JM et al. Radiation therapy as local treatment in Ewing's sarcoma. Results of the Cooperative Ewing's Sarcoma Studies CESS 81 and CESS 86. Cancer 1991;67:2818–2825. [DOI] [PubMed] [Google Scholar]
- 27. DuBois SG, Krailo MD, Gebhardt MC et al. Comparative evaluation of local control strategies in localized Ewing sarcoma of bone: A report from the Children's Oncology Group. Cancer 2015;121:467–475. [DOI] [PMC free article] [PubMed] [Google Scholar]