

Abstract
Passerines are the largest avian order, and the 6,000 species comprise more than half of all extant bird species. This successful radiation probably had its origin in the Australasian region, but dating this origin has been difficult due to a scarce fossil record and poor biogeographic assumptions. Many of New Zealand’s endemic passerines fall within the deeper branches of the passerine radiation, and a well resolved phylogeny for the modern New Zealand element in the deeper branches of the oscine lineage will help us understand both oscine and passerine biogeography. To this end we present complete mitochondrial genomes representing all families of New Zealand passerines in a phylogenetic framework of over 100 passerine species. Dating analyses of this robust phylogeny suggest Passeriformes originated in the early Paleocene, with the major lineages of oscines “escaping” from Australasia about 30 Ma, and radiating throughout the world during the Oligocene. This independently derived conclusion is consistent with the passerine fossil record.
Keywords: Passeriformes, mitochondrial genomes, oscine biogeography
Introduction
Passerines (perching birds) are the most speciose avian order, and oscine songbirds (the major clade within passerines) account for almost half of all bird species (Clements 2007). The DNA-DNA hybridization studies of Sibley and Ahlquist (1990) found oscines formed two sister groups (suborders Passerida and Corvida) that appeared broadly European and Australasian in composition. Further studies have shown that Passerida is nested within the paraphyletic Corvida (Barker et al. 2002, 2004; Ericson et al. 2002). “Corvida” is now largely superseded by “core Corvoidea” and “basal oscines” (fig. 1.). Sister to oscines, suboscines form two clades, broadly New World (NW) and Old World (OW) in composition (fig. 1). The species-rich New World clade originated in South America, and the relatively species-poor Old World lineages are mostly distributed through Africa, Madagascar, and Asia (Fjeldsa et al. 2003; Barker et al. 2004). Sister to all passerines is the small clade of New Zealand (NZ) wrens (Acanthisittidae). There is strong evidence that oscines arose in Australasia, with most deep lineages located in Australia, New Zealand, and Papua New Guinea (e.g., Jønsson et al. 2011). Because of Passeriformes incredibly speciose nature, both deep and shallow relationships within the passerines are still being resolved. The main hindrance to phylogenetic resolution within Passeriformes is the accessibility of DNA samples for some groups. Recent work suggests that as more groups within Passeriformes are sequenced, many nodes have high support (e.g., Aggerbeck et al. 2013).
Fig. 1.—

Summary of the main passerine lineages .
Dating Passeriformes
Biogeographic hypotheses regarding the worldwide distribution of passerines often begin by assuming the isolation of the NZ wrens on the Zealandian landmass since it rifted from Australia approximately 82 Ma (Barker et al. 2004), because extant and recently extinct NZ wrens are either flightless or poor fliers (Heather and Robertson 2005). Vicariance and the breakup of Gondwana are then often posited as the major influence on the diversity of passerines found today (reviewed in Ericson et al. 2003), although dispersal must then be inferred for many more recent distributions. This assumption is clearly unfounded—there is no good evidence one way or the other that the NZ wren lineage originated in Zealandia, nor that they have always been poor fliers. Despite this, almost all dating of passerines has used this vicariant calibration (e.g., Ericson et al. 2002, 2014; Barker et al. 2004; Shepherd and Lambert 2007; Jønsson, Irestedt, et al. 2008; Jønsson et al. 2011; Kennedy et al. 2012; Moyle et al. 2012), even though this date (82 Ma) significantly predates many modern estimations of the radiation of the wider Neoaves group (Neoaves contains the majority of modern bird species). Both fossil and molecular evidence suggests Neoaves is more likely to have radiated in the late Cretaceous (Ho and Phillips 2009; Jetz et al. 2012; Gibb et al. 2013; Mayr 2013; Jarvis et al. 2014). Recent molecular evidence suggests it is unlikely passerines were one of the first lineages to diverge within Neoaves (Hackett et al. 2008; Suh et al. 2011; Kimball et al. 2013; Jarvis et al. 2014), and if they were then they would need to have an extended hidden fossil record (Mayr 2013). Further geological evidence suggests the Zealandian continent was still closely associated with Australia at its northern end until more recently (∼55 Ma, e.g., Schellart et al. 2006). Ericson et al. (2014) use this broader range in their analysis of passerine evolution, but this does not resolve the underlying circular argument. This geographic date variation (82–55 Ma) does not change our assertion that the NZ wrens are a poor, though tempting, choice for passerine evolution calibration. Because the NZ wrens are a single isolated lineage (both phylogenetically and geographically), it is very hard to infer anything about their distribution and composition through time (Trewick and Gibb 2010). Other calibrations must be found to date passerine evolution.
So what calibrations can be used? The sometimes-used molecular clock rate of 2% per site per million years is not recommended for use in passerines (Nabholz et al. 2009). Dating lineages using fossil calibrations from within the passerines is also a difficult challenge. There are no good deep fossils for calibration, because most passerine fossils are found in Europe, belong to crown groups, and are relatively recent (Mourer-Chauviré et al. 1989). The only deep passerines are early Eocene fossils from Australia (∼54 Ma) reported by Boles (1995, 1997). Unfortunately, these Eocene fossils are likely to be stem passerines (Mayr 2009; Worthy et al. 2010) and cannot confidently be assigned to any modern lineage within passerines (such as oscines), so using this date for calibration is difficult. Nevertheless, these fossils support the biogeographic hypothesis of an Australasian origin of oscines, followed by a later radiation throughout the rest of the world.
Another suggestion would be to date passerines directly using external avian fossils, but this is also difficult because passerines are a relatively fast evolving group (e.g., Nabholz et al. 2009). The best deep avian fossil calibrations (see Benton and Donoghue 2007; Parham et al. 2012; Ksepka and Clarke 2015) are in Galloanserae (Clarke et al. 2005) and penguins (Slack et al. 2006), which are both relatively slowly evolving lineages, and not closely related to passerines. Evolution during the early neoavian radiation has resulted in many short and therefore difficult-to-resolve internodes (e.g., Hackett et al. 2008; Pratt et al. 2009; Pacheco et al. 2011; Gibb et al. 2013; Kimball et al. 2013). The root can easily be drawn towards passerines, as they are likely to have the longest relative internal branch lengths (e.g., Brown et al. 2008; Pacheco et al. 2011). A data set with calibrations on a few slowly evolving outgroups can lead to incorrect tree topology, with passerines artifactually drawn to the root of the tree through long branch attraction, and overestimation of passerine node ages, akin to what was seen with early studies of few taxa (e.g., Harlid and Arnason 1999). Recent studies from independent sources (nuclear and transposon, Hackett et al. 2008; Suh et al. 2011) using large numbers of taxa indicate passerines are part of the broad “higher land bird” radiation, and parrots are likely the closest extant relatives; however, these studies did not calculate a timeframe for passerine evolution. We have therefore chosen to calibrate passerine evolution using secondary calibrations derived from studies with broader taxon sampling using these deep fossils rather than biogeographic calibrations (Pratt et al. 2009; Schweizer et al. 2011; White et al. 2011).
Passerines have also been difficult to resolve morphologically, because convergent evolution in life history traits in this speciose group is common, and much more work is needed to define valid taxonomic constraints. Recent genetic studies have discovered convergence in a range of traits, such as the adaptation for nectivory of both New Zealand and Hawaiian honeyeaters (Ewen et al. 2006; Fleischer et al. 2008), evolution of toxicity in the paraphyletic Pitohui (Dumbacher et al. 2008; Jønsson, Bowie, et al. 2008), and behavior and plumage in birds of paradise (Cracraft and Feinstein 2000). In addition, passerines have a rather uniform skeletal morphology making the deeper nodes difficult to resolve as well (Mayr 2013). The large diversity within passerine families means molecular studies often find paraphyly and overturn previous taxonomic classifications (e.g., Moyle et al. 2012).
It is important to have agreement between independent lines of evidence such as mitochondrial, nuclear, and morphological data sets. Here, we provide a well-resolved mitochondrial phylogeny to complement existing molecular and morphological data sets. Mitochondrial genomes bridge the gap between exons (which perhaps evolve too slowly) and introns (which may not be alignable at deeper divergences). In addition it is relatively straightforward to sequence complete mitochondrial genomes from highly divergent avian species using conserved primers or high-throughput sequencing. A conservative alignment of protein and RNA genes provides over 13 kb of sequence after the removal of indels and noncoding sequence. Because the mitochondrion is a single molecule, all genes on it are considered to have the same gene tree, hopefully reducing the incidence of incongruence seen with Rag-1 (Irestedt and Ohlson 2008). Improved taxon sampling has been shown to provide greater phylogenetic resolution (Slack et al. 2007), and we expect that to be the case here also.
Until recently, very few passerine mitochondrial genomes were completed; only ten were included by Pacheco et al. (2011). However, since 2011 there has been a dramatic increase in oscine mitochondrial genome generation. We report 14 new oscine mitochondrial genomes, including one extinct species and at least one representative from all extant New Zealand families. New Zealand has 23 extant native passerine species, and most are endemic (Gill et al. 2010). A number of families are endemic (e.g., Acanthisittidae, Callaeidae, Notiomystidae, Mohouidae) and many fall within the deeper branches of the passerine radiation. With our robust phylogeny including over 100 passerines, we increase understanding of the composition of the New Zealand avifauna, strengthen deeper oscine phylogenetic resolution, and greatly improve the timing of biogeographic hypotheses regarding the worldwide radiation of this most successful order of birds.
Materials and Methods
DNA Extraction, Polymerase Chain Reaction, and Sequencing of Modern Samples
The species sampled are given in table 1, and sample collection details are in supplementary table S1, Supplementary Material online. DNA was extracted from blood or tissue using Roche High Pure PCR template preparation kit (Roche Applied Science, Mannheim, Germany) following the manufacturers’ instructions. Two approaches were used to sequence complete mitochondrial genomes (supplementary tables S2 and Supplementary Data, Supplementary Material online). The first approach was used for Rhipidura fuliginosa, Gerygone igata, Notiomystis cincta, Petroica australis, Philesturnus carunculatus, Turdus philomelos, Anthornis melanura, and Prosthemadera novaeseelandiae. The genomes were amplified in 2–3 overlapping segments using the Expand Long Template PCR System (Roche). These long-range polymerase chain reaction (PCR) products were used as templates for subsequent PCR of short overlapping fragments. This process is described in more detail in Gibb et al. (2007) and references therein. All short PCR products were amplified using primers identified from our database (available from the authors on request) employing standard protocols (e.g., Slack et al. 2007). Mitochondrial genomes were assembled using Sequencher 4.8 (Gene Codes Corp, Ann Arbour, MI) and electropherograms checked by eye.
Table 1.
Species and Accession Details of New Mitochondrial Genomes
| Common Name | Scientific Name | Family | GenBank Accession |
|---|---|---|---|
| Bellbird | Anthornis melanura | Meliphagidae | KC545408 |
| Browncreeper | Mohoua novaeseelandiae | Mohouidae | KC545409 |
| Fantail | Rhipidura fuliginosa | Rhipiduridae | KC545405 |
| Fernbird | Megalurus punctatus | Locustellidae | KC545398 |
| Grey warbler | Gerygone igata | Acanthizidae | KC545399 |
| Hihi | Notiomystis cincta | Notiomystidae | KC545400 |
| New Zealand robin | Petroica australis | Petroicidae | KC545401 |
| Piopio | Turnagra capensis | Turnagridae | KT894672 |
| Pipit | Anthus novaeseelandiae | Motacillidae | KC545397 |
| Saddleback | Philesturnus carunculatus | Callaeidae | KC545403 |
| Silvereye | Zosterops lateralis | Zosteropidae | KC545407 |
| Song thrush | Turdus philomelos | Turdidae | KC545406 |
| Tomtit | Petroica macrocephala | Petroicidae | KC545402 |
| Tui | Prosthemadera novaeseelandiae | Meliphagidae | KC545404 |
For the second approach, short paired reads were generated from either Illumina HiSeq or MiSeq runs following standard protocols. Samples were either run in their own lane (Anthus novaeseelandiae and Petroica macrocephala), indexed (Mohoua novaeseelandiae and Zosterops lateralis) or as part of a mixture (Megalurus punctatus, mixed with a frog, fish, and mollusc). De novo assembly of contigs from the paired reads run individually or indexed used Velvet v1.1.06 (Zerbino and Birney 2008) with a K-mer sweep to identify the highest N50 for each species (typically around k = 65). De novo assembly of the mitochondrial genome from a mixture of total genomic extracts followed a modified version of the pipeline described in McComish et al. (2010). Because of the presence of nuclear DNA, Velvet assemblies, as expected, produced much larger numbers of contigs than the assembly described in McComish et al. (2010). This meant that the assembly graphs could not be used to help separate contigs from the different samples, because the graphs consisted largely of isolated short contigs of nuclear origin. However, it was possible to identify almost all of the mitochondrial contigs by simply altering the script used to separate contigs so that it called Exonerate (v. 2.2.0, Slater and Birney 2005) with a score threshold of 150. This was sufficient to filter out all of the nuclear contigs, as these gave scores lower than 150 when aligned to the mitochondrial reference genomes. For all species, Geneious 6.1.7 (Biomatters Ltd, www.geneious.com, last accessed July 2015), was used to align the assembled contigs against reference genomes (supplementary tables S2 and Supplementary Data, Supplementary Material online), typically using “map to reference” with medium-low sensitivity. Contig overlaps were identified and contigs were combined into preliminary whole mitochondrial genome assemblies. Finished assemblies were verified by mapping the original sequence reads to the assembly using Geneious 6.1.7. Any small remaining gaps were filled by sequencing shorter PCR products as described above for the first approach. DNA from A. melanura and M. novaeseelandiae was of insufficient quantity to complete these genomes, resulting in partial genomes for these two species.
Extraction and Sequencing of Ancient DNA
DNA extraction from a toepad of the extinct Turnagra capensis (piopio) was performed using strict ancient DNA procedures at the Australian Centre for Ancient DNA, University of Adelaide, South Australia. Total genomic DNA was extracted using proteinase K digestion followed by phenol chloroform extraction. Subsequently, library preparation and indexing was performed using the TrueSeq Nano DNA sample preparation kit (Illumina Inc.) in a dedicated ancient DNA facility at Massey University, New Zealand. Because ancient DNA is already degraded, the protocol was modified to omit the fragmentation step. Magnetic bead size selection criteria were relaxed to capture a wider range of DNA fragments. DNA was sequenced as part of one lane on an Illumina HiSeq machine running a 100 bp paired end protocol by New Zealand Genomics Ltd. The resulting reads were trimmed of any 3′-adapter sequence using cutadapt v1.1 (Martin 2011) and de novo assembled using ABySS v1.3.0 (Simpson et al. 2009) with a kmer sweep of 45, 55, and 65, and parameters n (number of pairs) = 2, c (coverage) = 5, and e (erosion) = 5. The resulting contigs were imported into Geneious 6.1.7 and mitochondrial contigs were filtered by assembly to another passerine mitochondrial genome (supplementary tables S2 and Supplementary Data, Supplementary Material online). The finished assembly was checked by mapping the original sequence reads to the consensus using Geneious 6.1.7, and select short regions were independently verified by PCR. All genomes were checked to confirm protein regions translated and RNA stem loop structures matched appropriately.
In addition to the 14 new genomes reported here, a further 84 passerine genomes were included from GenBank (supplementary table S4, Supplementary Material online). Four genomes were also assembled from 454 transcriptomic data (from Nabholz et al. 2010), and rRNAs and some tRNAs were recoverable when the reads were mapped to reference genomes (see supplementary information and Supplementary Data, Supplementary Material online) using Geneious 6.1.7 as described above. Four diverse parrot species were also included as a more distant outgroup, as a number of recent studies place parrots as the closest extant outgroup to passerines, and use quite independent data sets to reach this conclusion (nuclear sequences [Hackett et al. 2008; Wang et al. 2012; Kimball et al. 2013], and retrotransposon data [Suh et al. 2011]). The details of all genomes used in these analyses can be found in supplementary table S4, Supplementary Material online.
Multiple genomes were manually aligned and checked in Geneious 6.1.7, at the amino acid level for protein-coding genes, and based on stem and loop secondary structure for RNA genes (Gutell et al. 1994; Springer and Douzery 1996), see supplementary tables S5 and Supplementary Data, Supplementary Material online, for RNA stem annotations. Gaps, ambiguous sites adjacent to gaps, the ND6 (light-strand encoded), noncoding regions, and stop codons (often incomplete in the DNA sequence), were excluded from the alignment. Conserved amino acids or stem columns were used to define the boundaries of ambiguous regions next to gaps. The third position of protein-coding genes have been coded as RY. RY recoding of third codons increases the proportion of observable changes on internal branches of the tree (tree-ness) and decreases the differences in nucleotide composition (relative compositional variability; e.g., Harrison et al. 2004; Gibb et al. 2007; Phillips et al. 2010). The full data set is 13,837 bp long and is separated into five partitions: three codon positions, and RNA stems and loops. The best partitioning and models were found using Bayesian information criterion model selection with a greedy search scheme and unlinked branch lengths in PartitionFinder (Lanfear et al. 2012), which recommended three final partitions: first codons plus loops, second codons plus stems, and third codons. These three partitions were used in all subsequent analyses. Alignments and trees can be downloaded from TreeBASE (TB2:15732).
Data were analyzed using maximum likelihood (ML) methods implemented in RAxML (Stamatakis et al. 2008), using a general time reversible model with gamma distribution (GTR + G). Bayesian analyses were performed using MrBayes (Huelsenbeck and Ronquist 2001) using GTR + I + G. Data sets were run for 10 million generations, and sampled every 2,500 generations after a burnin of 1,000,000 generations. Independent runs were checked for convergence, and trace files analyzed using Tracer (Rambaut and Drummond 2003) to ensure effective sample size (ESS) values greater than 200. Trees were viewed using Figtree v1.4 (http://tree.bio.ed.ac.uk/software/figtree/ [accessed June 2015]).
The data were also analyzed using the CAT–GTR mixture model of PhyloBayes-MPI 1.5a (Lartillot et al. 2013) with no RY coding, and constant sites removed. Two independent chains were run for 26,700 iterations and checked for convergence in likelihood and model parameters (tracecomp subprogram) and clade posterior probability (bpcomp subprogram). The first 10% of trees were discarded as burnin and a 50% majority rule Bayesian consensus tree with associated posterior probabilities was computed from the remaining trees using bpcomp.
Dating
Dated analyses were performed using BEAST and BEAUTI v1.8.0 (Drummond et al. 2012) with the data set partitioned as above. An uncorrelated relaxed clock model was used with rates among branches distributed according to a lognormal distribution. The tree prior used a speciation birth–death process. Nucleotide partitions used an estimated GTR + I + G model with the at-gc scale operators and delta exchange removed for the RY coded partition. To circumvent the use of the problematic Acanthisitta chloris biogeographic calibration, we chose indirect calibrations from recent published data sets that also included some passerines and used widely accepted fossil calibrations. The root prior had a normal distribution with mean 70 Ma, SD 9 Ma. All passerines excluding Ac. chloris had a normal distribution with mean 62 Ma, SD 10 Ma. These calibrations are based on the results of Schweizer et al. (2011) and are very similar to other recent publications (White et al. 2011; Jetz et al. 2012). Schweizer et al. (2011) used the penguin fossil Waimanu as a calibration for the split between Sphenisciformes and other seabirds (Slack et al. 2007). This calibration used a mean of 66 Ma (SD 3.06) with normal distribution. The lower bound takes into account potential dating errors of the fossil, and the upper bound allows for putative members of Gaviiformes (loons) from the late Cretaceous. In addition they tested uniform and lognormal priors, and alternate sister groups for penguins (Procellariformes, or a group consisting of Gaviiformes, Procellariiformes, Pelecaniformes, and Suliformes). They also used two additional fossil calibrations. First, a stem flamingo (Mayr 2005) as minimum age of the Podicipediformes–Phoenicoteriformes (grebe+flamingo) split with uniform prior and conservative upper bound (30–124 Ma). Second, a uniform distribution for the split between Galloanserae and Neoaves with the same 124 Ma upper bound and lower bound of 66 Ma based on the Vegavis fossil (Clarke et al. 2005). The results of Schweizer et al. (2011) were very similar with all calibration options.
Runs totaling 40,000,000 Markov chain Monte Carlo generations ensured ESS values greater than 200 (as estimated in Tracer v 1.5 Rambaut and Drummond 2003). Chains were sampled every 5000th generation after removing a burnin of 10%. Trees were viewed using Figtree v1.4 (http://tree.bio.ed.ac.uk/software/figtree/ [accessed June 2015]).
To compare alternative calibration schemes, the dating analysis was repeated using slightly younger calibrations based on the results of Pratt et al (2009). Four calibration priors were used, all with a normal distribution. These were: root prior, mean 61 Ma, SD 5 Ma, all passerines excluding Ac. chloris, mean 57.3 Ma, SD 5 Ma, all oscines 32.6 Ma, SD 6 Ma, all suboscines 46.3 Ma, SD 5.3 Ma. All other parameters were unaltered.
Results
Mitogenomic Features
Mitochondrial gene rearrangements are very common within birds (see Gibb et al. 2007), and passerines are no exception. The majority of the genomes reported here have the standard gene order found in the chicken (Desjardins and Morais 1990). Two species (Z. lateralis, silvereye and Me. punctatus, fernbird) have the duplicate control region (CR) gene order (a copy of the CR is located between tRNA Thr and tRNA Pro, see figure 1 in Gibb et al. 2007) also found in many other Sylvioidea. The two Petroica have the remnant CR 2 gene order as do other published Petroica (Cooke et al. 2012). The N. cincta (hihi) and T. philomelos (song thrush) genomes have independently evolved a novel gene order involving duplication of tRNA Pro-CR. This is contra to a previous report showing a standard gene order for these two genomes (Barker 2014). This gene rearrangement differs from previously recorded avian mitochondrial duplications, as tRNA Thr is not part of the duplicated segment (see fig. 1 in Gibb et al. 2007). As has been seen in other CR gene duplications, the duplicated segments are nearly identical (e.g., Gibb et al. 2007, 2013). It is surprising to note that all published genomes from Passeroidea and Muscicapoidea, except our thrush (T. philomelos), have only one CR. Given the regularity and repeated occurrence of gene rearrangements in passerines, it is likely researchers will soon uncover more species with duplicated regions in Passeroidea and Muscicapoidea. It is also very likely some of the genomes published by other groups actually have a hidden duplication not detected due to genome misassembly. This highlights the ongoing need for researchers to be aware of the possibility of hidden duplicate CRs when sequencing mitochondrial genomes (Gibb et al. 2007). Researchers need to actively check (as we do), using PCR or high throughput data, for “out of order” gene arrangements (such as the CR before tRNA Thr or tRNA Pro) and signs such as increased read coverage depth between cytb and 12S.
The majority of taxa are grouped as expected based on previous studies and taxonomic classification. We were, however, surprised by the placement of Pseudopodoces humilis (Tibetan ground-jay). Further analysis of this genome against a wider range of shorter mitochondrial genes indicates this genome may have been mislabeled and is likely to be Phoenicurus ochruros (black redstart, supplementary fig. S1, Supplementary Material online).
The complete genome of Turn. capensis (piopio) shows significant differences to a number of other published Turn. capensis mitochondrial gene sequences, and we suspect these other published sequences are incorrect. Portions of some sequences are more divergent than would be expected between sequences from the same order, let alone the same species. To elaborate, cytb (GenBank U51734) is 16% divergent, the second half of cytb (JN614879) is 10% divergent and ND3 (JN614657) is 20% divergent. Compare this with the correct ND2 (JN614707) where the sequences are less than 1% divergent. This highlights the challenge of accurately verifying novel sequences amplified from short PCRs of degraded ancient DNA. Generated by high throughput sequencing, our Turnagra mitochondrial genome has an average coverage of 171× (SD of 36×) with no ambiguous regions, giving us high confidence in the accuracy of the full sequence (supplementary table S2, Supplementary Material online). We find Turnagra to fall within the core Corvoidea, diverging from other orioles about 22 Ma. This is contra to the earlier cytb study of Christidis et al. (1996) which found Turnagra to be a basal bowerbird. Its position with orioles in the core Corvoidea agrees with our finding from a few short mtDNA regions (Gibb 2010) and recent nuclear studies (Johansson et al. 2011; Zuccon and Ericson 2012).
Phylogenetic Analyses
All ML and Bayesian analyses resulted in congruent phylogenies with the majority of branches having high bootstrap and Bayesian posterior probability support (fig. 2 and supplementary figs. S2–S5, Supplementary Material online). Those few branches that do differ between analyses have low support. As expected, passerines form three groups: oscines, suboscines, and the Ac. chloris (rifleman). Within oscines, the traditional Passerida forms three broad lineages: Paseroidea, Sylvioidea, and Muscicapoidea. We find Sylvioidea to diverge first, in agreement with some nuclear and mitochondrial studies (e.g., Barker et al. 2004; Treplin et al. 2008; Barker 2014), although alternative arrangements are also seen (e.g., Hackett et al. 2008; Johansson et al. 2008; Price et al. 2014). Support for the arrangement is still weak and the internodes are short, however, it is “locally stable” (Cooper and Penny 1997) in that the alternate arrangements are only one step away. The deepest split within Muscicapoidea has sometimes defined the separation of Muscicapoidea from Certhioidea (labeled in fig. 2, though it is a similar depth to the radiation of Sylvioidea). The core corvoids are monophyletic, and the majority of the New Zealand lineages fall within the basal oscine, core Corvoidea, and basal Passerida lineages. Menura novaehollandiae (lyrebird) is sister to all other oscines.
Fig. 2.—
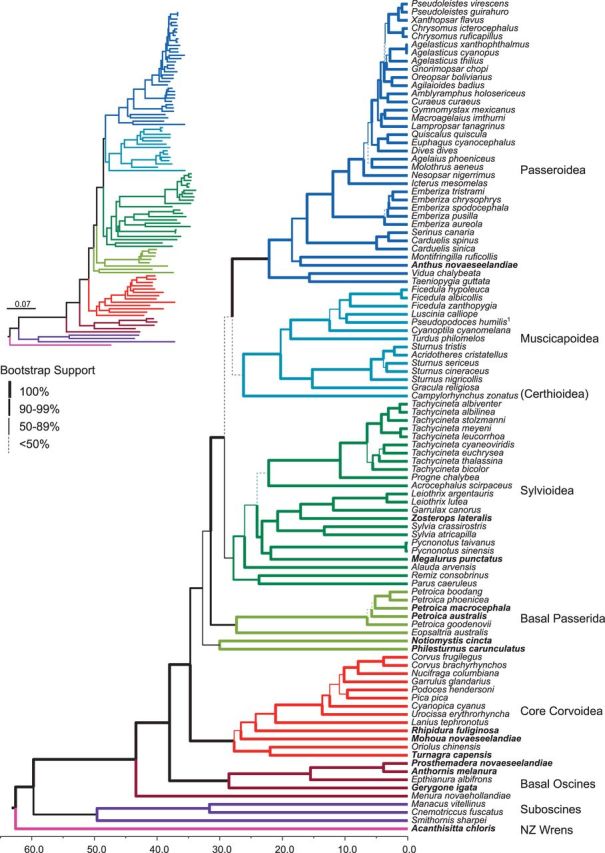
Beast chronogram of 102 passerine species derived from complete mitochondrial genomes. The 14 New Zealand native species are in bold. Branch thickness indicates ML bootstrap support, and the timescale is in millions of years. The insert ML tree shows relative branch length, and a full size version is shown in supplementary figure S2, Supplementary Material online. Colors are the same as in figure 1 for reference. 1Pseudopodoces humilis is likely Phoenicurus ochruros, see Results section and supplementary figure S1, Supplementary Material online.
Notiomystis cincta and Ph. carunculatus (saddleback) are deeply diverging sister taxa, as predicted from nuclear studies (Ewen et al. 2006; Driskell et al. 2007). Interestingly, they are sister to the rest of Passerida, near the Australasian robins (here, Petroica + Eopsaltria, fig. 2). This is contra to the results of studies including the Rag-1 gene (e.g., Barker et al. 2004) where Callaeidae (represented in our study by Ph. carunculatus) is sister to the core Corvoidea. That Callaeidae is sister to Passerida was also found by Irestedt and Ohlson (2008), who analyzed short introns and exons from a combination of Callaeas cinerea (kokako) and Ph. carunculatus sequences (both Callaeidae). The different results seen with some intron, exon, and mitochondrial data highlights the danger of simply combining different data sets without first checking for congruence between data sets. In the case of the Callaeidae, it appears Rag-1 overpowered the signal for an alternative topology now found independently by intron, exon, and mitochondrial data (fig. 1 and Irestedt and Ohlson 2008). This is akin to the case of β-fibrinogen intron 7 (Morgan-Richards et al. 2008) that appeared to provide signal supporting a Metaves/Coronaves split (Fain and Houde 2004) even in the presence of additional genes with alternate signal (Ericson et al. 2006; Kimball et al. 2013). However, given the agreement here from intron, exon, and mitochondrial data, we suggest these “basal passerids” should be included within the Passerida; this would simplify the formal classification of oscines.
Dating and Diversification
The two dating analyses resulted in trees with the same topology, as expected (fig. 2; supplementary figs. S6 and Supplementary Data, Supplementary Material online). The calibrations based on Schweizer et al. (2011) were slightly older than those of Pratt et al. (2009), and unsurprisingly, the resultant tree also has a deeper root node. While there was approximately 10 Ma difference in root calibration ages, the resultant trees only differ by 5 Ma at the deepest nodes (table 2), and differences between node ages in the two trees decrease as the nodes get younger. All node dates compared between the two trees fall within the 95% posterior probability intervals of the two trees, and we find these results very comparable. The broader, more conservative date ranges based on Schweizer et al. (2011) (fig. 2; supplementary fig. S6, Supplementary Material online) are referenced in the following paragraphs.
Table 2.
Passerine Radiation Dates and Calibrations Used in a Selection of Recent Studies
| Reference | Passerine Split Date | 95% Range | Suboscines and Oscines Split Date | 95% Range | Includes Waimanu and Vegavis Fossils in Calibrations? | Includes NZ Wrens as Biogeographic Calibration? | Comments |
|---|---|---|---|---|---|---|---|
| Avian Studies | |||||||
| Pratt et al. (2009) | 61.05 | 50–70a | 57.26 | 48–67a | Y | N | Focused on broad range of birds including passerines |
| Schweizer et al. (2011) | 70.36 | 52.6–88.13 | 62.4 | 45.5–79.7 | Y | N | Focused on parrots but uses broad range of birds for dating including passerines. |
| White et al. (2011) | 68.03 | 55–82a | 63.95 | 51–78a | Y | N | Focused on parrots but uses broad range of birds for dating including passerines. |
| Pacheco et al. (2011) | 78.5 | 71–85a | 74.9 | — | Y | Y | Includes broad range of birds. Dating includes NZ wrens calibration |
| Jetz et al. (2012) | 70a | — | 66.8 | — | Y | N | Supertree study. Soft maximum bounds on most calibrations of 110 Ma. |
| Jarvis et al. (2014) | 39a | 32–43a | 32a | 27–36a | Y | N | Primarily uses conservative (i.e. young) minimum bounds with no maximum. The surprisingly young passerine split requires further investigation. |
| Passerine Studies | |||||||
| Barker et al. (2004) | 82 | — | 77–76 | — | NA | Y | Dating primarily calibrated using NZ wrens split. |
| Jønsson et al. (2011) | — | — | 80a | — | NA | Y | Calibrations based on Barker et al. (2004). |
| Kennedy et al. (2012) | 44a | — | 42a | — | NA | Y | Root calibration has uniform prior of 85–0 Ma. All other calibrations are young (< 20 Ma) |
| Ericson et al. (2014) | 74a | 59–85a | 73a | 58–84a | NA | Y | Uses uniform root prior of 85–52 Ma. |
| Price et al. (2014) | — | — | 51 | — | Waimanu | Y | Many calibrations have broad uniform distribution with 80 Ma lower bound |
| This study | 62.5 | 48.5–76.3 | 59.7 | 40.5–73.2 | Y (indirectly) | N | Calibrations based on Schweizer et al. (2011) |
| This study | 57.61 | 51.0–64.4 | 54.75 | 48.9–60.8 | Y (indirectly) | N | Calibrations based on Pratt et al. (2009) |
aApproximate values inferred from figures.
We find the NZ wrens diverge from all other passerines around 62 Ma (with a range of 48.5–76.3 Ma, fig. 3), with oscines and suboscines diverging only a (geologically) short time later (fig. 2; supplementary fig. S3, Supplementary Material online). The major “backbone” branches of the oscines diverged from each other between 30 and 44 Ma. The deepest of these branches are all Australian-centric in origin, and it is around 30 Ma that the three speciose Passerida clades diverge. The subsequent extant radiation of all major oscine lineages, both Australasian corvoids and worldwide passerids, appears to have happened over a similar period of time (22–28 Ma), with the Australasian elements radiating slightly earlier than the Passerida lineages (figs. 2 and 3).
Fig. 3.—

Origin of major passerine clades and New Zealand endemic lineages, as summarized from the BEAST analysis. Horizontal lines show 95% posterior probability intervals for select lineages from the BEAST analysis. The two dashed lineages have age priors. Circle size indicates the number of species in that clade, and color indicates the centre of species diversity. Blue, New Zealand; green, Australasia; yellow, Africa and Eurasia; red, Americas. Key fossil locations and geological events are indicated (adapted from Barker et al. 2004; Mayr 2009; Worthy et al. 2010), and the timescale is in millions of years. For comparison with figure 2, the Meliphagoidea are basal oscines excluding Menura novaehollandiae, and Australasian robins are basal Passerida excluding Notiomystis and Philesturnus.1Note that for lineages where there is only one sampled taxon, the circle represents the time of clade divergence not the extant radiation.
We do not have enough information to comment on the timing of the radiation within the suboscines, with only one Old World suboscine representative. The extant New World suboscines appear to have begun radiating earlier than the oscines, as more than 30 Ma separates the two sampled NW suboscines, and the Furnarii (ovenbirds, antbirds and allies) probably diverged earlier (e.g., Barker et al. 2004). Greater sampling within suboscines will be needed to explore this further.
Discussion
It does appear that the addition of New Zealand lineages has helped clarify oscine radiation timing, for example, the deep split of G. igata in Meliphagoidea (28 Ma) and the separation of N. cincta and Ph. carunculatus around 30 Ma. Our finding of approximately 15 Ma for the split of Epithanura and Prosthemadera is a similar timeframe to Joseph et al. (2014) who used very different calibration methods for honeyeaters (a modified molecular rate estimation for ND2). Similar ages from different methods help increase our confidence in our findings. The extant core Corvoidea also began radiating around 28 Ma. BEAST and PhyloBayes analyses show Turnagra plus Oriolus as the deepest branch (fig. 2; supplementary figs. S4, Supplementary Data, and S7, Supplementary Material online); however, RAxML and MrBayes analyses slightly favored M. novaeseelandiae (brown creeper) as the deepest lineage in the core Corvoidea (supplementary figs. S2 and Supplementary Data, Supplementary Material online), in agreement with other recent work (Aggerbeck et al. 2013; Aidala et al. 2013). ML support for the two configurations is split around 50% for each topology (fig. 2 and supplementary fig. S2, Supplementary Material online), but they are locally stable.
Within the basal oscines there are a number of Australasian lineages that will be needed to further resolve our understanding of the basal oscine radiation, including catbirds, logrunners, babblers, berrycreepers and treecreepers, and others (work is currently underway on this project). These taxa will indicate whether more Australian lineages were diversifying before 30 Ma, or if all modern diversification of oscine lineages predominantly occurred 20–30 Ma.
Dating the Modern New Zealand Avifauna
Many of the New Zealand endemic lineages are quite deeply diverged and species poor (Acanthisitta, Turnagra, Mohoua, Notiomystis, Philesturnus). What we do know about the modern diversity within lineages is that it is very shallow—for example, the NZ Meliphagidae, Pr. novaeseelandiae (tui) and A. melanura (bellbird), diverged about 3.8 Ma, and P. australis and P. macrocephala (NZ robin and tomtit) are separate invasions of New Zealand approximately 5–6 Ma (figs. 2 and 3). It is tempting to infer the deeper lineages, (e.g., Mohoua) have been isolated in New Zealand as they diverged; however, the fossil record is able to tell us little about the geographic location of these lineages through time. From our phylogeny, Notiomystis and Philesturnus are the only hint of a deep split within Zealandia. Most, if not all, of Zealandia was submerged during the late Oligocene (22–25 Ma, Landis et al. 2008), which certainly caused great turnover in the New Zealand fauna. Many factors are involved in assembling a biota over time (Trewick and Gibb 2010; Bromham et al. 2012). The early Miocene fossil deposits at St Bathans (fig. 3, 16–19 Ma) show a great diversity of avian species (e.g., Worthy et al. 2009, 2007, 2011), some of which are still present today. These fossils include passerines from Cracticidae (currawongs and Australian magpies, not currently native NZ taxa, Worthy et al. 2007) and Acanthisittidae (Worthy et al. 2010), indicating at least the NZ wrens were present by this time. Further undescribed passerine fossils from St Bathans will improve our understanding of lineage assembly over time (Trevor Worthy, personal communication).
We do not expect this study to be the final word on the timing of passerine evolution. As our understanding of deep avian evolution improves, so will our calibrations for the timing of passerine evolution. The basic structure of the passerine tree appears to be consistent across many studies, and as different calibrations are applied to the tree, the branch lengths will shrink or expand proportionately. The recent work of Jarvis et al. (2014) used 1,156 clocklike exons to examine avian evolution, and found a much more recent timing for the radiation of Passerines (∼39 Ma, table 2). Jarvis et al. (2014) found oscines and suboscines diverged approximately 32 Ma, with the core Corvoidea and Passeriodea diverging approximately 20 Ma. If this is indeed the case, then late Oligocene fossil oscines from Europe (fig. 3 and Mayr 2009) would likely fall outside the modern oscine radiation. These surprisingly young divergence times for passerines will require further investigation.
In our study, two sets of secondary calibrations separated by approximately 10 Ma have resulted in trees approximately 5 Ma divergent at the root. The root divergence of around 57–62 Ma (with 95% posterior probabilities around 48–76 Ma) is much younger than the divergence of Zealandia and Australia around 82 Ma. It is certainly possible the ancestors of the modern NZ wrens were present on Zealandia around 55 Ma when the landmasses’ separation completed (Schellart et al. 2006). However, there is no physical evidence to indicate their location between the 57 and 62 Ma divergence inferred here and the Acanthisittidae fossils found in New Zealand during the Miocene 16–19 Ma (Worthy et al. 2010). Therefore we reiterate the use of NZ wrens as a vicariant biogeographic calibration should stop.
Many studies of passerines have been calibrated using the rifting of New Zealand and Australia around 82 Ma for the separation of Acanthisittidae from other passerines (table 2). We find no evidence to support this suggestion. Our study, which calibrates passerine evolution using secondary calibrations of the basal nodes from previous more widely sampled studies that used well-defined fossil calibrations, avoids this problem and allows us to more realistically begin to interpret passerine evolution and biogeography. We find crown group Passeriformes began diverging in the early Paleocene, with major expansion of the speciose oscine lineages during the Oligocene.
Our timescale for passerine evolution is concordant with the scant fossil record available for deep oscines (fig. 3 and Mayr 2013). The oldest “passerine” fossils are found in the early Eocene of Australia, 45–55 Ma (Boles 1997). From our results these fossils are older than the modern oscine radiation, which would be concordant with the stem passerine status suggested for them (Mayr 2009; Worthy et al. 2010). Early Oligocene fossils from Europe are not crown oscines and could be suboscines or near passerines, but oscine passerines are known from the late Oligocene in Europe (fig. 3 and Mayr 2009). This is in agreement with our timescale of a radiation of the modern passerid lineages beginning in the late Oligocene (22–28 Ma). The timing of this “escape” of passerines from the Australasian region into the rest of the world is coincident with the collision of the Australian and South East Asian plates and the emergence of Wallacea 20–30 Ma (fig. 3, Hall 2009). How much influence this increased SE Asian land area had on lineages that were already volant is still to be determined. Much denser sampling of these incredibly speciose lineages will be needed to fully understand the worldwide radiation of Passerida.
Supplementary Material
Acknowledgments
The authors thank many people for providing tissue and bone samples for this study including John Ewen and Phill Cassey (samples from Tiritiri Matangi), Dick Gill (Waikanae Department of Conservation), Lara Shepherd and Gillian Stone (Museum of New Zealand Te Papa Tongarewa), and Sandra Burles (Wairarapa DoC). Thanks to Philippa Horton and Trevor Worthy for access to the piopio specimen at the South Australian Museum, and to Trevor Worthy, Dorothee Huchon and anonymous referees for helpful comments on the manuscript. This work was supported by the Allan Wilson Centre for Molecular Ecology and Evolution, the New Zealand Marsden Fund, and Massey University.
Literature Cited
- Aggerbeck M, et al. 2013. Resolving deep lineage divergences in core corvoid passerine birds supports a proto-Papuan island origin. Mol Phylogenet Evol. 70:272–285. [DOI] [PubMed] [Google Scholar]
- Aidala Z, et al. 2013. Phylogenetic relationships of the genus Mohoua, endemic hosts of New Zealand’s obligate brood parasitic long-tailed cuckoo (Eudynamys taitensis). J Ornithol. 154:1127–1133. [Google Scholar]
- Barker FK. 2014. Mitogenomic data resolve basal relationships among passeriform and passeridan birds. Mol Phylogenet Evol. 79:313–324. [DOI] [PubMed] [Google Scholar]
- Barker FK, Barrowclough GF, Groth JG. 2002. A phylogenetic hypothesis for passerine birds: taxonomic and biogeographic implications of an analysis of nuclear DNA sequence data. Proc R Soc Lond B Biol Sci. 269:295–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barker FK, et al. 2004. Phylogeny and diversification of the largest avian radiation. Proc Natl Acad Sci U S A. 101:11040–11045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benton MJ, Donoghue PCJ. 2007. Paleontological evidence to date the tree of life. Mol Biol Evol. 24:26–53. [DOI] [PubMed] [Google Scholar]
- Boles WE. 1995. The world’s oldest songbird. Nature 374:21–22. [Google Scholar]
- Boles WE. 1997. Fossil songbirds (Passeriformes) from the early Eocene of Australia. Emu 97:43–50. [Google Scholar]
- Bromham L, et al. 2012. Reconstructing past species assemblages reveals the changing patterns and drivers of extinction through time. Proc R Soc Lond B Biol Sci. 279:4024–4032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown JW, et al. 2008. Strong mitochondrial DNA support for a Cretaceous origin of modern avian lineages. BMC Biol. 6:6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christidis L, Leeton PR, Westerman M. 1996. Were bowerbirds part of the New Zealand fauna? Proc Natl Acad Sci U S A. 93:3898–3901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke JA, et al. 2005. Definitive fossil evidence for the extant avian radiation in the Cretaceous. Nature 433:305–308. [DOI] [PubMed] [Google Scholar]
- Clements JF. 2007. The clements checklist of birds of the world. Ithaca (NY): Cornell University Press. [Google Scholar]
- Cooke GM, et al. 2012. Rapid characterization of mitochondrial genome rearrangements in Australian songbirds using next-generation sequencing technology. J Hered. 103:882–886. [DOI] [PubMed] [Google Scholar]
- Cooper A, Penny D. 1997. Mass survival of birds across the cretaceous-tertiary boundary: molecular evidence. Science 275:1109–1113. [DOI] [PubMed] [Google Scholar]
- Cracraft J, Feinstein J. 2000. What is not a bird of paradise? Molecular and morphological evidence places Macgregoria in the Meliphagidae and the Cnemophilinae near the base of the corvoid tree. Proc R Soc Lond B Biol Sci. 267:233–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desjardins P, Morais R. 1990. Sequence and gene organization of the chicken mitochondrial genome: a novel gene order in higher vertebrates. J Mol Biol. 212:599–634. [DOI] [PubMed] [Google Scholar]
- Driskell AC, et al. 2007. A new endemic family of New Zealand passerine birds: adding heat to a biodiversity hotspot. Aust J Zool. 55:73–78. [Google Scholar]
- Drummond AJ, Suchard MA, Xie D, Rambaut A. 2012. Bayesian phylogenetics with BEAUti and the BEAST 1.7. Mol Biol Evol. 29:1969–1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dumbacher JP, Deiner K, Thompson L, Fleischer RC. 2008. Phylogeny of the avian genus Pitohui and the evolution of toxicity in birds. Mol Phylogenet Evol. 49:774–781. [DOI] [PubMed] [Google Scholar]
- Ericson P, et al. 2006. Diversification of Neoaves: integration of molecular sequence data and fossils. Biol Lett. 2:543–547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ericson PGP, et al. 2002. A Gondwanan origin of passerine birds supported by DNA sequences of the endemic New Zealand wrens. Proc R Soc Lond B Biol Sci. 269:235–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ericson PGP, et al. 2014. Dating the diversification of the major lineages of Passeriformes (Aves). BMC Evol Biol. 14:8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ericson PGP, Irestedt M, Johansson US. 2003. Evolution, biogeography, and patterns of diversification in passerine birds. J Avian Biol. 34:3–15. [Google Scholar]
- Ewen JG, Flux I, Ericson PGP. 2006. Systematic affinities of two enigmatic New Zealand passerines of high conservation priority, the Hihi or Stitchbird Notiomystis cincta and the Kokako Callaeas cinerea. Mol Phylogenet Evol. 40:281–284. [DOI] [PubMed] [Google Scholar]
- Fain MG, Houde P. 2004. Parallel radiations in the primary clades of birds. Evolution 58:2558–2573. [DOI] [PubMed] [Google Scholar]
- Fjeldsa J, et al. 2003. Sapayoa aenigma: a New World representative of ‘Old World suboscines’. Proc R Soc Lond B Biol Sci. 270(Suppl 2):S238–S241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleischer R, James H, Olson SL. 2008. Convergent evolution of Hawaiian and Australo-Pacific honeyeaters from distant songbird ancestors. Cur Biol. 18:1927–1931. [DOI] [PubMed] [Google Scholar]
- Gibb G. 2010. Birds in a tree: a journey through avian phylogeny, with particular emphasis on the birds of New Zealand [PhD thesis]. [Palmerston North, New Zealand]: Massey University. [Google Scholar]
- Gibb GC, et al. 2007. Mitochondrial genomes and avian phylogeny: complex characters and resolvability without explosive radiations. Mol Biol Evol. 24:269–280. [DOI] [PubMed] [Google Scholar]
- Gibb GC, Kennedy M, Penny D. 2013. Beyond phylogeny: pelecaniform and ciconiiform birds, and long-term niche stability. Mol Phylogenet Evol. 68:229–238. [DOI] [PubMed] [Google Scholar]
- Gill BJ, et al. 2010. Checklist of the birds of New Zealand, Norfolk and Macquarie Islands, and the Ross Dependency, Antarctica. Wellington: Te Papa Press. [Google Scholar]
- Gutell RR, Larsen N, Woese C. 1994. Lessons from an evolving rRNA: 16S and 23S rRNA structures from a comparative perspective. Microbiol Rev. 58:10–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hackett SJ, et al. 2008. A phylogenomic study of birds reveals their evolutionary history. Science 320:1763–1768. [DOI] [PubMed] [Google Scholar]
- Hall R. 2009. Southeast Asia's changing palaeogeography. Blumea 54:148–161. [Google Scholar]
- Harlid A, Arnason U. 1999. Analyses of mitochondrial DNA nest ratite birds within the Neognathae: supporting a neotenous origin of ratite morphological characters. Proc R Soc Lond B Biol Sci. 266:305–309. [Google Scholar]
- Harrison GL, et al. 2004. Four new avian mitochondrial genomes help get to basic evolutionary questions in the late Cretaceous. Mol Biol Evol. 21:974–983. [DOI] [PubMed] [Google Scholar]
- Heather B, Robertson HA. 2005. The field guide to the birds of New Zealand. Auckland: Penguin Books. [Google Scholar]
- Ho SYW, Phillips MJ. 2009. Accounting for calibration uncertainty in phylogenetic estimation of evolutionary divergence times. Syst Biol. 58:367–380. [DOI] [PubMed] [Google Scholar]
- Huelsenbeck JP, Ronquist F. 2001. MrBayes: Bayesian inference on phylogenetic trees. Bioinformatics 17:754–755. [DOI] [PubMed] [Google Scholar]
- Irestedt M, Ohlson J. 2008. The division of the major songbird radiation into Passerida and ‘core Corvoidea’ (Aves: Passeriformes)—the species tree vs. gene trees. Zool Script. 37:305–313. [Google Scholar]
- Jarvis ED, et al. 2014. Whole-genome analyses resolve early branches in the tree of life of modern birds. Science 346:1320–1331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jetz W, et al. 2012. The global diversity of birds in space and time. Nature 491:444–448. [DOI] [PubMed] [Google Scholar]
- Johansson US, Fjeldsa J, Bowie RCK. 2008. Phylogenetic relationships within Passerida (Aves: Passeriformes): a review and a new molecular phylogeny based on three nuclear intron markers. Mol Phylogenet Evol. 48:858–876. [DOI] [PubMed] [Google Scholar]
- Johansson US, Pasquet E, Irestedt M. 2011. The New Zealand thrush: an extinct oriole. PLoS One 6:e24317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jønsson KA, Bowie RCK, Norman JA, Christidis L, Fjeldså J. 2008. Polyphyletic origin of toxic Pitohui birds suggests widespread occurrence of toxicity in corvoid birds. Biol Lett. 4:71–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jønsson KA, Fabre P-H, Ricklefs RE, Fjeldså J. 2011. Major global radiation of corvoid birds originated in the proto-Papuan archipelago. Proc Natl Acad Sci U S A. 108:2328–2333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jønsson KA, Irestedt M, et al. 2008. Explosive avian radiations and multi-directional dispersal across Wallacea: evidence from the Campephagidae and other Crown Corvida (Aves). Mol Phylogenet Evol. 47:221–236. [DOI] [PubMed] [Google Scholar]
- Joseph L, et al. 2014. A new synthesis of the molecular systematics and biogeography of honeyeaters (Passeriformes: Meliphagidae) highlights biogeographical and ecological complexity of a spectacular avian radiation. Zool Script. 43:235–248. [Google Scholar]
- Kennedy JD, et al. 2012. Ecological limits on diversification of the Himalayan core Corvoidea. Evolution 66:2599–2613. [DOI] [PubMed] [Google Scholar]
- Kimball RT, et al. 2013. Identifying localized biases in large datasets: a case study using the avian tree of life. Mol Phylogenet Evol. 69:1021–1032. [DOI] [PubMed] [Google Scholar]
- Ksepka DT, Clarke JA. 2015. Phylogenetically vetted and stratigraphically constrained fossil calibrations within Aves. Palaeontol Electron. 18.1.3FC:1–25. [Google Scholar]
- Landis C, et al. 2008. The Waipounamu erosion surface: questioning the antiquity of the New Zealand land surface and terrestrial fauna and flora. Geol Mag. 145:173–197. [Google Scholar]
- Lanfear R, Calcott B, Ho SYW, Guindon S. 2012. PartitionFinder: combined selection of partitioning schemes and substitution models for phylogenetic analyses. Mol Biol Evol. 29:1695–1701. [DOI] [PubMed] [Google Scholar]
- Lartillot N, Rodrigue N, Stubbs D, Richer J. 2013. PhyloBayes MPI: phylogenetic reconstruction with infinite mixtures of profiles in a parallel environment. Syst Biol. 62:611–615. [DOI] [PubMed] [Google Scholar]
- Martin M. 2011. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 17:10–12. [Google Scholar]
- Mayr G. 2005. The Paleogene fossil record of birds in Europe. Biol Rev. 80:515–542. [DOI] [PubMed] [Google Scholar]
- Mayr G. 2009. Paleogene fossil birds. Berlin: Springer. [Google Scholar]
- Mayr G. 2013. The age of the crown group of passerine birds and its evolutionary significance—molecular calibrations versus the fossil record. Syst Biodivers. 11:7–13. [Google Scholar]
- McComish BJ, Hills SFK, Biggs PJ, Penny D. 2010. Index-free de novo assembly and deconvolution of mixed mitochondrial genomes. Genome Biol Evol. 2:410–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan-Richards M, et al. 2008. Bird evolution: testing the Metaves clade with six new mitochondrial genomes. BMC Evol Biol. 8:20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mourer-Chauviré C, Hugueney M, Jonet P. 1989. Découverte de Passeriformes dans l’Oligocène Supérieur de France. Comp Rend Acad des Sci Paris Ser 2. 309:843–849. [Google Scholar]
- Moyle RG, et al. 2012. Phylogeny and biogeography of the core babblers (Aves: Timaliidae). Syst Biol. 61:631–651. [DOI] [PubMed] [Google Scholar]
- Nabholz B, Glemin S, Galtier N. 2009. The erratic mitochondrial clock: variations of mutation rate, not population size, affect mtDNA diversity across birds and mammals. BMC Evol Biol. 9:13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nabholz B, Jarvis ED, Ellegren H. 2010. Obtaining mtDNA genomes from next-generation transcriptome sequencing: a case study on the basal Passerida (Aves: Passeriformes) phylogeny. Mol Phylogenet Evol. 57:466–470. [DOI] [PubMed] [Google Scholar]
- Pacheco M, et al. 2011. Evolution of modern birds revealed by mitogenomics: timing the radiation and origin of major orders. Mol Biol Evol. 28:1927–1942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parham JF, et al. 2012. Best practices for justifying fossil calibrations. Syst Biol. 61:346–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips MJ, Gibb GC, Crimp EA, Penny D. 2010. Tinamous and moa flock together: mitochondrial genome sequence analysis reveals independent losses of flight among ratites. Syst Biol. 59:90–107. [DOI] [PubMed] [Google Scholar]
- Pratt RC, et al. 2009. Toward resolving deep Neoaves phylogeny: data, signal enhancement, and priors. Mol Biol Evol. 26:313–326. [DOI] [PubMed] [Google Scholar]
- Price TD, et al. 2014. Niche filling slows the diversification of Himalayan songbirds. Nature 509:222–225. [DOI] [PubMed] [Google Scholar]
- Rambaut A, Drummond AJ. 2003. Tracer: MCMC trace analysis tool. Version 1.5.0. Available from: http://tree.bio.ed.ac.uk/software/tracer/. [Google Scholar]
- Schellart WP, Lister GS, Toy VG. 2006. A Late Cretaceous and Cenozoic reconstruction of the Southwest Pacific region: tectonics controlled by subduction and slab rollback processes. Earth Sci Rev. 76:191–233. [Google Scholar]
- Schweizer M, Seehausen O, Hertwig ST. 2011. Macroevolutionary patterns in the diversification of parrots: effects of climate change, geological events and key innovations. J Biogeogr. 38:2176–2194. [Google Scholar]
- Shepherd LD, Lambert DM. 2007. The relationships and origins of the New Zealand wattlebirds (Passeriformes, Callaeatidae) from DNA sequence analyses. Mol Phylogenet Evol. 43:480–492. [DOI] [PubMed] [Google Scholar]
- Sibley CG, Ahlquist JE. 1990. Phylogeny and classification of birds: a study in molecular evolution. New Haven (CT): Yale University Press. [Google Scholar]
- Simpson JT, et al. 2009. ABySS: a parallel assembler for short read sequence data. Genome Res. 19:1117–1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slack KE, et al. 2006. Early penguin fossils, plus mitochondrial genomes, calibrate avian evolution. Mol Biol Evol. 23:1144–1155. [DOI] [PubMed] [Google Scholar]
- Slack KE, et al. 2007. Resolving the root of the avian mitogenomic tree by breaking up long branches. Mol Phylogenet Evol. 42:1–13. [DOI] [PubMed] [Google Scholar]
- Slater GS, Birney E. 2005. Automated generation of heuristics for biological sequence comparison. BMC Bioinformatics 6:31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Springer M, Douzery E. 1996. Secondary structure and patterns of evolution among mammalian mitochondrial 12S rRNA molecules. J Mol Evol. 43:357–373. [DOI] [PubMed] [Google Scholar]
- Stamatakis A, Hoover P, Rougemont J. 2008. A rapid bootstrap algorithm for the RAxML web servers. Syst Biol. 57:758–771. [DOI] [PubMed] [Google Scholar]
- Suh A, et al. 2011. Mesozoic retroposons reveal parrots as the closest living relatives of passerine birds. Nat Commun. 2:443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Treplin S, et al. 2008. Molecular phylogeny of songbirds (Aves: Passeriformes) and the relative utility of common nuclear marker loci. Cladistics 24:328–349. [Google Scholar]
- Trewick SA, Gibb GC. 2010. Vicars, tramps and assembly of the New Zealand avifauna: a review of molecular phylogenetic evidence. Ibis 152:226–253. [Google Scholar]
- Wang N, Braun EL, Kimball RT. 2012. Testing hypotheses about the sister group of the Passeriformes using an independent 30 locus dataset. Mol Biol Evol. 29:737–750. [DOI] [PubMed] [Google Scholar]
- White NE, et al. 2011. The evolutionary history of cockatoos (Aves: Psittaciformes: Cacatuidae). Mol Phylogenet Evol. 59:615–622. [DOI] [PubMed] [Google Scholar]
- Worthy TH, et al. 2007. Miocene waterfowl and other birds from central Otago, New Zealand. J Syst Palaeontol. 5:1–39. [Google Scholar]
- Worthy TH, et al. 2009. A large fruit pigeon (Columbidae) from the early Miocene of New Zealand. Auk 126:649–656. [Google Scholar]
- Worthy TH, et al. 2010. Biogeographical and phylogenetic implications of an early Miocene wren (Aves: Passeriformes: Acanthisittidae) from New Zealand. J Vert Paleo. 30:479–498. [Google Scholar]
- Worthy TH, Tennyson AJD, Scofield RP. 2011. An early Miocene diversity of parrots (Aves, Strigopidae, Nestorinae) from New Zealand. J Vert Paleo. 31:1102–1116. [Google Scholar]
- Zerbino DR, Birney E. 2008. Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 18:821–829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuccon D, Ericson PGP. 2012. Molecular and morphological evidences place the extinct New Zealand endemic Turnagra capensis in the Oriolidae. Mol Phylogenet Evol. 62:414–426 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.