

Abstract
Fanconi Anemia (FA) is a rare, inherited genomic instability disorder, caused by mutations in genes involved in the repair of interstrand DNA crosslinks (ICLs). The FA signaling network contains a unique nuclear protein complex that mediates the monoubiquitylation of the FANCD2 and FANCI heterodimer, and coordinates activities of the downstream DNA repair pathway including nucleotide excision repair, translesion synthesis, and homologous recombination. FA proteins act at different steps of ICL repair in sensing, recognition and processing of DNA lesions. The multi-protein network is tightly regulated by complex mechanisms, such as ubiquitination, phosphorylation, and degradation signals that are critical for the maintenance of genome integrity and suppressing tumorigenesis. Here, we discuss recent advances in our understanding of how the FA proteins participate in ICL repair and regulation of the FA signaling network that assures the safeguard of the genome. We further discuss the potential application of designing small molecule inhibitors that inhibit the FA pathway and are synthetic lethal with DNA repair enzymes that can be used for cancer therapeutics.
Keywords: DNA repair, Fanconi Anemia (FA) signaling network, DNA damage response, Cancer therapeutics, Synthetic lethality, Combination Therapy Genomic instability, Interstrand crosslink (ICL), Homologous recombination, Translesion synthesis
Background
Fanconi Anemia (FA), a rare genetic cancer-susceptibility syndrome is a recessive autosomal or X-linked genetic disease [1–3]. FA is characterized by genomic instability, bone marrow failure leading to progressive aplastic anemia, chromosomal fragility and heightened susceptibility to cancer, particularly acute myelogenous leukemia (AML) [1, 4]. With an incidence of ~1–5 per 1,000,000 births, many FA patients suffer from developmental disorders and physical abnormalities ranging from short stature, abnormal skin pigmentation, organ malformation, hypogonadism, and developmental delay [5]. Patients are often diagnosed with early onset of solid tumors including squamous cell carcinomas of the head and neck, cervical cancer and liver tumors [6, 7]. FA was first described by the Swiss pediatrician Guido Fanconi in 1927 while treating a family of five siblings, three of whom presented with developmental birth defects and died from an early-onset of clinical features resembling pernicious anemia [8]. Additional clinical features included microcephaly, vitiligo and hypoplasia of the testes [8]. After nearly four decades another article reported an accumulation of large number of chromatid breaks in the blood lymphocytes of FA patients [9]. Due to high frequencies of chromosomal abnormalities, predominantly chromatid breaks during S-phase of the cell cycle, researchers concluded that FA patients have impaired double strand break repair (DSBR) [10]. Also despite the varied clinical phenotypes of the disease, a defining characteristic of FA cells is the cellular hypersensitivity to DNA crosslinking agents such as mitomycin C (MMC), chemotherapeutic agent cisplatin (CDDP), and diepoxybutane (DEB) [9, 11–15]. These crosslinks block ongoing DNA replication, DNA transcription, and if left unrepaired, activate cell apoptosis [16]. The observation that a functional FA pathway is required for processing damage after exposure to crosslinking agents has led to a great deal of research implicating the FA pathway in crosslink repair and the maintenance of genomic stability [17, 18]. Additionally, since the FA pathway has also been associated with cancer susceptibility, a better understanding of the mechanisms and roles of this pathway will enable the development of better-targeted cancer therapeutics.
In this review will we will focus on the repair of DNA interstrand crosslinks (ICLs) by the FA network of proteins. We aim to summarize our current understanding of ICL repair largely based on studies in the mammalian system. We will discuss the etiology of ICLs, the DNA repair pathways involved in the repair of ICLs, FA proteins, FA-DNA repair network and conclude with a perspective on targeting the FA pathway to identify anticancer therapeutic strategies.
Interstrand crosslinks
ICLs are highly toxic DNA lesions that prevent the separation of the Watson and Crick strands of the double helix by covalently linking the two DNA strands. In doing so ICLs block critical cellular processes such as transcription and replication. ICLs can lead to gross-chromosomal aberrations like chromosome deletion, chromosome loss and DNA breaks [19]. The ability of ICLs to impede DNA replication and thereby block cell proliferation is used in chemotherapy to treat various cancers [20]. Chemotherapeutic drugs like cisplatin and its derivatives, carboplatin and oxaliplatin are bifunctional alkylating agents that form ICLs [21]. Although ICL repair remains poorly understood, factors involved in nucleotide excision repair (NER), homologous recombination (HR), and translesion synthesis (TLS) have been implicated in ICL removal and subsequent repair [22]. In non-proliferating cells such as quiescent cells, NER plays an important role in ICL recognition and removal [23, 24]. In contrast, in cells undergoing genome duplication, the DNA replication machinery serves as a sensor for ICLs. This subsequently triggers DNA damage checkpoint activation and initiates repair. In these S-phase cells, HR and TLS are the DSBR pathways employed for ICL repair [24]. In the past several years the role of FA network of proteins in the detection and repair of ICLs by promoting HR has been much better understood.
Mechanistic insights into replication-dependent ICL repair
ICL repair is initiated when a traveling replication fork is stalled due to collision with a lesion on the DNA that triggers the activation of the DNA repair machinery [12, 22, 25]. Structure-specific endonucleases generate incisions on either side of the ICL, followed by TLS and then HR-mediated replication fork restart allows for the rescue of such stalled forks [12] (Fig. 1). It is important to note that majority of ICL repair in dividing cells is coupled to DNA replication. In mammalian cells, irrespective of the cell-cycle phase where the ICL is formed, the repair occurs exclusively during S-phase i.e., replication-dependent ICL repair [26].
Fig. 1.
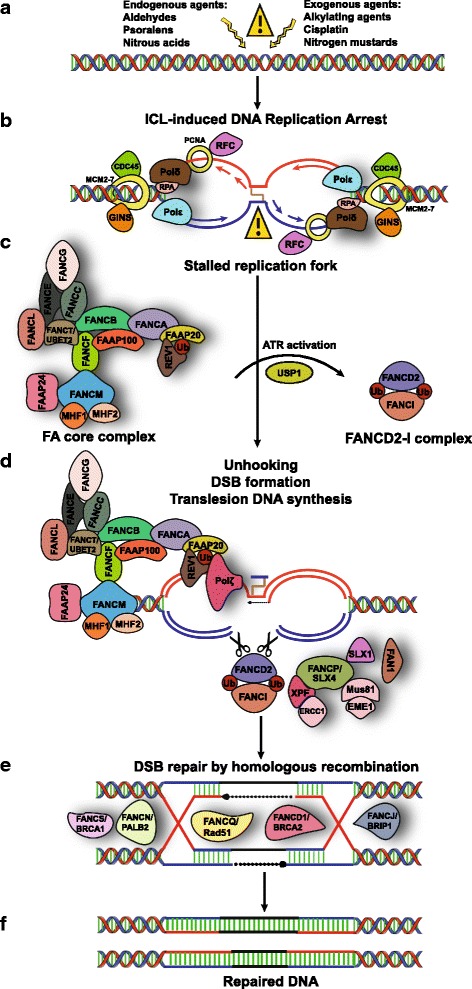
A model for the DNA interstrand crosslink (ICL) repair: Crosstalk between the Fanconi Anemia (FA) pathway, translesion synthesis (TLS) and homologous recombination (HR). a Certain endogenous, environmental sources and chemotherapeutic agents inflict damage to the DNA forming adducts between each DNA strands creating inter-strand crosslinks. b Two replication forks converge at the DNA ICL covalently linking the Watson and Crick strands of the DNA. The replication machinery encounters the DNA lesion at the fork leading to fork stalling. c The FA core complex detects the stalled replication fork, assembles on the DNA lesion and initiates checkpoint response by activating ATR, which in turn phosphorylates multiple FA proteins. This triggers the ubiquitin ligase activity of FANCL resulting in monoubiquitination of FANCD2 and FANCI. d The FANCD2-FANCI heterodimeric complex is recruited to the ICL site. This further recruits downstream nucleases, in particular structure specific endonucleases like SLX4 (FANCP), ERCC1-XPF, FAN1 and MUS81-EME1 to coordinate nucleolytic incisions flanking the ICL. The incisions unhook the ICL leaving crosslinked nucleotides tethered to the complementary strand. FAAP20 interacts with the FA core complex and binds to monoubiquitinated REV1. This catalyze TLS-dependent lesion bypass across the adduct, mediated by specialized TLS polymerases such as REV1 and Polζ. This restores the integrity of the template strand required for the progression of the nascent leading strand. e DSB generated after nucleolytic incisions serves as a suitable substrate for repair by the HR pathway. Downstream FA proteins promote RAD51-dependent strand invasion forming the synaptic filament. Branch migration and intermediates containing Holliday junctions are formed. f The resulting double Holliday junction is resolved by HR specific nucleases, HR repair is completed and the integrity of the DNA is restored
Mechanistic details of replication-dependent ICL repair emerged from studies in Xenopus egg extracts where replication-coupled ICL repair was reconstituted in vitro by using site-specific ICL templates [27]. When a plasmid containing a site-specific ICL is incubated in this cell-free system, replication initiates at multiple origins of replication sites on the plasmid with two replication forks converging on the ICL. Initially, the leading strand polymerases stall ~20 nucleotides from the crosslink due to steric hindrance by the replisome (replicative helicase complex consisting of Cdc45, MCM2-7 and the GINS, collectively referred to as the CMG complex, and the replication polymerase) [27–29] which travels along the leading strand template and pauses at the lesion [30] (Fig. 1). After the initial fork pause, the stalled CMGs are unloaded and lesion bypass is initiated when the leading strand of a single fork is extended to within 1 nucleotide of the ICL lesion [30, 31]. Concurrent with this, the structure-specific endonucleases localize to the site of the ICL and promote dual incisions on either side of the ICL, a process also referred to as “unhooking” of the ICL [32]. A number of endonucleases have been implicated in the incision events of ICL repair including the 3′ flap endonuclease XPF-ERCC1, MUS81-EME1, FAN1, the 5′ flap endonuclease SLX1 and the scaffolding protein SLX4 [33–44]. TLS polymerases then fill in the gap at the site of the DNA incision. TLS incorporates a nucleotide across the ICL lesion by utilizing the error-prone DNA polymerase ζ. This allows the leading strand to be extended and ligated to the first downstream Okazaki fragment [12, 45, 46]. Finally, the broken sister chromatids generated by incision generates a DSB in the DNA that is repaired by RAD51-mediated HR utilizing the intact sister chromatid as a homology donor [47, 48] (Fig. 1).
In recent years the role of FA network of proteins in replication-dependent ICL repair has been the subject of intense research in many laboratories. In this section, we summarize the functions of the FA network of proteins in ICL repair and discuss the mechanisms by which they function in the repair of ICLs by promoting HR.
Overview of the Fanconi Anemia DNA damage response pathway
The FA pathway is a nuclear multi-protein network comprised of 20 complementation groups and associated genes. Interestingly, 19 of the 20 genes of this network are autosomally inherited with the notable exception of FANCB. FANCB is localized on the X chromosome and its mutation has only been observed in males [2]. The genes were identified by methods such as, complementation analysis of cell lines from different FA patients, positional cloning, biochemical purification, and by sequencing candidate genes [49, 50]. The proteins encoded by these genes make up the FA network of proteins that cooperate in the DNA damage response (DDR) for the cellular resistance to ICLs (Fig. 1). These proteins have been placed into three groups based on the stage of ICL repair they participate in [15]. Group I, also referred to as the FA core complex consists of FANCA, FANCB, FANCC, FANCE, FANCF, FANCG, FANCL, FANCM and FANCT (UBET2) along with five additional proteins that associate with the FA core complex, including FAAP100, FAAP24, FAAP20, and the histone fold dimer proteins MHF1 and MHF2 [51–61]. Group II also referred to as the ID complex consists of FANCD2 and FANCI proteins [62–64]. Group III proteins include the DNA repair factors including HR proteins BRCA2 (FANCD1), BRIP1 (FANCJ), PALB2 (FANCN), RAD51C (FANCO), RAD51 (FANCR), SLX4 (FANCP), BRCA1 (FANCS), and XRCC2 (FANCU), TLS gene REV7 (FANCV) and DNA endonuclease XPF (FANCQ) [60, 65, 66]. Some patients with FA-like cellular phenotypes are yet to be assigned a FA-subtype indicating that additional FA or FA-associated genes are yet to be identified [11].
The FA Core complex
FANCM is a DNA translocase which together with Fanconi anemia-associated protein 24 (FAAP24), FAAP 100 and the histone fold proteins MHF1 (FAAP16 or CENPS) and MHF2 (FAAP10 or CENPX) is responsible for lesion recognition and recruitment of the core complex which comprises of FANCA, FANCB, FANCC, FANCE, FANCF, FANCG, FANCL, FANCT, and FAAP20 to the ICL site [56, 67–69] (Fig. 1). It is important to note that recruitment of FANCM to ICLs is dependent on its phosphorylation by the ataxia telangiectasia and RAD3-related (ATR) checkpoint kinase [70]. Once recruited to the site of damage, the FA core complex serves as a multi-subunit ubiquitin E3 ligase for two other FA proteins, FANCD2 and FANCI [71]. FANCD2 is phosphorylated in an ATR-dependent manner which is essential for FANCD2 monoubiquitination and the establishment of the intra-S-phase checkpoint response [72]. Phosphorylation of FANCI is also essential for the monoubiquitination and localization of the FANCD2–I heterodimeric complex to DNA damage sites [73]. The phosphorylated FANCD2–I complex is subsequently monoubiquitinated by the FA core complex through its catalytic subunits, FANCL (the E3 ligase) and UBE2T (the ubiquitin E2 ligase also known as FANCT) [74–77]. Ubiquitinated PCNA also stimulates FANCD2 and FANCI monoubiquitination in vitro [78–80]. The ubiquitinated FANCD2–I complex is then recruited to chromatin by UHRF1 (ubiquitin-like with PHD and RING finger domains 1) protein that is involved in ICL sensing [81, 82].
Ubiquitination of FANCD2–I is a reversible regulatory modification. Deubiquitination of the FANCD2–I complex is required to release FANCD2 from the DNA repair complex crucial for subsequent repair steps to complete ICL repair [83–85]. The deubiquitination of FANCD2–I relies on USP1 (ubiquitin carboxy-terminal hydrolase 1) in conjunction with UAF1 (USP1-associated factor 1) [83, 86].
DNA incision and Translesion repair
Ubiquination of the FANCD2–I complex is crucial for the recruitment of nucleases to the site of the ICL to orchestrate nucleolytic incision of the ICL. This facilitates ‘unhooking’ of the ICL from one of the two parental DNA strands to uncouple one sister chromatid from the other [32] (Fig. 1). FANCD2-Ub recruits the nuclease scaffold protein SLX4 (FANCP) by an interaction with ubiquitin-recognizing UBZ4 motif [35, 36]. SLX4 (FANCP) functions as a molecular platform to coordinate, recruit and activate other structure-specific endonucleases like XPF-ERCC1, MUS81-EME1 and SLX1 to aid ICL repair [87–90]. Interestingly, in vitro studies have shown that XPF–ERCC1–SLX4 complex is the essential nuclease for ICL unhooking whereas MUS81-EME1, SLX1 and FAN1 (Fanconi-associated nuclease 1, another structure-specific nuclease that acts in a FANCP independent manner) possess redundant ICL processing activities [44, 91]. It is important to note that in human cells, the recruitment of XPF at sites of ICL damage is dependent on the structural protein nonerythroid αspectrin (αIISp) during the S-phase of the cell cycle [92–94]. After unhooking of the ICL lesion, ubiquitinated PCNA and the FA core complex recruit translesion synthesis polymerases to coordinate the next step of ICL repair. Translesion DNA polymerases such as REV7 (FANCV), polymerase ζ and polymerase η fill the single-strand DNA (ssDNA) gaps resulting from ICL unhooking. Translesion DNA polymerases have larger binding pockets compared to replicative polymerases and can accommodate bulky ICL adducts thereby incorporating nucleotides opposite to the ICL and filling the DNA gap [95, 96].
Downstream Effector complex
In addition to ssDNA gaps formed in one strand of the double helix, unhooking results in the formation of DSB afflicting both strands. Repair of DSBs relies on the HR pathway (Fig. 1). Consistent with this, cells deficient in HR proteins display hypersensitivity to ICL agents [47, 97]. FA proteins involved in HR are not required for FANCD2–I monoubiquitination suggesting they function downstream of the FANCD2–I complex. Several FA factors have been shown to promote different stages of HR [60]. BRCA2 (FANCD1), FANCO (RAD51C) and PALB2 (FANCN) help load RAD51 onto ssDNA by displacing RPA, which specifically promotes RAD51-dependent nucleofilament formation and also stimulates RAD51-dependent strand invasion of a homologous DNA template [98–100]. End resection is a key step in DSBR and initiates HR. FANCD2 and BRCA1 (FANCS) promote the recruitment of the resection factor CtIP at the site of DSBs to initiate HR [101–104]. FANCC has been implicated in inhibiting non-homologous end joining (NHEJ) factors from accessing the DSB ends thus preventing NHEJ and thereby promoting HR [105]. FANCJ’s (BRIP) 5′ to 3′ helicase activity has been shown to unwind D-loops and may be involved in resolving RAD51 nucleofilaments [106].
Regulation of the FA network of proteins
ICL repair is a highly complex process involving the FA pathway as well as other repair pathways that needs to be tightly controlled. Post-translational modifications (PTMs) and protein-protein interactions are crucial for the regulation of this process. ATR plays a major regulatory role in the activation of the FA pathway. This kinase is responsible for the phosphorylation of the FANCD2-I heterodimer in the S-phase, which is indispensible for efficient FANCD2 ubiquitination and focus formation [72, 107, 108]. ATR also phosphorylates FANCA, FANCG and FANCM to promote efficient crosslink repair [109–113]. Chk1 also negatively regulates the FA pathway by phosphorylating FANCE to trigger its proteasomal degradation [114]. Ubiquitination of various FANC proteins is crucial for the regulation of the FA pathway. Monoubiquitination of the FANCD2-I complex by the FANCL-UBE2T is crucial for recruitment of the core complex to damaged DNA [115, 116]. Additionally, ubiquitination of effector proteins like FANCN, FANCS and FANCG have been implicated in the regulation of ICL repair [117, 118]. Deubiquitination of FANCD2 and FANCI by the constitutively active deubiquitinating complex UAF1-USP1 keeps the pathway turned off unless required [86]. Upon DNA damage, the activity of UAF1-USP1 is repressed either by proteosomal degradation of USP1 or by transcription repression of the USP1 gene [86]. Finally, SUMOylation plays a pivotal role in the regulation to FA-mediated ICL repair [119]. SUMOylation of FANCD2 and FANCI by PIAS1/4 and UBC9 promotes polyubiquitination of the complex, which in turn promotes dissociation of FANCD2 and FANCI from chromatin [120].
FA factors as therapeutic targets in cancer
A hallmark of cancer cells is genome instability. This can be attributed to a failure of the DNA repair machinery, which essentially acts as a tumor suppressor network to preserve genome integrity and prevent malignancy. The link between FA and cancer predisposition has been well established with FA patient populations exhibiting a wide range of cancers [121]. Almost 25% of FA patients develop malignancies [121]. Although the most common malignancies are either hematologic, like myelodysplastic syndrome and AML or solid tumors, particularly squamous cell carcinomas of the head and neck [121], recently FA proteins mutations have been reported in familial and sporadic cancers outside the FA patient population [121]. For instance, FANCD1 mutations have been associated with ovarian, breast, prostate, stomach and pancreatic cancers [122]. FANCL mutations have been associated with lung cancer, pancreatic cancer, breast cancer and leukemia [123, 124]. FANCD2 mutations have been associated with breast cancer [125]. FANCN mutations have been reported in prostate and breast cancer [126]. FANCC and FANCG have also been implicated in pancreatic cancer, breast cancer and leukemia [124, 127, 128].
Leveraging synthetic lethal interactions with the FA pathway for cancer therapeutics
A major drawback of chemotherapy lies in the fact that it is not selective, i.e., it kills both cancer cells and normal cells indiscriminately. However, inactivation/defects in DNA repair pathways can make cancer cells over-dependent on a compensatory DNA repair pathway for survival. Current approaches for cancer therapy that rely on inhibiting the intact functional DNA repair pathways by using a synthetic lethal approach can provide a therapeutic strategy for specific killing of such tumors. Two genes are said to be in a synthetic lethal relationship if a mutation in either gene alone is not lethal but simultaneous mutations are lethal [48, 129]. A new approach is directed at exploiting the synthetic lethality of cancer cells that are defective in the FA pathway [130].
The best example of the therapeutic potential of the synthetic lethality approach is development of poly(adenosine diphosphate [ADP]–ribose) polymerase 1 (PARP1) inhibitors to treat breast and ovarian cancers carrying mutations in the tumor-suppressor genes BRCA1 or BRCA2 [131, 132] (Fig. 2). Recognition of DNA breaks by PARP1 is one of the earliest events in DSBR. Once a DNA strand break is formed, PARP1 binds to the broken DNA ends and facilitates chromatin decondensation at the break site [133]. This allows repair enzymes to access the damaged DNA sites [133]. Inhibition or deletion of PARP1 leads to inactivation of the single strand break repair (SSBR) pathways including NER, base excision repair (BER), mismatch repair (MMR) which leads to the accumulation SSBs which may subsequently lead to the formation of DSBs [133]. BRCA1 and BRCA2 are also key participants in HR. In normal cells, loss of activity of PARP1 enzyme induces high levels of DSBR through the HR pathway during the S-phase of the cell cycle. Cancer cells that are defective in HR are selectively sensitive to PARP inhibition due to the simultaneous loss of two DNA repair pathways. Thus, treating cells carrying BRCA1 or BRCA2 mutations with small-molecule inhibitors of PARP1 are lethal as the cells are deficient in DSBR. This results in targeted killing of the cancerous cells, while cells with intact HR can repair the damage and survive [134] (Fig. 2).
Fig. 2.
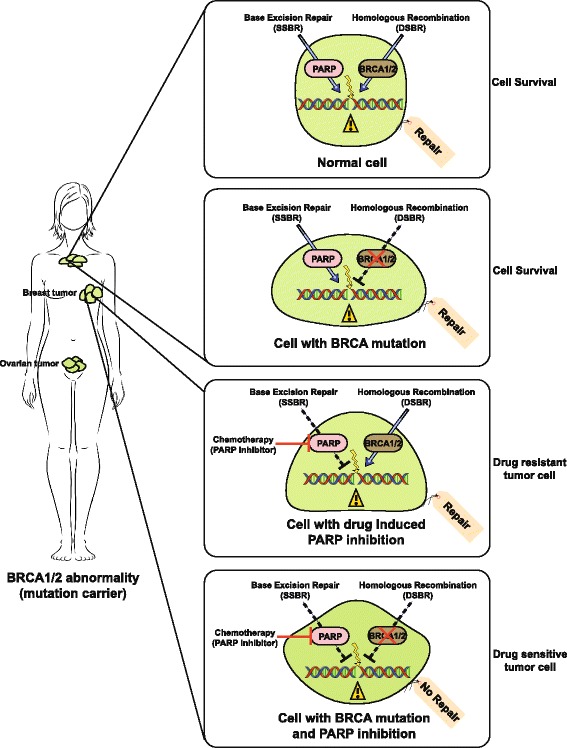
Synthetic lethal interactions to identify molecular targets for cancer therapy: Sensitizing genetically defined tumor cells by targeted inhibition of DNA damage repair pathways. A model for synthetic lethality using PARP inhibitors. In breast/ovarian tumor cells, mutation in BRCA1/2 leaves the cancer cell vulnerable to chemotherapeutic drugs against single strand break repair (SSBR). In contrast, cells with functional BRCA1/2 genes are spared as they can repair the lesions on the DNA using double strand break repair (DSBR) pathway. Compromised base excision repair (BER) pathway combined with homologous recombination (HR) deficiency leads to tumor cell death
Synthetic lethal interactions with the FA pathway for the development of inhibitors have been explored. A siRNA-based synthetic lethal screening identified several genes including ATM, PARP1, CDK1, NBS1, and PLK1 that showed synthetic lethal interactions with FANCG, indicating that these genes could be targeted concomitant with a FA pathway inhibitor [135]. Since ATM deficiency has been reported in triple- negative breast cancer and several types of hematological malignancies like mantle cell lymphoma, chronic lymphocytic leukemia, and acute lymphoblastic leukemia [136, 137], the FA pathway inhibitor could have immense therapeutic potential. CHK1 inhibition has also been shown to be synthetically lethal with FANCA deficiency following cisplatin treatment [138].
Several small molecule inhibitors have been identified that inhibit specific components of the FA pathway. This in turn leads to inhibition of FANCD2 foci formation and abrogation of the FA pathway. For example, wortmannin (inhibits ATR kinase), H-9 (inhibits several kinases including protein kinase A, G, and C), alsterpaullone (inhibits cyclin-dependent kinase 1 and 5), phenylbutyrate (inhibits FANCS) and curcumin (inhibits FANCF) are some of the small-molecule inhibitors of the FA/BRCA pathway that have already been identified by high-throughput screen using human cells and are now in various stages of subsequent validation [139, 140]. Bortezomib, the natural compound curcumin and its analogs such as EF24 and 4H-TTD and MLN4924 have been shown to impair FANCD2 activation and sensitize cancer cells to ICL-inducing agents [18, 139, 141]. USP1 inhibitors like C527, pimozide and GW7647 affect the ubiquitin-deubiquitination cycle of FANCD2 leading to the selective inhibition of the FA pathway [142–144]. Understanding the mechanism by which these compounds chemically inhibit the FA/BRCA2 pathway is crucial for translating this research from the laboratory to the clinic. For instance, phenylbutyrate sensitizes head and neck cancer cells to cisplatin by specifically attenuating FANCS thereby inhibiting FANCD2 foci formation and abrogating the FA/BRCA pathway [140]. This observation makes phenylbutyrate an excellent candidate for sensitizing cisplatin-resistant head and neck tumors in a clinical setting [140]. Curcumin (diferuloylmethane), a low-molecular-weight polyphenol and a component in the spice turmeric inhibits FANCF [139]. Since FANCF acts upstream in the FA/BRCA pathway, inhibition of FANCF attenuates monoubiquitination of FANCD2 and FANCD2 foci formation [139]. In ovarian and breast tumor cell lines, curcumin-mediated inhibition of the FA/BRCA pathway sensitizes tumor cells to cisplatin by inducing apoptotic cell death. This opens up the possibility that curcumin could be used to sensitize cisplatin-resistant ovarian and breast tumors in the clinic. The precise inhibition of the FA pathway in combination with DNA repair inhibitors could increase the efficacy of chemotherapy and improve current cancer treatment regimens.
Conclusion
Understanding the molecular details of the DNA damage response is essential for advancing cancer research. Due to the critical importance of the FA network in maintaining genome stability and the current limitations in treating FA patients in the clinic, a large body of research has been directed to this subject. The FA pathway plays a central role in ICL repair during which the FA proteins function to coordinate NER factors, TLS polymerase, HR factors and checkpoint kinases to ensure genome stability. In the absence of a functional FA pathway, cells are predisposed to spontaneous and DNA damage-induced chromosomal breaks. More research into the FA DNA repair pathway will identify novel factors that can be specifically inhibited. Such targeted modulation of the FA pathway by exploiting synthetic lethal relationships may play an important role for the development of new cancer treatments and potential development of personalized therapies.
Acknowledgements
We apologize to those colleagues whose work has not been cited due to space limitation.
Funding
Not applicable
Availability of data and materials
Not applicable
Abbreviations
- AML
Acute myelogenous leukemia
- ATR
Ataxia telangiectasia and RAD3-related
- CDDP
Chemotherapeutic agent cisplatin
- DDR
DNA damage response
- DEB
Diepoxybutane
- DSB
Double strand break
- DSBR
Double strand break repair
- dsDNA
Double-strand DNA
- FA
Fanconi Anemia
- FAN1
Fanconi-associated nuclease 1
- HR
Homologous recombination
- ICLs
Interstrand DNA crosslinks
- MMC
Mitomycin C
- NER
Nucleotide excision repair
- PTMs
Post-translational modifications
- ssDNA
Single-strand DNA
- TLS
Translesion synthesis
- UAF1
USP1-associated factor 1
- UHRF1
Ubiquitin-like with PHD and RING finger domains 1
- USP1
Ubiquitin carboxy-terminal hydrolase 1
Authors’ contributions
SB and SN were involved in the conception, design and drafting of the manuscript. Both authors read and approved the final manuscript.
Ethics approval and consent to participate
Not applicable
Consent for publication
Not applicable
Competing interests
The authors declare that they have no competing interest.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Sonali Bhattacharjee, Email: bhattacharjee@cshl.edu.
Saikat Nandi, Email: snandi@cshl.edu.
References
- 1.Mathew CG. Fanconi anaemia genes and susceptibility to cancer. Oncogene. 2006;25(43):5875–5884. doi: 10.1038/sj.onc.1209878. [DOI] [PubMed] [Google Scholar]
- 2.Meetei AR, et al. X-linked inheritance of Fanconi anemia complementation group B. Nat Genet. 2004;36(11):1219–1224. doi: 10.1038/ng1458. [DOI] [PubMed] [Google Scholar]
- 3.Su XY, Huang J. The Fanconi anemia pathway and DNA interstrand cross-link repair. Protein Cell. 2011;2(9):704–711. doi: 10.1007/s13238-011-1098-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Alter BP. Fanconi anemia and the development of leukemia. Best Pract Res Clin Haematol. 2014;27(3-4):214–221. doi: 10.1016/j.beha.2014.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rosenberg PS, Tamary H, Alter BP. How high are carrier frequencies of rare recessive syndromes? Contemporary estimates for Fanconi Anemia in the United States and Israel. Am J Med Genet A. 2011;155A(8):1877–1883. doi: 10.1002/ajmg.a.34087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Garaycoechea JI, et al. Genotoxic consequences of endogenous aldehydes on mouse haematopoietic stem cell function. Nature. 2012;489(7417):571–575. doi: 10.1038/nature11368. [DOI] [PubMed] [Google Scholar]
- 7.Ceccaldi R, et al. Bone marrow failure in Fanconi Anemia is triggered by an exacerbated p53/p21 DNA damage response that impairs hematopoietic stem and progenitor cells. Cell Stem Cell. 2012;11(1):36–49. doi: 10.1016/j.stem.2012.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lobitz S, Velleuer E. Guido Fanconi (1892-1979): a jack of all trades. Nat Rev Cancer. 2006;6(11):893–898. doi: 10.1038/nrc2009. [DOI] [PubMed] [Google Scholar]
- 9.Schroeder TM, Kurth R. Spontaneous chromosomal breakage and high incidence of leukemia in inherited disease. Blood. 1971;37(1):96–112. [PubMed] [Google Scholar]
- 10.German J. Genes which increase chromosomal instability in somatic cells and predispose to cancer. Prog Med Gen. 1972;8:61. [PubMed] [Google Scholar]
- 11.Moldovan GL, D'Andrea AD. How the fanconi anemia pathway guards the genome. Annu Rev Genet. 2009;43:223–249. doi: 10.1146/annurev-genet-102108-134222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Niedernhofer LJ, Lalai AS, Hoeijmakers JH. Fanconi anemia (cross)linked to DNA repair. Cell. 2005;123(7):1191–1198. doi: 10.1016/j.cell.2005.12.009. [DOI] [PubMed] [Google Scholar]
- 13.Patel KJ, Joenje H. Fanconi anemia and DNA replication repair. DNA Repair (Amst) 2007;6(7):885–890. doi: 10.1016/j.dnarep.2007.02.002. [DOI] [PubMed] [Google Scholar]
- 14.Thompson LH, Hinz JM. Cellular and molecular consequences of defective Fanconi anemia proteins in replication-coupled DNA repair: mechanistic insights. Mutat Res. 2009;668(1-2):54–72. doi: 10.1016/j.mrfmmm.2009.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang W. Emergence of a DNA-damage response network consisting of Fanconi anaemia and BRCA proteins. Nat Rev Genet. 2007;8(10):735–748. doi: 10.1038/nrg2159. [DOI] [PubMed] [Google Scholar]
- 16.Jung Y, Lippard SJ. Direct cellular responses to platinum-induced DNA damage. Chem Rev. 2007;107(5):1387–1407. doi: 10.1021/cr068207j. [DOI] [PubMed] [Google Scholar]
- 17.Taniguchi T, et al. Disruption of the Fanconi anemia–BRCA pathway in cisplatin-sensitive ovarian tumors. Nat Med. 2003;9(5):568–574. doi: 10.1038/nm852. [DOI] [PubMed] [Google Scholar]
- 18.Landais I, et al. A novel cell-free screen identifies a potent inhibitor of the Fanconi anemia pathway. Int J Cancer. 2009;124(4):783–792. doi: 10.1002/ijc.24039. [DOI] [PubMed] [Google Scholar]
- 19.Vogel EW, et al. Heritable and cancer risks of exposures to anticancer drugs: inter-species comparisons of covalent deoxyribonucleic acid-binding agents. Mutat Res. 1998;400(1-2):509–540. doi: 10.1016/S0027-5107(98)00060-8. [DOI] [PubMed] [Google Scholar]
- 20.Stern RS. Psoralen and ultraviolet a light therapy for psoriasis. N Engl J Med. 2007;357(7):682–690. doi: 10.1056/NEJMct072317. [DOI] [PubMed] [Google Scholar]
- 21.Clauson C, Scharer OD, Niedernhofer L. Advances in understanding the complex mechanisms of DNA interstrand cross-link repair. Cold Spring Harb Perspect Biol. 2013;5(10):a012732. doi: 10.1101/cshperspect.a012732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.McHugh PJ, Spanswick VJ, Hartley JA. Repair of DNA interstrand crosslinks: molecular mechanisms and clinical relevance. Lancet Oncol. 2001;2(8):483–490. doi: 10.1016/S1470-2045(01)00454-5. [DOI] [PubMed] [Google Scholar]
- 23.Nojima K, et al. Multiple repair pathways mediate tolerance to chemotherapeutic cross-linking agents in vertebrate cells. Cancer Res. 2005;65(24):11704–11711. doi: 10.1158/0008-5472.CAN-05-1214. [DOI] [PubMed] [Google Scholar]
- 24.Ben-Yehoyada M, et al. Checkpoint signaling from a single DNA interstrand crosslink. Mol Cell. 2009;35(5):704–715. doi: 10.1016/j.molcel.2009.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Thompson LH, et al. How Fanconi anemia proteins promote the four Rs: replication, recombination, repair, and recovery. Environ Mol Mutagen. 2005;45(2-3):128–142. doi: 10.1002/em.20109. [DOI] [PubMed] [Google Scholar]
- 26.Akkari YM, et al. DNA replication is required to elicit cellular responses to Psoralen-induced DNA Interstrand cross-links. Mol Cell Biol. 2000;20(21):8283–8289. doi: 10.1128/MCB.20.21.8283-8289.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Raschle M, et al. Mechanism of replication-coupled DNA interstrand crosslink repair. Cell. 2008;134(6):969–980. doi: 10.1016/j.cell.2008.08.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhu W, Abbas T, Dutta A. DNA replication and genomic instability. Adv Exp Med Biol. 2005;570:249–279. doi: 10.1007/1-4020-3764-3_9. [DOI] [PubMed] [Google Scholar]
- 29.Sclafani RA, Holzen TM. Cell cycle regulation of DNA replication. Annu Rev Genet. 2007;41:237–280. doi: 10.1146/annurev.genet.41.110306.130308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fu YV, et al. Selective bypass of a lagging strand roadblock by the eukaryotic replicative DNA helicase. Cell. 2011;146(6):931–941. doi: 10.1016/j.cell.2011.07.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Long DT, et al. BRCA1 promotes unloading of the CMG helicase from a stalled DNA replication fork. Mol Cell. 2014;56(1):174–185. doi: 10.1016/j.molcel.2014.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Knipscheer P, et al. The Fanconi anemia pathway promotes replication-dependent DNA interstrand cross-link repair. Science. 2009;326(5960):1698–1701. doi: 10.1126/science.1182372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ciccia A, McDonald N, West SC. Structural and functional relationships of the XPF/MUS81 family of proteins. Annu Rev Biochem. 2008;77:259–287. doi: 10.1146/annurev.biochem.77.070306.102408. [DOI] [PubMed] [Google Scholar]
- 34.Sengerova B, Wang AT, McHugh PJ. Orchestrating the nucleases involved in DNA interstrand cross-link (ICL) repair. Cell Cycle. 2011;10(23):3999–4008. doi: 10.4161/cc.10.23.18385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yamamoto KN, et al. Involvement of SLX4 in interstrand cross-link repair is regulated by the Fanconi anemia pathway. Proc Natl Acad Sci U S A. 2011;108(16):6492–6496. doi: 10.1073/pnas.1018487108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Crossan GP, et al. Disruption of mouse Slx4, a regulator of structure-specific nucleases, phenocopies Fanconi anemia. Nat Genet. 2011;43(2):147–U99. doi: 10.1038/ng.752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hodskinson MRG, et al. Mouse SLX4 is a tumor suppressor that stimulates the activity of the nuclease XPF-ERCC1 in DNA crosslink repair. Mol Cell. 2014;54(3):472–484. doi: 10.1016/j.molcel.2014.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Douwel DK, et al. XPF-ERCC1 acts in unhooking DNA Interstrand Crosslinks in cooperation with FANCD2 and FANCP/SLX4. Mol Cell. 2014;54(3):460–471. doi: 10.1016/j.molcel.2014.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Guervilly JH, et al. The SLX4 complex is a SUMO E3 Ligase that impacts on replication stress outcome and genome stability. Mol Cell. 2015;57(1):123–137. doi: 10.1016/j.molcel.2014.11.014. [DOI] [PubMed] [Google Scholar]
- 40.Liu T, et al. FAN1 acts with FANCI-FANCD2 to promote DNA Interstrand cross-link repair. Science. 2010;329(5992):693–696. doi: 10.1126/science.1192656. [DOI] [PubMed] [Google Scholar]
- 41.Smogorzewska A, et al. A genetic screen identifies FAN1, a Fanconi Anemia-associated nuclease necessary for DNA Interstrand crosslink repair. Mol Cell. 2010;39(1):36–47. doi: 10.1016/j.molcel.2010.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhao Q, et al. Structural insights into 5 ‘ flap DNA unwinding and incision by the human FAN1 dimer. Nat Commun. 2014;5:5726. [DOI] [PMC free article] [PubMed]
- 43.Takahashi D, et al. Human FAN1 promotes strand incision in 5 ‘-flapped DNA complexed with RPA. J Biochem. 2015;158(3):263–270. doi: 10.1093/jb/mvv043. [DOI] [PubMed] [Google Scholar]
- 44.Pizzolato J, et al. FANCD2-associated nuclease 1, but not exonuclease 1 or flap Endonuclease 1, is able to unhook DNA Interstrand cross-links in vitro. J Biol Chem. 2015;290(37):22602–22611. doi: 10.1074/jbc.M115.663666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mirchandani KD, D'Andrea AD. The Fanconi anemia/BRCA pathway: a coordinator of cross-link repair. Exp Cell Res. 2006;312(14):2647–2653. doi: 10.1016/j.yexcr.2006.06.014. [DOI] [PubMed] [Google Scholar]
- 46.Prakash S, Johnson RE, Prakash L. Eukaryotic translesion synthesis DNA polymerases: specificity of structure and function. Annu Rev Biochem. 2005;74:317–353. doi: 10.1146/annurev.biochem.74.082803.133250. [DOI] [PubMed] [Google Scholar]
- 47.Long DT, et al. Mechanism of RAD51-dependent DNA interstrand cross-link repair. Science. 2011;333(6038):84–87. doi: 10.1126/science.1204258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bhattacharjee S, Nandi S. Choices have consequences: the nexus between DNA repair pathways and genomic instability in cancer. Clin Transl Med. 2016;5(1):45. doi: 10.1186/s40169-016-0128-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kee Y, D'Andrea AD. Expanded roles of the Fanconi anemia pathway in preserving genomic stability. Genes Dev. 2010;24(16):1680–1694. doi: 10.1101/gad.1955310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wang LC, Gautier J. The Fanconi anemia pathway and ICL repair: implications for cancer therapy. Crit Rev Biochem Mol Biol. 2010;45(5):424–439. doi: 10.3109/10409238.2010.502166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sun W, et al. The FANCM Ortholog Fml1 promotes recombination at stalled replication forks and limits crossing over during DNA double-strand break repair. Mol Cell. 2008;32(1):118–128. doi: 10.1016/j.molcel.2008.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Nandi S, Whitby MC. The ATPase activity of Fml1 is essential for its roles in homologous recombination and DNA repair. Nucleic Acids Res. 2012;40(19):9584–9595. doi: 10.1093/nar/gks715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bhattacharjee S, et al. MHF1-2/CENP-S-X performs distinct roles in centromere metabolism and genetic recombination. Open Biology. 2013:3(9):130102. [DOI] [PMC free article] [PubMed] [Retracted]
- 54.Dong HB, et al. Update of the human and mouse Fanconi anemia genes. Human Genomics. 2015;9:32. [DOI] [PMC free article] [PubMed]
- 55.Chen H, Zhang S, Wu Z. Fanconi anemia pathway defects in inherited and sporadic cancers. Transl Pediatr. 2014;3(4):300–4. doi: 10.3978/j.issn.2224-4336.2014.07.05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ciccia A, et al. Identification of FAAP24, a Fanconi anemia core complex protein that interacts with FANCM. Mol Cell. 2007;25(3):331–343. doi: 10.1016/j.molcel.2007.01.003. [DOI] [PubMed] [Google Scholar]
- 57.Ling C, et al. FAAP100 is essential for activation of the Fanconi anemia-associated DNA damage response pathway. EMBO J. 2007;26(8):2104–2114. doi: 10.1038/sj.emboj.7601666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ali AM, et al. FAAP20: a novel ubiquitin-binding FA nuclear core-complex protein required for functional integrity of the FA-BRCA DNA repair pathway. Blood. 2012;119(14):3285–3294. doi: 10.1182/blood-2011-10-385963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kim H, et al. Regulation of Rev1 by the Fanconi anemia core complex. Nat Struct Mol Biol. 2012;19(2):164–170. doi: 10.1038/nsmb.2222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ceccaldi R, Sarangi P, D'Andrea AD. The Fanconi anaemia pathway: new players and new functions. Nat Rev Mol Cell Biol. 2016;17(6):337–349. doi: 10.1038/nrm.2016.48. [DOI] [PubMed] [Google Scholar]
- 61.Cheung RS, et al. Ubiquitination-linked Phosphorylation of the FANCI S/TQ cluster contributes to activation of the Fanconi anemia I/D2 complex. Cell Rep. 2017;19(12):2432–2440. doi: 10.1016/j.celrep.2017.05.081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.de Oca RM, et al. Regulated interaction of the Fanconi anemia protein, FANCD2, with chromatin. Blood. 2005;105(3):1003–1009. doi: 10.1182/blood-2003-11-3997. [DOI] [PubMed] [Google Scholar]
- 63.Dorsman JC, et al. Identification of the Fanconi anemia complementation group I gene. FANCI Cell Oncol. 2007;29(3):211–218. doi: 10.1155/2007/151968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Smogorzewska A, et al. Identification of the FANCI protein, a monoubiquitinated FANCD2 paralog required for DNA repair. Cell. 2007;129(2):289–301. doi: 10.1016/j.cell.2007.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Duxin JP, Walter JC. What is the DNA repair defect underlying Fanconi anemia? Curr Opin Cell Biol. 2015;37:49–60. doi: 10.1016/j.ceb.2015.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Mamrak NE, Shimamura A, Howlett NG. Recent discoveries in the molecular pathogenesis of the inherited bone marrow failure syndrome Fanconi anemia. Blood Rev. 2017;31(3):93–99. doi: 10.1016/j.blre.2016.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kim JM, et al. Cell cycle-dependent chromatin loading of the Fanconi anemia core complex by FANCM/FAAP24. Blood. 2008;111(10):5215–5222. doi: 10.1182/blood-2007-09-113092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Singh TR, et al. MHF1-MHF2, a histone-fold-containing protein complex, participates in the Fanconi anemia pathway via FANCM. Mol Cell. 2010;37(6):879–886. doi: 10.1016/j.molcel.2010.01.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Yan Z, et al. A histone-fold complex and FANCM form a conserved DNA-remodeling complex to maintain genome stability. Mol Cell. 2010;37(6):865–878. doi: 10.1016/j.molcel.2010.01.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ling C, et al. Bloom syndrome complex promotes FANCM recruitment to stalled replication forks and facilitates both repair and traverse of DNA interstrand crosslinks. Cell Discov. 2016;2:16047. doi: 10.1038/celldisc.2016.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kennedy RD, D'andrea AD. The Fanconi Anemia/BRCA pathway: new faces in the crowd. Genes Dev. 2005;19(24):2925–2940. doi: 10.1101/gad.1370505. [DOI] [PubMed] [Google Scholar]
- 72.Ho GPH, et al. Phosphorylation of FANCD2 on two novel sites is required for mitomycin C resistance. Mol Cell Biol. 2006;26(18):7005–7015. doi: 10.1128/MCB.02018-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Ishiai M, et al. FANCI phosphorylation functions as a molecular switch to turn on the Fanconi anemia pathway. Nat Struct Mol Biol. 2008;15(11):1138–1146. doi: 10.1038/nsmb.1504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Hira A, et al. Mutations in the gene encoding the E2 conjugating enzyme UBE2T cause Fanconi anemia. Am J Hum Genet. 2015;96(6):1001–1007. doi: 10.1016/j.ajhg.2015.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Miles JA, et al. The Fanconi Anemia DNA repair pathway is regulated by an interaction between Ubiquitin and the E2-like fold domain of FANCL. J Biol Chem. 2015;290(34):20995–21006. doi: 10.1074/jbc.M115.675835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Rickman KA, et al. Deficiency of UBE2T, the E2 Ubiquitin Ligase necessary for FANCD2 and FANCI Ubiquitination, causes FA-T subtype of Fanconi anemia. Cell Rep. 2015;12(1):35–41. doi: 10.1016/j.celrep.2015.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Virts EL, et al. AluY-mediated germline deletion, duplication and somatic stem cell reversion in UBE2T defines a new subtype of Fanconi anemia. Hum Mol Genet. 2015;24(18):5093–5108. doi: 10.1093/hmg/ddv227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Howlett NG, et al. Functional interaction between the Fanconi Anemia D2 protein and proliferating cell nuclear antigen (PCNA) via a conserved putative PCNA interaction motif. J Biol Chem. 2009;284(42):28935–28942. doi: 10.1074/jbc.M109.016352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Geng LY, Huntoon CJ, Karnitz LM. RAD18-mediated ubiquitination of PCNA activates the Fanconi anemia DNA repair network. J Cell Biol. 2010;191(2):249–257. doi: 10.1083/jcb.201005101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Williams SA, et al. The E3 ubiquitin ligase RAD18 regulates ubiquitylation and chromatin loading of FANCD2 and FANCI. Blood. 2011;117(19):5078–5087. doi: 10.1182/blood-2010-10-311761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Liang CC, et al. UHRF1 is a sensor for DNA Interstrand Crosslinks and recruits FANCD2 to initiate the Fanconi Anemia pathway. Cell Rep. 2015;10(12):1947–1956. doi: 10.1016/j.celrep.2015.02.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Tian YY, et al. UHRF1 contributes to DNA damage repair as a lesion recognition factor and nuclease scaffold. Cell Rep. 2015;10(12):1957–1966. doi: 10.1016/j.celrep.2015.03.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Nijman SMB, et al. The deubiquitinating enzyme USP1 regulates the Fanconi anemia pathway. Mol Cell. 2005;17(3):331–339. doi: 10.1016/j.molcel.2005.01.008. [DOI] [PubMed] [Google Scholar]
- 84.Oestergaard VH, et al. Deubiquitination of FANCD2 is required for DNA crosslink repair. Mol Cell. 2007;28(5):798–809. doi: 10.1016/j.molcel.2007.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Kim JM, et al. Inactivation of Murine Usp1 results in genomic instability and a Fanconi Anemia phenotype. Dev Cell. 2009;16(2):314–320. doi: 10.1016/j.devcel.2009.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Cohn MA, et al. A UAF1-containing multisubunit protein complex regulates the Fanconi anemia pathway. Mol Cell. 2007;28(5):786–797. doi: 10.1016/j.molcel.2007.09.031. [DOI] [PubMed] [Google Scholar]
- 87.Kim Y, et al. Regulation of multiple DNA repair pathways by the Fanconi anemia protein SLX4. Blood. 2013;121(1):54–63. doi: 10.1182/blood-2012-07-441212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Klein Douwel D, et al. XPF-ERCC1 acts in unhooking DNA interstrand crosslinks in cooperation with FANCD2 and FANCP/SLX4. Mol Cell. 2014;54(3):460–471. doi: 10.1016/j.molcel.2014.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Svendsen JM, et al. Mammalian BTBD12/SLX4 assembles a Holliday junction resolvase and is required for DNA repair. Cell. 2009;138(1):63–77. doi: 10.1016/j.cell.2009.06.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Munoz IM, et al. Coordination of structure-specific nucleases by human SLX4/BTBD12 is required for DNA repair. Mol Cell. 2009;35(1):116–127. doi: 10.1016/j.molcel.2009.06.020. [DOI] [PubMed] [Google Scholar]
- 91.Zhang J, Walter JC. Mechanism and regulation of incisions during DNA interstrand cross-link repair. DNA Repair (Amst) 2014;19:135–142. doi: 10.1016/j.dnarep.2014.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Lambert MW. Nuclear alpha spectrin: critical roles in DNA interstrand cross-link repair and genomic stability. Exp Biol Med (Maywood) 2016;241(15):1621–1638. doi: 10.1177/1535370216662714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.McMahon LW, et al. Human alpha spectrin II and the FANCA, FANCC, and FANCG proteins bind to DNA containing psoralen interstrand cross-links. Biochemistry. 2001;40(24):7025–7034. doi: 10.1021/bi002917g. [DOI] [PubMed] [Google Scholar]
- 94.Sridharan D. Nonerythroid alphaII spectrin is required for recruitment of FANCA and XPF to nuclear foci induced by DNA interstrand cross-links. J Cell Sci. 2003;116(5):823–835. doi: 10.1242/jcs.00294. [DOI] [PubMed] [Google Scholar]
- 95.Waters LS, et al. Eukaryotic Translesion polymerases and their roles and regulation in DNA damage tolerance. Microbiol Mol Biol Rev. 2009;73(1):134. doi: 10.1128/MMBR.00034-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.de Groote FH, et al. The Rev1 translesion synthesis polymerase has multiple distinct DNA binding modes. DNA Repair. 2011;10(9):915–925. doi: 10.1016/j.dnarep.2011.04.033. [DOI] [PubMed] [Google Scholar]
- 97.Vaz F, et al. Mutation of the RAD51C gene in a Fanconi anemia-like disorder. Nat Genet. 2010;42(5):406–409. doi: 10.1038/ng.570. [DOI] [PubMed] [Google Scholar]
- 98.Prakash R, et al. Homologous recombination and human health: the roles of BRCA1, BRCA2, and associated proteins. Cold Spring Harb Perspect Biol. 2015;7(4):a016600. doi: 10.1101/cshperspect.a016600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Yang H, et al. The BRCA2 homologue Brh2 nucleates RAD51 filament formation at a dsDNA-ssDNA junction. Nature. 2005;433(7026):653–657. doi: 10.1038/nature03234. [DOI] [PubMed] [Google Scholar]
- 100.Somyajit K, Subramanya S, Nagaraju G. RAD51C: a novel cancer susceptibility gene is linked to Fanconi anemia and breast cancer. Carcinogenesis. 2010;31(12):2031–2038. doi: 10.1093/carcin/bgq210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Zhong Q, et al. Association of BRCA1 with the hRad50-hMre11-p95 complex and the DNA damage response. Science. 1999;285(5428):747–750. doi: 10.1126/science.285.5428.747. [DOI] [PubMed] [Google Scholar]
- 102.Yu X, et al. The C-terminal (BRCT) domains of BRCA1 interact in vivo with CtIP, a protein implicated in the CtBP pathway of transcriptional repression. J Biol Chem. 1998;273(39):25388–25392. doi: 10.1074/jbc.273.39.25388. [DOI] [PubMed] [Google Scholar]
- 103.Wong AK, et al. Characterization of a carboxy-terminal BRCA1 interacting protein. Oncogene. 1998;17(18):2279–2285. doi: 10.1038/sj.onc.1202150. [DOI] [PubMed] [Google Scholar]
- 104.Sartori AA, et al. Human CtIP promotes DNA end resection. Nature. 2007;450(7169):509–514. doi: 10.1038/nature06337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Bunting SF, Nussenzweig A. Dangerous liaisons: Fanconi anemia and toxic nonhomologous end joining in DNA crosslink repair. Mol Cell. 2010;39(2):164–166. doi: 10.1016/j.molcel.2010.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Gupta R, et al. Analysis of the DNA substrate specificity of the human BACH1 helicase associated with breast cancer. J Biol Chem. 2005;280(27):25450–25460. doi: 10.1074/jbc.M501995200. [DOI] [PubMed] [Google Scholar]
- 107.Andreassen PR, D'Andrea AD, Taniguchi T. ATR couples FANCD2 monoubiquitination to the DNA-damage response. Genes Dev. 2004;18(16):1958–1963. doi: 10.1101/gad.1196104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Pichierri P, Rosselli F. The DNA crosslink-induced S-phase checkpoint depends on ATR-CHK1 and ATR-NBS1-FANCD2 pathways. EMBO J. 2004;23(5):1178–1187. doi: 10.1038/sj.emboj.7600113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Collins NB, et al. ATR-dependent phosphorylation of FANCA on serine 1449 after DNA damage is important for FA pathway function. Blood. 2009;113(10):2181–2190. doi: 10.1182/blood-2008-05-154294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Qiao FY, et al. Phosphorylation of Fanconi anemia (FA) complementation group G protein, FANCG, at serine 7 is important for function of the FA pathway. J Biol Chem. 2004;279(44):46035–46045. doi: 10.1074/jbc.M408323200. [DOI] [PubMed] [Google Scholar]
- 111.Wilson JB, et al. FANCG promotes formation of a newly identified protein complex containing BRCA2, FANCD2 and XRCC3. Oncogene. 2008;27(26):3641–3652. doi: 10.1038/sj.onc.1211034. [DOI] [PubMed] [Google Scholar]
- 112.Xia B, et al. Fanconi anemia is associated with a defect in the BRCA2 partner PALB2. Nat Genet. 2007;39(2):159–161. doi: 10.1038/ng1942. [DOI] [PubMed] [Google Scholar]
- 113.Kee Y, Kim JM, D'Andrea A. Regulated degradation of FANCM in the Fanconi anemia pathway during mitosis. Genes Dev. 2009;23(5):555–560. doi: 10.1101/gad.1761309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Wang XZ, et al. Chk1-mediated phosphorylation of FANCE is required for the Fanconi anemia/BRCA pathway. Mol Cell Biol. 2007;27(8):3098–3108. doi: 10.1128/MCB.02357-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Meetei AR, et al. A novel ubiquitin ligase is deficient in Fanconi anemia. Nat Genet. 2003;35(2):165–170. doi: 10.1038/ng1241. [DOI] [PubMed] [Google Scholar]
- 116.Machida YJ, et al. UBE2T is the E2 in the Fanconi anemia pathway and undergoes negative autoregulation. Mol Cell. 2006;23(4):589–596. doi: 10.1016/j.molcel.2006.06.024. [DOI] [PubMed] [Google Scholar]
- 117.Orthwein A, et al. A mechanism for the suppression of homologous recombination in G1 cells. Nature. 2015;528(7582):422. doi: 10.1038/nature16142. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 118.Zhu B, et al. K63-linked ubiquitination of FANCG is required for its association with the Rap80-BRCA1 complex to modulate homologous recombination repair of DNA interstand crosslinks. Oncogene. 2015;34(22):2867–2878. doi: 10.1038/onc.2014.229. [DOI] [PubMed] [Google Scholar]
- 119.Gibbs-Seymour I, et al. Ubiquitin-SUMO circuitry controls activated Fanconi Anemia ID complex dosage in response to DNA damage. Mol Cell. 2015;57(1):150–164. doi: 10.1016/j.molcel.2014.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.He HJ, et al. DNA replication components as regulators of epigenetic inheritance-lesson from fission yeast centromere. Protein Cell. 2014;5(6):411–419. doi: 10.1007/s13238-014-0049-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Kutler DI, et al. A 20-year perspective on the international Fanconi Anemia registry (IFAR) Blood. 2003;101(4):1249–1256. doi: 10.1182/blood-2002-07-2170. [DOI] [PubMed] [Google Scholar]
- 122.Friedenson B. BRCA1 and BRCA2 pathways and the risk of cancers other than breast or ovarian.pdf. MedGenMed. 2005;29;7(2):60. [PMC free article] [PubMed]
- 123.van der Heijden MS, et al. Fanconi anemia gene mutations in young-onset pancreatic cancer. Cancer Res. 2003;63(10):2585–2588. [PubMed] [Google Scholar]
- 124.Hess CJ, et al. Hypermethylation of the FANCC and FANCL promoter regions in sporadic acute leukaemia. Cell Oncol. 2008;30(4):299–306. doi: 10.3233/CLO-2008-0426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Barroso E, et al. FANCD2 associated with sporadic breast cancer risk. Carcinogenesis. 2006;27(9):1930–1937. doi: 10.1093/carcin/bgl062. [DOI] [PubMed] [Google Scholar]
- 126.Erkko H, et al. A recurrent mutation in PALB2 in Finnish cancer families. Nature. 2007;446(7133):316–319. doi: 10.1038/nature05609. [DOI] [PubMed] [Google Scholar]
- 127.Couch FJ, et al. Germ line Fanconi anemia complementation group C mutations and pancreatic cancer. Cancer Res. 2005;65(2):383–386. [PubMed] [Google Scholar]
- 128.Rogers CD, et al. The genetics of FANCC and FANCG in familial pancreatic cancer. Cancer Biol Ther. 2004;3(2):167–169. doi: 10.4161/cbt.3.2.609. [DOI] [PubMed] [Google Scholar]
- 129.Helleday T, et al. DNA repair pathways as targets for cancer therapy. Nat Rev Cancer. 2008;8(3):193–204. doi: 10.1038/nrc2342. [DOI] [PubMed] [Google Scholar]
- 130.Jenkins C, Kan J, Hoatlin ME. Targeting the fanconi anemia pathway to identify tailored anticancer therapeutics. Anemia. 2012;2012:481583. doi: 10.1155/2012/481583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Bryant HE, et al. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature. 2005;434(7035):913–917. doi: 10.1038/nature03443. [DOI] [PubMed] [Google Scholar]
- 132.Farmer H, et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature. 2005;434(7035):917–921. doi: 10.1038/nature03445. [DOI] [PubMed] [Google Scholar]
- 133.Malyuchenko NV, et al. PARP1 Inhibitors: antitumor drug design. Acta Nat. 2015;7(3):27–37. [PMC free article] [PubMed] [Google Scholar]
- 134.Johnson N, et al. Compromised CDK1 activity sensitizes BRCA-proficient cancers to PARP inhibition. Nat Med. 2011;17(7):875–882. doi: 10.1038/nm.2377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Kennedy RD, et al. Fanconi anemia pathway-deficient tumor cells are hypersensitive to inhibition of ataxia telangiectasia mutated. J Clin Investig. 2007;117(5):1440–1449. doi: 10.1172/JCI31245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Fang NY, et al. Oligonucleotide microarrays demonstrate the highest frequency of ATM mutations in the mantle cell subtype of lymphoma. Proc Natl Acad Sci U S A. 2003;100(9):5372–5377. doi: 10.1073/pnas.0831102100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Haidar MA, et al. ATM gene deletion in patients with adult acute lymphoblastic leukemia. Cancer. 2000;88(5):1057–1062. doi: 10.1002/(SICI)1097-0142(20000301)88:5<1057::AID-CNCR16>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- 138.Chen CC, et al. CHK1 inhibition as a strategy for targeting fanconi anemia (FA) DNA repair pathway deficient tumors. Mol Cancer. 2009;8:24. [DOI] [PMC free article] [PubMed]
- 139.Chirnomas D, et al. Chemosensitization to cisplatin by inhibitors of the Fanconi anemia/BRCA pathway. Mol Cancer Ther. 2006;5(4):952–961. doi: 10.1158/1535-7163.MCT-05-0493. [DOI] [PubMed] [Google Scholar]
- 140.Burkitt K, Ljungman M. Phenylbutyrate interferes with the Fanconi anemia and BRCA pathway and sensitizes head and neck cancer cells to cisplatin. Mol Cancer. 2008;7:24. doi: 10.1186/1476-4598-7-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Kee Y, et al. Inhibition of the Nedd8 system sensitizes cells to DNA interstrand cross-linking agents. Mol Cancer Res. 2012;10(3):369–377. doi: 10.1158/1541-7786.MCR-11-0497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Chen J, et al. Selective and cell-active inhibitors of the USP1/ UAF1 deubiquitinase complex reverse cisplatin resistance in non-small cell lung cancer cells. Chem Biol. 2011;18(11):1390–1400. doi: 10.1016/j.chembiol.2011.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Liang Q, et al. A selective USP1-UAF1 inhibitor links deubiquitination to DNA damage responses. Nat Chem Biol. 2014;10(4):298–304. doi: 10.1038/nchembio.1455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Mistry H, et al. Small-molecule inhibitors of USP1 target ID1 degradation in leukemic cells. Mol Cancer Ther. 2013;12(12):2651–2662. doi: 10.1158/1535-7163.MCT-13-0103-T. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Not applicable