

Abstract
There is limited understanding of how signaling pathways are altered by oncogenic fusion transcription factors that drive leukemogenesis. To address this, we interrogated activated signaling pathways in a comparative analysis of mouse and human leukemias expressing the fusion protein E2A-PBX1, which is present in 5-7% of pediatric and 50% of pre-B-cell receptor (preBCR+) acute lymphocytic leukemia (ALL). In this study, we describe remodeling of signaling networks by E2A-PBX1 in pre-B-ALL which result in hyperactivation of the key oncogenic effector enzyme PLCγ2. Depletion of PLCγ2 reduced proliferation of mouse and human ALLs, including E2A-PBX1 leukemias, and increased disease-free survival after secondary transplantation. Mechanistically, E2A-PBX1 bound promoter regulatory regions and activated the transcription of its key target genes ZAP70, SYK, and LCK, which encode kinases upstream of PLCγ2. Depletion of the respective upstream kinases decreased cell proliferation and phosphorylated levels of PLCγ2 (pPLCγ2). Pairwise silencing of ZAP70, SYK or LCK showed additive effects on cell growth inhibition, providing a rationale for combination therapy with inhibitors of these kinases. Accordingly, inhibitors such as the SRC family kinase (SFK) inhibitor dasatinib reduced pPLCγ2 and inhibited proliferation of human and mouse preBCR+/E2A-PBX1+ leukemias in vitro and in vivo. Further, combining small-molecule inhibition of SYK, LCK and SFK showed synergistic interactions and preclinical efficacy in the same setting. Our results show how the oncogenic fusion protein E2A-PBX1 perturbs signaling pathways upstream of PLCγ2 and renders leukemias amenable to targeted therapeutic inhibition.
Keywords: Acute lymphoblastic leukemia, E2A-PBX1, pre-BCR, signaling pathways, genetic interactions
Introduction
Acute lymphoblastic leukemia (ALL) is the most common childhood cancer and one of the main causes of cancer death in children and young adults. ALL is a heterogeneous disease and can be sub-classified by karyotype, cell type and immunophenotype. One distinctive immunophenotype is characterized by expression and tonic functional activity of the pre-B cell receptor (preBCR). Recent studies indicate that half of preBCR+ ALLs harbor the chromosomal translocation t(1:19), which codes for the chimeric transcription factor E2A-PBX1 (1,2). Although E2A-PBX1+ ALLs are associated with an intermediate risk, about 20% of younger patients and 70% of adult patients with E2A-PBX1+ ALL die five years after diagnosis, with relapse being one of the main causes of death (3,4). Although the treatment and prognosis of patients with pediatric ALL have improved during the last decades, there is a clinical need for more effective/selective and less toxic therapies, particularly in this distinctive ALL subset and in preBCR+ ALL in general.
To address this, we employed a comparative approach based on human and mouse E2A-PBX1 leukemias using biochemical and genetic functional assays. We identified activated signaling pathways that lead to higher phosphorylation levels of PLCγ2, which plays a crucial role in cell survival and proliferation. The chimeric transcription factor E2A-PBX1 transcriptionally regulates genes that code for key kinases upstream of PLCγ2, resulting in signaling network remodeling in pre-B-ALLs. Small molecule inhibitors targeting specifically these pathways show promising preclinical efficacy in vitro and in vivo and should be considered in future clinical trials to treat E2A-PBX1+/preBCR+ ALLs.
Material and Methods
Mice
Conditional E2A-PBX1 transgenic mice were reported previously (5). Transgenic CD19.Cre (Jackson laboratory, (6)), Mb1.Cre (provided by Dr. David Allman, University of Pennsylvania, Philadelphia, PA, USA; and by Dr. Michael Reth, University of Freiburg, Germany (7)) and Mx1.Cre (The Jackson Laboratory (8)) mice were intercrossed to generate E2A-PBX1/CD19.Cre, E2A-PBX1/Mb1.Cre and E2A-PBX1/Mx1.Cre mice respectively, on a C57BL/6 background. Leukemia cells derived from E2A-PBX1/CD19.Cre and E2A-PBX1/Mx1.Cre mice, which were preBCR+ as seen by cytoplasmic μ chain, were used for in vitro and in vivo experiments. Leukemia cells derived from E2A-PBX1/Mb1.Cre mice, which were preBCR-, were used for in vitro experiments. Disease-free survival was defined when showing signs of illness including general lymphadenopathy, lethargy, weight loss and shivering. Moribund mice were euthanized.
Human cells
Human leukemia cell lines 697, RCH-ACV, Kasumi-2, SEM, RS4;11, REH and HAL-01 (obtained from DSMZ, Braunschweig, Germany) were cultured in RPMI 1640 medium supplemented with 10 % FBS, 100 U/ml penicillin/streptomycin, and 0.29 mg/ml L-glutamine. SUP-B15 cells (obtained from ATCC, Manasas, VA) were cultured in RPMI 1640 medium supplemented with 20 % FBS. E2A-PBX1 positive cell lines (697, RCH-ACV, Kasumi-2) were authenticated using western blot for E2A-PBX1 fusion protein expression. E2A-PBX1 negative cell lines (SEM, RS4;11, REH, HAL-01) were obtained from DSMZ in 2013 and not further authenticated. PreBCR status was assessed by flow cytometry for surface VPREB. Primary human ALL samples were obtained from the Tissue Bank of the Department of Pediatrics, Stanford University.
Flow cytometry and fluorescence activated cell sorting (FACS)
Bone marrow cells from transgenic mice were prepared as described previously (5). Flow cytometry was performed in an LSR Fortessa (BD Biosciences, San Jose, CA) and FACS sorting in a FACS Aria (BD Biosciences) using FACS DIVA software (BD Biosciences) and FlowJo (Treestar, Ashland, OR) for analysis. Antibodies used for flow cytometry analysis and FACS sorting are listed in Supplementary Table S1. Lineage negative (Lin-) cells were detected with a cocktail of antibodies including anti-CD3, CD4, CD8, Mac1, Gr1, NK1.1, and Ter119.
Phospho-flow analysis
Murine and human leukemia cells pre-incubated in DMEM high glucose medium (Thermo Scientific) containing 10 % FBS for 1 hour at 37 °C, then treated for 30 minutes at 37°C using following small molecule inhibitors: dasatinib (LC laboratories, Woburn, MA), saracatinib (Selleckchem, Houston,TX), bosutinib (Selleckchem), p505-15 (Selleckchem), R406 (Selleckchem), R778 (fostamatinib, Selleckchem), RK24466 (Cayman Chemical, Ann Harbor, MI), LCKi-II (Fisher Scientific, Waltham, MA), A770041 (Axon Medchem, Reston,VA), ibrunitinib (PCI-32765, Selleckchem), buparlisib (Selleckchem), or trametinib (LC laboratories).
For intracellular staining, cells were fixed with 1.5% formaldehyde for 10 minutes at room temperature, permeabilized with 100% ice-cold methanol for 20 minutes on ice, and washed twice with staining buffer as described previously (9,10), and stained with conjugated antibodies to intracellular phospho-proteins (Supplementary Table S2). Data were analyzed using Flowjo software.
Bone marrow transplantation assays and in vivo drug treatment
Leukemia cells were transduced with lentiviral vectors containing shRNA for luciferase (control) or indicated genes and mCherry fluorescence reporter. mCherry+ cells were sorted 7 days after transduction. Secondary bone marrow transplantation assay using 1000 mouse preBCR+/E2A-PBX1+ leukemia cells per recipient was described elsewhere (5). For in vivo treatment, mice were treated daily, intra-peritoneally, for 21 days, starting the day after transplantation with vehicle (30% PEG1500, 1% Tween 80, 2.5 % DMSO dissolved in PBS), 10 mg/kg b.w. p505-15 (Selleckchem) (11) or 5 mg/kg b.w. A770041 (Axon Medchem) (12).
Colony-forming assays and in vitro drug treatment
Mouse leukemia cells (1000/well) were cultured in methylcellulose medium (M3234, Stem Cell Technologies, Vancouver, Canada) supplemented with 10 ng/ml IL7 (Miltenyi Biotec, Auburn, CA). Human leukemia cells (2000/well) were cultured in methylcellulose medium (H4230, Stem Cell Technologies). Colonies from mouse leukemias were counted after 7 days, and from human leukemia cell lines after 14 days. Leukemia cells were cultured in the presence of dasatinib, saracatinib, bosutinib, p505-15, R406, R778, RK24466, LCKi-II, A770041, ibrunitinib, buparlisib and trametinib at the concentrations indicated.
RT-qPCR
RNA was isolated using the RNeasy Mini Kit (Qiagen) and cDNA was synthesized using Superscript reverse transcriptase III kit (Life Technologies) following the manufacturers recommendations. Relative expression was quantified using an ABI 7900HT Thermocycler with an annealing temperature of 60°C. Taqman Master mix (Applied Biosystems, Carlsbad, CA) following Taqman gene expression assays from Life Technologies (Supplementary Table S3). All signals were quantified using the ΔΔCt method and normalized to ΔΔCt-values of the Actb gene expression levels.
Gene expression microarray
Bone marrow cells from mouse E2A-PBX1 leukemic mice and B-cell progenitors (lin-CD19+CD43+) from wild-type and preleukemic mice (GFP- or GFP+) were used. Healthy preleukemic mice were defined as 3-month-old transgenic mice without any sign of disease. RNA used for microarray analysis was prepared using an RNeasy Mini kit (QIAGEN). Gene 1.0 ST arrays (Affymetrix) were used according to the manufacturer's instructions. Normalizations of CEL file data, comparison between groups and generation of heatmaps were performed using software Nexus Expression (Biodiscover, Hawthorne, CA, USA). The Gene Expression Omnibus (http://ncbi.nlm.nih.gov/geo) accession number for the microarray raw data reported in this article is GSE81010.
shRNA knock-down vectors and transduction
For knockdown studies, the shRNAs were designed using a commercial web tool (Invitrogen). Individual shRNA sequences (Supplementary Table S4) were cloned into the BstXI site of the p309 lentiviral vector (13,14), for stable transduction in mouse E2A-PBX1 leukemia cells and human ALL cell lines.
Lentivirus generation and transduction of mouse and human cells are described elsewere (15). The sorted mCherry-positive cells were cultured for 7 days for colony forming assay or used in secondary bone marrow transplantation experiments (1000 cells each secondary recipient).
Chromatin immunoprecipitation and genomic qPCR
ChIP assays were performed as described elsewhere (16). Briefly, cells were harvested and fixed with 1% fresh formaldehyde and the immunocomplexes were precipitated using GFP-Trap or control magnetic agarose beads (gtma-20 and bmab-20, respectively, ChromoTek Inc., Germany). Quantitative real-time PCR (qRT-PCR) was performed on the precipitated DNA using primers flanking the ZAP70, SYK, LCK, TBP and NCAPD2 genes. Previously published ChIP seq peaks for E2A-PBX1 (1) within regulatory regions described in immortalized cell lines analyzed in the ENCODE project (17) were chosen for validation as well as control sequences in promoter or intergenic regions. The relative values to input were determined using genomic qPCR with SYBR Green as fluorescence dye. Primers are listed in Supplementary Table S5.
Western blot analysis
Western blot for proteins quantification was performed using a modified RIPA lysis buffer as previously described (5). Following antibodies were used for immunodetection: rat anti-E2A (clone 826927; R&D Systems), rabbit anti-PLCγ2 (#3872S, Cell Signaling Technology, Danvers, MA), mouse anti-ZAP70 (clone 1E7.2, Merck Millipore, Billerica, MA), mouse anti-SYK (clone SYK-01, Biolegend, San Diego, CA), rabbit anti-LCK (#2752S, Cell Signaling), rabbit anti histone H3 (#Ab1791, Abcam, Cambridge, UK) mouse anti-Tubulin (GenScript, Piscataway, NJ) or rabbit anti-Gapdh (#G9545, Sigma-Aldrich, St. Louis, MO) antibodies. Secondary antibodies mouse anti-rat ((#HAF005, R&D Systems), rabbit anti-mouse (#816729, Life technologies, Carlsbad, CA) and goat anti-rabbit (#G21234, Life Technologies) coupled to HRP were used. Quantification by densitometry was performed using ImageJ software (NIH) (18).
Statistics
Statistical differences between two groups were analyzed by two-sided Mann Whitney U test or by Student's t-test. Dose-response curves were compared using the extra sum of squares F test. The Bliss independence model (19) was used to evaluate synergy between drug combinations. A p-value below 0.05 was considered statistically significant. Statistical differences from Kaplan-Meier curves were analyzed by log-rank (Mantel Cox) test. Statistical analysis and graphs were performed using Graphpad prism software (Graphpad Inc., La Jolla, CA).
Study approval
All experiments on mice were performed with the approval of and in accordance with Stanford's Administrative Panel on Laboratory Animal Care (APLAC, Protocol 9839).
Results
Oncogenic role for hyper-phosphorylated pPLCγ2 in pre-B-ALL
Various signaling pathways were interrogated in engineered mouse strains that conditionally activate and express E2A-PBX1 and develop ALL (5). Phospho-flow analysis revealed hyper-phosphorylation of PLCγ2 in B-cell progenitors from E2A-PBX1 bone marrow leukemia cells compared to healthy wild-type mice (about 7 fold difference of median fluorescence intensity (MFI)), consistent with constitutive hyper-activation of upstream signaling pathways. (Fig. 1A-B).
Figure 1. PLCγ2 is hyper-phosphorylated in mouse E2A-PBX1+ leukemias and is essential for proliferation.
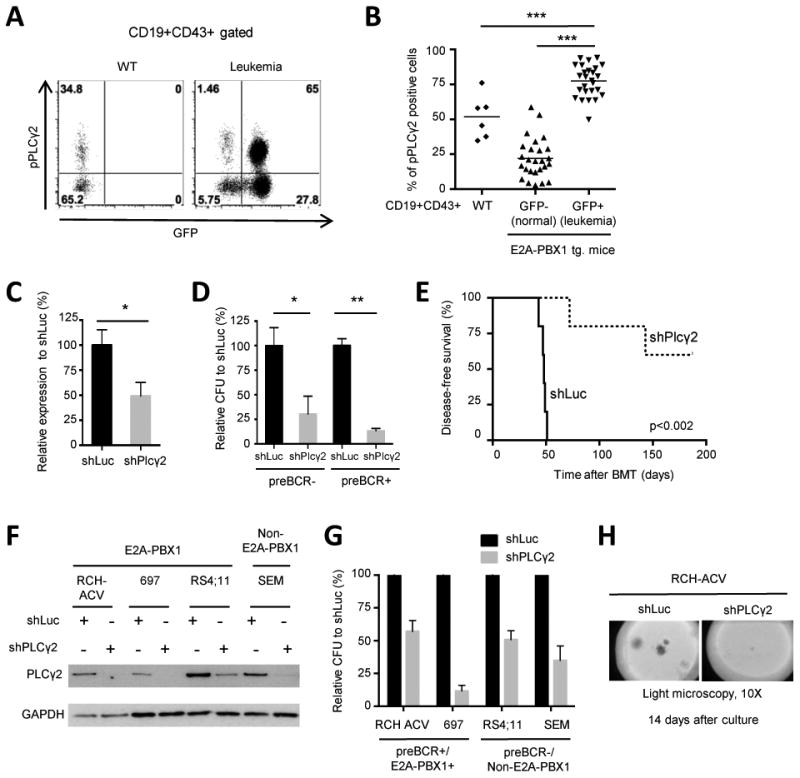
(A) Dot plots show phospho-flow analysis of pPLCγ2 in CD19+CD43+ B-cell progenitors from bone marrow of representative wild-type (WT), and leukemic mice. GFP is activated in mouse cells expressing the E2A-PBX1 transgene. CD19+CD43+GFP- cells from leukemia mice are B-cell progenitors, which did not recombine E2A with the PBX1 transgene. (B) Diagram shows percentages of pPLCγ2 positive cells in WT (n=6) and E2A-PBX1 transgenic/leukemic mice (n=26) of the indicated subpopulations. Each dot represents a bone marrow sample. Statistical analysis performed by Mann-Whitney U test, *** p-value <0.001. Tg., transgenic. (C) Knockdown efficiency of PLCγ2 in mouse E2A-PBX1+ leukemias was quantified by RT-qPCR after 7 days of transduction. Data represent mean ± SEM (n=5 independent experiments). (D) Colony forming units were enumerated 7 days after FACS-sorting of mCherry+ mouse leukemia cells (pre-BCR- and pre-BCR+ leukemias derived from different E2A-PBX1 transgenic mice) with lentiviral vectors containing indicated shRNAs (n=3 independent experiments). Statistical analysis performed by Student's t-test in (C) and (D). ** p-value <0.01, * p-value <0.05. (E) Mouse preBCR+/E2A-PBX1+ leukemia cells were transduced with control (shLuc) and PLCγ2 shRNAs and transplanted into secondary mouse recipients (n=5 mice in each group). Kaplan-Meier curve shows percentage of disease-free survival after secondary transplantation. Statistical analysis was performed using the log-rank (Mantel-Cox) test. (F) Western blot shows shRNA-mediated knock-down of PLCγ2 in human ALL cell lines. GAPDH was used as loading control. (G) Colony forming assay of human ALL cell lines after shRNA-mediated knockdown of PLCγ2 or luciferase. Data represent mean ± SEM (n=3 independent experiments). (H) Colony morphologies of transduced cells after 14-day culture as visualized by light microscopy (original magnification 10×).
The role of PLCγ2 has been extensively characterized as a key enzyme in B cell receptor signaling in mature B cells (20), however its role in B-cell progenitors and pre-B cell leukemogenesis is less well known. To study the functional role of PLCγ2 in mouse and human E2A-PBX1 leukemias, an shRNA knock-down approach was employed using lentiviral transduction (13,14). Efficient shRNA-mediated knockdown of PLCγ2 (Fig. 1C) reduced clonogenicity in mouse E2A-PBX1+ leukemia cells, independently of the presence or absence of the pre-B cell receptor (Fig. 1D). Although E2A-PBX1+/preBCR+ leukemias have increased clonogenicity compared to E2A-PBX1+/preBCR- (p-value= 0.029), no differences in pPLCγ2 were observed consistent with shRNA-knockdown experiments (data not shown). In secondary bone marrow transplantation experiments recipients of PLCγ2-depleted mouse leukemia cells showed an extended disease-free survival compared to controls (Fig. 1E). Similarly, efficient depletion of PLCγ2 by shRNA in human ALL cell lines (Fig. 1F) correlated with reduced clonogenicity (Fig. 1G-H), suggesting a pathogenic role of hyper-activated PLCγ2 not only in E2A-PBX1+ but also in B-ALL leukemogenesis in general.
Identification of E2A-PBX1 target genes upstream of PLCγ2
Transcriptional and bioinformatics analyses were employed to identify candidate signaling proteins that may serve key roles in hyper-activation of PLCγ2 and leukemia pathogenesis. Gene ontology analysis of E2A-PBX1 target genes in human ALLs (1) revealed several biological processes that lead to the activation of PLCγ2 including leukocyte and lymphocyte activation pathways (Fig. 2A). From the gene lists associated with these gene ontology terms, we identified three candidate kinases that are upstream of PLCγ2: the cytoplasmic kinases ZAP70 (zeta chain associated protein) and SYK (spleen tyrosine kinase), and the SRC-family kinase LCK (lymphocyte-specific protein tyrosine kinase).
Figure 2. E2A-PBX1 binds target genes encoding kinases ZAP70, LCK and SYK that function upstream of PLCγ2.
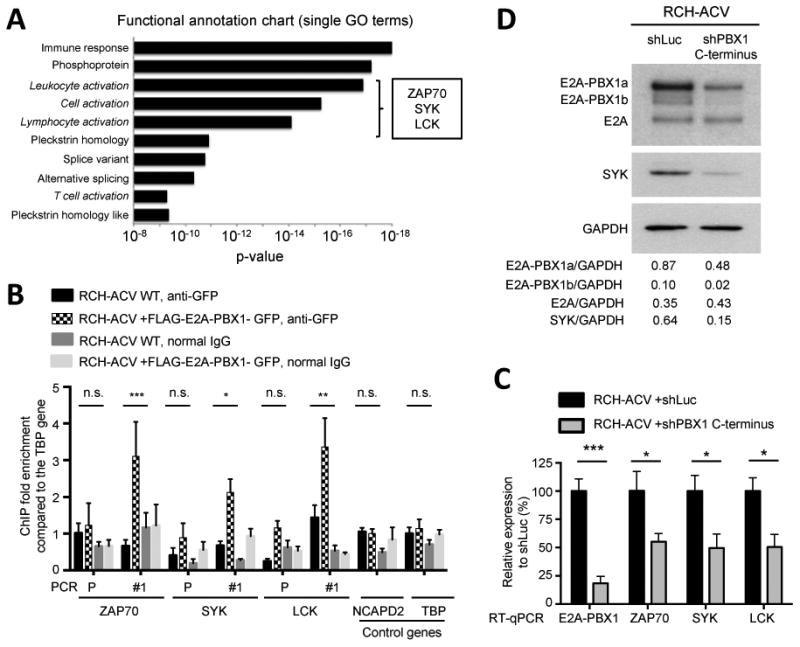
(A) Functional annotation clustering of enriched pathways and biological processes of previously published E2A-PBX1 target genes by ChIP-seq (1) was performed using DAVID functional annotation tool. In italic, signaling pathways identified upstream of PLCγ2. Candidate kinases were identified from the list of genes of each gene ontology term. (B) Chromatin occupancy determined by ChIP-qPCR of exogenous GFP-tagged E2A-PBX1 in RCH-ACV cells is shown. Primers were designed for identified ChIP-seq peaks (1) (Supplementary Fig. S1). Promoter regions of ZAP70 and SYK and intergenic region of LCK not bound by E2A-PBX1 were used as controls. Immunoprecipitations with normal IgG were used as controls for unspecific binding. Data were analyzed using the ΔΔCT method and normalized to 1% input. Diagram shows fold enrichment relative to the TBP control gene and data represent mean ± SEM (n=3 independent experiments). Statistical analysis was performed by Student's t-test. *** p-value <0.001, ** p-value <0.01, * p-value <0.05 and n.s., not significant. (C) RCH-ACV cells were transduced with lentiviral vectors expressing shRNA for luciferase (control) or PBX1 C-terminus (sequence contained in the fusion gene). Of note, wild type PBX1 is not expressed in RCH-ACV cells (data not shown). Expression of indicated transcripts was quantified by RT-qPCR 7 days after transduction and normalized to control cells. ACTB was used as housekeeping gene. Data represent mean ± SEM (n=4 independent experiments). Statistical analysis performed by Student's t-test, * p-value <0.05, *** p-value <0.001. (D) Western blot of a representative experiment from (C) showing the expression of E2A, E2A-PBX1 and SYK. GAPDH was used as loading control. The relative ratio of protein expression (shown below) was determined by densitometry and normalized to GAPDH.
The genes for all three of these kinases showed consistent enrichment for proteins reactive with E2A and PBX1 antibodies in publicly available ChIP-seq datasets (1) (Supplementary Fig. S1). Using ChIP-PCR assays, we confirmed that E2A-PBX1 localizes to sites identified by ChIP-seq in the vicinity of these genes (Fig. 2B) within regions that display chromatin features of gene regulatory elements defined by DNaseI hypersensitivity peaks in immortalized cell lines from the ENCODE project (17) (Supplementary Fig. S1). Furthermore, shRNA-mediated E2A-PBX1-depletion resulted in decreased levels of SYK, LCK and ZAP70 (Fig. 2C-D), indicating that E2A-PBX1 binds and activates their transcription in pre-B-ALL.
Consistent upregulation of ZAP70, SYK and LCK in E2A-PBX1 transformation
Gene expression patterns in mouse leukemias and preleukemic transgenic B cell progenitors, as well as in human primary ALLs (21) induced by the E2A-PBX1 oncogene were compared to the transcriptional profile of normal B-cell progenitors. Intersection of the microarray datasets revealed ZAP70 as the only gene, other than E2A-PBX1, that is upregulated in all E2A-PBX1-expressing cells (Fig. 3A-B, mouse E2A-PBX1 leukemias vs normal B-cell progenitors (p<0.001); preleukemic B-cell progenitors GFP+ vs GFP- (p<0.05); human E2A-PBX1 vs normal B-cell progenitors (p<0.01)). These data suggest that ZAP70 is one of the earliest and most consistently up-regulated genes during E2A-PBX1 leukemogenesis. Increased ZAP70 expression does not reflect merely differentiation stage after comparison to closely related (lin-CD19+CD43+) progenitor B cells (Fig. 3C). Increased ZAP70 protein was also observed in human E2A-PBX1 primary ALL cells and cell lines (Fig. 3C) consistent with previous observations (22).
Figure 3. Consistent upregulation of ZAP70, SYK and LCK in E2A-PBX1 transformation.

(A) Heat map shows differential intensity values for probes in microarray analysis comparing mouse E2A-PBX1 leukemias (n=18) and sorted Lin-CD19+CD43+ wild-type progenitor B cells (n=9). Each row represents a gene and each column a sample. Red: lower expression, green: higher expression. Analyses were performed using an FDR < 0.05 and fold differential expression >1.2. Probes with low intensity values were filtered. (B) Venn diagram shows shared upregulated genes across E2A-PBX1 cells compared to normal mouse (Lin-CD19+CD43+) and human (CD19+CD10+) B-cell progenitors. Gene expression from human E2A-PBX1 patients and normal B-cell progenitors were obtained from a publicly available database (21). (C) Western blot analysis of Zap70 in E2A-PBX1-expressing cells from mouse E2A-PBX1+ leukemias and preleukemias, human primary leukemias and cell lines. Tubulin and Gapdh served as loading controls. The ratio of ZAP70/Tubulin and ZAP70/GAPDH levels (shown below) was determined by densitometry. (D) Published data sets (21) were analyzed for ZAP70, SYK and LCK expression levels in E2A-PBX1 (n=22) versus non-E2A-PBX1 (n=216) human primary B-ALL. Statistical analysis was performed by Mann-Whitney U test.
Expression of ZAP70, SYK and LCK genes was assessed in human primary ALLs using a publicly available dataset (Figure 3D, Supplementary Fig. S2A) and the Oncomine database (23) (Supplementary Fig. S2B). E2A-PBX1+ primary ALLs showed higher expression levels of all three kinases compared to non-E2A-PBX1 primary ALLs in both cohorts of patients, reinforcing further our conclusion that E2A-PBX1 activates transcription of these kinase genes. Their higher-level expression in human and mouse E2A-PBX1-expressing cells strongly suggests a pathogenic role.
Oncogenic roles of the E2A-PBX1 target genes ZAP70, SYK and LCK in pre-B-ALLs
The functional roles of ZAP70, SYK and LCK in E2A-PBX1 leukemias were studied using an shRNA knock-down approach. Efficient depletion of each kinase in human (Fig. 4A) and mouse (Supplementary Fig. S3A) E2A-PBX1 leukemia cells correlated with decreased clonogenicity (Fig 4B; Supplementary Fig. S3B). Sub-lethally irradiated mice transplanted with kinase-depleted mouse leukemia cells showed extended disease-free survival, demonstrating an important in vivo role during leukemogenesis (Supplementary Fig. S3C). Interestingly, E2A-PBX1+ cells were more susceptible to ZAP70-depletion as shown by reduced cell proliferation compared to non-E2A-PBX1 cells (Figure 4C). Phospho-flow analysis of shRNA-transduced cells showed that depletion of ZAP70, LCK or SYK resulted in decreased pPLCγ2 in E2A-PBX1+ cells (Fig. 4D) and in E2A-PBX1- cells (data not shown). These results confirmed that the kinases are upstream of PLCγ2 and may serve as possible pharmacological targets to decrease cellular levels of pPLCγ2 in pre-B-ALL.
Figure 4. Oncogenic role of ZAP70, SYK and LCK in human ALL cell lines.

(A) Western blots show protein levels after shRNA-mediated knock-down of the indicated genes. GAPDH and histone H3 were used as loading controls. (B) Colonies were enumerated 14 days after FACS sorting and normalized to the number of colonies in control cells. Data represent mean ± SEM (n=4 independent experiments). CFU, colony-forming units. Statistical analysis performed by Student's t-test, * p-value <0.05, n.s., not significant. (C) E2A-PBX1 leukemia cell lines (n=3) and non-E2A-PBX1 cell lines (n=3) were depleted for ZAP70 and cells were enumerated in liquid culture using trypan blue exclusion assay 4 days after FACS sorting and compared to control transduced cells (shLuc). Data represent mean ± SEM (n=3 independent experiments). Statistical analysis between groups was performed using two-way ANOVA. (D) RCH-ACV cells were interrogated for phosphorylated PLCγ2 after shRNA-mediated knock-down of E2A-PBX1 target genes compared to control transduced cells (shLuc). mCherry positive cells (transduced cells) were gated for the analysis. Left panel, histogram from a representative experiment after LCK-depletion is shown. Right panel, data represent mean ± SEM (n=4 independent experiments). RCH-ACV cells transduced with shRNAs for FYN or BCL6 genes served as controls. MFI, median fluorescence intensity. Statistical analysis by Student's t-test, *** p-value <0.001, * p-value<0.05, n.s. not significant.
Pharmacological inhibition of PLCγ2 upstream signaling pathways in E2A-PBX1 leukemias
Several small molecule inhibitors were evaluated for their effects on PLCγ2 upstream pathways in E2A-PBX1 leukemia cells. SRC-family kinase (SFK) inhibitors including dasatinib as well as inhibitors of SYK and LCK were effective in reducing pPLCγ2 in ALL cells, consistent with shRNA knock-down studies (Fig. 5A). The effect of SFK-, SYK- and LCK-inhibitors was specific to pPLCγ2 decrease as compared to stable pSTAT5 levels using the same conditions (data not shown). Preclinical efficacy was tested in vitro comparing IC50 concentrations in colony forming assays of mouse preBCR+ and preBCR- leukemias (Fig. 5B). PreBCR+ leukemias were more sensitive to the SFK inhibitor dasatinib, as previously described (5), as well as to SYK inhibitors p505-15 (Fig. 5C-D) and R406 (data not shown) and LCK inhibitors A770041 (Fig. 5C-D) and RK2446 (data not shown). The preclinical efficacy of p505-15 and A770041 was tested in vivo after secondary bone marrow transplantation of preBCR+ leukemias (11,12). As expected, in vivo treatment with SYK and LCK inhibitors prolonged disease-free survival of mice (Fig. 5E). These data identified novel compounds, which inhibit specifically the proliferation of preBCR+/E2A-PBX1+ leukemias compared to their genetically similar preBCR-/E2A-PBX1+ leukemias and show promising preclinical efficacy.
Figure 5. Pharmacological inhibition of signaling pathways upstream of PLCγ2 in E2A-PBX1 leukemias.

(A) Bars denote relative pPLCγ2 levels determined by phospho-flow after incubation of human RCH-ACV and SEM leukemia cell lines with the indicated compounds. Data represent mean ± SEM (n=3 independent experiments). Statistical analysis by Mann Whitney U test. * p-value<0.05. MFI, median fluorescence intensity. (B) Half maximal inhibitory concentration (IC50) of compounds targeting PLCγ2 upstream signaling pathways are shown for E2A-PBX1+/preBCR+ (n=3) and E2A-PBX1+/preBCR- (n=3) mouse leukemias in colony forming assays. No differences were found regarding pPLCγ2 or Zap70 status. Statistical analysis was performed using F test comparing titration curve of each compound. * p-value <0.05. (C) Images show number and morphology of colonies from preBCR+ and preBCR- mouse leukemia cells treated with p505-15 (SYKi, upper panel) and A770041 (LCKi, lower panel) (original magnification 4×). (D) Dose response curves of preBCR+ (n=3) and preBCR- (n=3) mouse leukemia cells treated with p505-15 (upper panel) and A770041 (lower panel) in colony forming assays. Statistical analysis performed by F test. (E) Kaplan-Meier curve shows disease-free survival after secondary bone marrow transplantation of mouse preBCR+ leukemia cells (n=5 in each cohort) and treatment with P505-15 (upper panel) and A770041 (lower panel), for 21 days. Statistical analysis was performed using the log-rank (Mantel-Cox) test.
Genetic interactions among E2A-PBX1 target genes reveal effective combination therapies
The partial decrease in pPLCγ2 after pharmacological inhibition and shRNA-mediated knock-down targeting individually ZAP70, SYK or LCK, and the modest effects on cell proliferation suggest that compensatory effects might be occurring in these pathways. Therefore, genetic interactions between E2A-PBX1 target genes in human ALL cells were studied using vectors co-expressing shRNAs with mCherry or GFP as fluorescence markers (Supplementary Fig. S4A). Co-transduced cells were monitored using fluorescence microscopy or flow cytometry (Supplementary Fig. S4B-C). Co-suppression of ZAP70 and LCK in RCH-ACV and 697 cells showed a statistically significant additive inhibition of cell growth (Fig. 6A). A trend for additive inhibition was observed following co-suppression of ZAP70 and SYK (Supplementary Fig S4D) as well as LCK and SYK (Supplementary Fig S4E), suggesting that genetic inactivation of a specific kinase might increase the efficacy of inhibitors targeting compensatory kinases.
Figure 6. Genetic interactions among E2A-PBX1 target genes reveal effective combination therapies.
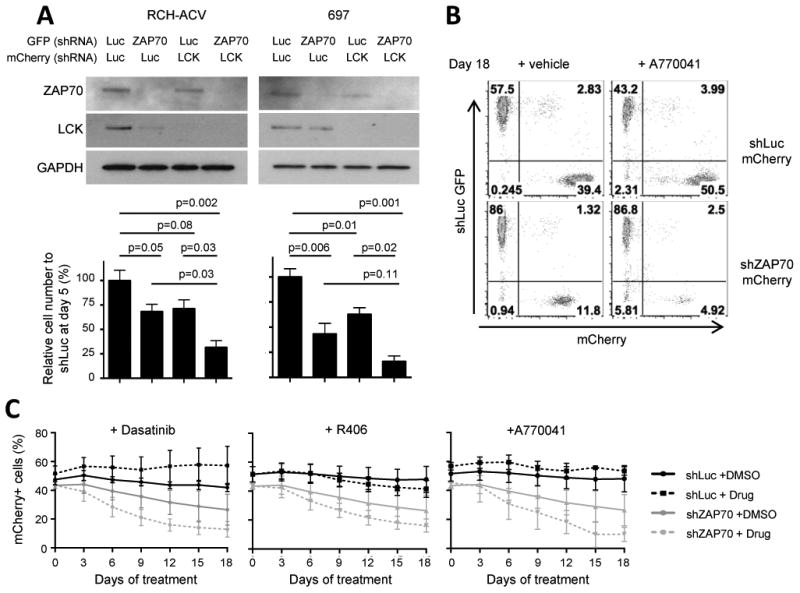
(A) Upper panel, western blot shows the expression of indicated proteins. GAPDH was used as loading control. Lower panel, cell concentration of shRNA transduced cells relative to control transduced cells (shLuc) 5 days after sorting. Cell numbers were quantified by trypan blue exclusion assay. Data represent mean ± SEM (n=4 independent experiments). Statistical analysis by Student's t-test. (B) Dot plots show the proportion of GFP+ and mCherry+ cells at day 18 in flow cytometry from a representative experiment in which RCH-ACV cells were transduced with shRNA for luciferase (GFP) or ZAP70 (mCherry) and mixed 50/50 at day 0. Cells were cultured in the presence of vehicle or A770041 (500 nM) for 18 days. (C) Diagrams showing % of mCherry+ cells transduced from foregoing experiment and treated with the indicated compounds for 18 days: dasatinib (20 nM), R406 (500 nM) and A770041 (500 nM). Data represent mean ± SEM (n=3 independent experiments).
Thus, competition assays of shRNA-transduced cells were performed (Supplementary Fig S5A). Co-cultured mCherry+ (shRNA for gene of interest) and GFP+ (control shRNA) transduced cells were monitored over time by flow cytometry in the presence of small molecule inhibitors (Fig. 6B). Compared to control cells, cells depleted for ZAP70 showed a decreased competitive fitness, that was further reduced by dasatinib (SFKi), R406 (SYKi) or A770041 (LCKi) (Fig. 6C). Similar results were observed for cells depleted of SYK (Supplementary Fig. S5B) or LCK (Supplementary Fig S5C), confirming genetic interactions between E2A-PBX1 target genes. Of note, SYK-depleted human cells showed markedly impaired cell proliferation consistent with reduced leukemogenesis of SYK-depleted mouse leukemias in secondary bone marrow transplantation (Supplementary Fig. S3C). These results provide a rationale for combination therapy to block the hyper-activated PLCγ2 signaling pathway at different levels in preBCR+ leukemias.
Preclinical efficacy of combination treatment with small molecules in preBCR+ E2A-PBX1+ leukemias
Finally, the ability of combination targeted therapy to block signaling pathways upstream of PLCγ2 was tested in human and mouse ALL cells. Human cell lines RCH-ACV (preBCR+/E2A-PBX1+) and SEM (preBCR-/E2A-PBX1-) were treated with A770041 (LCKi) and either p505-15 or R406 (SYKi). Combination therapy inhibited synergistically the clonogenicity of RCH-ACV cells using the Bliss independence model (19) (p-value <0.05, Fig. 7A-B, Supplementary Fig. S6A). However, the addition of A770041 (LCKi) to the SYK inhibitors did not affect the colony forming capacity of SEM cells (Fig. 7B, and data not shown), suggesting that the inhibitory effect of the combination therapy is specific for preBCR+/E2A-PBX1+ cells. We also tested whether SYK inhibitors might increase the sensitivity of human preBCR+/E2A-PBX1+ leukemias to dasatinib. Indeed, combination therapies with P505-15 (SYKi) and dasatinib (SFKi) inhibited synergistically clonogenicity of RCH-ACV compared to SEM cells (p-value<0.001, Fig. 7B).
Figure 7. Preclinical efficacy of combination treatment with small molecules in preBCR+ E2A-PBX1+ leukemias.
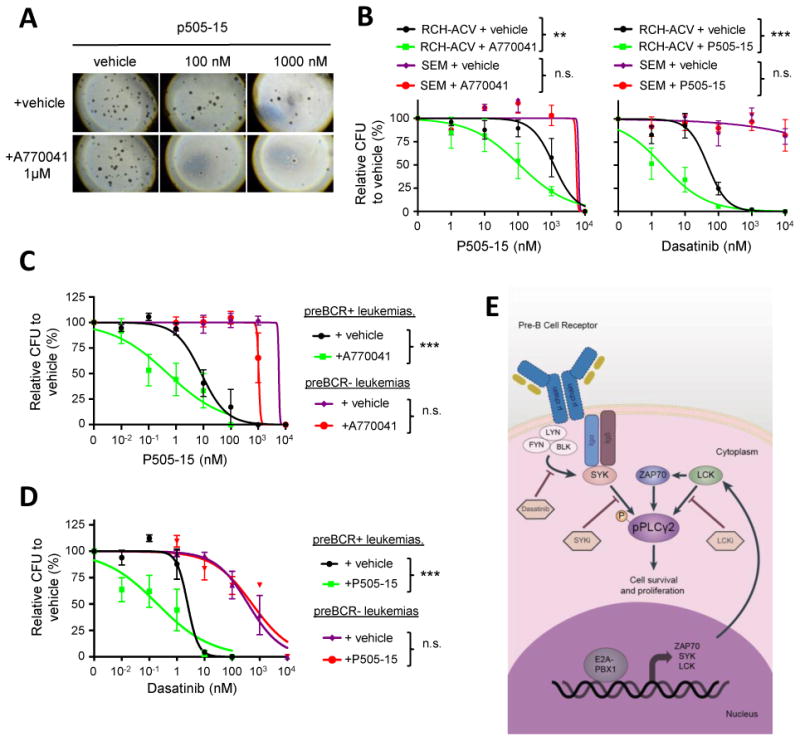
(A) Images show number and morphology of colonies from RCH-ACV cells treated with p505-15 and either vehicle or A770041 in combination (original magnification 4×). (B) Titration growth curves for RCH-ACV and SEM cells cultured in increasing concentrations of p505-15 in combination with either vehicle or A770041 (1μM) (left panel) and increasing concentrations of dasatinib with either vehicle or p505-15 (2 μM) (right panel). Colonies were enumerated after 14 days. Data represent mean ± SEM (n=3 independent experiments). CFU, colony forming units. (C) Titration curves are shown for mouse preBCR+ (n=3) and preBCR- (n=3) leukemia cells cultured in increasing concentrations of p505-15 in combination with either vehicle or A770041 (200 nM). (D) Titration curves are shown for mouse preBCR+ (n=3) and preBCR- (n=3) leukemia cells cultured in methylcellulose at increasing concentrations of dasatinib in combination with either vehicle or p505-15 (50 nM). Colonies were enumerated after 7 days. Statistical analysis performed in (B), (C) and (D) by F test, *** p-value<0.001, ** p-value<0.01. (E) Schematic representation of signaling network remodeling by E2A-PBX1+ in pre-BCR+ ALLs.
To elucidate whether the enhanced effect of combination treatment is due to the presence of the preBCR, mouse preBCR+/E2A-PBX1+ and preBCR-/E2A-PBX1+ leukemias that arose on similar genetic backgrounds were assessed in vitro in drug titration experiments. Mouse preBCR+/E2A-PBX1+ leukemias were more sensitive to the combination therapy of A770041 (LCKi) with either SYK inhibitor P505-15 (Fig. 7C,) or R406 (Supplementary Fig. S6B). Similarly, cell growth inhibition by dasatinib (SFKi) was enhanced by P505-15 (Fig. 7D), A770041 or R406 (Supplementary Fig. S6C) in preBCR+/E2A-PBX1+ but not in preBCR-/E2A-PBX1+ leukemias. The combination therapy of SYK inhibitor P505-15 with A770041 (LCKi, p-value <0.05) exhibited synergy as did the co-treatment with P505-15 and dasatinib (SFKi, p-value<0.001).
Taken together, these data reveal novel genetic interactions of E2A-PBX1 target genes that form the rationale for combination therapies, which show promising in vitro and in vivo preclinical efficacy.
Discussion
Aberrant activation of signaling pathways has been linked to leukemogenesis, however little is known about cell signaling perturbations induced by fusion transcription factors. Using a cross-species comparative approach of human and mouse leukemias from novel engineered transgenic mice, we describe here signaling network remodeling by the chimeric fusion protein E2A-PBX1 in pre-B-ALL, which results in hyperactivation of PLCγ2. E2A-PBX1 binds regulatory elements and activates the transcription of its target genes ZAP70, SYK and LCK, upstream of PLCγ2, which are essential for cell survival and proliferation. Consequently, several kinase inhibitors targeting signaling pathways upstream PLCγ2 display promising preclinical efficacy in preBCR+/E2A-PBX1+ leukemias (Fig. 7E).
A direct link of E2A-PBX1 with de-regulated signaling pathways upstream of PLCγ2 is supported by multiple lines of evidence: E2A-PBX1 binds genomic regions of ZAP70, SYK and LCK genes, respectively, that display chromatin features of gene regulatory elements; depletion of E2A-PBX1 by shRNA reduces the expression of these kinases; E2A-PBX1 expression in mouse and human primary ALLs as well as in human ALL cell lines positively correlates with elevated ZAP70, SYK and LCK expression; and E2A-PBX1+ ALL cells are strongly dependent on expression of these kinases as demonstrated by knockdown and pharmacologic inhibition. Since their expression was not absolutely restricted to E2A-PBX1+ ALLs, alternative mechanisms may drive their oncogenic roles in other ALL subtypes, and variable dependence on the kinases among individual E2A-PBX1+ ALLs likely reflects pathway redundancies, sample heterogeneity, or acquisition of secondary mutations affecting transcription or epigenetic factors involved in E2A-PBX1 target gene regulation.
ZAP70, SYK and LCK have been previously implicated in hematological malignancies other than B-cell precursor ALL. Expression of ZAP70 is associated with elevated downstream signaling (24,25) and is an adverse prognostic marker in B-chronic lymphocytic leukemia (B-CLL) (26,27). LCK overexpression results from chromosomal translocation in T-cell leukemias (28-30). SYK overexpression has been described in peripheral T-cell lymphomas (31) as well as in chromosomal translocations of peripheral T cell lymphomas and myelodysplastic syndromes (32-34). Our studies describe a novel mechanism of kinase overexpression by a chimeric fusion protein in B-cell precursor ALL. Using a comparative approach, we revealed the functional consequences as well as the in vitro and in vivo dependencies on overexpressed kinases. Hence, our findings significantly extend the spectrum of hematological malignancies in which ZAP70, SYK and LCK play important roles to enhance oncogenic signaling in malignant lymphoid cells.
Our pharmacologic screen identified inhibitors of SFKs, SYK and LCK as compounds that inhibit activated signaling pathways upstream of pPLCγ2 in E2A-PBX1+ leukemia cells. Hence, dasatinib, SYK-inhibitors and LCK-inhibitors specifically inhibit cell proliferation of preBCR+/E2A-PBX1+ leukemias. Dasatinib is a tyrosine kinase inhibitor (TKI) that is approved by the FDA for treatment of BCR-ABL+ CML and ALL (35). Recent preclinical studies have suggested that dasatinib may also be a candidate therapy for additional ALL subtypes including BCR-ABL-like ALL (36) and preBCR+ ALL, which includes E2A-PBX1+ pre-B-ALLs (1,5,37,38). The SYK inhibitor R406/R778 (fostamatinib) is FDA-approved for patients with immune thrombocytopenic purpura and P505-15 is currently in phase 2 clinical trial for rheumatoid arthritis. SYK inhibition also showed preclinical efficacy in preBCR+ xenografts in previous studies (1,2). Our data confirm and extend these previous observations, and for the first time demonstrate LCK inhibitors as small molecules with promising preclinical efficacy in preBCR+/E2A-PBX1+ ALL. In addition, we observed enhanced efficacy in combination therapies employing dasatinib, SYK- and LCK-inhibitors, which might be relevant to prevent or ameliorate drug resistance in future clinical applications. Although LCK inhibitors have only been used as a research tool, the translation of the findings regarding dasatinib and SYK inhibitors for the treatment of preBCR+ ALL patients might be accelerated in future clinical trials.
Our genetic studies of individual kinase knockdowns showed a modest decrease in cell growth inhibition (Fig. 4B), which is enhanced by double shRNA knockdown (Fig. 6A). shRNA-mediated knockdown of individual kinases decreases pPLCγ2 (Fig. 4D). However, double shRNA knockdown does not further reduce pPLCγ2 levels (data not shown), suggesting some redundancy of PLCγ2 upstream kinases. Small molecules show a much more robust effect on cell proliferation and decrease of pPLCγ2 (Fig. 5A-B). We hypothesize that small molecules have broader inhibitory effects compared to shRNA knock-downs, targeting several kinases simultaneously. Indeed, dasatinib inhibits SRC kinases and LCK (39) and the SYK inhibitors R406/R778 have significant off-target effects including LCK (40,41). Alternatively, shRNA knock-down vectors reduce the transcript levels of targeted genes without inhibiting the kinase activity directly and low kinase activity might be sufficient to sustain cell survival and proliferation. Hence, leukemia cells are more dependent on SYK than on LCK and ZAP70 as shown in vivo in mouse transplantation assays (Supplementary Fig S3C) as well as in vitro in human cell lines (Supplementary Fig. S5B), suggesting different hierarchical dependencies among E2A-PBX1 target genes.
The cross-species comparative studied using genetic inactivation by shRNAs and pharmacological inhibition by small molecules strongly suggests that ZAP70, SYK, LCK as well as PLCγ2 play a pathogenic role in E2A-PBX1 leukemogenesis. Our studies indicate that PLCγ2 is a key downstream protein, which mediates cell proliferation not only in E2A-PBX1+/preBCR+ but also in E2A-PBX1+/preBCR- and non-E2A-PBX1 ALLs. Although E2A-PBX1 leukemia cells have different levels of pPLCγ2, we did not correlate any characteristic with the activation of pPLCγ2. Particularly, we did not find any difference regarding PAX5 status, mouse Cre line employed or preBCR expression. We hypothesize that preBCR- ALL might activate alternative pathways involved in proliferation and survival. Indeed, we described the acquisition of mutations leading to increased activation of the JAK/STAT and RAS/MAPK pathway in previous studies (5). Although ZAP70, LCK and SYK are expressed in E2A-PBX1+/preBCR- leukemias, they maintain pPLCγ2 levels through alternative pathways that might result in resistance to the inhibitors as shown for human cell lines (Fig 5A).
Our data suggest that the presence of the preBCR increases the dependence on the enzymatic activity of ZAP70, LCK and SYK in leukemia cells without increasing substantially pPLCγ2 levels. We elucidated pathways upstream of PLCγ2 that are activated in E2A-PBX1+/preBCR+ leukemias and confer susceptibility to small molecule inhibitors. Future studies are necessary to characterize activated pathways upstream of PLCγ2 in E2A-PBX1+/preBCR- as well as in B-ALLs driven by different oncogenes, which might reveal novel pathways for targeted therapies. Alternative pathways may be involved in PLCγ2 activation including receptor tyrosine kinases (42), Tec family kinases as BTK (43) and PI3 kinases (44).
In previous studies, PLCγ2 and ZAP70 were described as predictive markers for dasatinib response in CLL(45,46) and PLCγ2 in DLBCL (47). Our data suggest that PLCγ2 may be used as a predictive marker for dasatinib as well as SYK- and LCK-inhibitor responses in preBCR+ ALL, although prospective studies in primary ALLs are needed. ZAP70 shows promise as a possible therapeutic target in several hematological malignancies, including CLL and preBCR+ ALL, however ZAP70 inhibitors are currently not available. We suggest that the development of ZAP70 inhibitors will have an impact in preBCR+ ALL treatment, as single and also combination therapy.
In summary, signaling network remodeling by E2A-PBX1 induces aberrant phosphorylation of PLCγ2, consistent with activation of upstream signaling pathways. The E2A-PBX1 target genes ZAP70, SYK and LCK encode kinases upstream of PLCγ2 and play an oncogenic role in pre-B-ALL. Small molecules including dasatinib, as well as SYK and LCK inhibitors target signaling pathways upstream of PLCγ2 and show promising activity in vitro and in vivo for the treatment of preBCR+ ALLs.
Supplementary Material
Acknowledgments
We thank Maria Ambrus and Cita Nicolas for technical assistance, members of the Cleary laboratory for helpful discussions and Carlos Duque-Afonso for graphic design.
Financial support: This work was supported in part by grants from William Lawrence and Blanche Hughes Foundation (M.L. Cleary), the German Research Foundation (Deutsche Forschungsgemeinschaft, ref. DU 1287/2-1) (J. Duque-Afonso), the Lucile Packard Foundation for Children's Health, the Child Health Research Institute and the Stanford NIH-NCATS-CTSA grant #UL1 TR001085 (M.L. Cleary, J. Duque-Afonso, C.-H. Lin, J. Feng), Alex's Lemonade Stand Foundation for Childhood Cancer (S. H.-K. Wong) and Dr. Mildred Scheel Fellowship of the German Cancer Aid (C. Schneidawind). K. Han was supported by The Walter V. and Idun Berry Postdoctoral Fellowship Program and M.C. Bassik was supported by a grant from Stanford ChEM-H and an NIH Directors New Innovator Award (1DP2HD08406901).
Footnotes
Conflict of Interest Disclosure: The authors declare no competing financial interests.
References
- 1.Geng H, Hurtz C, Lenz KB, Chen Z, Baumjohann D, Thompson S, et al. Self-enforcing feedback activation between BCL6 and pre-B cell receptor signaling defines a distinct subtype of acute lymphoblastic leukemia. Cancer Cell. 2015;27(3):409–25. doi: 10.1016/j.ccell.2015.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Köhrer S, Havranek O, Seyfried F, Hurtz C, Coffey GP, Kim E, et al. PreBCR signaling in precursor B-cell acute lymphoblastic leukemia regulates PI3K/AKT, FOXO1 and MYC, and can be targeted by SYK inhibition. Leukemia. 2016 doi: 10.1038/leu.2016.9. (in press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pui CH, Sandlund JT, Pei D, Campana D, Rivera GK, Ribeiro RC, et al. Improved outcome for children with acute lymphoblastic leukemia: results of Total Therapy Study XIIIB at St Jude Children's Research Hospital. Blood. 2004;104(9):2690–6. doi: 10.1182/blood-2004-04-1616. [DOI] [PubMed] [Google Scholar]
- 4.Moorman AV, Harrison CJ, Buck GA, Richards SM, Secker-Walker LM, Martineau M, et al. Karyotype is an independent prognostic factor in adult acute lymphoblastic leukemia (ALL): analysis of cytogenetic data from patients treated on the Medical Research Council (MRC) UKALLXII/Eastern Cooperative Oncology Group (ECOG) 2993 trial. Blood. 2007;109(8):3189–97. doi: 10.1182/blood-2006-10-051912. [DOI] [PubMed] [Google Scholar]
- 5.Duque-Afonso J, Feng J, Scherer F, Lin CH, Wong SH, Wang Z, et al. Comparative genomics reveals multistep pathogenesis of E2A-PBX1 acute lymphoblastic leukemia. J Clin Invest. 2015;125(9):3667–80. doi: 10.1172/JCI81158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rickert RC, Roes J, Rajewsky K. B lymphocyte-specific, Cre-mediated mutagenesis in mice. Nucleic Acids Res. 1997;25(6):1317–8. doi: 10.1093/nar/25.6.1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hobeika E, Thiemann S, Storch B, Jumaa H, Nielsen PJ, Pelanda R, et al. Testing gene function early in the B cell lineage in mb1-cre mice. Proc Natl Acad Sci U S A. 2006;103(37):13789–94. doi: 10.1073/pnas.0605944103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kühn R, Schwenk F, Aguet M, Rajewsky K. Inducible gene targeting in mice. Science. 1995;269(5229):1427–1429. doi: 10.1126/science.7660125. [DOI] [PubMed] [Google Scholar]
- 9.Krutzik PO, Nolan GP. Intracellular phospho-protein staining techniques for flow cytometry: monitoring single cell signaling events. Cytometry A. 2003;55(2):61–70. doi: 10.1002/cyto.a.10072. [DOI] [PubMed] [Google Scholar]
- 10.Kuo HP, Wang Z, Lee DF, Iwasaki M, Duque-Afonso J, Wong SH, et al. Epigenetic roles of MLL oncoproteins are dependent on NF-κB. Cancer Cell. 2013;24(4):423–37. doi: 10.1016/j.ccr.2013.08.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Coffey G, DeGuzman F, Inagaki M, Pak Y, Delaney SM, Ives D, et al. Specific inhibition of spleen tyrosine kinase suppresses leukocyte immune function and inflammation in animal models of rheumatoid arthritis. J Pharmacol Exp Ther. 2012;340(2):350–9. doi: 10.1124/jpet.111.188441. [DOI] [PubMed] [Google Scholar]
- 12.Stachlewitz RF, Hart MA, Bettencourt B, Kebede T, Schwartz A, Ratnofsky SE, et al. A-770041, a novel and selective small-molecule inhibitor of Lck, prevents heart allograft rejection. J Pharmacol Exp Ther. 2005;315(1):36–41. doi: 10.1124/jpet.105.089169. [DOI] [PubMed] [Google Scholar]
- 13.Bassik MC, Kampmann M, Lebbink RJ, Wang S, Hein MY, Poser I, et al. A systematic mammalian genetic interaction map reveals pathways underlying ricin susceptibility. Cell. 2013;152(4):909–22. doi: 10.1016/j.cell.2013.01.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Matheny CJ, Wei MC, Bassik MC, Donnelly AJ, Kampmann M, Iwasaki M, et al. Next-generation NAMPT inhibitors identified by sequential high-throughput phenotypic chemical and functional genomic screens. Chem Biol. 2013;20(11):1352–63. doi: 10.1016/j.chembiol.2013.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wong SH, Goode DL, Iwasaki M, Wei MC, Kuo HP, Zhu L, et al. The H3K4-Methyl epigenome regulates leukemia stem cell oncogenic potential. Cancer Cell. 2015;28(2):198–209. doi: 10.1016/j.ccell.2015.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yokoyama A, Somervaille TC, Smith KS, Rozenblatt-Rosen O, Meyerson M, Cleary ML. The menin tumor suppressor protein is an essential oncogenic cofactor for MLL-associated leukemogenesis. Cell. 2005;123(2):207–18. doi: 10.1016/j.cell.2005.09.025. [DOI] [PubMed] [Google Scholar]
- 17.The ENCODE Project Consortium. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489(7414):57–74. doi: 10.1038/nature11247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nat Methods. 2012;9(7):671–675. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bliss CI. The toxicity of poisons applied jointly. Ann Appl Biol. 1939;26:585–615. [Google Scholar]
- 20.Hempel WM, Schatzman RC, DeFranco AL. Tyrosine phosphorylation of phospholipase C-gamma 2 upon cross-linking of membrane Ig on murine B lymphocytes. J Immunol. 1992;148(10):3021–7. [PubMed] [Google Scholar]
- 21.Coustan-Smith E, Song G, Clark C, Key L, Liu P, Mehrpooya M, et al. New markers for minimal residual disease detection in acute lymphoblastic leukemia. Blood. 2011;117(23):6267–76. doi: 10.1182/blood-2010-12-324004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chiaretti S, Guarini A, De Propris MS, Tavolaro S, Intoppa S, Vitale A, et al. ZAP-70 expression in acute lymphoblastic leukemia: association with the E2A/PBX1 rearrangement and the pre-B stage of differentiation and prognostic implications. Blood. 2006;107(1):197–204. doi: 10.1182/blood-2005-04-1755. [DOI] [PubMed] [Google Scholar]
- 23.Haferlach T, Kohlmann A, Wieczorek L, Basso G, Kronnie GT, Béné MC, et al. Clinical utility of microarray-based gene expression profiling in the diagnosis and subclassification of leukemia: report from the International Microarray Innovations in Leukemia Study Group. J Clin Oncol. 2010;28(15):2529–37. doi: 10.1200/JCO.2009.23.4732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chen L, Widhopf G, Huynh L, Rassenti L, Rai KR, Weiss A, et al. Expression of ZAP-70 is associated with increased B-cell receptor signaling in chronic lymphocytic leukemia. Blood. 2002;100(13):4609–14. doi: 10.1182/blood-2002-06-1683. [DOI] [PubMed] [Google Scholar]
- 25.Chen L, Apgar J, Huynh L, Dicker F, Giago-McGahan T, Rassenti L, et al. ZAP-70 directly enhances IgM signaling in chronic lymphocytic leukemia. Blood. 2005;105(5):2036–41. doi: 10.1182/blood-2004-05-1715. [DOI] [PubMed] [Google Scholar]
- 26.Crespo M, Bosch F, Villamor N, Bellosillo B, Colomer D, Rozman M, et al. ZAP-70 expression as a surrogate for immunoglobulin-variable-region mutations in chronic lymphocytic leukemia. N Engl J Med. 2003;348(18):1764–75. doi: 10.1056/NEJMoa023143. [DOI] [PubMed] [Google Scholar]
- 27.Rassenti LZ, Huynh L, Toy TL, Chen L, Keating MJ, Gribben JG, et al. ZAP-70 compared with immunoglobulin heavy-chain gene mutation status as a predictor of disease progression in chronic lymphocytic leukemia. N Engl J Med. 2004;351(9):893–901. doi: 10.1056/NEJMoa040857. 26. [DOI] [PubMed] [Google Scholar]
- 28.Tycko B, Smith SD, Sklar J. Chromosomal translocations joining LCK and TCRB loci in human T cell leukemia. J Exp Med. 1991;174(4):867–73. doi: 10.1084/jem.174.4.867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Burnett RC, David JC, Harden AM, Le Beau MM, Rowley JD, Diaz MO. The LCK gene is involved in the t(1;7)(p34;q34) in the T-cell acute lymphoblastic leukemia derived cell line, HSB-2. Genes Chromosomes Cancer. 1991;3(6):461–7. doi: 10.1002/gcc.2870030608. [DOI] [PubMed] [Google Scholar]
- 30.Wright DD, Sefton BM, Kamps MP. Oncogenic activation of the Lck protein accompanies translocation of the LCK gene in the human HSB2 T-cell leukemia. Mol Cell Biol. 1994;14(4):2429–37. doi: 10.1128/mcb.14.4.2429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Feldman AL, Sun DX, Law ME, Novak AJ, Attygalle AD, Thorland EC, et al. Overexpression of Syk tyrosine kinase in peripheral T-cell lymphomas. Leukemia. 2008;22(6):1139–43. doi: 10.1038/leu.2008.77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Streubel B, Vinatzer U, Willheim M, Raderer M, Chott A. Novel t(5;9)(q33;q22) fuses ITK to SYK in unspecified peripheral T-cell lymphoma. Leukemia. 2006;20(2):313–8. doi: 10.1038/sj.leu.2404045. [DOI] [PubMed] [Google Scholar]
- 33.Dierks C, Adrian F, Fisch P, Ma H, Maurer H, Herchenbach D, et al. The ITK-SYK fusion oncogene induces a T-cell lymphoproliferative disease in mice mimicking human disease. Cancer Res. 2010;70(15):6193–204. doi: 10.1158/0008-5472.CAN-08-3719. [DOI] [PubMed] [Google Scholar]
- 34.Kuno Y, Abe A, Emi N, Iida M, Yokozawa T, Towatari M, et al. Constitutive kinase activation of the TEL-Syk fusion gene in myelodysplastic syndrome with t(9;12)(q22;p12) Blood. 2001;97(4):1050–5. doi: 10.1182/blood.v97.4.1050. [DOI] [PubMed] [Google Scholar]
- 35.Brave M, Goodman V, Kaminskas E, Farrell A, Timmer W, Pope S, et al. Sprycel for chronic myeloid leukemia and Philadelphia chromosome-positive acute lymphoblastic leukemia resistant to or intolerant of imatinib mesylate. Clin Cancer Res. 2008;14(2):352–9. doi: 10.1158/1078-0432.CCR-07-4175. [DOI] [PubMed] [Google Scholar]
- 36.Roberts KG, Li Y, Payne-Turner D, Harvey RC, Yang YL, Pei D, et al. Targetable kinase-activating lesions in Ph-like acute lymphoblastic leukemia. N Engl J Med. 2014;371(11):1005–15. doi: 10.1056/NEJMoa1403088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fischer U, Forster M, Rinaldi A, Risch T, Sungalee S, Warnatz HJ, et al. Genomics and drug profiling of fatal TCF3-HLF-positive acute lymphoblastic leukemia identifies recurrent mutation patterns and therapeutic options. Nat Genet. 2015;47(9):1020–9. doi: 10.1038/ng.3362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bicocca VT, Chang BH, Masouleh BK, Muschen M, Loriaux MM, Druker BJ, et al. Crosstalk between ROR1 and the Pre-B cell receptor promotes survival of t(1;19) acute lymphoblastic leukemia. Cancer Cell. 2012;22(5):656–67. doi: 10.1016/j.ccr.2012.08.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lee KC, Ouwehand I, Giannini AL, Thomas NS, Dibb NJ, Bijlmakers MJ. Lck is a key target of imatinib and dasatinib in T-cell activation. Leukemia. 2010;24(4):896–900. doi: 10.1038/leu.2010.11. [DOI] [PubMed] [Google Scholar]
- 40.Braselmann S, Taylor V, Zhao H, Wang S, Sylvain C, Baluom M, et al. R406, an orally available spleen tyrosine kinase inhibitor blocks fc receptor signaling and reduces immune complex-mediated inflammation. J Pharmacol Exp Ther. 2006;319(3):998–1008. doi: 10.1124/jpet.106.109058. [DOI] [PubMed] [Google Scholar]
- 41.Davis RE, Ngo VN, Lenz G, Tolar P, Young RM, Romesser PB, et al. Chronic active B-cell-receptor signalling in diffuse large B-cell lymphoma. Nature. 2010;463(7277):88–92. doi: 10.1038/nature08638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Montoye T, Lemmens I, Catteeuw D, Eyckerman S, Tavernier J. A systematic scan of interactions with tyrosine motifs in the erythropoietin receptor using a mammalian 2-hybrid approach. Blood. 2005;105(11):4264–71. doi: 10.1182/blood-2004-07-2733. [DOI] [PubMed] [Google Scholar]
- 43.Takata M, Kurosaki T. A role for Bruton's tyrosine kinase in B cell antigen receptor-mediated activation of phospholipase C-gamma 2. J Exp Med. 1996;184(1):31–40. doi: 10.1084/jem.184.1.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Boudot C, Kadri Z, Petitfrère E, Lambert E, Chrétien S, Mayeux P, et al. Phosphatidylinositol 3-kinase regulates glycosylphosphatidylinositol hydrolysis through PLC-gamma(2) activation in erythropoietin-stimulated cells. Cell Signal. 2002;14(10):869–78. doi: 10.1016/s0898-6568(02)00036-0. [DOI] [PubMed] [Google Scholar]
- 45.Veldurthy A, Patz M, Hagist S, Pallasch CP, Wendtner CM, Hallek M, et al. The kinase inhibitor dasatinib induces apoptosis in chronic lymphocytic leukemia cells in vitro with preference for a subgroup of patients with unmutated IgVH genes. Blood. 2008;112(4):1443–52. doi: 10.1182/blood-2007-11-123984. [DOI] [PubMed] [Google Scholar]
- 46.Song Z, Lu P, Furman RR, Leonard JP, Martin P, Tyrell L, et al. Activities of SYK and PLCgamma2 predict apoptotic response of CLL cells to SRC tyrosine kinase inhibitor dasatinib. Clin Cancer Res. 2010;16(2):587–99. doi: 10.1158/1078-0432.CCR-09-1519. [DOI] [PubMed] [Google Scholar]
- 47.Yang C, Lu P, Lee FY, Chadburn A, Barrientos JC, Leonard JP, et al. Tyrosine kinase inhibition in diffuse large B-cell lymphoma: molecular basis for antitumor activity and drug resistance of dasatinib. Leukemia. 2008;22(9):1755–66. doi: 10.1038/leu.2008.163. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.