

Abstract
Here we report an efficient CRISPR-Cas9 knock-in strategy to activate silent biosynthetic gene clusters (BGCs) in streptomycetes. We applied this one-step strategy to activate multiple BGCs of different classes in five Streptomyces species and triggered the production of unique metabolites, including a novel pentangular type II polyketide in Streptomyces viridochromogenes. This potentially scalable strategy complements existing activation approaches and facilitates discovery efforts to uncover new compounds with interesting bioactivities.
Graphical abstract
Microbial natural products are a rich source of pharmaceutical agents and current advances in genomics have unveiled a vast source of potential unexplored BGCs. Because majority of encoded metabolites of these BGCs are undetectable using current analytical methods due to minimal or zero BGC expression under laboratory conditions (such BGCs are commonly defined as silent BGCs1), strategies to activate BGC expression and trigger metabolite production are critical to realize the full potential of Nature’s chemical répertoire.1 While heterologous expression bypass native regulation networks and can be engineered rationally,2 entire biosynthetic pathways often spanning large areas of genomes will have to be cloned and refactored.3,4 Additionally, heterologous hosts may lack regulatory, enzymatic or metabolic requirements necessary for product biosynthesis. Inducing cluster expression in native hosts circumvents these limitations but may be hindered by low homologous recombination efficiencies. Technologies that improve genetic manipulation of streptomycetes will expedite discovery and characterization of BGCs in their native contexts as well as guide the improvement of heterologous systems.5
CRISPR technology has enabled the genetic manipulation of many genetically recalcitrant organisms.6,7 The Streptococcus pyogenes CRISPR-Cas9 system was recently reconstituted in model streptomycetes to delete genes and entire BGCs as well as perform site-directed mutagenesis and gene replacement at significantly improved efficiencies.8–11 Here we extend this CRISPR-Cas9 technology to perform strategic promoter knock-in for the activation of silent BGCs in native Streptomyces hosts (Fig. 1a).
Figure 1. CRISPR-Cas9 based promoter knock-in strategy to activate silent biosynthetic gene clusters in streptomycetes.

(a) Using CRISPR-Cas9, efficient and precise introduction of promoter cassettes (red arrows) drive expression of biosynthetic genes (blue) and trigger the production of unique metabolites (*) that are not detected for the parent strain. (b) Knock-in efficiencies with and without use of CRISPR-Cas9 in different Streptomyces species, namely 1) S. albus, 2) S. lividans, 3) S. roseosporus, and 4) S. venezuelae. See Supplementary Table 1 for actual values. For S. albus, the knock-in efficiencies for different sized inserts (100 bp vs 1 kb) using editing templates with different homology lengths (1 kb vs 2 kb) were examined. No knock-in was observed for S. roseosporus without CRISPR-Cas9. n.d., not determined. (c) Wild type (wt) or indicated engineered S. albus strains on MGY or MGY+thiostrepton plates. (d) Wild type and engineered S. lividans strains with activated RED (left panel) or ACT (right panel) clusters on MGY plates. Ammonia fuming confirmed production of pH-sensitive actinorhodin-related pigments (Supplementary Fig. 4).
To demonstrate that CRISPR-Cas9 can be used to efficiently and precisely introduce heterologous promoters into Streptomyces genomes for BGC activation, we selected well-characterized pigment BGCs, namely the indigoidine cluster in Streptomyces albus,12 as well as the actinorhodin (ACT) and undecylprodigiosin (RED) clusters in Streptomyces lividans.13,14 Using CRISPR-Cas9 mediated knock-in, we replaced upstream promoter regions of main biosynthetic operons or pathway-specific activators with constitutive promoters that are stronger than the commonly used ermE* promoter and work in multiple Streptomyces species (Supplementary Results, Supplementary Fig. 1).15,16 In S. albus, CRISPR-Cas9 increased knock-in efficiency of the kasO* promoter upstream of the indC-like indigoidine synthase gene compared to without CRISPR-Cas9 (Fig. 1b). Higher knock-in efficiency observed with 2 kb homologous arms as compared to 1 kb arms is consistent with homology-directed repair of Cas9-induced double stranded breaks. Co-introduction of longer inserts such as the ~1 kb thiostrepton-resistance cassette (tsr) with kasO*p was achieved at lower efficiencies but it was still higher than that of inserting kasO*p alone without CRISPR-Cas9. Selected solely on apramycin, tsr-kasO*p knock-in strains grew on thiostrepton plates while maintaining pigment production as expected of an activated indigoidine synthase cluster (Fig. 1c, Supplementary Fig. 2). Similar to S. albus, recovery of desired kasO*p knock-in strains in S. lividans was greatly enhanced with the use of CRISPR-Cas9 (Fig. 1b). Confirming successful activation of the RED and ACT clusters, the engineered strains produced red undecylprodigiosin and pH-responsive actinorhodin-related metabolites respectively (Fig. 1d, Supplementary Fig. 3–5).
Together, these results demonstrated that CRISPR-Cas9 can be used to precisely introduce heterologous genetic elements into Streptomyces genomes at relatively high efficiencies for secondary metabolite production from silent BGCs. The enhanced knock-in efficiencies allowed use of donor DNA with shorter homology flanks as well as the introduction of larger genetic elements, both of which will be challenging without CRISPR-Cas9. While homologous recombination occurs efficiently in model strains like S. lividans and S. albus without CRISPR-Cas9, for other strains like Streptomyces roseosporus, the increase in efficiency afforded by CRISPR-Cas9 is critical and allows genetic manipulation of otherwise challenging strains (Fig. 1b).
Next, we employed this strategy to activate two silent unexplored BGCs from S. roseosporus with relatively high homology to known BGCs. The S. roseosporus NRRL15998 genome contains 29 predicted BGCs,17 the majority of which are yet to be characterized (Supplementary Table 2).18 One of the predicted BGCs showed >90% sequence identity to the polycyclic tetramate macrolactam (PTM) cluster in Streptomyces griseus (Supplementary Table 3), which was refactored for expression in S. lividans by introducing individual promoters in front of each of the six genes.19 Notably, insertion of a single strong promoter failed to drive cluster expression in the heterologous system.19 Here in the native S. roseosporus host, knock-in of kasO*p upstream of the first open reading frame (ORF) was sufficient to drive expression of PTM biosynthetic genes (Supplementary Fig. 6) and yielded the production of photocyclized alteramide A (1, m/z 511.2808, [M+H]+) and a second PTM 2 (m/z 513.2961, [M+H]+) with the same planar structure as dihydromaltophilin (Fig. 2a, Supplementary Note). Since alteramide A photocyclization is spontaneous,20 we surmised that alteramide A was the original metabolite produced by S. roseosporus. Interestingly, 2 was not identified in the previous study involving the almost identical S. griseus cluster,19 suggesting possible host-dependent factors or differences between native and heterologous hosts.
Figure 2. Activation of biosynthetic gene clusters in multiple streptomycetes.
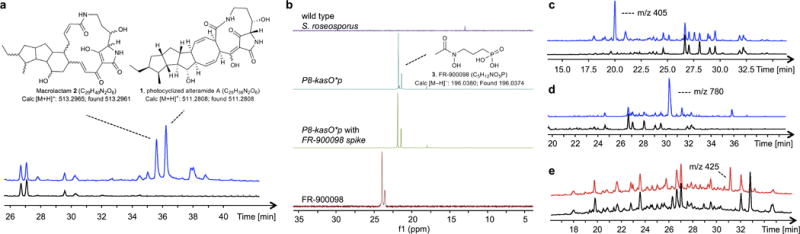
(a) HPLC analysis of ethyl acetate extracts from wild type S. roseosporus and an engineered strain in which kasO*p was introduced into cluster 24. Indicated are the two major PTM compounds that are isolated from the engineered strain. Stereochemistry was not assigned for 2. See Supplementary Fig. 7 and Supplementary Note for full chromatograms and chemical characterization data. (b) 31P-NMR spectra of methanol extracts from wild type S. roseosporus and an engineered strain in which a bidirectional P8-kasO*p cassette was introduced into the phosphonate cluster 10. Also shown are the 31P-NMR spectra of authentic FR-900098 sample and FR-900098 spiked into the extract of the P8-kasO*p strain. HRMS and HMBC analyses confirmed FR-900098 production (Supplementary Note). Difference in chemical shifts in authentic and spiked FR-900098 is due to difference in sample pH.22 (c–e) LC-MS analyses of culture extracts from wild type (black) and engineered S. roseosporus (blue), S. venezuelae (red) strains in which kasO*p was introduced into the respective clusters. Major metabolites uniquely produced by the engineered strains and not observed for the wild type strains are highlighted with their indicated m/z values. We note that there are additional differences between the metabolic profiles of the engineered and the respective wildtype strains. For details, full chromatograms and mass spectra, see Supplementary Fig. 12–14.
S. roseosporus also possesses a phosphonate BGC with genes showing high homology and synteny to the Streptomyces rubellomurinus FR-900098 BGC (Supplementary Table 4).21,22 Intriguingly, BLASTP search within ~2000 NCBI-deposited actinobacteria assemblies for FR-900098 biosynthetic enzymes did not uncover similar BGCs (accessed in May 2016), suggesting that S. roseosporus has the uncommon biosynthetic potential to synthesize the antimalarial compound, which to date has been attributed to S. rubellomurinus and Streptomyces lavendulae.21 To determine if S. roseosporus can produce FR-900098, we introduced a bidirectional P8-kasO*p promoter cassette to drive expression of the putative frbD operon and frbC homolog (Supplementary Fig. 8, 9). The engineered strain produced 3 with 31P-NMR, HMBC and mass values consistent with FR-900098 (Fig. 2b, Supplementary Note), validating the inherent ability of S. roseosporus to make FR-900098. The estimated FR-900098 titer of 6–10 mg/L in the engineered strain, while lower than the 22.5 mg/L reported for S. rubellomurinus,21 is ~1000-fold higher than the minimum inhibitory concentration against the malarial parasite,23 suggesting that this activation strategy may be applied for bioactivity-guided discovery.
We next asked whether this activation strategy can be generally applied to uncharacterized BGCs of different classes in multiple Streptomyces species, namely S. roseosporus (Supplementary Table 2), Streptomyces venezuelae (Supplementary Table 5) and Streptomyces viridochromogenes (Supplementary Table 6). For pathway-specific activation, we targeted single and bidirectional promoter cassettes to the first ORF(s) of the main biosynthetic operon(s) predicted based on gene directionality alone or if available, predicted transcriptional activators (Supplementary Fig. 10, 11). Introduction of single or bidirectional promoter cassettes into additional clusters in S. roseosporus and S. venezuelae yielded production of unique compounds that were not observed for the parent strains (Fig. 2c–e). For example, cluster 3 in S. roseosporus was predicted to be a nucleoside-type I PKS with biosynthetic enzymes for incorporation of a 3-amino-5-hydroxybenzoic acid starter unit and naphthalene ring formation. Insertion of kasO*p upstream of the main synthase gene encoding a loading domain and three PKS modules triggered the production of a major metabolite with m/z 405 (Fig. 2c). A distinct compound with m/z 780 was observed for another engineered S. roseosporus strain, in which kasO*p was introduced upstream of a predicted LuxR-type regulator within a type I PKS cluster (Fig. 2d). In S. venezuelae, insertion of a bidirectional promoter cassette between a type III PKS gene encoding an RppA synthase and a cytochrome P450 gene resulted in production of pigmented products (Fig. 2e). Production of these newly observed metabolites was independently validated at least three times in solid and liquid MGY media to be unique to the respective knock-in strains and detected in the parent wild type strains.
Identification of a novel compound using CRISPR-Cas9 based promoter knock-in further demonstrates the potential of this strategy for natural product discovery. We isolated and characterized the major product selectively produced by an engineered S. viridochromogenes strain, in which kasO*p was inserted in front of the main biosynthetic operon SSQG_RS26895-26920 of an uncharacterized type II PKS gene cluster NZ_GG657757 (Supplementary Fig. 15). Except for an additional cytochrome P450, NZ_GG657757 has high homology and similar gene arrangement as a spore pigment BGC in Streptomyces avermitilis (Accession number: AB070937.1). The engineered S. viridochromogenes strain produced an obvious brown pigment in liquid and solid medium before sporulation with a major unique metabolite 4 observed by HPLC (Fig. 3a, b). HRMS of the 4 predicted a molecular formula C23H16O8. 1H NMR, 13C NMR, COSY/TOCSY, HSQC and HMBC analyses of 4 revealed a novel polyketide with a dihydrobenzo[α]naphthacenequinone core that is shared by a family of polyketides including frankiamycin, benastatin and pradimicin (Fig. 3c, Supplementary Note).24 The cyclohexanone (ring E) in 4 is atypical and has not been observed for pentangular aromatic polyketides.25 Further mechanistic studies will elucidate the contributions of each enzyme in the biosynthesis of this pentangular polyketide.
Figure 3. Activation of type II PKS biosynthetic gene cluster in S. viridochromogenes yields a novel pigmented compound.

(a) Production of brown pigment by the engineered strain but not wild type (wt) S. viridochromogenes on MGY medium. (b) HPLC analysis of extracts from an engineered S. viridochromogenes strain harbouring a kasO*p knock-in in front of SSQG_RS26895 (red) and the parent wild type strain (black). Indicated is the major metabolite 4 that is uniquely produced by the engineered strain. Here we focus on the major distinct metabolite produced by the engineered strain but we note that there are additional differences between the engineered and wild type strain (Supplementary Fig. 15). (c) Structure of 4. The five rings are labelled A–E.
In this study, we showed that relatively small genome perturbations in the form of strategically introduced promoters using the CRISPR-Cas9 technology, are sufficient to activate BGCs of different classes in multiple Streptomyces species, including type I, II and III PKSs, NRPS, hybrid PKS-NRPS and phosphonate clusters. Our current efforts focus on BGCs with 1–2 major predicted biosynthetic operons and we do not expect this strategy to work for all BGCs, especially if global transcriptional changes are required or if there are multiple major operons within the target BGC. For activation of these BGCs, more complex engineering involving knock-in of multiple promoters, manipulation of pathway/global regulators or enzyme/domain swaps and mutations, all of which may also be performed using CRISPR-Cas9. An obvious limitation of this strategy is that it requires introduction of recombinant DNA, which may be challenging for some strains, especially natural isolates. Nonetheless, the activation of multiple clusters in different Streptomyces species highlights the potential of this approach to complement existing strategies, including heterologous expression, to discover, characterize and reengineer BGCs.1 This strategy should be generally applicable and potentially scalable to better explore the biosynthetic potential of streptomycetes.
ONLINE METHODS
Reagents and Media
Unless otherwise indicated, all reagents are obtained from Sigma. 1 L of MGY medium contains 10 g malt extract broth, 4 g Bacto yeast extract (BD Biosciences), 4 g glucose (1st Base, Axil Scientific) and for MGY agar plates, an additional 20 g of Bacto agar (BD Biosciences). Conjugation experiments involving WM6026 and WM3780 E. coli strains were performed on R2 agar without sucrose: 0.25 g K2SO4, 10.12 g MgCl2•6H2O, 10 g glucose, 0.1 g Bacto casamino acids (BD Biosciences), 5.73 g TES, 20 g agar in 1 L water, autoclaved, after which 1 mL filter-sterilized 50 mg/mL KH2PO4 solution and filter-sterilized 2.94 g CaCl2•2H2O and 3 g L-proline in 5 mL 1 N NaOH were added to the medium.
Strains and Growth conditions
Strains and plasmids used in this study are listed in Supplementary Table 7. Unless otherwise indicated, strains are propagated in MGY medium at 30 °C. Spore preparations and conjugation protocols were similar to those described by Keiser and Bibb.26 For spore preparations, 1:1000 of a spore preparation or 1:100 dilution of a saturated seed culture is plated on MGY plates and incubated at 30 °C until thick spores are observed. Spores were removed from the plate using 5 mm glass beads (Sigma) and resuspended in sterile TX buffer (50 mM Tris pH 7.4, 0.001% (v/v) Triton X) by vigorous vortexing for 30 s. The eluant containing free spores were pelleted by spinning at maximum speed in an Eppendorf 5810R centrifuge for 10 min, resuspended in 1 mL sterile water and repelleted. The spores were then resuspended in water and stored at –80 °C. A typical spore prep contains ~107–109 spores/mL as determined by serial dilution plating.
Construction of genome editing plasmids
All DNA manipulations were carried out in Escherichia coli DH5α or OmniMAX™ (Thermo Fisher). Primers used in this study are listed in Supplementary Table 8. Restriction enzymes were obtained from New England Biolabs. Helper pCRISPomyces-2 plasmids for making promoter knock-in constructs were made by ligating adapter sequences, containing restriction sites flanking the promoter of choice (Supplementary Fig. 16) to facilitate insertion of homology arms, at the XbaI site of pCRISPomyces-2.8 The protospacer of a target cluster was first inserted via BbsI-mediated Golden Gate Assembly as previously described.8 The helper plasmid (pCRISPomyces-2-kasO*p, pCRISPomyces-2-P8-kasO*p) was linearized using SpeI and assembled with the downstream homology arm (2 kb unless otherwise indicated) by Gibson assembly (New England Biolabs). The second upstream homology arm (2 kb unless otherwise indicated) was subsequently inserted by Gibson assembly using HindIII or NheI linearized construct containing the first homology arm. See Supplementary Fig. 17 for workflow to construct genome editing plasmids.
Interspecies conjugation
Promoter knock-in constructs were used to transform conjugating E. coli strains and colonies with the appropriate antibiotic resistance (e.g. 50 mg/L apramycin) were picked into LB with antibiotics. WM6026 requires diaminopimelic acid in LB for growth and it was added to LB for subsequent wash and resuspension steps. Overnight cultures were diluted 1:100 into fresh LB with antibiotics and grown to an OD600 of 0.4–0.6. 400 μL of the culture was pelleted, washed twice and resuspended in LB without antibiotics. The washed E. coli cells were then mixed with spores at 1:5 volume ratio and spotted on R2 without sucrose plates. After incubation for 16–20 h at 30 °C, the plates were flooded with nalidixic acid and apramycin and incubated until exconjugants appear. Exconjugants were streaked onto MGY plates containing apramycin at 30 °C followed by restreaking to MGY plates at 37 °C to cure the CRISPR-Cas9 plasmid containing a temperature-sensitive origin of replication. Apramycin-sensitive clones growing at 37 °C were then subjected to validation of promoter knock-in and genome editing as described below.
Validation of promoter knock-in and genome editing
Genomic DNA from wild type and exconjugants from the indicated strains were isolated from liquid cultures using the Blood and Tissue DNeasy kit (Qiagen) after pretreating the cells with 20 mg/mL lysozyme for 0.5–1 h at 30 °C. PCR was performed using control primers beyond the homology regions or knock-in specific primers (Supplementary Table 8) with KODXtreme Taq polymerase (Millipore). Where indicated, PCR products were subjected to digest with specific restriction enzymes to differentiate between PCR products of wild type genomic sequences and successful genome editing by knock-ins. Positive samples were purified using Qiaquick PCR purification kit (Qiagen) and validated by Sanger sequencing.
RNA isolation and Real-time quantitative PCR (RT-qPCR)
RNA from wild type and engineered S. roseosporus were isolated using RNAsy Midi Kit (Qiagen) 72 h after seed cultures were diluted 1:100 into 50 mL of MGY broth in 250 mL baffled flasks containing ~30–40 5 mm glass beads and growth at 30 °C. Isolated RNA was treated with DNase (Qiagen) before being reverse transcribed with random hexamers using SuperScript III (Invitrogen). RT-qPCR was performed on a Roche LightCycler 480 using SYBR FAST qPCR master mix (KAPA). The housekeeping rpsL gene of S. roseosporus was used as constitutive reference to normalize gene expression of each target gene such that rpsL expression = 1.27 Technical triplicates of three biological repeats were performed per condition. Gene-specific primers are listed in Supplementary Table 8.
Fermentation, ethyl acetate extraction and LC-MS analysis metabolites from wild type and engineered Streptomyces strains
Liquid seed cultures (2 mL MGY) of wild type and engineered S. roseosporus and S. venezuelae strains were inoculated from a plate or spore stock in 14 mL culture tubes. Seed cultures were incubated at 30 °C with 250 rpm shaking until achieving turbidity or high particle density (typically 2–3 days). Seed cultures were diluted 1:100 into 50 mL of MGY broth in 250 mL baffled flasks containing ~30–40 5 mm glass beads and incubated at 30 °C with 250 rpm shaking (10–14 days for S. roseosporus, 5–7 days for S. venezuelae). The cultures were harvested by pelleting at maximum speed in an Eppendorf 5810R centrifuge for 10 min. The cell pellet was stored at –80 °C while the supernatants were split into two 50 mL falcon tubes. Culture supernatants were extracted three times with equal volume ethyl acetate. For solid-state cultures, the strains were grown on MGY plates at 30 °C for 10 days. The plates were chopped into small pieces and extracted twice with ethyl acetate. Extracts were dried and resuspended in methanol, and analysed by LCMS using ESI source in positive ion mode (Bruker, Amazon SL Ion Trap) equipped with a Kinetex 2.6 μm XB-C18 100 Å (Phenomenex). HPLC parameters were as follows: solvent A, 0.1% trifluoroacetic acid in water; solvent B, 0.1% trifluoroacetic acid in acetonitrile; gradient at a constant flow rate of 0.2 mL/min, 10% B for 5 min, 10% to 100% B in 35 min, maintain at 100% B for 10 min, return to 10% B in 1 min and finally maintain at 10% B for 10 min; detection by ultraviolet spectroscopy at 210, 254, 280, 320 nm.
Extraction and NMR analysis of phosphonate compounds
Liquid seed cultures (2 mL MGY) of wild type and engineered S. roseosporus strains were inoculated from a plate or spore stock into 14 mL culture tubes. Seed cultures were incubated at 30 °C with 250 rpm shaking until achieving turbidity or high particle density (typically 2–3 days). Seed cultures were diluted 1:100 into 50 mL of MGY broth in 250 mL baffled flasks containing ~30–40 5 mm glass beads and incubated at 30 °C with 250 rpm shaking for 10–14 days. The cultures were harvested by pelleting at maximum speed in an Eppendorf 5810R centrifuge for 10 min. The cell pellet was stored at –80 °C while the supernatants were split into two 50 mL falcon tubes, flash frozen liquid nitrogen and lyophilized to dryness. 25 and 10 mL of methanol was added to each tube containing dried supernatant and frozen cell pellets respectively. The methanol mixtures were vortexed for 1 min each and incubated on a platform shaker at 4 °C for 2 h. Samples were clarified by spinning at maximum speed in an Eppendorf 5810R centrifuge for 10 min twice and pooling the methanol extracts from the respective pellets and lyophilized culture supernatants. A generous amount of anhydrous sodium sulfate was added to the extracts and stirred. The extracts were decanted, concentrated to dryness and resuspended in 700 μL deuterium oxide added in two 350 μL aliquots. A spatula-full of Chelex-100 resin (Bio-Rad) was added to each sample in a 1.7 mL centrifuge tube, which was incubated for 30 min at room temperature with agitation on a Thermo microplate shaker. The samples were clarified twice by centrifuging at maximum speed in an Eppendorf benchtop centrifuge for 1 min each time. The supernatants were then filtered using a 10 kDa Vivaspin column (GE Healthcare) and the filtrates were transferred to a 5 mm NMR tube for NMR analysis. 31P-NMR has been acquired using a Bruker DRX-600 spectrometer equipped with a 5mm BBFO cryoprobe. Proton decoupled 31P-NMR spectra are referenced to an external H3PO4 (aq) standard (δ 0.0 ppm). All samples have been acquired for 6000 scans. Identity of FR-900098 was confirmed by 1) spiking with the sample with authentic FR-900098, 2) 31P HMBC data comparison; 3) HRMS data. Production titers were estimated by spiking in known amounts of FR-900098.
Isolation and NMR analysis of PTM compounds
The crude extract was fractionated using silica gel flash chromatography and generated 11 fractions: F1 (5% ethyl acetate with 95% hexanes), F2 (15% ethyl acetate with 85% hexanes), F3 (20% ethyl acetate with 80% hexanes), F4 (30% ethyl acetate with 70% hexanes), F5 (40% ethyl acetate with 60% hexanes), F6 (50% ethyl acetate with 50% hexanes), F7 (60% ethyl acetate with 40% hexanes), F8 (80% ethyl acetate with 20% hexanes), F9 (100% ethyl acetate), F10 (100% acetone), F11 (100% methanol). PTM was eluted in F11 according to LCMS analysis. F11 was subjected to semi-prep HPLC using a C18 column (Phenomenex, 250 × 10 mm) with the following gradient: 5~40 min 5%~20% acetonitrile in water with 0.1% formic acid; 40~60 min 20%~50% acetonitrile in water with 0.1% formic acid; 60~70 min 50%~60% acetonitrile in water with 0.1% formic acid. 2 was eluted at 62 min. 1 was eluted at 61 min. NMR analysis was performed on an Agilent 600 MHz NMR spectrometer.
Isolation and NMR analysis of type II polyketide from S. viridochromogenes
Large-scale cultivation on solid plates (equivalent to 5 L liquid culture) of the knock-in strain was carried out to obtain sufficient amounts of potential new compound. 10-day growth solid plates were soaked in equal volume ethyl acetate overnight. The extract was fractionated using C18 flash column chromatography and the fraction containing the target compound was further subjected to silica gel flash column chromatography. The column elution was monitored by TLC and the fractions containing the target compound were further confirmed by HPLC. NMR analysis was performed on an Agilent 600 MHz NMR spectrometer.
Fermentation, extraction and LC-MS analysis of RED, ACT and indigoidine from wild type and engineered Streptomyces strains
Liquid seed cultures (2 mL MGY) of wild type and engineered S. lividans and S. albus strains were inoculated from a plate or spore stock in 14 mL culture tubes. Seed cultures were incubated at 30 °C with 250 rpm shaking until achieving turbidity or high particle density (typically 1–2 days). For S. lividans, seed cultures were diluted 1:100 and plated onto MGY plates and grown at 30 °C for 3–4 days. The plates were chopped into small pieces and extracted with methanol (RED) or acidified methanol (ACT). For S. albus, seed cultures were diluted 1:100 into 50 mL of MGY broth in 250 mL baffled flasks and grown at 25 °C with 250 rpm shaking for 2–3 days. Culture supernatants of wild type and engineered S. albus strains were extracted twice with equal volume ethyl acetate containing 1% (v/v) formic acid. Extracts were dried and resuspended in methanol prior to analysis by LCMS using ESI source in positive ion mode (Bruker, Amazon SL Ion Trap) equipped with a Kinetex 2.6 μm XB-C18 100 Å (Phenomenex). HPLC parameters were as follows: solvent A, 0.1% trifluoroacetic acid in water; solvent B, 0.1% trifluoroacetic acid in acetonitrile; gradient at a constant flow rate of 1 mL/min, 5% B for 2 min, 5% to 100% B in 15 min, maintain at 100% B for 2 min, return to 5% B and maintain for 2 min; detection by ultraviolet spectroscopy at 500 nm (RED, ACT) or 600 nm (indigoidine). MS/MS was performed in positive auto MS(n) mode with scan range m/z 100–1000.
Supplementary Material
Acknowledgments
We gratefully acknowledge financial support from the National Institutes of Health (GM077596) (H.Z.), the A*STAR Visiting Investigator Program (H.Z.), and the National Research Foundation, Singapore (NRF2013-THE001-094) (M.M.Z., F.T.W., Y.H.L., E.L.A. and H.Z.). NMR data collection at the UIUC IGB Core was funded by NIH (S10-RR028833). Conjugative donor strains of E. coli were gifted by Prof. William Metcalf at UIUC. We thank members of the Zhao laboratory in UIUC, coworkers in MEL and MERL in A*STAR for constructive comments, Dr. Xudong Guan and Dr. Lingyang Zhu from UIUC for assisting with NMR data acquisition, and Dr. Ying Swan Ho from BTI, A*STAR for HRMS data acquisition.
Footnotes
Data Availability
The authors declare that the data supporting the findings of this study are available within the article (and its Supplementary Information files).
AUTHOR CONTRIBUTIONS
M.M.Z., F.T.W. and H.Z. conceived and designed the research. M.M.Z., F.T.W., Y.W., E.H., W.L.Y., R.E.C., and B.E. performed the molecular biology, conjugation and fermentation experiments. S.L. and Y.H.L performed structure elucidation of compounds. M.M.Z., F.T.W., E.L.A. and H.Z. analyzed the data. M.M.Z., F.T.W. and H.Z. wrote the manuscript.
COMPETING FINANCIAL INTERESTS
The authors declare no competing financial interests.
ACCESSION CODES
Referenced Accession code: AB070937.1
References
- 1.Rutledge PJ, Challis GL. Discovery of microbial natural products by activation of silent biosynthetic gene clusters. Nat Rev Microbiol. 2015;13:509–23. doi: 10.1038/nrmicro3496. [DOI] [PubMed] [Google Scholar]
- 2.Luo Y, Cobb RE, Zhao H. Recent advances in natural product discovery. Curr Opin Biotechnol. 2014;30:230–7. doi: 10.1016/j.copbio.2014.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kang HS, Charlop-Powers Z, Brady SF. Multiplexed CRISPR/Cas9- and TAR-mediated promoter engineering of natural product biosynthetic gene clusters in yeast. ACS Synth Biol. 2016;5:1002–10. doi: 10.1021/acssynbio.6b00080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cobb RE, Zhao H. Direct cloning of large genomic sequences. Nat Biotechnol. 2012;30:405–6. doi: 10.1038/nbt.2207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhang MM, Wang Y, Ang EL, Zhao H. Engineering microbial hosts for production of bacterial natural products. Nat Prod Rep. 2016;33:963–87. doi: 10.1039/c6np00017g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Doudna JA, Charpentier E. Genome editing. The new frontier of genome engineering with CRISPR-Cas9. Science. 2014;346:1258096. doi: 10.1126/science.1258096. [DOI] [PubMed] [Google Scholar]
- 7.Sander JD, Joung JK. CRISPR-Cas systems for editing, regulating and targeting genomes. Nat Biotechnol. 2014;32:347–55. doi: 10.1038/nbt.2842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cobb RE, Wang Y, Zhao H. High-efficiency multiplex genome editing of Streptomyces species using an engineered CRISPR/Cas system. ACS Synth Biol. 2015;4:723–8. doi: 10.1021/sb500351f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Huang H, Zheng G, Jiang W, Hu H, Lu Y. One-step high-efficiency CRISPR/Cas9-mediated genome editing in Streptomyces. Acta Biochim Biophys Sin (Shanghai) 2015;47:231–43. doi: 10.1093/abbs/gmv007. [DOI] [PubMed] [Google Scholar]
- 10.Tong Y, Charusanti P, Zhang L, Weber T, Lee SY. CRISPR-Cas9 based engineering of actinomycetal genomes. ACS Synth Biol. 2015;4:1020–9. doi: 10.1021/acssynbio.5b00038. [DOI] [PubMed] [Google Scholar]
- 11.Zeng H, et al. Highly efficient editing of the actinorhodin polyketide chain length factor gene in Streptomyces coelicolor M145 using CRISPR/Cas9-CodA(sm) combined system. Appl Microbiol Biotechnol. 2015;99:10575–85. doi: 10.1007/s00253-015-6931-4. [DOI] [PubMed] [Google Scholar]
- 12.Olano C, et al. Activation and identification of five clusters for secondary metabolites in Streptomyces albus J1074. Microbial biotechnology. 2014;7:242–256. doi: 10.1111/1751-7915.12116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bruheim P, Sletta H, Bibb MJ, White J, Levine DW. High-yield actinorhodin production in fed-batch culture by a Streptomyces lividans strain overexpressing the pathway-specific activator gene actll-ORF4. J Ind Microbiol Biotechnol. 2002;28:103–11. doi: 10.1038/sj/jim/7000219. [DOI] [PubMed] [Google Scholar]
- 14.Malpartida F, Niemi J, Navarrete R, Hopwood DA. Cloning and expression in a heterologous host of the complete set of genes for biosynthesis of the Streptomyces coelicolor antibiotic undecylprodigiosin. Gene. 1990;93:91–9. doi: 10.1016/0378-1119(90)90141-d. [DOI] [PubMed] [Google Scholar]
- 15.Wang W, et al. An engineered strong promoter for streptomycetes. Appl Environ Microbiol. 2013;79:4484–92. doi: 10.1128/AEM.00985-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Luo Y, Zhang L, Barton KW, Zhao H. Systematic identification of a panel of strong constitutive promoters from Streptomyces albus. ACS Synth Biol. 2015;4:1001–10. doi: 10.1021/acssynbio.5b00016. [DOI] [PubMed] [Google Scholar]
- 17.Weber T, et al. antiSMASH 3.0-a comprehensive resource for the genome mining of biosynthetic gene clusters. Nucleic Acids Res. 2015;43:237–43. doi: 10.1093/nar/gkv437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liu WT, et al. MS/MS-based networking and peptidogenomics guided genome mining revealed the stenothricin gene cluster in Streptomyces roseosporus. J Antibiot (Tokyo) 2014;67:99–104. doi: 10.1038/ja.2013.99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Luo Y, et al. Activation and characterization of a cryptic polycyclic tetramate macrolactam biosynthetic gene cluster. Nat Commun. 2013;4:2894. doi: 10.1038/ncomms3894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Moree WJ, et al. Microbiota of healthy corals are active against fungi in a light-dependent manner. ACS Chem Biol. 2014;9:2300–8. doi: 10.1021/cb500432j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Okuhara M, et al. Studies on new phosphonic acid antibiotics. I. FR-900098, isolation and characterization. J Antibiot (Tokyo) 1980;33:13–7. doi: 10.7164/antibiotics.33.13. [DOI] [PubMed] [Google Scholar]
- 22.Eliot AC, et al. Biosynthesis of the potent antimalarial compound FR900098. Chem Biol. 2008;15:765. doi: 10.1016/j.chembiol.2008.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jomaa H, et al. Inhibitors of the nonmevalonate pathway of isoprenoid biosynthesis as antimalarial drugs. Science. 1999;285:1573–1576. doi: 10.1126/science.285.5433.1573. [DOI] [PubMed] [Google Scholar]
- 24.Lackner G, et al. Biosynthesis of pentangular polyphenols: deductions from the benastatin and griseorhodin pathways. J Am Chem Soc. 2007;129:9306–12. doi: 10.1021/ja0718624. [DOI] [PubMed] [Google Scholar]
- 25.Hillenmeyer ME, Vandova GA, Berlew EE, Charkoudian LK. Evolution of chemical diversity by coordinated gene swaps in type II polyketide gene clusters. Proc Natl Acad Sci U S A. 2015;112:13952–7. doi: 10.1073/pnas.1511688112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kieser T, Bibb MJ, Buttner MJ, Chater KF, Hopwood DA. Practical Streptomyces Genetics. John Innes Foundation; Norwich, U.K.: 2000. [Google Scholar]
- 27.Rhee KH, Davies J. Transcription analysis of daptomycin biosynthetic genes in Streptomyces roseosporus. J Microbiol Biotechnol. 2006;16:1841–1848. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.