

Advances in sequencing technologies and improved algorithms for detecting specific molecular aberrations make it possible to perform biomarker‐driven clinical trials. The analytical sensitivity and clinical validity have not yet been demonstrated in a large cohort. This article describes the implementation of a targeted‐sequencing panel in a precision oncology clinic and the feasibility of a clinic‐based molecular screening program.
Keywords: Next‐generation sequencing, Molecular screening, Clinical trials, Metastatic cancer
Abstract
Molecular profiling of actionable mutations in refractory cancer patients has the potential to enable “precision medicine,” wherein individualized therapies are guided based on genomic profiling. The molecular‐screening program was intended to route participants to different candidate drugs in trials based on clinical‐sequencing reports. In this screening program, we used a custom target‐enrichment panel consisting of cancer‐related genes to interrogate single‐nucleotide variants, insertions and deletions, copy number variants, and a subset of gene fusions. From August 2014 through April 2015, 654 patients consented to participate in the program at Samsung Medical Center. Of these patients, 588 passed the quality control process for the 381‐gene cancer‐panel test, and 418 patients were included in the final analysis as being eligible for any anticancer treatment (127 gastric cancer, 122 colorectal cancer, 62 pancreatic/biliary tract cancer, 67 sarcoma/other cancer, and 40 genitourinary cancer patients). Of the 418 patients, 55 (12%) harbored a biomarker that guided them to a biomarker‐selected clinical trial, and 184 (44%) patients harbored at least one genomic alteration that was potentially targetable. This study demonstrated that the panel‐based sequencing program resulted in an increased rate of trial enrollment of metastatic cancer patients into biomarker‐selected clinical trials. Given the expanding list of biomarker‐selected trials, the guidance percentage to matched trials is anticipated to increase.
Implications for Practice.
This study demonstrated that the panel‐based sequencing program resulted in an increased rate of trial enrollment of metastatic cancer patients into biomarker‐selected clinical trials. Given the expanding list of biomarker‐selected trials, the guidance percentage to matched trials is anticipated to increase.
摘要
难治性癌症患者中可诉性突变(actionable mutation)的分子表达谱有助于实现”精准医疗”(根据基因组表达谱指导个体化疗法)。分子筛选计划拟根据临床测序报告将试验参与者分配至不同的候选药物组。本筛选计划使用了包括癌症相关基因的定制靶向富集面板, 以分析单核苷酸变体、插入和删除, 拷贝数目变异(CNV)和基因融合子集。2014年8月至2015年4月期间, 654名患者表示同意参与在三星医疗中心开展的筛选项目。在这些患者中, 588名通过了381个基因癌症面板检测的质控过程, 418名患者由于适合使用任何抗癌治疗被纳入最终分析(127名胃癌患者, 122名结直肠癌患者, 62名胰腺/胆道癌患者, 67名肉瘤/其它癌症患者, 40名泌尿生殖肿瘤患者)。在这418名患者中, 55名(12%)有使其适合参加选定生物标志物临床试验的生物标志物, 184名(44%)有至少一种潜在靶向的基因组变异。本研究证实, 基于面板的测序计划增加了选定生物标志物临床试验中转移性肿瘤患者的入组率。由于选定生物标志物试验增多, 预计被分配到匹配试验的患者百分比会升高。
对临床实践的提示:本研究证实, 基于面板的测序计划增加了选定生物标志物临床试验中转移性肿瘤患者的入组率。由于选定生物标志物试验增多, 预计被分配到匹配试验的患者百分比会升高。
Introduction
Rapid advances in sequencing technologies together with improved algorithms for detecting specific molecular aberrations have made it feasible to perform biomarker‐driven clinical trials [1]. Many anecdotal cases highlighted the potential implementation of “precision medicine” based on routine cancer‐genome profiling in clinical practice [2]; however, the analytical sensitivity, as well as the clinical validity, of molecular testing must be demonstrated through a trial in a large cohort [3], [4], [5], [6]. Recently, low incidences of candidate biomarkers, such as anaplastic lymphoma kinase (ALK) rearrangement [7], phosphatidylinositol‐4,5‐bisphosphate 3‐kinase catalytic subunit alpha (PIK3CA) mutation [8], [9], [10], and human epidermal growth factor receptor 2 (HER2) amplification in solid tumors [11], were reported. Therefore, the need to find a sufficient number of eligible patients harboring specific biomarkers for biomarker‐driven clinical trials is becoming more important in the era of precision medicine [12]. To meet this need, a recently emerged type of umbrella or basket trial was proposed called a master‐protocol clinical trial. Under the molecular‐screening program, metastatic cancer patients are screened for various biomarkers upfront and assigned to trials for drugs with the highest likelihood of being effective [12], [13], [14].
Although diagnostic assays using immunohistochemistry (IHC), fluorescence in situ hybridization (FISH), or polymerase chain reaction sequencing have been used as standard molecular assays in the clinic, they are not scalable to the growing list of actionable molecular targets, thereby limiting the number of patients who can benefit from a more comprehensive approach, such as next‐generation sequencing (NGS). This has led to several clinical trials using NGS to evaluate clinical benefit in larger cohorts and over a diverse set of targets in the form of a master‐protocol clinical trial [15], [16], [17].
The preliminary results of initial feasibility testing previously reported high accrual and high success rates for genomic profiling using clinical specimens [18], which aligns with other trials [15], [19]. To accommodate the expanding list of potential target genes, we designed a target‐enrichment sequencing panel by including a larger number of cancer‐related genes in order to screen a set of single‐nucleotide variants (SNVs), copy number variants (CNVs), and translocations. This platform provided a very high depth of coverage (∼800×), resulting in high sensitivity and specificity.
We implemented a targeted‐sequencing panel in the precision oncology clinic at Samsung Medical Center and conducted the molecular‐screening program for oncology patients. The aim of this molecular‐screening program in metastatic cancer patients was to sequence patient tumor specimens and facilitate enrollment in 13 ongoing biomarker‐guided trials. Each biomarker‐selected clinical trial operated independently, with separate consenting procedures following biomarker identification through the master program. Here, we described the feasibility of this clinic‐based molecular‐screening program and reported on the clinical trial‐enrollment rate based on use of the molecular‐screening program.
Materials and Methods
Ethics Statement
This investigation was conducted in accordance with the ethical standards of the Declaration of Helsinki and national and international guidelines and was approved by the Institutional Review Board at Samsung Medical Center.
The NEXT Screening Program Design and Available Clinical Trials
This study was designed as a molecular‐screening program for oncology patients upon patient consent, followed by guidance to biomarker‐selected clinical trials based on genomic alterations (Fig. 1). Within the program, there were five cohorts according to cancer type: Cohort 1, gastric cancer (GC); Cohort 2, colorectal cancer (CRC); Cohort 3, pancreatic/biliary tract cancer; Cohort 4, sarcoma/others; and Cohort 5, genitourinary cancer. Statistical assumptions and specific lists of available targeted agents were designed independently for each cohort. Each drug trial was designed independently from this screening protocol. The primary endpoint was response rate of the matched therapy group versus a conventional‐chemotherapy group for each cohort. The study (www.clinicaltrials.gov, NCT#02141152) was approved by the institutional review board of the Samsung Medical Center, and all participants provided written informed consent before study entry. The time of treatment decision for the trial was as follows: (a) for GC, at the point beyond second‐line treatment; (b) for CRC, after failure of oxaliplatin‐based treatment, irinotecan‐based treatment, approved biologics (bevacizumab and/or cetuximab), and/or regorafenib treatment; (c) for pancreatic‐biliary tract cancer, after failure of gemcitabine‐based first‐line treatment; and (d) for rare cancer, after failure of first‐line treatment. All molecular‐targeted drugs approved by the regulatory authority in Korea at the time of analysis (i.e., trastuzumab for HER2‐amplified GC and cetuximab for Kirsten rat sarcoma viral oncogene homolog [KRAS] wild‐type CRC) were not considered as biomarker‐selected treatments in this study.
Figure 1.

Overall scheme of the NEXT‐1 molecular‐screening program. (A): Quality control test for molecular tests. (B): Patients for analysis: pancreatic/biliary tract cancer included 28 pancreatic adenocarcinoma patients (*), 11 gall bladder cancer patients, 13 biliary tract cancer patients, and 10 ampulla of Vater cancer patients; others included 21 sarcoma patients, 21 melanoma patients, 4 gastrointestinal stromal tumor patients, 2 duodenal cancer patients, 1 anal squamous cell cancer patient, 1 adenocortical carcinoma patient, 3 patients with metastases of unknown origin, 1 anaplastic astrocytoma patient, 2 small‐bowel cancer patients, and 9 neuroendocrine carcinoma patients (**).
Abbreviations: QC, quality control; FFPE, formalin‐fixed paraffin‐embedded.
Available Clinical Trials
The biomarker‐selected basket trials that were predefined at the time of this study included sirolimus (NCT#02449564), gefitinib (NCT#02447419), lapatinib (NCT#02342587), sunitinib (NCT#02450123), crizotinib (NCT#02435108), everolimus (NCT#02449538), imatinib (NCT#02461849), MET‐antibody (NCT#02296879), hepatocyte growth factor antibody (NCT#02499224), cetuximab/LGX818/BYL719 (NCT#01719380), dabrafenib/trametinib expanded access program (EAP), HM95573 (NCT#02405065), and vemurafenib (NCT#02318329). Vemurafinib or dabrafenib/trametinib EAP were considered as matched therapies because these drugs are not approved for any cancer types, including melanoma, in Korea. If tumors harbored multiple molecular alterations, prioritization was determined by the tumor board based on the following criteria: (a) first, known oncogenic mutations/amplifications approved by regulatory authority in Korea or outside of Korea (i.e., BRAF V600E mutation) were considered as category 1 (high priority); (b) second, oncogenic mutations/amplifications being studied in clinical trials (i.e., TP53 mutation, epidermal growth factor receptor [EGFR] amplification, and/or phosphatase and tensin homolog [PTEN] loss) were considered as category 2; (c) third, if genomic aberrations were identified at the experimental stage with cell‐line data only, these were categorized as category 3.
Targeted Sequencing and Detection of Somatic Alterations
Genomic DNA was extracted from the unstained slides of formalin‐fixed paraffin‐embedded (FFPE) tumor blocks using the Qiagen DNA FFPE tissue kit or from fresh frozen tissue using the QIAamp DNA mini kit (Qiagen, Valencia, CA, https://www.qiagen.com) according to manufacturer instructions. Tumor areas (>20% of cancer cells) were dissected under microscopy from 4‐μm thick unstained paraffin‐embedded sections or from fresh frozen tissues by comparison with a hematoxylin and eosin‐stained slide. After manual microdissection, tumor areas reached to >70% in most cases. The IHC protocol for PTEN, MET (MET proto‐oncogene, receptor tyrosine kinase), and HER2 for the trial were previously reported [18]. Somatic alterations were detected by targeted deep sequencing by CancerSCAN, as previously reported [20]. From August 2014 through January 2015, we applied CancerSCAN version 1 consisting of 83 cancer‐related genes (n = 167), and from February 2015, CancerSCAN version 2 was applied, encompassing 381 cancer‐related genes (n = 251). Briefly, extracted genomic DNA was sheared to 150 bp to 200 bp using Covaris S220 (Covaris Inc., Woburn, MA, https://covarisinc.com/) and targeted genes were captured using a custom‐panel capture library (Agilent Technologies, Santa Clara, CA, http://www.agilent.com/), covering 2.5 Mb of exonic regions for the Illumina paired‐end sequencing library kit (Illumina, San Diego, CA, https://www.illumina.com/). We performed DNA sequencing of 100‐bp or 101‐bp paired‐end reads using an Illumina HiSeq 2500 sequencer (Illumina, San Diego, CA, https://www.illuminacom/) and aligned the sequencing reads to the human reference genome (GRCh37/hg19), excluded duplicated reads, and extracted uniquely mapped and properly paired reads with an insert size. Actionable variants included in this panel were selected based on publicly available databases, such as My Cancer Genome (http://www.mycancergenome.org/).
Statistical Considerations
At the time of study design, we presumed that the reference overall response rate (ORR) for matched therapy would be in the range of what is known for targeted agents for each disease cohort (i.e., HER2 amplification with trastuzumab in GC). For the GC cohort, we assumed that ∼30% of patients would harbor a genomic aberration with matched therapy available. The ORR was expected to be ∼25% for the matched‐therapy group and ∼5% for the nonmatched‐therapy group. With 130 GC patients enrolled, 39 patients were expected to be allocated into the matched group and 91 patients into the non‐matched group. The two‐sided chi‐square test with a 5% alpha had an 89% power to detect. For the CRC cohort, the planned sample size was 130 CRC patients, with ∼5% of these assumed to harbor genomic aberrations with a target drug available. The median ORR was assumed to be ∼25% for the targeted group and ∼5% for the nonmatched‐therapy group. With 130 CRC patients, ∼33 patients were expected to be assigned to the targeted group and ∼97 would belong to the nonmatched‐therapy group. The two‐sided chi‐square test with a 5% alpha had an 87% power to detect. For the pancreatic/biliary tract cancer cohort, a total of 78 patients were anticipated, with ∼20% of these assumed to harbor a mutation with a target drug available. ORRs were compared between the targeted group and the nonmatched‐therapy group. The ORR was expected to be ∼35% for the targeted group and ∼5% for the nonmatched‐therapy group (16 patients in the targeted group and ∼62 in the nonmatched‐therapy group). The two‐sided chi‐square test with a 5% alpha had an 87% power to detect. For the sarcoma/others cohort, a total of 87 patients were anticipated, with ∼25% of these expected to harbor a mutation with a target drug available. The ORR was expected to be ∼30% for the targeted group and ∼5% for the nonmatched‐therapy group (22 patients in the targeted group and ∼65 in the nonmatched‐therapy group). The two‐sided chi‐square test with a 5% alpha had an 86% power to detect. The incidence of each mutation was assumed to be low (mostly <5%), allowing the different types of mutations to be assumed as mutually exclusive for statistical‐design purposes. The interim analysis was preplanned to assess feasibility and the rate of clinical‐trial enrollment based on genomic sequencing.
Results
The Feasibility of the Molecular‐Screening Program in the Oncology Clinic
From the 654 patients who consented to participate in the molecular‐screening program from August 2014 through April 2015, 588 (89.9%) tumor tissues consisting of 247 (37.8%) fresh tumors and 407 (62.2%) archival FFPE specimens were prepared for genomic molecular‐screening tests (Fig. 1A). For fresh tissues, 208 (84.2%) were from primary tumors and 39 (15.8%) from metastatic sites. For FFPE specimens (n = 407), 330 (81.1%) were from primary tumors and 77 (18.9%) from metastatic sites. Thirty‐eight (5.8%) specimens did not proceed to sequencing due to no or very low tumor content. Of the 616 tissues available for sequencing, 28 (4.5%) samples were further excluded from the analysis due to an insufficient amount of DNA. Therefore, tissue from 588 (89.9%) of 654 consenting patients was successfully sequenced using targeted sequencing. The average number of 4‐μm slides successfully entered into sequencing was 9.8 (range: 3–60). For fresh tissues, the smallest size of the biopsied tissue was 0.2 × 0.2 × 0.2 cm in 30 cases, from which >1 μg DNA was extracted, with all tissues showing high levels of DNA purity (260 nm/280 nm ratio > 1.86). Finally, 99.2% (245/247) of freshly obtained tissues passed the quality control (QC) process, whereas 91.2% (371/407) of FFPE specimens passed QC. The QC pass rate was slightly lower in metastatic FFPE tissues (67/77; 87%) when compared with that of primary tumor FFPE tissues (304/330; 92.1%) (Fig. 1A).
To analyze accurate rates of biomarker‐selected clinical‐trial enrollment based on this program, we excluded 170 patients who were not eligible to be considered for any type of anticancer treatment regardless of molecular tests, mostly due to deteriorating performance status. The median turnaround time from the time of consent to genomic test report to clinician was 38 days (range: 16–49 days). The genomic profiling report was reviewed by the tumor board at the time of standard‐of‐care failure, and a genome‐guided matched trial was identified by the board. The board meeting was held every 2 weeks to review all genomic data available for the enrolled patients planning to be seen in the outpatient clinic in the upcoming 2 weeks.
The analysis cohort of 418 patients consisted of 127 GC patients, 122 CRC patients, 62 pancreatic/biliary tract cancer patients, 67 sarcoma/rare patients, and 40 genitourinary cancer patients. Patients who were treated with chemotherapy due to a lack of genomic aberration or unavailability of specific trials for the detected genomic alteration were considered as the conventional‐chemotherapy group for this study.
Identification of Actionable Mutations
Analysis of the targeted‐sequencing data revealed at least one clinically actionable variant in 187 (44.7%) of 418 patients. The frequency of each actionable variant detected per cohort is shown in Figure 2. The most frequent mutations in refractory cancer patients were as follows: KRAS codons 12 and 13, BRAF V600E, PIK3CA E545K, PIK3CA E542K, PIK3CA H1047, KIT, ERBB2 V842I, NRAS; and amplifications of ERBB2, EGFR, MDM2, fibroblast growth factor receptor 2 (FGFR2), CDK4, KIT, MET, and KRAS. The landscape of the genomic aberration profile in each cohort is shown in Figure 2. Notably, there was no significant difference in the number of actionable variants between the 80‐ (n = 167) and the 381‐gene (n = 251) panels used in this cohort, although it was speculated that this was dependent upon the availability of clinical trials.
Figure 2.

Mutations in key genes and pathways. Variants are shown in association with the RAF/MEK, PI3K/AKT/mTOR, and DNA damage and repair pathways, as well as others. The upper panel shows patients guided toward matched therapy (blue) or conventional chemotherapy (olive green) and their responses. The right panel indicates a frequency of the variants in each gene.
Enrollment of Patients onto Biomarker‐selected Clinical Trials Based on NGS
In the GC cohort, 127 patients had genomic profiling available for treatment. Based on the sequencing‐based clinical report, 33 (26.7%) of 127 patients were enrolled into clinical trials and 94 (74.0%) patients received conventional chemotherapy. Of the 33 patients enrolled into clinical trials, 9 harboring a tumor protein p53 (TP53) mutation, 8 harboring a PIK3CA mutation, 3 harboring a KRAS mutation, 4 with a PTEN loss, 2 exhibiting 3‐fold MET overexpression, 2 exhibiting EGFR amplification, 1 exhibiting FGFR2 amplification, and 1 exhibiting MET amplification were enrolled into biomarker‐selected clinical trials. Notably, genomic aberrations involving the DNA damage and repair (DDR) pathway were frequently observed in the GC cohort. Additionally, there were nine AT‐rich interaction domain 1A (ARID1A) mutations and one mothers against decapentaplegic homolog 4 deletion. Another significantly deranged pathway in GC patients involved the PIK/AKT/mTOR pathway, with mutations in tuberous sclerosis complex (TSC)2, TSC1, and serine/threonine kinase 11 and amplifications in RAC‐alpha serine/threonine‐protein kinase (AKT)1, AKT2, and regulatory‐associated protein of mTOR. In this trial, there were no directed therapies against these genes; however, they can be considered as potential candidates for future trials. For the conventional‐chemotherapy group, 67 patients received taxane‐based chemotherapy (n = 41), irinotecan‐based therapy (n = 25), or 5‐fluorouracil‐based chemotherapy (n = 2) as salvage therapy. There was no significant difference between matched group versus conventional‐chemotherapy group in terms of performance status, number of metastatic sites, or age (Table 1).
Table 1. Patient characteristics.
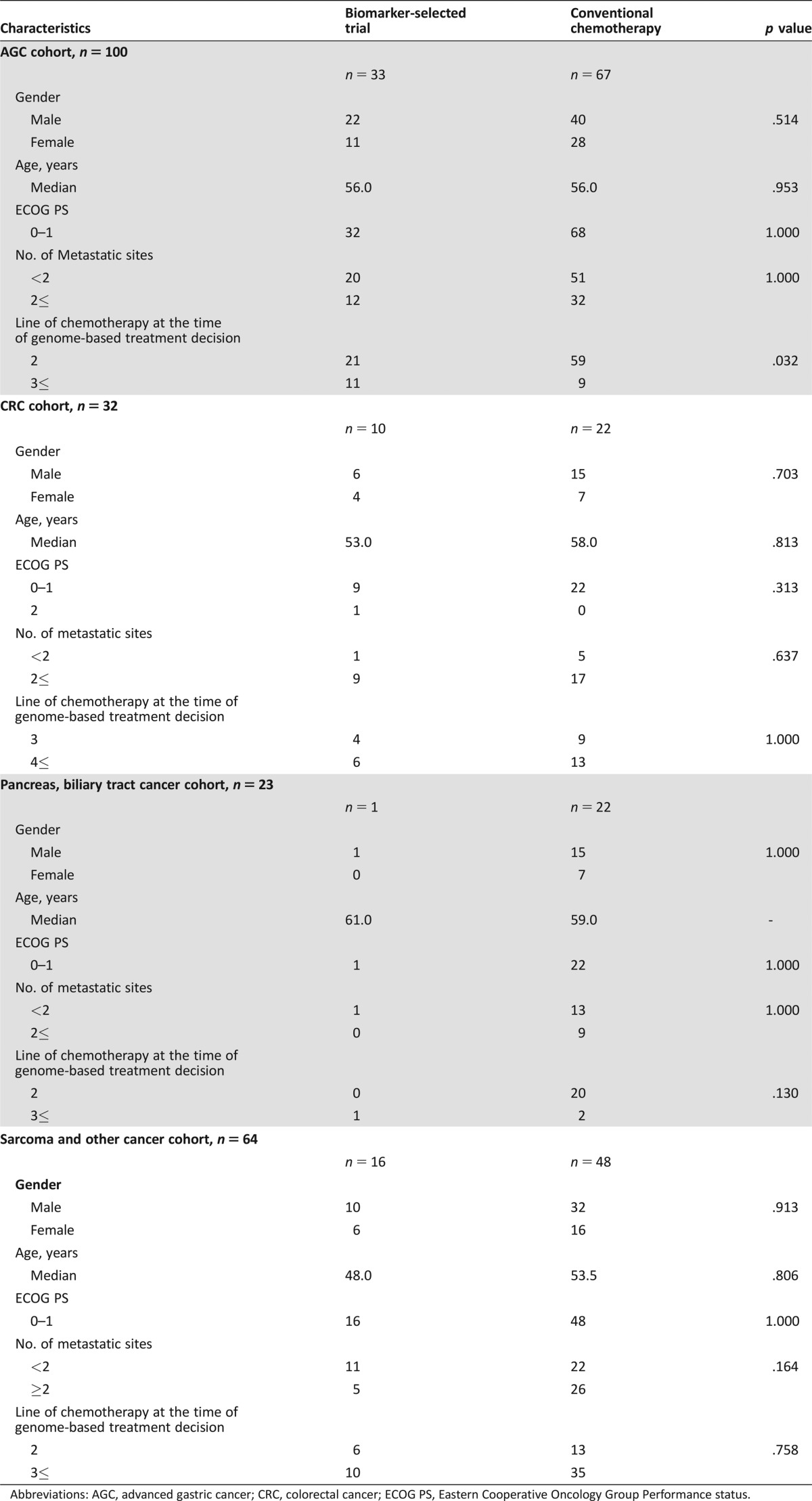
Abbreviations: AGC, advanced gastric cancer; CRC, colorectal cancer; ECOG PS, Eastern Cooperative Oncology Group Performance status.
Among the 122 CRC patients who failed all available standard chemotherapy treatments, including oxaliplatin‐ and irinotecan‐based therapies and biologics, 10 (8.2%) were enrolled onto biomarker‐selected trials. Of these 10 patients, 2 harboring a BRAF V600E mutation were enrolled into an ongoing trial with cetuximab/LGX818/BYL179, 5 exhibiting MET overexpression according to IHC (without MET amplification) received an anti‐MET monoclonal antibody, and 3 patients exhibiting FISH‐confirmed HER2 amplification were enrolled into a lapatinib‐monotherapy trial. In the conventional‐chemotherapy group, we identified patients harboring RET‐NCOA4 fusion (n = 2), NTRK1‐TPM fusion (n = 1), and EML4‐ALK fusion (n = 1) events and who were potentially treatable with targeted agents; however, all of these patients died of the disease due to a rapidly progressive clinical course while on standard first‐ or second‐line chemotherapy. Additionally, a significant portion of CRC patients exhibited DDR‐pathway gene aberrations, including TP53, ATR, and ATM mutations, and received conventional chemotherapy, and two patients exhibiting EGFR amplifications were not successfully routed to matched therapy. Another potentially treatable genomic alteration in the CRC cohort included an isocitrate dehydrogenase 1 (IDH1) mutation (Fig. 2). Therefore, a substantial percentage of CRC patients who might have been assigned to matched therapy were treated with cytotoxic therapy due to either patient performance or the unavailability of biomarker‐driven trials.
For the pancreatic/biliary tract cancer cohort, relevant genomic alterations included point mutations in TP53, KRAS, adenomatous polyposis coli (APC), neurofibromatosis type 1 (NF1), and HER2, as well as EGFR amplification. However, only one patient harboring a PIK3CA mutation and PTEN loss according to IHC results was enrolled into the sirolimus trial, but they continued to exhibit progressive disease after two cycles. The presumed proportion of the sample size for targeted therapy was not met in the trial, and, therefore, the ORR was not analyzable for this cohort.
From the sarcoma/others cohort, 16 patients were enrolled into biomarker‐selected clinical trials. One gastrointestinal stromal tumor (GIST) patient harboring a BRAFV600E mutation was enrolled into the RAF‐inhibitor trial after failure to respond to imatinib, sunitinib, and regorafenib treatment. Two patients with melanoma or non‐GIST sarcoma and harboring a KIT SNV mutation had been previously enrolled into a phase II imatinib basket trial. Three patients exhibiting PTEN loss/PIK3CA SNV mutations had been previously enrolled into the sirolimus basket trial.
Taken together, we reviewed 418 patients with molecular‐profiling data, with 55 (12.0%) patients enrolled into clinical trials based on genomic sequencing data. Of these 55 patients, 37 (7.3%) were enrolled into predefined biomarker‐selected clinical trials, and 23 (41.8%) were directed to other available biomarker‐selected trials.
Discussion
We designed a genomic profiling program for metastatic solid‐tumor patients with the goal of enrolling patients into biomarker‐driven trials based on the variants identified. Independent biomarker‐selected clinical trials were aligned to the screening program as phase I or II trials, with independent patient‐consent forms and statistical design. In total, 55 (12.0%) of 418 patients harbored a biomarker that allowed subsequent guidance to a clinical trial. Additionally, 184 (44.4%) patients, including the 55 who were guided to clinical trials, harbored at least one potentially targetable genomic aberration detected through molecular testing (Fig. 3).
Figure 3.

Therapy distribution for 418 patients and the recurrently mutated genes. (A): The inner circle represents the patients undergoing different therapies, and the outer circle shows mutation‐variant distributions: potentially treatable variants (blue), any variants (light blue), and no variants (grey) among the three groups. The right panel shows frequently detected genes representing (B) key genes in the matched‐therapy group and (C) genes with potentially treatable variants in all three groups. The number of patients is indicated in brackets.
Abbreviations: CRC, colorectal cancer; GC, gastric cancer; GUC, genitourinary cancer; PC/BTC, pancreatic/biliary tract cancer.
We adopted a genome‐profiling method comprising cancer‐related genes to detect SNVs/insertions or deletions (Indels), CNVs, and gene fusions with high accuracy. The average sequencing coverage reached 774.5× in read depth, and each patient harbored an average of 2.8 or 5.4 genomic alterations based on the use of 83‐ or 381‐gene panels, respectively (supplemental online Table 1). The failure rate for entry into sequencing from either fresh tumor tissue or an FFPE sample was ∼10%, 38 samples were excluded due to no or very low tumor content upon pathologic examination, and 28 samples were excluded due to insufficient DNA quantity (<500 ng) or quality.
Of 184 patients with at least one genomic alteration, the most commonly detected SNV/Indel/CNV variants occurred in PIK3CA (n = 35), BRAF (n = 21), ERBB2 (n = 13), TSC1 (n = 11), TP53 (n = 9), MET (n = 8), and EGFR (n = 8) or involved PTEN deletion/mutation (n = 9). It is anticipated that expansions of biomarker‐driven trials will result in the rates of enrollment into such trials increasing accordingly. One of the trials that we plan to open is a phase II AZD2014 (TORC1/2 inhibitor) trial associated with TSC1/TSC2‐null tumors. Currently, there is no significant difference in the number of actionable variants that can be directly associated with biomarker‐driven trials between 80‐ and 381‐gene panels.
In this study, we reported results of an interim feasibility analysis. We designed this trial to allow continuous addition of precision‐medicine trials as they become available based on a general assumption that clinical‐sequencing screening programs may benefit refractory cancer patients having few treatment options remaining. With the rapid expansion of precision‐medicine programs in recent years [12], this type of master‐screening program should be expanded to route these patients into appropriately matched trials. There are currently similar ongoing studies, such as NCI‐MATCH [21], AURORA [22], SPECTAColor [23], SPECTALung [24], and CRUK SMP1 [25], that serve as screening programs to guide patients to precision‐medicine trials.
Our results showed that despite their low incidence in CRC, gene fusions, such as RET‐NCOA4, NTRK1‐TPM, and EML4‐ALK, represented potential therapeutic targets. Although ERBB2 amplification was present in some CRC patients, they did not respond to lapatinib treatment. For the CRC cohort, our results indicated that aside from point mutations in the well‐known oncogenes KRAS, APC, BRAF, and PIK3CA, the identification rate of targets capable of being routed to matched therapy was relatively low when compared with other cohorts. By contrast, the pancreatic/biliary tract cancer cohort revealed several interesting targets, including HER2 amplification, EGFR amplification, BRAF mutation, and IDH1 mutation. Therefore, because the number of patients in the matched groups varied (n = 10 for the CRC cohort and n = 1 for pancreatic/biliary tract cancer cohort) mostly due to an insufficient number of available trials addressing frequently detected genomic alterations (i.e., APC mutation, IDH1 mutation, RET fusion, NF1 mutation, ARID1A mutation, or NRAS mutation), it may be premature to conclude that clinical sequencing does not benefit CRC or pancreatic/biliary tract cancer cohorts.
The major limitations of the study were as follows: (a) this study was not a randomized trial, such as that of the SHIVA trial [19], which renders the clinical benefit in terms of antitumor efficacy of matched trials inconclusive; (b) the trial was not a large trial, considering the limited variety of tumor types included in the trial; and (c) other limitations were rooted in the statistical assumptions incorporated in the design of the study (i.e., all major mutations were mutually exclusive). Nevertheless, this study has value in that the prospective, planned sequencing of refractory cancer patients actually guided an acceptable proportion of them into clinical trials. Our results supported the feasibility of the use of targeted‐sequencing panels for refractory cancer patients, with high patient‐consent rates indicating a high demand on the part of refractory cancer patients. We also revealed that up to 44% of the patients were able to be guided to targeted agents depending on the availability of clinical trials and other factors, such as patient medical condition, with 14.4% of the patients subsequently treated with targeted agents based on genomic data. Recently, a similar prescreening molecular study (the ATTACC trial) accompanied by companion clinical trials [26] enrolled 484 CRC patients, with 19% (92 patients) ultimately enrolled in biomarker‐selected clinical trials.
Conclusion
Guidelines for clinical sequencing with optimized classifications based on clinical implication for genomic variants should be established. Based on our findings, upfront molecular tests consisting of >300 genes might guide refractory cancer patients into ongoing biomarker‐selected clinical trials in a timely and effective manner.
See http://www.TheOncologist.com for supplemental material available online.
Supplementary Material
Acknowledgments
This work was supported by a grant from the Korean Health Technology R&D Project, Ministry of Health & Welfare, Republic of Korea (HI13C2096, HI14C0072, HI14C2750, and HI14C3418). Support was also provided by a grant from the 20 by 20 project sponsored by Samsung Medical Center (GF01140111). Trial Registration: ClinicalTrials.gov (no. NCT02141152).
Contributed equally
Footnotes
For Further Reading: Kim M. Hirshfield, Denis Tolkunov, Hua Zhong et al. Clinical Actionability of Comprehensive Genomic Profiling for Management of Rare or Refractory Cancers. The Oncologist 2016;21:1315‐1325; first published on August 26, 2016.
Implications for Practice: Identification of key factors that facilitate use of genomic tumor testing results and implementation of genomically guided therapy may lead to enhanced benefit for patients with rare or difficult to treat cancers. Clinical use of a targeted next‐generation sequencing assay in the setting of an institutional molecular tumor board led to implementable clinical action in over one third of patients with rare and poor prognosis cancers. The major barriers to implementation of genomically guided therapy were clinical status of the patient and drug access both on trial and off label. Approaches to increase actionability include early and serial sequencing in the clinical course and expanded access to genomically guided early phase clinical trials and targeted agents.
Contributor Information
Woong‐Yang Park, Email: woongyang@skku.edu.
Jeeyun Lee, Email: jyunlee@skku.edu.
Author Contributions
Conception/design: Jeeyun Lee, Seung Tae Kim, Kyoung‐Mee Kim, Woong‐Yang Park
Administrative support: Jae‐Won Yun
Provision of study material or patients: Joon Oh Park, Won Ki Kang, Ho Yeong Lim, Young Suk Park, Se Hoon Park, Jin Seok Heo, Jong Ho Cho, Hee Cheol Kim, Dong Il Choi, Byung‐Hoon Min, Hyuk Lee, Sung No Hong, Tae Sung Sohn
Collection and/or assembly of data: Soomin Ahn, Kyoung‐Mee Kim
Data analysis and interpretation: Kyoung‐Mee Kim, Seung Tae Kim, Nayoung K. D. Kim, Jeeyun Lee, Woong‐Yang Park
Manuscript writing: Jeeyun Lee, Seung Tae Kim, Kyoung‐Mee Kim, Woong‐Yang Park, Kyu‐Tae Kim, Peter J. Park, Nayoung K. D. Kim
Final approval of manuscript: Seung Tae Kim, Kyoung‐Mee Kim, Nayoung K. D. Kim, Joon Oh Park, Soomin Ahn, Jae‐Won Yun, Kyu‐Tae Kim, Se Hoon Park, Peter J. Park, Hee Cheol Kim, Tae Sung Sohn, Dong Il Choi, Jong Ho Cho, Jin Seok Heo, Wooil Kwon, Hyuk Lee, Byung‐Hoon Min, Sung No Hong, Young Suk Park, Ho Yeong Lim, Won Ki Kang, Woong‐Yang Park, Jeeyun Lee
Disclosures
The authors indicated no financial relationships.
References
- 1. Meyerson M, Gabriel S, Getz G. Advances in understanding cancer genomes through second‐generation sequencing. Nat Rev Genet 2010;11:685–696. [DOI] [PubMed] [Google Scholar]
- 2. Collins FS, Varmus H. A new initiative on precision medicine. N Engl J Med 2015;372:793–795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Lander ES. Cutting the gordian helix–regulating genomic testing in the era of precision medicine. N Engl J Med 2015;372:1185–1186. [DOI] [PubMed] [Google Scholar]
- 4. Evans JP, Watson MS. Genetic testing and FDA regulation: Overregulation threatens the emergence of genomic medicine. JAMA 2015;313:669–670. [DOI] [PubMed] [Google Scholar]
- 5. Evans BJ, Burke W, Jarvik GP. The FDA and genomic tests–getting regulation right. N Engl J Med 2015;372:2258–2264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Robson ME, Bradbury AR, Arun B et al. American society of clinical oncology policy statement update: Genetic and genomic testing for cancer susceptibility. J Clin Oncol 2015;33:3660–3667. [DOI] [PubMed] [Google Scholar]
- 7. Gerber DE, Minna JD. Alk inhibition for non‐small cell lung cancer: From discovery to therapy in record time. Cancer Cell 2010;18:548–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kim S, Lee J, Hong ME et al. High‐throughput sequencing and copy number variation detection using formalin fixed embedded tissue in metastatic gastric cancer. PLoS One 2014;9:e111693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Lee J, van Hummelen P, Go C et al. High‐throughput mutation profiling identifies frequent somatic mutations in advanced gastric adenocarcinoma. PLoS One 2012;7:e38892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Cancer Genome Atlas Research Network. Comprehensive molecular characterization of gastric adenocarcinoma. Nature 2014;513:202–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Cancer Genome Atlas Research Network. Comprehensive molecular characterization of human colon and rectal cancer. Nature 2012;487:330–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Biankin AV, Piantadosi S, Hollingsworth SJ. Patient‐centric trials for therapeutic development in precision oncology. Nature 2015;526:361–370. [DOI] [PubMed] [Google Scholar]
- 13. Ledford H. 'Master protocol' aims to revamp cancer trials. Nature 2013;498:146–147. [DOI] [PubMed] [Google Scholar]
- 14. Hollingsworth SJ. Precision medicine in oncology drug development: A pharma perspective. Drug Discov Today 2015;20:1455–1463. [DOI] [PubMed] [Google Scholar]
- 15. Le Tourneau C, Paoletti X, Servant N et al. Randomised proof‐of‐concept phase II trial comparing targeted therapy based on tumour molecular profiling vs conventional therapy in patients with refractory cancer: Results of the feasibility part of the SHIVA trial. Br J Cancer 2014;111:17–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Andre F, Bachelot T, Commo F et al. Comparative genomic hybridisation array and DNA sequencing to direct treatment of metastatic breast cancer: A multicentre, prospective trial (safir01/unicancer). Lancet Oncol 2014;15:267–274. [DOI] [PubMed] [Google Scholar]
- 17. Geoerger B, Deschamps F, Puget S et al. Molecular screening for cancer treatment optimization (moscato 01) in pediatric patients: First feasibility results of a prospective molecular stratification trial. J Clin Oncol 2014;32:abstract 10050. [Google Scholar]
- 18. Kim ST, Lee J, Hong M et al. The NEXT‐1 (Next generation pErsonalized tX with mulTi‐omics and preclinical model) trial: Prospective molecular screening trial of metastatic solid cancer patients, a feasibility analysis. Oncotarget 2015;6:33358–33368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Le Tourneau C, Delord JP, Goncalves A et al. Molecularly targeted therapy based on tumour molecular profiling versus conventional therapy for advanced cancer (SHIVA): A multicentre, open‐label, proof‐of‐concept, randomised, controlled phase 2 trial. Lancet Oncol 2015;16:1324–1334. [DOI] [PubMed] [Google Scholar]
- 20. Chang MY, Kim AR, Kim NK et al. Identification and clinical implications of novel MYO15A mutations in a non‐consanguineous Korean family by targeted exome sequencing. Mol Cells 2015;38:781–788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Lei Z, Tan IB, Das K et al. Identification of molecular subtypes of gastric cancer with different responses to PI3‐kinase inhibitors and 5‐fluorouracil. Gastroenterology 2013;145:554–565. [DOI] [PubMed] [Google Scholar]
- 22. Zardavas D, Maetens M, Irrthum A et al. The aurora initiative for metastatic breast cancer. Br J Cancer 2014;111:1881–1887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Grimes DA, Schulz KF. Compared to what? Finding controls for case‐control studies. Lancet 2005;365:1429–1433. [DOI] [PubMed] [Google Scholar]
- 24. Schwaederle M, Zhao M, Lee JJ et al. Impact of precision medicine in diverse cancers: A meta‐analysis of phase II clinical trials. J Clin Oncol 2015;33:3817–3825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Lindsay CR, Shaw E, Walker I et al. Lessons for molecular diagnostics in oncology from the Cancer Research UK Stratified Medicine Programme. Expert Rev Mol Diagn 2015;15:287–289. [DOI] [PubMed] [Google Scholar]
- 26. Overman MJ, Morris V, Kee B et al. Utility of a molecular prescreening program in advanced colorectal cancer for enrollment on biomarker‐selected clinical trials. Ann Oncol 2016;27:1068–1074. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.