

Abstract
The green tea polyphenol epigallocatechin-3-gallate (EGCG) is widely consumed as a dietary supplement. Its potential properties include slowing aging and extending lifespan, although how exactly this is achieved remains unclear. Here, we report that EGCG promoted healthy lifespan in Caenorhabditis elegans when administered throughout or only at early-to-mid adulthood. Specifically, EGCG extended lifespan in an inverted U-shaped dose-response manner. The life-extending mechanism was stimulated by EGCG-induced production of reactive oxygen species (ROS). Additionally, EGCG triggered mitochondrial biogenesis to restore mitochondrial function. The EGCG-induced increase in lifespan depends on known energy sensors such as AMPK/AAK-2, as well as SIRT1/SIR-2.1 and FOXO/DAF-16. Interestingly, aging decreased the response to EGCG and progressively neutralized its beneficial effects on longevity. Collectively, our findings link EGCG to the process of mitohormesis and suggest an inducible, AMPK/SIRT1/FOXO-dependent redox signaling module that could be invoked in different contexts to extend healthy lifespan. Its effectiveness is higher in younger adults and declines with age.
Keywords: Epigallocatechin-3-gallate, Healthy lifespan, Mitohormesis, Early-to-mid adulthood, AMPK, C. elegans
Graphical abstract
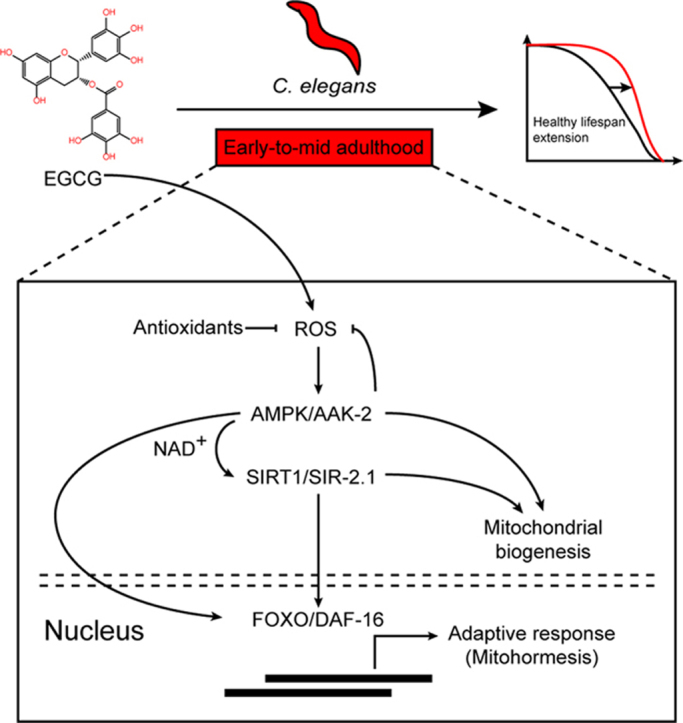
Highlights
-
•
EGCG promoted healthy lifespan at early-to-mid adulthood.
-
•
The life-extending mechanism of EGCG involved the process of mitohormesis.
-
•
EGCG triggered mitochondrial biogenesis to restore mitochondrial function.
-
•
Promotion of longevity due to EGCG occur dependent on AAK-2/SIR-2.1/DAF-16.
-
•
Aging progressively blunted the EGCG beneficial effects to longevity.
1. Introduction
Epigallocatechin-3-gallate (EGCG) is the major bioactive polyphenol in green tea (Camellia sinensis L.). EGCG has been shown to participate in the regulation of various metabolic processes [1] and has lifespan-extending properties in non-disease animal models [2], [3], [4], [5]. It has also been shown to improve disease phenotypes in animal models of cancer (reviewed in [6]), neurodegeneration (reviewed in [7]), and metabolic syndrome (reviewed in [8]). This suggests that EGCG acts on common pathways involved in a variety of aging-associated processes. Because of its demonstrated health benefits on lifespan in invertebrates, such as Drosophila melanogaster [2] and Caenorhabditis elegans [4], [9], as well as in the mammal Rattus norvegicus [3], EGCG may be a bona fide anti-aging phytochemical.
It should be noted that previous studies on the impact of EGCG on C. elegans lifespan have been inconclusive, as they claimed either a positive or no effect on survival under normal conditions [4], [9], [10], [11]. These discrepancies may be attributed to experimental design and EGCG dosage. Longevity has been extensively correlated with resistance to stress, and studies in C. elegans revealed that EGCG could protect against several stressors [5], [11]. In particular, the beneficial effect of EGCG is thought to be directly related to its intrinsic antioxidant properties, however other redox effects have also been documented (reviewed in [12]). Whereas numerous specific targets of EGCG have been identified and characterized [1], [6], [8], [12], the exact mechanism by which EGCG affects longevity remains poorly understood.
The energy-sensing AMP-activated protein kinase (AMPK) is a well-established longevity factor [13] and an attractive candidate to mediate EGCG's effect on lifespan in C. elegans. The induction of AMPK by EGCG has been demonstrated both in vivo and in vitro (reviewed in [8]). Recently, EGCG has been proposed to inhibit gluconeogenesis by activating AMPK and in this way mediate lifespan extension in D. melanogaster [2]. Additionally, EGCG may promote longevity also through other energy sensors, such as sirtuin 1 (SIRT1) and the forkhead box O proteins (FOXOs) [3], [4]. However, whether AMPK, SIRT1, or FOXO mediate directly lifespan extension by EGCG is still unknown and the link between EGCG and these three important regulators is not well established.
In this work, we report that EGCG affected longevity in an inverted U-shaped dose-response manner. A moderate level of EGCG extended healthy lifespan mainly during early-to-mid adulthood. This occurred somewhat counterintuitively through the transient induction of reactive oxygen species (ROS), and subsequent activation of an antioxidant response. We also show that AAK-2, the homolog of AMPK in C. elegans, acted as a feedback regulator that modulated longevity in response to transiently increased ROS levels. This triggered SIRT1/SIR-2.1- NAD+- and FOXO/DAF-16-dependent activation. Moreover, EGCG improved mitochondrial biogenesis and restored mitochondrial function in a way that was dependent on AMPK/AAK-2 and SIRT1/SIR-2.1. Finally, EGCG-induced benefits declined with age. Overall, these findings provide new insights on the role of EGCG or other plant-derived polyphenols in healthy aging.
2. Materials and methods
2.1. Chemicals
EGCG (98% pure) was purchased from Sigma-Aldrich (St. Louis, MO, USA) and stored in water solution at −20 °C. All other chemicals and reagents were also purchased from Sigma-Aldrich unless otherwise stated. For all experiments, stock solutions were freshly prepared in distilled water, unless specified, and sterilized by filtration through 0.2-μm pore size membranes prior to administration.
2.2. C. elegans strains
The following strains were obtained from the Caenorhabditis Genetics Center (University of Minnesota, MN, USA): N2 (Bristol, wild type), BA17 (fem-1(hc17) IV), DA465 (eat-2(ad465) II), RB754 (aak-2(ok524) X), MR507 (aak-2(rr48) X), VC199 (sir-2.1(ok434) IV), IU7 (rrf-3(pk1426) II; sir-2.1(ok434) IV), MIR13 (sir-2.1(ok434) IV; aak-2(ok524) X), CF1038 (daf-16(mu86) I), CB1370 (daf-2(e1370) III), CF1588 (daf-16(mu86) I; daf-2(e1370) III), TJ1052 (age-1(hx546) II), MQ130 (clk-1(qm130) III), CW152 (gas-1(fc21) X), MQ887 (isp-1(qm150) IV), MQ1333 (nuo-6(qm200) I), TK22 (mev-1(kn1) III), SJ4100 (hsp-6::GFP), SJ4058 (hsp-60::GFP+lin-15(+)) SJ4103 (myo-3::GFP(mit)), and TJ356 (daf-16p::daf-16a/b::GFP + rol-6(su1006)). Strains were cultivated on standard nematode growth medium (NGM) seeded with Escherichia coli OP50. All experiments were performed at 20 °C unless stated otherwise.
2.3. Lifespan experiments and oxidative stress resistance assays
Lifespan assays were performed according to standard protocols unless otherwise indicated. In brief, at the prefertile young adult stage (Day 0), age-synchronized nematodes were transferred to NGM plates with 50 μM 5-fluoro-2′-deoxyuridine (FUdR) and 100 μg/mL ampicillin.
For oxidative stress experiments, prefertile (Day 0) age-synchronized worms were transferred to NGM plates (with or without EGCG) containing 50 μM FUdR for 2 days (or 6 days) and subsequently to plates containing 5 mM paraquat (PQ).
Surviving and dead animals were counted daily until all individuals had died. Worms that failed to respond to a gentle touch were scored as dead. All lifespan experiments were conducted in a double-blind manner. The SPSS 18.0 (Demo version, Armonk, NY, USA) statistical analysis package was used for all lifespan and stress resistance statistics. Lifespan assays and stress resistance experiments were analyzed using the Kaplan-Meir test, and P values were calculated with the log-rank test.
2.4. Quantitative RT-PCR for mRNA and DNA quantification
Total RNA was isolated using Trizol (Invitrogen, Carlsbad, CA, USA), and cDNA was synthesized using a cDNA synthesis kit (TaKaRa Bio, Dalian, China). Quantitative RT-PCR was carried out using SYBR Premix Ex Taq (TaKaRa Bio, Kyoto, Japan) and the Rotor Gene Q real-time PCR cycler (Qiagen, Hilden, Germany). Expression of the ama-1 and act-1 genes was used as endogenous control to normalize the amount of mRNA obtained from a target gene. Samples were run in triplicate, and primers are listed in Table S1.
Quantification of the mtDNA copy number in worms was performed by real-time PCR as previously described. Briefly, relative values for nd-1 and act-3 (or 18S) were compared within each sample to generate a ratio representing the relative level of mtDNA per nuclear genome. The results obtained were confirmed with a second mitochondrial gene MTCE.26. The average of at least three technical repeats was used for each biological data point. Primer sequences are listed in Table S1.
2.5. Quantification of ROS production
ROS formation was quantified with dichlorofluorescein diacetate (DCF-DA) (Beyotime Institute of Biotechnology, Haimen, China) (Ex 488 nm/Em 525 nm). Briefly, worms were maintained and treated as described above. After exposure to EGCG, worms were washed off the plates with cold M9 buffer. OP50 were removed by three to five repeated washes. Hydrogen peroxide production was quantified using the Amplex Red hydrogen peroxide kit (Invitrogen) (Ex 550 nm/Em 590 nm).
2.6. Superoxide dismutase and catalase activity assay
Superoxide dismutase (SOD) and catalase (CAT) activity was measured spectrophotometrically using commercially available kits (Nanjing Jiancheng Bioengineering Institute, Nanjing, China) according to the manufacturer's instructions. Protein content was determined using a commercially available kit (BCA; Auragene, Changsha, China).
2.7. Determination of ATP
Worms were harvested and immediately shock-frozen in liquid nitrogen. The frozen pellet was ground in a nitrogen-chilled mortar. The ATP level was analyzed using a commercially available kit (Promega, Madison, WI, USA) according to the manufacturer's instructions. For normalization of the luminescence signal, protein content was determined as described above.
2.8. Oxygen consumption assays
Oxygen consumption was measured using the Seahorse XF96 apparatus (Seahorse Bioscience, North Billerica, MA, USA) as described previously [14]. Respiration rates were normalized to the number of worms in each individual well. Each experiment was repeated at least twice.
2.9. NAD+ measurement
For NAD+ quantification, worms were collected in M9 buffer, washed five times to remove residual bacteria, flash-frozen in liquid nitrogen, and stored at −80 °C until analysis. NAD+ levels were determined using a commercial kit (Enzychrom, BioAssays Systems, Hayward, CA, USA) following the manufacturer's instructions.
2.10. Confocal microscopy and image processing
Worms were immobilized with 6 mM solution of tetramisole hydrochloride in M9 and mounted on 6% agarose pads on glass slides. Images were acquired using a Zeiss LSM 710 upright confocal microscope (Carl Zeiss AG, Jena, Germany) under non-saturating exposure conditions. For each condition, multiple worms were observed and imaged. Image processing was performed using ImageJ software (NIH, Bethesda, MD, USA).
2.11. Statistical analyses
All experiments were repeated at least twice with identical or similar results. Data represent biological replicates. Appropriate statistical tests were used for every figure. Data were expressed as means ± standard deviation (SD). All comparisons for differences among two and more than two data sets were performed by applying Student's t-test and One-way Analysis of Variance (ANOVA), respectively. Statistical analyses were performed using SPSS 18.0 and Origin 8.0 (Demo version, Northampton, MA, USA) software. Significance was established at P < 0.05.
3. Results
3.1. EGCG extends C. elegans healthy lifespan mainly during early-to-mid adulthood
Previously published studies indicated an unclear role of EGCG in lifespan extension of C. elegans under basal, unstressed conditions [4], [5], [10], [11]. To assess the role of EGCG in C. elegans aging, we treated adult worms with various concentrations of EGCG and measured lifespan. EGCG treatment in the range of 50–300 μM resulted in increased longevity, with 200 μM EGCG achieving maximal mean lifespan extension (Fig. 1A, B, and Table S2). Therefore, 200 μM was the concentration used in all subsequent experiments unless stated otherwise. Interestingly, higher doses (800–1000 μM) shortened mean lifespan (Fig. 1A and Table S2). The effect of EGCG on lifespan extension was confirmed in an independent genetic background (fem-1(hc17)) and at different temperatures (Fig. 1B, Fig. S1A, B, and Table S2). Thus, EGCG-mediated lifespan extension in C. elegans depended on concentration but not on genetic background or temperature.
Fig. 1.

Timing requirement of EGCG for the lifespan extension in C.elegans. (A) Percent changes in the lifespan of N2 worms treated with various concentrations of EGCG were shown. (B) EGCG extended the lifespan of worms fed live or dead bacteria. (C) Schematic of lifespan analysis by EGCG transfer. (D) EGCG treatment during the first 6 days only of the adulthood extended lifespan. (E) EGCG treatment from 6th to 12th days only of the adulthood both extended lifespan. (F-H) The longevity capacity of EGCG from Day 6 (F), 12 (G) and/or 18 (H) of adulthood was progressively decreased for the remaining lifespan. (I) EGCG treatment for 15 days early in adulthood only extended lifespan.
Interventions altering or killing E. coli can affect C. elegans lifespan [15]. To test this possibility, we first assessed the effect of EGCG on growth of E. coli (OP50). We report that, at the maximal life-extension dose, EGCG had a very modest effect on the growth of this strain [16] (Fig. S1C and Table S2). We then raised OP50 in the presence of EGCG and transferred them to EGCG-free NGM. EGCG-pretreated OP50 did not extend worm lifespan (Fig. S1D and Table S2). Worms also did not view EGCG-treated food as less favorable, and there was only a slight increase (P > 0.05) in food intake (Fig. S1E, F, and G). Notably, EGCG increased worm lifespan when they were fed dead bacteria (Fig. 1B and Table S2). In addition, the effect of EGCG on lifespan was not FUdR- or ampicillin-dependent (Fig. S1H, I, and Table S2). Altogether, these data demonstrated that EGCG-mediated lifespan extension occurred by a direct effect on C. elegans rather than indirectly through ingested bacteria or other unrelated compounds.
Next, we evaluated the effect of EGCG on worm activity by assessing pharyngeal pumping rate and mobility. Both parameters were robustly improved by EGCG treatment during aging as compared with vehicle (Fig. S2A and B) [10]. Interestingly, we observed that, unlike other pharmacological interventions (e.g., rapamycin, metformin), 200 μM EGCG did not interfere with body length, hatching rate, brood size, or larval development (Fig. S2C, D, E, and F). Thus, EGCG promoted healthy lifespan in adult worms with no side effects.
In fact, EGCG supplementation during adulthood was sufficient to improve longevity, resembling life-long exposure (Fig. S3A and Table S3). Additionally, EGCG supplementation during development had no effect on adult lifespan (Fig. S3B and Table S3), not even in the second (F2) or sixth (F6) generation (following EGCG treatment over five successive generations during the larval stage) (Fig. S3C, D, and Table S3). Notably, supplementation with EGCG during adulthood increased only the mean lifespan, but not maximum lifespan (Fig. 1 and Table S2). Given this result, we wondered whether the beneficial effect of EGCG coincided with a specific timing during the worms’ adult life. Specifically, we tested whether EGCG supplementation increased lifespan in wild type worms continuously throughout adulthood or at discrete life windows. To this end, worms were transferred from vehicle plates to EGCG plates at different stages (Fig. 1C and Table S4). Populations of N2 worms subjected to EGCG treatment during the first 6 days of early adult life or during days 6–12 of adulthood exhibited prolonged lifespan (Fig. 1D, E, and Table S4), however the increase was not as pronounced as in animals treated over their entire adult life. We then assessed the effect of commencing EGCG treatment at older ages. Worms that were switched onto plates containing EGCG at 6 or 12 days of adult age showed an extension of their remaining lifespan (Fig. 1F, G, and Table S4). In contrast, animals transferred at 18 days of adult age lived a normal lifespan when compared to vehicle-treated worms (Fig. 1H and Table S4). To demonstrate that the progressive shift in longevity with late-onset administration of EGCG was not due to its lower intake during C. elegans aging, we increased EGCG concentration to 400 μM and found that EGCG still elicited this kind of shift (Fig. S3E, F, G, and Table S4). Lastly, we found that exposing the worms to EGCG during the first 15 days of adulthood was sufficient to extend lifespan to the same extent as lifelong exposure (Fig. 1I and Table S4). Taken together, these results demonstrated that EGCG prolonged healthy lifespan mostly during early-to-mid adulthood.
3.2. EGCG-induced longevity is redox regulated
EGCG is a natural plant product that has been widely studied for its potential beneficial effects on the redox balance [1], [6], [8], [12]. To determine whether EGCG-induced lifespan extension involved ROS, we first measured ROS levels in worms following exposure to EGCG at various time points. Interestingly, when we exposed wild type worms to EGCG for 12 h, we observed a transient increase in ROS formation, which was undetectable at 3 or 4 days after initiation of exposure. Notably, at 5 days and beyond, i.e. in the steady state, a persistent reduction in ROS was observed following exposure to EGCG (Fig. 2A). In contrast to the elevated levels of ROS at the early stage, EGCG-treated worms exhibited much lower levels of ROS accumulation later in life than their age-matched controls (Fig. S4A). ROS levels were consistent with those of hydrogen peroxide (Fig. S4B). Notably, this transient increase in ROS seems to be an integral part of EGCG-mediated lifespan benefits, because the latter were abolished in the presence of the potent antioxidants N-acetylcysteine (NAC) (5 mM) or butylated hydroxyanisole (BHA) (25 μM) (Fig. 2B and Table S5). To test whether the benefits derived from the transiently higher ROS levels induced by EGCG were analogous to those conferred by low levels of the superoxide generator PQ, we assessed whether lifespan of EGCG-treated worms could be further prolonged by addition of 200 μM PQ, which is known to increase lifespan. Results showed that EGCG and PQ appeared to act independently (Fig. 2C and Table S6). Thus, while ROS induction was required to extend lifespan, EGCG and PQ might achieve this, at least in part, through different ROS pathways. A similar result was obtained when worms were co-treated with EGCG and 2-deoxy-D-glucose (2-DG) (Fig. S4C and Table S6), which also transiently increased ROS content. Finally, we did not observe any difference in resistance to PQ (5 mM) after 2 days of EGCG exposure (Fig. S4D and Table S7). However, following a 6-day exposure to EGCG, worms became resistant to oxidative stress. Notably, the effect was abolished by NAC co-treatment (Fig. 2D and Table S7).
Fig. 2.

EGCG improves ROS homeostasis that is crucial for C. elegans healthspan extension. (A) EGCG induced a transient ROS formation early and exhibited lower ROS levels later in life, 2 h 5 mM paraquat treatment (PQ) was the positive control. (B) The antioxidant NAC or BHA abolished the lifespan-extending effect of EGCG. (C) The mix of EGCG and PQ (200 μM) achieved a robust increase in lifespan, substantially greater than those observed in any experiment with a single compound. (D) EGCG increased 5 mM PQ resistance following 6 d treatment. (E) EGCG attenuated age-associated changes in antioxidative genes. Heat map depicting log2 changes in gene expression for antioxidative genes elicited by EGCG. EGCG was added on day 0 of adulthood and RNA samples were harvested on Day 0 (Vehicle treatment), Day 2, Day 6, and Day 10. (F) EGCG increased SOD and CAT activities after 6 days treatment. Bar graphs are expressed as mean ± SEM, *P < 0.05; **P < 0.01; ***P < 0.001.
To better understand the dynamic change in ROS levels and increased oxidative stress resistance, we quantified the transcriptional levels of antioxidant enzymes, such as SOD, CAT, peroxiredoxin (PRDX), and thioredoxin (TRX). As expected, antioxidant capacity declined with aging, and the mRNA levels of most antioxidant genes were higher in EGCG-treated worms compared to age-matched controls (Fig. 2E). Only the mRNA levels of sod-5 and trx-3 were lower upon EGCG treatment (Fig. 2E). In addition, we found that SOD and CAT activities were significantly increased following EGCG treatment for 6 days, but interestingly no significant alterations at earlier time points (2 days) could be detected (Fig. 2F). The relatively late induction of SOD and CAT was preceded by increases in ROS. These findings suggested that adaptive induction of both SOD and CAT efficiently counteracted the initial rise in ROS levels and culminated in decreased ROS exposure after EGCG treatment for 6 days. This pattern is consistent with the increased oxidative stress resistance observed after 6 but not after 2 days of EGCG treatment.
Taken together, these findings indicated that early ROS induction by EGCG stimulated an endogenous response that enabled detoxification of ROS through defense systems, which served to reduce the levels of ROS and potentially contributed to healthy lifespan promotion.
3.3. EGCG restores mitochondrial function to extend healthy lifespan
Mitochondria are major sites of intracellular ROS. mtROS such as superoxide (O2-) are generated as by-products of the electron transport chain (ETC) in the mitochondrial matrix or intermembrane space [17], [18]. mtROS produced by PQ can activate the mitochondrial unfolded protein response (UPRmt) [19], [20]. However, EGCG appeared to act via a different mechanism, because we did not observe up-regulation of the UPRmt marker hsp-6 or hsp-60 in worms exposed to EGCG (Fig. 3A, Fig. S5A, and B). To further understand the mechanism by which EGCG regulated ROS, we monitored worms at different ages for both mitochondrial mass and function. In each of two experiments, we observed a significant increase in the ratio of mitochondrial to nuclear DNA (mtDNA/nDNA) in Day 2 and Day 6 adult worms subjected to EGCG treatment as compared to vehicle controls (Fig. 3B). We also examined expression of the nuclear gene cts-1, which encodes mitochondrial citrate synthase and is a proxy for mitochondrial biogenesis. cts-1 expression increased in Day 2 and Day 6 adults treated with EGCG (Fig. S5C). Interestingly, both mtDNA/nDNA ratio and cts-1 expression were partially affected by NAC (Fig. 3B and Fig. S5C), indicating that mitochondrial biogenesis occurred after ROS generation.
Fig. 3.

EGCG restores mitochondrial function. (A) EGCG did not induce the mitochondrial unfolded protein response (UPRmt) (hsp-6 reporter) at Day 2 worms. (B) EGCG increased mitochondrial biogenesis, partially inhibited by NAC, at Day 2 and Day 6 of adulthood, as evidenced by the increased mtDNA/nDNA ratio. (C) EGCG decreased worm ATP level at Day 2 and Day 6, and maintained it at Day 10 of adulthood. (D-E) Oxygen consumption at basal (D) or after short-term incubation with 10 µM FCCP (E) was restored in wild type worms after EGCG treatment. (F) The alteration of mitochondrial network morphology was postponed by EGCG, representative images are shown. (G-K) EGCG decreased or did not affect lifespan of ETC mutants. Bar graphs are expressed as mean ± SEM, *P < 0.05; **P < 0.01; ***P < 0.001.
Next, we observed that EGCG treatment was associated with a significantly lower ATP level during early-to-mid adulthood, which was surprisingly not affected by NAC (Fig. 3C). Worms exposed to EGCG had a constant basal or carbonilcyanide p-triflouromethoxyphenylhydrazone (FCCP)-uncoupled mitochondrial respiration (Fig. 3D and E). In addition, ATP and respiration capacity were both greater in older (Day 10) worms treated with EGCG than in age-matched controls (Fig. 3C, D, and E). Furthermore, the mitochondrial network of body wall muscle in EGCG-treated worms was well-organized compared to vehicle-treated worms, in which it was fragmented and disorganized (Fig. 3F). Accordingly, the benefits of the EGCG treatment correlated with a better maintenance of mitochondrial function.
Two types of ETC mutants that cause elevated ROS and alter mitochondrial function have been described to affect aging in C. elegans: those with a shorter lifespan, such as gas-1 and mev-1, and those with a longer lifespan, such as clk-1, nuo-6, and isp-1. We found that several such ETC mutants displayed unchanged or shorter lifespan following EGCG treatment (Fig. 3G, H, I, J, K, and Table S8). These results confirmed that EGCG's healthy lifespan extension depended on mitochondrial function.
Thus, EGCG activates mitochondrial biogenesis in early-to-mid adult worms and restores mitochondrial function.
3.4. AAK-2 is required for the NAD+/SIR-2.1 pathway to extend healthy lifespan via EGCG
EGCG has been previously reported to be an AMPK activator [8]. We examined the effect of EGCG on various mutants defective for AMPK/AAK-2, and found that AAK-2 was essential for EGCG-induced longevity (Fig. 4A, Fig. S6A, and Table S9). We also observed that EGCG did not affect mobility (Fig. S6B), although it improved the pharyngeal pumping rate (Fig. S6C) of aak-2(ok524) worms. Because ROS could activate AAK-2 to mediate longevity [13], [21], we wondered whether AAK-2 was involved in the transient increase in ROS induced by EGCG. To explore this possibility, we directly measured ROS in wild type and aak-2(ok524) worms. We found that total ROS and hydrogen peroxide content were elevated in aak-2(ok524) mutants after 2 days of EGCG treatment (Fig. 4B and Fig. S6D), suggesting that AAK-2 played a role in reducing ROS and possibly contributed to EGCG-induced longevity. Moreover, no up-regulation of sod-1/sod-2/sod-3, CAT (ctl-1/ctl-2/ctl-3), or prdx-2 genes was observed in aak-2(ok524) mutants (Fig. 4C). In addition, EGCG did not increase resistance to PQ in aak-2(ok524) mutants (Fig. 4D and Table S9). Next, we sought to determine whether AAK-2 had any effect on EGCG-restored mitochondrial function. Measurements of the mtDNA/nDNA ratio and cts-1 mRNA level revealed that EGCG did not increase mitochondrial content in aak-2(ok524) worms (Fig. 4E and Fig. S6E). Together, these results indicated that EGCG-induced longevity required AAK-2.
Fig. 4.

EGCG-induced longevity requires AAK-2, the C.elegans AMPK homolog, and SIR-2.1, the C.elegans SIRT1 homolog. (A) EGCG did not extend lifespan in the aak-2(ok524) mutant. (B) Total ROS level was increased by EGCG treatment and by aak-2(ok524) mutations for 2 days. (C) EGCG did not up-regulate antioxidative gene expression in the aak-2(ok524) mutant compare to age-matched Vehicle-treated worms. (D) EGCG treatment for 6 d did not increase resistance to oxidative stress in the aak-2(ok524) mutant. (E) AAK-2/AMPK was required for EGCG-induced mitochondrial biogenesis. (F) Aging decreased worm NAD+ level in Vehicle- and EGCG-treated worms, however, EGCG supplementation increased NAD+ level. PQ (5 mM) treatment was the negative control. (G) EGCG did not extend lifespan in the sir-2.1(ok434) mutant. (H) EGCG increased NAD+ level in the sir-2.1(ok434) mutant, not aak-2(ok524) or aak-2(ok524), sir-2.1(ok434) mutant. (I) EGCG did not increase lifespan of the aak-2(ok524), sir-2.1(ok434) mutant. Bar graphs are expressed as mean ± SEM, *P < 0.05; **P < 0.01; ***P < 0.001.
Given that AMPK increases the NAD+-to-NADH ratio and activates SIRT1 [22], we measured NAD+ in wild type worms and lifespan in two genetically different strains lacking SIRT1/SIR-2.1. NAD+/SIR-2.1 signaling appeared to be required for lifespan extension following EGCG treatment (Fig. 4F, G, Fig. S6F, and Table S9). Interestingly, EGCG increased NAD+ levels during the aging process both in wild type and sir-2.1(ok434) worms, but not in aak-2(ok524) worms (Fig. 4H). These results demonstrated that the EGCG-mediated increase in NAD+ depended on AMPK/AAK-2, but not on SIRT1/SIR-2.1. To further confirm this finding, we measured the NAD+ level and lifespan in aak-2(ok524), sir-2.1(ok434) double mutants, and found that EGCG had no effect on either parameter (Fig. 4H, I, and Table S9). However, we observed that EGCG-induced mitochondrial biogenesis required SIRT1/SIR-2.1 based on the expression of cts-1 (Fig. S6G). Thus, the NAD+/SIR-2.1 pathway might function downstream of AAK-2.
3.5. FOXO/DAF-16 mediates EGCG-induced longevity
Consistent with a previous report [4], EGCG required FOXO/DAF-16 to extend lifespan (Fig. 5A and Table S10). DAF-16's role in EGCG-induced increase in lifespan may be dependent on the insulin signaling pathway, a well-known regulator of DAF-16 activity. To test this possibility, we investigated whether the insulin receptor DAF-2 mediated the beneficial effect of EGCG. In contrast to daf-16, the long lifespan of daf-2(e1370) and age-1(hx546) mutants was further extended by EGCG (Fig. 5B, C, and Table S10), suggesting that EGCG might regulate DAF-16 independently of the insulin receptor pathway. EGCG triggered nuclear localization of DAF-16 (Fig. 5D and E), without causing any changes to daf-16 expression (Fig. 5F), and activated its antioxidant target genes (Figs. 2E and 5F). Furthermore, we confirmed that induction of sod-3 was dependent on the presence of daf-16 (Fig. 5F), suggesting that DAF-16, like AAK-2, was involved in the transient increase in ROS induced by EGCG. To this end, we report that EGCG did not increase lifespan in daf-16(mu86), daf-2(e1370) double mutants (Fig. 5G and Table S10). These results suggested that DAF-16 might be involved in EGCG-induced lifespan extension via the AAK-2/SIR-2.1 pathway [23]. However, because the daf-2 mutation was not a null mutation, it was still possible that part of the EGCG-induced increase in lifespan might depend on the insulin receptor pathway [23].
Fig. 5.

The lifespan-extending effect of EGCG requires DAF-16, not DAF-2 or AGE-1. (A) EGCG did not extend lifespan in the daf-16(mu86) mutant. (B-C) EGCG extended lifespan in the daf-2(e1370) (B) or age-1(hx546) (C) mutants. (D) Representative images of daf-16::GFP reporter worms treated with Vehicle or EGCG, showing nuclear accumulation of daf-16 following treatment. (E) Quantification of daf-16 nuclear translocation following treatment with EGCG. Localization was shown as percentage of worms that shows nuclear or cytosolic localization. (F) EGCG did not affect daf-16 mRNA levels in the wild type worms, and up-regulates sod-3 expression dependent on daf-16. (G) EGCG did not extend lifespan in the daf-2(e1370), daf-16(mu86) mutant. Bar graphs are expressed as mean ± SEM, *P < 0.05; **P < 0.01; ***P < 0.001.
3.6. Response to EGCG declines with age in C. elegans
Based on the above observations, we found that EGCG did not extent the maximum lifespan (Fig. 1 and Table S2) and induced a progressive shift to the left in longevity following late-onset administration of EGCG (Fig. 1F, G, and H). When a 2-day EGCG treatment was initiated in 0-, 6- and 10-day-old worms, their antioxidant response to EGCG was progressively lost with increasing age (Fig. 6A). We also found that early adult worms exhibited a stronger ability to generate the transient increase in ROS, which then declined with age (Fig. 6B). Moreover, oxidative stress resistance was weaker in Day 6 worms than in Day 0 worms, even if both were exposed to EGCG for 6 days (Fig. 6C and Table S11). When mtDNA/nDNA and cts-1 mRNA levels were tested, similar results were obtained: young worms exhibited faster mitochondrial biogenesis than old worms (Fig. 6D and Fig. S7). Consistent with these results, EGCG treatment for 6 days caused a larger increase in lifespan in Day 0 worms than in Day 6 adult worms (Fig. 1D and E). Altogether, these findings indicated that aging caused a decreased response to EGCG and progressively blunted the beneficial effect of EGCG on longevity.
Fig. 6.

EGCG induces beneficial effects declined with age. (A) Aging caused decreased antioxidative response ability to EGCG. EGCG was added on Day 0, Day 6 and Day 10 of adulthood and RNA samples were harvested after 2 days treatment. (B) The transient ROS induced by EGCG was decreased during the aging process. (C) EGCG increased 5 mM PQ resistance stronger on Day 0 worms than Day 6 worms for 6 days EGCG treatment. (D) EGCG induced mtDNA/nDNA ratio declined with age. (E) Model of EGCG-mediated longevity.
4. Discussion
In this study, we have confirmed that EGCG, whose role in regulating the lifespan of C. elegans was somewhat uncertain, promoted longevity in a concentration-dependent way. Mechanistically, we suggest that EGCG extends lifespan during early-to-mid adulthood via AMPK/SIRT1/FOXO signaling and mitohormesis (Fig. 6E).
4.1. EGCG acts via mitohormesis as a pro-longevity xenohormetic chemical
In experiments using pharmacological agents, it is being gradually recognized that there are complex dose-response relationships for diet-derived agents. Instead of a linear dose response, hormesis is defined as a biphasic dose response, characterized by a low-dose stimulation and a high-dose inhibition [24]. Here, we show that the effect of EGCG on C. elegans lifespan reflected a nonlinear or inverted U-shaped dose-response relationship. Our study indicates that concentration might be the reason why previous reports were inconclusive [4], [5], [10], [11]. For example, EGCG did not extend lifespan at concentrations below 25 μM [10], [11] and, as in rats [3], would only achieve such effect above a certain dosage. However, high doses of EGCG shortened the lifespan of C. elegans. In fact, some reports suggested that excessive amounts of EGCG triggered adverse reactions, mainly hepatotoxicity, in humans and rodents [25], [26], [27], [28]. Thus, EGCG stimulated a hormetic response in aging. Hormesis in aging is defined as an adaptive response to a short-term or nonlethal stress, which promotes healthy lifespan [29], [30]. In our research, we found that treatment with EGCG commencing from adulthood onward or during the first few days of adulthood was sufficient to extend lifespan (Fig. 1D, E, G and Fig. S3A), resembling the critical phase for other hormetic compounds (e.g. metformin, 2-DG) and supporting the hormetic role of EGCG.
EGCG, a classical plant polyphenol, has been widely perceived as a strong antioxidant [6], [8]. However, the bioavailability of EGCG in tea or other dietary forms, is generally far too low to explain a direct antioxidant effect in living systems. At the same time, EGCG is also prone to auto-oxidation and ROS production in vitro, which is unlikely to occur in internal organs because of the lower oxygen partial pressure (than in solution in vitro) and the antioxidant defenses in animal tissues [6], [8]. In vivo, EGCG may induce ROS formation in mitochondria [1], [31], [32], [33] which, in turn, may activate antioxidant systems and other cytoprotective enzymes [8]. This “indirect antioxidant” effect implies that EGCG may induce a particular form of hormesis, named mitochondrial hormesis (or mitohormesis) [29], [30]. We were able to fully reproduce the mitohormetic phenomenon in EGCG-treated worms and show that EGCG transiently increased the generation of ROS. The latter subsequently acted as messengers to induce further stress defenses and promote a healthy lifespan. Inhibition of ROS signaling abolished lifespan-extension by EGCG, proving its dependence on the mitohormetic process. However, the EGCG-mediated mitohormetic pathway might be at least partially distinct from that induced by PQ [21], [34] (or 2-DG [35]), because EGCG and PQ (or 2-DG) co-treatment further increased lifespan compared to each single compound only. These results also suggest that EGCG might not simply act as an antioxidant in aging, because the lifespan extension brought by 2-DG and EGCG was abolished by administration of the antioxidant NAC. Alternatively, EGCG and PQ (or 2-DG) might not act on the same cell types or in the same cellular compartments or sites. Taken together, the antioxidant activity of EGCG does not appear to be the primary source of health benefits and increased longevity in C. elegans. We found that EGCG induced ROS in a time-resolved manner, and EGCG-treated worms exhibited much lower levels of ROS accumulation later in life than did age-matched controls, which was consistent with previous reports [5], [11]. In addition, our observations did not conflict with previous reports in terms of experimental design.
In nature, plants have developed specific biological systems that can help them adapt to and counteract various forms of stress. Phytochemicals, one such category of compounds, provide health benefits to animals, including humans. In theory, these chemicals are supposed to have no nutritional value and, in fact, most phytochemicals are actively detoxified and excreted. In line with the hormesis hypothesis, these foreign compounds must essentially remain in the lower range to have any beneficial effect, otherwise adverse outcomes would become dominant. EGCG is synthesized when tea plants face a variety of environmental stresses. This inter-species hormesis is known as xenohormesis [36] and might help explain how EGCG exerts a beneficial effect on longevity in C. elegans.
4.2. Period-specific response to EGCG determines healthy lifespan extension induced by EGCG
We report here that if the expression of an antioxidant gene increased with age, EGCG treatment prevented the increase and if the expression of an antioxidant gene decreased with age, EGCG prevented the decrease. These observations are in line with the “transcriptional drift”, a phenomenon that describes how aging causes genes within functional groups to change expression in opposite directions [37]. Accordingly, EGCG preserved homeostatic capacity of the redox function via attenuated transcriptional drift. EGCG treatment for 6 days greatly improved stress resistance to PQ, suggesting that the preserved redox homeostatic ability improved the response to environmental stimuli. Interestingly, suppression of transcriptional drift might occur during early adulthood and delay aging [37]. Consistent with this concept, exposure to EGCG for the first 15 days during C. elegans adulthood was sufficient to extend lifespan to the same extent as lifelong exposure. Further, in accordance with the improved redox homeostatic capacity, we found that EGCG restored mitochondrial function, resulting in an overall improvement of multiple cellular and organismal phenotypes in aging worms. In addition, administration of EGCG increased mitochondrial content while maintaining respiratory capacity. This supports the hypothesis that, despite a reduction in ATP, EGCG treatment might produce very efficient mitochondria, which could attenuate molecular and cellular damage resulting from oxidative stress and then promote healthy lifespan at cellular and organismal levels, in young worms. Thus, early-to-mid adulthood was critical for improving redox homeostatic capacity and mitochondrial function by EGCG.
Another interesting finding was that the ability to respond to EGCG declined with age. Thus, EGCG treatment later on in adult life (Day 12 or Day 18) had no impact on lifespan and did not slow the rate of aging. This was in line with a previous observation in mice, where green tea polyphenol administered to mice beginning at 13 months of age, but not at 19 months, successfully increased lifespan [38]. Notably, both redox response ability and mitochondrial function induced by EGCG were progressively lost during the aging process. These findings demonstrate that the benefits of EGCG on longevity depend on the response capacity of the organism. EGCG lost its lifespan extension capacity when the organism lost the ability to respond to EGCG with progressing age.
In summary, it is possible that the health benefits brought by EGCG may be effective only within a period during young adulthood, in which the rate of age-associated physiological change is specifically low.
4.3. AMPK/SIRT1/FOXO are required for EGCG-induced longevity
AMPK and SIRT1 have been shown to play similar roles in stress response, induction of mitochondrial biogenesis, and regulation of energy homeostasis [13], [39]. Similarly, the longevity-beneficial effects of EGCG have been suggested to involve activation of AMPK and SIRT1 [3], [8]. Based on our data, AMPK/AAK-2 might be activated by the EGCG-induced transient increase in ROS, which modulated EGCG-induced longevity by reducing internal levels of ROS. AMPK/AAK-2 has been shown to alter the metabolism of NAD+, which activates SIRT1/SIR-2.1 [22]. These lines of evidence suggest that the activation of SIRT1/SIR-2.1 by EGCG was indirect, and was mediated by activation of AMPK/AAK-2 through a mechanism that relied on maintaining the NAD+ concentration [22]. As a consequence, SIRT1/SIR-2.1 activated the FOXO/DAF-16 transcription factor, which up-regulated various antioxidant genes. It remains to be determined whether transiently increased ROS were generated through inhibition of ETC complex I or ATP synthase [1], which might also contribute to AMPK activation in C. elegans via a decrease in ATP. Although the anti-aging effect of EGCG is likely to involve multiple modes of action, our observations reinforce the potential of targeting metabolic pathways as a way of improving healthy aging.
The complex nature of the effect EGCG exerts on longevity was demonstrated by our observation that even though EGCG prolonged lifespan in a DAF-16-dependent manner, it further improved lifespan also in DAF-2 or AGE-1 mutants, which are involved in mitohormetic lifespan extension [40]. Moreover, co-treatment with 2-DG, another compound related to mitohormetic lifespan extension dependent on AMPK/AAK-2 [35], increased lifespan compared to EGCG or 2-DG treatment alone.
In summary, this work provides novel insights on the aging process and shows that EGCG extends lifespan through mitohormesis and, specifically, a mechanism that depends on the AMPK/SIRT1/FOXO pathway. Interestingly, our findings provide a solid proof that EGCG might exert its beneficial role mainly in young or middle-aged adults. Our data validate a link between mitochondria, antioxidants, and lifespan control, which should warrant further research into EGCG or green tea (plant polyphenols) and ways of applying these compounds to improve healthy aging or for other therapeutic indications.
Fundings
The research is supported by the National Natural Science Foundation of China (No. 31270733), the National Tea Industry Technical System Project of China (CARS-23-11B) and the Synergistic and Innovative Research Projects for Utilization of Botanical Functional Ingredients (HNCR-2014003). Some C. elegans strains were provided by CGC, which is funded by the NIH Office of Research Infrastructure Programs (P40 OD010440)
Author contributions
L-G.X., Y-S.G. and Z-H.L. designed the research; L-G.X. conducted the main research and analyzed the data; Y-J C and J-W.T. contributes reagents and mediums; L-G.X. and J-A.H. wrote the manuscript.
Conflict of interest
The authors declare no conflict of interest.
Footnotes
Supplementary data associated with this article can be found in the online version at doi:10.1016/j.redox.2017.09.019.
Contributor Information
Yu-Shun Gong, Email: gongyushun@hunau.net.
Jian-An Huang, Email: jian7513@sina.com.
Zhong-Hua Liu, Email: larkin-liu@163.com.
Appendix A. Supplementary material
Supplementary material
References
- 1.de Oliveira M.R., Nabavi S.F., Daglia M., Rastrelli L., Nabavi S.M. Epigallocatechin gallate and mitochondria – a story of life and death. Pharmacol. Res. 2016;104:70–85. doi: 10.1016/j.phrs.2015.12.027. [DOI] [PubMed] [Google Scholar]
- 2.Wagner A.E., Piegholdt S., Rabe D., Baenas N., Schloesser A., Eggersdorfer M., Stocker A., Rimbach G. Epigallocatechin gallate affects glucose metabolism and increases fitness and lifespan in Drosophila melanogaster. Oncotarget. 2015;6:30568–30578. doi: 10.18632/oncotarget.5215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Niu Y., Na L., Feng R., Gong L., Zhao Y., Li Q., Li Y., Sun C. The phytochemical, EGCG, extends lifespan by reducing liver and kidney function damage and improving age‐associated inflammation and oxidative stress in healthy rats. Aging Cell. 2013;12:1041–1049. doi: 10.1111/acel.12133. [DOI] [PubMed] [Google Scholar]
- 4.Bartholome A., Kampkötter A., Tanner S., Sies H., Klotz L.-O. Epigallocatechin gallate-induced modulation of FoxO signaling in mammalian cells and C. elegans: FoxO stimulation is masked via PI3K/Akt activation by hydrogen peroxide formed in cell culture. Archiv. Biochem. Biophys. 2010;501:58–64. doi: 10.1016/j.abb.2010.05.024. [DOI] [PubMed] [Google Scholar]
- 5.Abbas S., Wink M. Epigallocatechin gallate from green tea (Camellia sinensis) increases lifespan and stress resistance in Caenorhabditis elegans. Planta Med. 2009;75:216. doi: 10.1055/s-0028-1088378. [DOI] [PubMed] [Google Scholar]
- 6.Yang C.S., Wang X., Lu G., Picinich S.C. Cancer prevention by tea: animal studies, molecular mechanisms and human relevance. Nat. Rev. Cancer. 2009;9:429–439. doi: 10.1038/nrc2641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yang C.S., Hong J. Prevention of chronic diseases by tea: possible mechanisms and human relevance. Annu. Rev. Nutr. 2013;33:161–181. doi: 10.1146/annurev-nutr-071811-150717. [DOI] [PubMed] [Google Scholar]
- 8.Yang C.S., Zhang J., Zhang L., Huang J., Wang Y. Mechanisms of body weight reduction and metabolic syndrome alleviation by tea. Mol. Nutr. Food Res. 2016;60:160–174. doi: 10.1002/mnfr.201500428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Abbas S., Wink M. Epigallocatechin gallate inhibits beta amyloid oligomerization in Caenorhabditis elegans and affects the daf-2/insulin-like signaling pathway. Phytomedicine. 2010;17:902–909. doi: 10.1016/j.phymed.2010.03.008. [DOI] [PubMed] [Google Scholar]
- 10.Brown M.K., Evans J.L., Luo Y. Beneficial effects of natural antioxidants EGCG and α-lipoic acid on life span and age-dependent behavioral declines in Caenorhabditis elegans. Pharmacol. Biochem. Behav. 2006;85:620–628. doi: 10.1016/j.pbb.2006.10.017. [DOI] [PubMed] [Google Scholar]
- 11.Zhang L., Jie G., Zhang J., Zhao B. Significant longevity-extending effects of EGCG on Caenorhabditis elegans under stress. Free Radic. Biol. Med. 2009;46:414–421. doi: 10.1016/j.freeradbiomed.2008.10.041. [DOI] [PubMed] [Google Scholar]
- 12.Kim H.-S., Quon M.J., Kim J.-a. New insights into the mechanisms of polyphenols beyond antioxidant properties; lessons from the green tea polyphenol, epigallocatechin 3-gallate. Redox Biol. 2014;2:187–195. doi: 10.1016/j.redox.2013.12.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Burkewitz K., Zhang Y., Mair W.B. AMPK at the nexus of energetics and aging. Cell Metab. 2014;20:10–25. doi: 10.1016/j.cmet.2014.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chin R.M., Fu X., Pai M.Y., Vergnes L., Hwang H., Deng G., Diep S., Lomenick B., Meli V.S., Monsalve G.C. The metabolite [agr]-ketoglutarate extends lifespan by inhibiting ATP synthase and TOR. Nature. 2014;510:397–401. doi: 10.1038/nature13264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cabreiro F., Au C., Leung K.-Y., Vergara-Irigaray N., Cochemé H.M., Noori T., Weinkove D., Schuster E., Greene N.D., Gems D. Metformin retards aging in C. elegans by altering microbial folate and methionine metabolism. Cell. 2013;153:228–239. doi: 10.1016/j.cell.2013.02.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Xiong L.-G., Chen Y.-J., Tong J.-W., Huang J.-A., Li J., Gong Y.-S., Liu Z.-H. Tea polyphenol epigallocatechin gallate inhibits Escherichia coli by increasing endogenous oxidative stress. Food Chem. 2017;217:196–204. doi: 10.1016/j.foodchem.2016.08.098. [DOI] [PubMed] [Google Scholar]
- 17.Bratic A., Larsson N.-G. The role of mitochondria in aging. J. Clin. Investig. 2013;123:951–957. doi: 10.1172/JCI64125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Murphy M.P. How mitochondria produce reactive oxygen species. Biochem. J. 2009;417:1–13. doi: 10.1042/BJ20081386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Runkel E.D., Liu S., Baumeister R., Schulze E. Surveillance-activated defenses block the ROS – induced mitochondrial unfolded protein response. PLoS Genet. 2013;9:e1003346. doi: 10.1371/journal.pgen.1003346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Haynes C.M., Ron D. The mitochondrial UPR–protecting organelle protein homeostasis. J. Cell Sci. 2010;123:3849–3855. doi: 10.1242/jcs.075119. [DOI] [PubMed] [Google Scholar]
- 21.Hwang A.B., Ryu E.-A., Artan M., Chang H.-W., Kabir M.H., Nam H.-J., Lee D., Yang J.-S., Kim S., Mair W.B. Feedback regulation via AMPK and HIF-1 mediates ROS-dependent longevity in Caenorhabditis elegans. Proc. Natl. Acad. Sci. 2014;111:E4458–E4467. doi: 10.1073/pnas.1411199111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cantó C., Gerhart-Hines Z., Feige J.N., Lagouge M., Noriega L., Milne J.C., Elliott P.J., Puigserver P., Auwerx J. AMPK regulates energy expenditure by modulating NAD+ metabolism and SIRT1 activity. Nature. 2009;458:1056–1060. doi: 10.1038/nature07813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Greer E.L., Dowlatshahi D., Banko M.R., Villen J., Hoang K., Blanchard D., Gygi S.P., Brunet A. An AMPK-FOXO pathway mediates longevity induced by a novel method of dietary restriction in C. elegans. Curr. Biol. 2007;17:1646–1656. doi: 10.1016/j.cub.2007.08.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Calabrese E.J., Baldwin L.A. Defining hormesis. Hum. Exp. Toxicol. 2002;21:91–97. doi: 10.1191/0960327102ht217oa. [DOI] [PubMed] [Google Scholar]
- 25.Wang D., Wei Y., Wang T., Wan X., Yang C.S., Reiter R.J., Zhang J. Melatonin attenuates (‐)‐epigallocatehin‐3‐gallate‐triggered hepatotoxicity without compromising its downregulation of hepatic gluconeogenic and lipogenic genes in mice. J. Pineal Res. 2015;59:497–507. doi: 10.1111/jpi.12281. [DOI] [PubMed] [Google Scholar]
- 26.Schönthal A.H. Adverse effects of concentrated green tea extracts. Mol. Nutri. Food Res. 2011;55:874–885. doi: 10.1002/mnfr.201000644. [DOI] [PubMed] [Google Scholar]
- 27.Mazzanti G., Menniti-Ippolito F., Moro P.A., Cassetti F., Raschetti R., Santuccio C., Mastrangelo S. Hepatotoxicity from green tea: a review of the literature and two unpublished cases. Eur. J. Clin. Pharmacol. 2009;65:331–341. doi: 10.1007/s00228-008-0610-7. [DOI] [PubMed] [Google Scholar]
- 28.Sarma D.N., Barrett M.L., Chavez M.L., Gardiner P., Ko R., Mahady G.B., Marles R.J., Pellicore L.S., Giancaspro G.I., Dog T.L. Safety of green tea extracts. Drug Saf. 2008;31:469–484. doi: 10.2165/00002018-200831060-00003. [DOI] [PubMed] [Google Scholar]
- 29.Ristow M. Unraveling the truth about antioxidants: mitohormesis explains ROS-induced health benefits. Nat. Med. 2014;20:709–711. doi: 10.1038/nm.3624. [DOI] [PubMed] [Google Scholar]
- 30.Yun J., Finkel T. Mitohormesis. Cell Metab. 2014;19:757–766. doi: 10.1016/j.cmet.2014.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li G.-X., Chen Y.-K., Hou Z., Xiao H., Jin H., Lu G., Lee M.-J., Liu B., Guan F., Yang Z. Pro-oxidative activities and dose–response relationship of (−)-epigallocatechin-3-gallate in the inhibition of lung cancer cell growth: a comparative study in vivo and in vitro. Carcinogenesis. 2010;31:902–910. doi: 10.1093/carcin/bgq039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tao L., Forester S.C., Lambert J.D. The role of the mitochondrial oxidative stress in the cytotoxic effects of the green tea catechin,(–)‐epigallocatechin‐3‐gallate, in oral cells. Mol. Nutr. Food Res. 2014;58:665–676. doi: 10.1002/mnfr.201300427. [DOI] [PubMed] [Google Scholar]
- 33.Schroeder E.K., Kelsey N.A., Doyle J., Breed E., Bouchard R.J., Loucks F.A., Harbison R.A., Linseman D.A. Green tea epigallocatechin 3-gallate accumulates in mitochondria and displays a selective antiapoptotic effect against inducers of mitochondrial oxidative stress in neurons. Antioxid. Redox Signal. 2009;11:469–480. doi: 10.1089/ars.2008.2215. [DOI] [PubMed] [Google Scholar]
- 34.Yang W., Hekimi S. A mitochondrial superoxide signal triggers increased longevity in Caenorhabditis elegans. PLoS Biol. 2010;8:e1000556. doi: 10.1371/journal.pbio.1000556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schulz T.J., Zarse K., Voigt A., Urban N., Birringer M., Ristow M. Glucose restriction extends Caenorhabditis elegans life span by inducing mitochondrial respiration and increasing oxidative stress. Cell Metab. 2007;6:280–293. doi: 10.1016/j.cmet.2007.08.011. [DOI] [PubMed] [Google Scholar]
- 36.Howitz K.T., Sinclair D.A. Xenohormesis: sensing the chemical cues of other species. Cell. 2008;133:387–391. doi: 10.1016/j.cell.2008.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rangaraju S., Solis G.M., Thompson R.C., Gomez-Amaro R.L., Kurian L., Encalada S.E., Niculescu A.B., III, Salomon D.R., Petrascheck M. Suppression of transcriptional drift extends C. elegans lifespan by postponing the onset of mortality. Elife. 2015;4:e08833. doi: 10.7554/eLife.08833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kitani K., Osawa T., Yokozawa T. The effects of tetrahydrocurcumin and green tea polyphenol on the survival of male C57BL/6 mice. Biogerontology. 2007;8:567–573. doi: 10.1007/s10522-007-9100-z. [DOI] [PubMed] [Google Scholar]
- 39.Haigis M.C., Guarente L.P. Mammalian sirtuins – emerging roles in physiology, aging, and calorie restriction. Genes Dev. 2006;20:2913–2921. doi: 10.1101/gad.1467506. [DOI] [PubMed] [Google Scholar]
- 40.Zarse K., Schmeisser S., Groth M., Priebe S., Beuster G., Kuhlow D., Guthke R., Platzer M., Kahn C.R., Ristow M. Impaired insulin/IGF1 signaling extends life span by promoting mitochondrial L-proline catabolism to induce a transient ROS signal. Cell Metab. 2012;15:451–465. doi: 10.1016/j.cmet.2012.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material