

Abstract
Sponges harbor a complex consortium of microbial communities living in symbiotic relationship benefiting each other through the integration of metabolites. The mechanisms influencing a successful microbial association with a sponge partner are yet to be fully understood. Here, we sequenced the genome of Pseudovibrio sp. POLY-S9 strain isolated from the intertidal marine sponge Polymastia penicillus sampled from the Atlantic coast of Portugal to identify the genomic features favoring the symbiotic relationship. The draft genome revealed an exceptionally large genome size of 6.6 Mbp compared with the previously reported genomes of the genus Pseudovibrio isolated from a coral and a sponge larva. Our genomic study detected the presence of several biosynthetic gene clusters—polyketide synthase, nonribosomal peptide synthetase and siderophore—affirming the potential ability of the genus Pseudovibrio to produce a wide variety of metabolic compounds. Moreover, we identified a repertoire of genes encoding adaptive symbioses factors (eukaryotic-like proteins), such as the ankyrin repeats, tetratrico peptide repeats, and Sel1 repeats that improve the attachment to the eukaryotic hosts and the avoidance of the host’s immune response. The genome also harbored a large number of mobile elements (∼5%) and gene transfer agents, which explains the massive genome expansion and suggests a possible mechanism of horizontal gene transfer. In conclusion, the genome of POLY-S9 exhibited an increase in size, number of mobile DNA, multiple metabolite gene clusters, and secretion systems, likely to influence the genome diversification and the evolvability.
Keywords: symbiont, marine sponge, adaptation, mobile elements, biosynthetic gene clusters, comparative genomics
Introduction
Marine sponges (phylum Porifera) harbor dense and diverse microbial communities. Different microbial lineages have been detected in sponges, representing a specific and stable association with its hosts that are distinct from the surrounding seawater (Simister et al. 2012; Webster and Taylor 2012; Naim et al. 2014). This specific nature of association indicates the strict vertical transmission of microbes through larvae (de Caralt et al. 2007; Lee et al. 2009; Gloeckner et al. 2013). Amplicon sequencing of sponge-dwelling microbes has shown that a complex consortium of microbes live on sponges in various ocean gradients (Lee et al. 2011; Schmitt et al. 2012; Gao et al. 2014; Alex and Antunes 2015). Factors regulating the interactions between microbes and their sponge hosts could be revealed by whole-genome sequencing.
Metagenomic (Thomas et al. 2010; Liu et al. 2011; Fan et al. 2012) and metaproteogenomic (Liu et al. 2012) approaches of studying environmental samples provide significant knowledge about the functional role of uncultured symbiotic microorganisms. For example, metagenomic analyses of the bacterial community in the sponge Cymbastela concentrica revealed the presence of a large number of transposable insertion elements crucial for the evolution of symbiotic bacterial genomes (Thomas et al. 2010). Genomic factors involved in the sponge-microbe symbioses, like adhesion-related proteins, ankyrin repeat (ANK) proteins, and tetratricopeptide repeats domain-encoding proteins (TPR), were frequently detected in C. concentrica metagenome (Thomas et al. 2010) and in the genome of Deltaproteobacteria from C. concentrica (Liu et al. 2012). Recent whole genome and metabolic analyses of two Pseudovibrio strains isolated from a scleractinian coral and from the sponge Mycale laxissima also revealed symbiont-supporting metabolic adaptations (Bondarev et al. 2013).
The genus Pseudovibrio is found in many marine habitats and is generally associated with marine invertebrates, such as tunicates and sponges (reviewed in Crowley et al. 2014). The Pseudovibrio-related bacteria dominated the majority of the sponge bacterial isolates and those of the M. laxissima sponge larvae (Enticknap et al. 2006) suggesting that the Pseudovibrio sp. is vertical transmitted. Many isolated strains of the genus Pseudovibrio are known to produce active secondary metabolites with potential antimicrobial activity (Dupont et al. 2013), namely heptylprodigiosin from Pseudovibrio denitrificans (Sertan-de Guzman et al. 2007), tropodithietic acid from an epiphytic Pseudovibrio strain isolated from the red algae Delisea pulchra (Penesyan et al. 2011), and pseudovibriocin from coral-associated Pseudovibrio sp. (Vizcaino 2011). In addition to antimicrobial activity, a recent study revealed a role in heavy metal detoxification of the Pseudovibrio sp. isolated from the sponge Spongia officinalis (Bauvais et al. 2015).
The widespread association of Pseudovibrio sp. with various marine animals and its expanded genomic repertoire enabling the production of secondary metabolites prompted us to investigate the genome content of Pseudovibrio sp. from the intertidal sponge Polymastia penicillus (class Demospongiae). Here, we sequenced the genome of Pseudovibrio sp. POLY-S9, sampled from the Atlantic coast of Portugal. The genomic repertoire of Pseudovibrio sp. POLY-S9 (genes for protein translocation, attachment, and invasion; secretion system (SS) apparatus and its effector molecules) suggests that it has the ability to interfere with host cell function and regulate bacterial survival in the community. Genome analyses also identified the presence of various secondary metabolic gene clusters. The large number of mobile transfer agents/gene transfer agents (GTAs) detected suggests the genome plasticity of this bacterium, which could also act as a possible mechanism of gene procurement through horizontal gene transfer.
Materials and Methods
Isolation of Pseudovibrio sp.
We used Pseudovibrio sp. isolated from the intertidal marine sponge Po. penicillus reported earlier (Alex et al. 2013). Briefly, 1 cm3 of sponge tissue was dissected and grinded using sterile seawater. Serially diluted homogenate was plated on DifcoTM Marine Agar 2216 medium containing amphotericin B (1 ml/100 ml). After repeated streaking for obtaining pure culture, bacterial colonies were inoculated in 2 ml tube containing DifcoTM Marine broth 2216 medium and maintained at 28 °C with constant shaking until obtaining the adequate biomass. Genomic DNA was isolated using PureLinkTM Genomic DNA kit (Invitrogen) following the manufactures instructions for bacterial DNA isolation.
Genome Sequencing, Assembly, and Annotation of Sponge Symbiont
Whole genome sequencing was performed using the Illumina’s HiSeq 2000 Sequencing System. Paired-end read library (2 ×100 nt) construction with an insert size of ∼300 bp and sequencing were performed at Macrogen, South Korea. The sequencing reads were assembled using de novo short read sequence assembler, Velvet 1.2.10 (Zerbino and Birney 2008). Illumina reads were quality filtered by discarding the reads with a Phred quality score below 30. Adapter sequences were trimmed off raw reads with cutadapt (Martin 2011) retrieving a total of 77492810 reads. The best possible k-mer coverage of reads (k = 99) for the current genome assembly was opted from VelevetK. Overall, 271 contigs of >1,400-fold coverage were retrieved and annotated using the Prokka v1.10 (Seemann 2014) annotation pipeline. This Whole Genome Shotgun project has been deposited at DDBJ/EMBL/GenBank under the accession LCWX00000000. The version described in this article is version LCWX01000000.
Prokka depends on many external feature prediction tools for genome annotation. Coding sequence (CDS) predication was carried out by Prodigal v2.60 (Hyatt et al. 2010). We compiled a core database for assigning the function to the predicted CDS using a set of trustworthy curated data set comprising annotated bacterial proteins obtained from the current release of UniProt Knowledgebase, UniProtKB Release 2014_10 (Magrane and Consortium 2011). Ribosomal RNA (rRNA), transfer RNA (tRNA) genes, and noncoding RNA were predicted using RNAmmer v1.2 (Lagesen et al. 2007), ARAGORN v1.2.36 (Laslett and Canback 2004), and Infernal v1.1 (Kolbe and Eddy 2011), respectively. SignalP v4.1 was used for the prediction of signal peptides (Petersen et al. 2011).
Other genomes from the genus Pseudovibrio (FO-BEG and JE062) used for comparative purposes in this study were downloaded from NCBI ftp (ftp://ftp.ncbi.nih.gov/genomes/). The predicted ORFs were assigned to Clusters of Orthologous Groups of proteins (COGs) using WebMGA server (Wu et al. 2011) with RPSBLAST v2.2.15 (Altschul et al. 1990).
Metabolic Gene Cluster Prediction and Phage Identification
The secondary metabolite biosynthesis gene clusters from the Pseudovibrio sp. POLY-S9 isolated in this study and from previously sequenced genomes of other Pseudovibrio strains (FO-BEG and JE062) were analyzed using antiSMASH v3.0.3. (Weber et al. 2015). Prophage gene sequences within the bacterial genomes analyzed were identified using PHAST (Zhou et al. 2011).
Results and Discussion
General Genome Features
The isolated Pseudovibrio sp. POLY-S9 from the intertidal marine sponge Po. penicillus showed 100% SSU (small subunit) rRNA similarity to the Pseudovibrio ascidiaceicola strain NBRC 100514 (AB681198), which has been previously isolated from the ascidian Polycitor proliferus (Fukunaga et al. 2006). Phylogenetic analyses further revealed the clustering of POLY-S9 strain with Ps. ascidiaceicola isolated from tunicates (AB681198, AB175663, AB210280) suggesting the ability of the genus Pseudovibrio to infect a wide range of marine hosts (supplementary fig. S1, Supplementary Material online).
The genome analyses of the strain Pseudovibrio sp. POLY-S9 isolated from the intertidal marine sponge Po. penicillus revealed the chromosomal genome size of 6.6 Mbp with a G + C (Guanine + Cytosine) content of 51.26% (fig. 1 and table 1) consistent with previously described values for the Ps. ascidiaceicola strain (Fukunaga et al. 2006). The genome assembly of the Pseudovibrio sp. POLY-S9 resulted in 271 contigs and the annotation with Prokka revealed the presence of 6171 CDSs, greater than the estimated CDS from the genomes of Pseudovibrio sp. FO-BEG1 and Pseudovibrio sp. JE062 (table 1). The abundance of mobile elements in the genome of POLY-S9 caused difficulty for the assembly, which explains the persistence of multiple contigs. However, the obtained genome assembly allowed us to understand the genomic features of the sponge-associated bacteria with a greater confidence.
Fig. 1.—
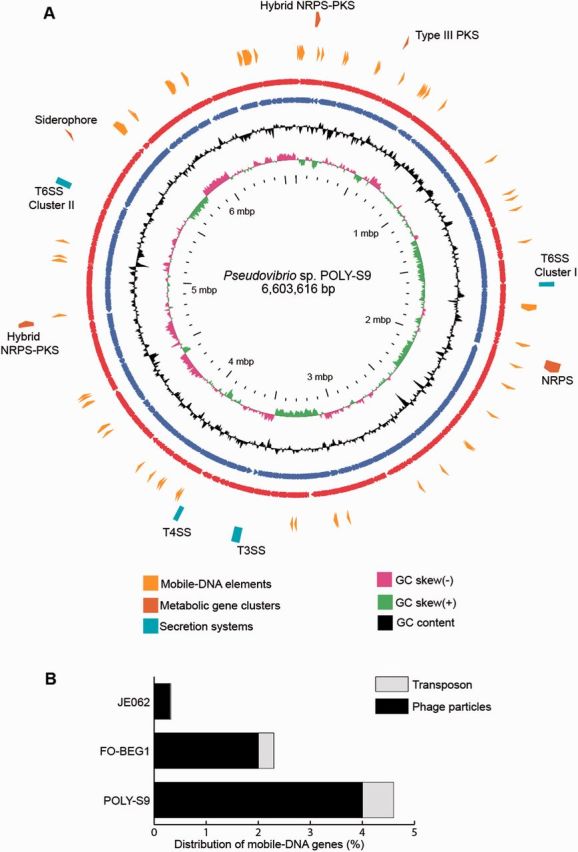
Circular view of the Pseudovibrio sp. POLY-S9 genome highlighting the proportion of the main genomic features like mobile elements, SSs, and metabolic gene clusters. (A) The red and blue ring represents the sense and antisense strand of POLY-S9 chromosome. The black ring represents the GC content followed by GC skew (purple/green color). GView was used to generate the circular view of the bacterial chromosome (Petkau et al. 2010). The distribution of mobile elements and GTAs are shown as yellow clockwise arrows. Aqua colored block shows the location of SSs in the genome of POLY-S9. Brown (outermost ting) represents the distribution of various secondary metabolic gene clusters. (B) Stacked graph representing the percentage of mobile DNA elements detected in the genus Pseudovibrio.
Table 1.
Genome Characteristics of the Three Fully Sequenced Pseudovibrio species
| Pseudovibrio sp. POLY-S9 | Pseudovibrio sp. FO-BEG1 | Pseudovibrio sp. JE062 | |
|---|---|---|---|
| Genome size | 6,603,616 bp | 5,916,782 bp | 5,726,521 bp |
| No. of contigs | 271 | 2 | 19 |
| GC content | 51.26% | 52.50% | 52.40% |
| No. of CDS | 6171 | 5478 | 5225 |
| No. of rRNA | 3 | 6 | 7 |
| No. of tRNA | 59 | 69 | 72 |
| Mobile DNA | ∼5% | ∼2.5% | ∼0.3% |
Note.—POLY-S9 was sequenced in this study. FO-BEG1 and JE-062 were obtained from Bondarev et al. (2013).
Comparison of gene contents based on the COG categories revealed mostly commonality across three genomes of Pseudovibrio sp. However, the POLY-S9 showed overabundance of strain-specific gene content, especially the COG categories related to the following: 1) Secondary metabolite biosynthesis, transport, and catabolism (COG ‘Q’); 2) replication, recombination, and repair (COG ‘L’); 3) intracellular trafficking, secretion, and vesicular transport (COG ‘U’); 4) posttranslational modification, protein turnover, and chaperones (COG ‘O’); 5) energy production and conversion (COG ‘C’); and 6) lipid metabolism (COG ‘I’), which are likely to encode specialized niche-specific physiological functions (fig. 2). Some of the relevant genes from the above-mentioned categories are discussed in the following sections.
Fig. 2.—

Distribution of the categories of functional genes present in three Pseudovibrio strains based on the COGs of proteins. The alphabetic code for the COG categories is as follows: B, chromatin structure and dynamics; C, energy production and conversion; D, cell cycle control, cell division, chromosome partitioning; E, amino acid transport and metabolism; F, nucleotide transport and metabolism; G, carbohydrate transport and metabolism; H, coenzyme transport and metabolism; I, lipid transport and metabolism; J, translation, ribosomal structure, and biogenesis; K, transcription; L, replication, recombination, and repair; M, cell wall/membrane/envelope biogenesis; N, cell motility; O, posttranslational modification, protein turnover, chaperones; P, inorganic ion transport and metabolism; Q, secondary metabolite biosynthesis, transport, and catabolism; R, general function prediction only; S, function unknown; T, signal transduction mechanisms; U, intracellular trafficking, secretion, and vesicular transport; V, defense mechanisms.
Genome Mining of Secondary Metabolites
Polyketide Synthase and Nonribosomal Peptide Synthetase Gene Clusters
Sponges are proven to be a rich source of bioactive compounds with pharmacological application and in recent years it has been shown that their symbiotic bacteria produce potential novel metabolites (Fuerst 2014). Most bioactive compounds are produced by polyketide synthase (PKS) and nonribosomal peptide synthetase (NRPS) (Staunton and Weissman 2001; Schwarzer et al. 2003). Genomic mining using antiSMASH (Weber et al. 2015) detected one gene cluster belonging to NRPS (40 kb), T3PKS (5.2 kb), and hybrid NRPS–PKS (42 kb), and two bacteriocin gene clusters in the genome of Pseudovibrio sp. POLY-S9, likely involved in the bioactive compound production (supplementary table S1, Supplementary Material online). The hybrid NRPS–PKS system (42 kb) similar to Pseudovibrio sp. FO-BEG1 strain was observed as a major metabolic gene cluster in POLY-S9 distributed in two different contigs (fig. 3). According to antiSMASH analyses, this gene cluster is known to be a colibactin biosynthetic gene cluster, which is common among Escherichia coli and induces the DNA double-strand break in eukaryotic cells (Nougayrède et al. 2006). In addition, among the sequenced genomes of the genus Pseudovibrio, the POLY-S9 genome encoded a 40 kb NRPS gene cluster exhibiting similarity with the distantly related bacteria Pseudomonas fluorescens strain SF39a belonging to the class Alphaproteobacteria (supplementary fig. S2, Supplementary Material online). This suggests that the NRPS gene cluster was likely obtained through horizontal gene transfer and future detailed methodological analyses should validate this hypothesis.
Fig. 3.—

Biosynthetic gene cluster detected in the genome of Pseudovibrio sp. POLY-S9. The hybrid NRPS–PKS system responsible for colibactin production in the POLY-S9 genome sequenced in this study and the previously sequenced FO-BEG1 genome. Different color codes represent detected ORFs responsible for biosynthesis, hypothetical proteins, and transport proteins.
The potential ability of the genus Pseudovibrio to produce bioactive compounds has been widely studied (Crowley et al. 2014). For instance, the Pseudivibrio strains isolated from the sponge Haliclona simulans were reported to contain putative PKS and NRPS genes (Kennedy et al. 2008), and exhibited antimicrobial activity against methycillin-resistant Staphylococcus aureus, Bacillus cereus, E. coli, and Bacillus subtilus (Kennedy et al. 2009). Subsequent studies further demonstrated the antimicrobial activity of the genus Pseudovibrio isolated from other marine sponge species, like Erylus discophorus (Graça et al. 2013), Amphilectus fucorum, and Eurypon major (Margassery et al. 2012).
Siderophore Biosynthetic Gene Cluster
Interestingly, a gene cluster coding for NRPS-independent siderophore biosynthetic pathway was detected in the POLY-S9 genome. Siderophores are iron chelators synthesized and excreted by many pathogenic/saprophytic microorganisms in response to iron limitation (Drechsel and Jung 1998). Detailed analyses revealed that these siderophore gene cluster harbored four genes (iucABCD) coding for aerobactin biosynthesis enzymes. In many virulent E. coli strains, aerobactin orchestrates iron acquisition, playing a key role in pathogenesis (Dho and Lafont 1984; Lafont et al. 1987; Knöbl et al. 2001; Ling et al. 2013). Reanalysis of FO-BEG1 and JE062 genomes using antiSMASH could not detect the presence of siderophore gene clusters. However, PKS/NRPS proteins similar to predicted siderophore biosynthetic gene has been previously reported from various Pseudovibrio spp. isolated from the Irish marine sponges (O’Halloran et al. 2011) using a PCR (polymerase chain reaction) screening approach. Whole genome sequencing of Actinokineospora sp. strain EG49 isolated from the Red Sea sponge Spheciospongia vagabunda identified the presence of several metabolic gene clusters coding for PKS, NRPS, hybrid NRPS–PKS, and siderophore (Harjes et al. 2014), revealing the importance of searching novel bioactive products in the sponge-associated microbes.
Genes in the COG ‘Q’ category are overrepresented in Pseudovibrio POLY-S9 when compared with FO-BEG1 and JE062 (161, 118, and 124 genes, respectively) (fig. 2). Among these overrepresented COGs are genes associated with NRPS and PKS, indicating the capacity of Pseudovibrio sp. POLY-S9 isolated from the marine sponge Po. penicillus to produce novel secondary metabolites. We also detected the overabundance of COG ‘I’ category in POLY-S9 involved in lipid transport and metabolism, which might suggest the improved ATP utilization derived from fats, relatively to carbohydrates.
Role of Secretion Systems in Sponge–Bacteria Interaction
T3SS/T6SS Involved in the Successful Bacterial Colonization
The successful establishment of symbiosis depends on the ability of the microbes to interact with its hosts. Such interactions are mediated by delivering virulence factors, proteins, or toxins (effector molecules) to gain control of the host response through several mechanisms (table 2). Gram-negative bacteria use specialized translocation systems called SSs to transport proteins across the outer membrane and to facilitate the bacterial invasion (Cossart and Sansonetti 2004). Type III SS (T3SS), type VI SS (T6SS) (figs. 4A and 5A), and multiple copies of homologues of effector molecules (figs. 4B and 5B) were detected in the genome of Pseudovibrio sp. POLY-S9. The presence of T3SSs/T6SSs in POLY-S9 and other Pseudovibrio strains indicates the likely function of these SS proteins in the sponge–bacteria interaction in various habitats.
Table 2.
Predicted Major Genomic Features of Pseudovibrio sp
| Predicted Gene/Gene Category | Functional Category |
|---|---|
| T3SSs | Involved in flagellar assembly, secretion of extracellular proteins and effector molecules into the host cell (Dale and Moran 2006; Preston 2007) |
| T3SS effector proteins | Responsible for the disruption of host cytoskeletal system and phagocytotic activity (Niebuhr et al. 2000; Mukherjee et al. 2006; Wiley et al. 2006) |
| T6SSs | Widespread secretion system responsible for protein translocation, enhancing the pathogenicity, and biofilm formation (Aschtgen et al. 2008; Filloux et al. 2008; Kapitein and Mogk 2013; Sheng et al. 2013) |
| T6SS effector proteins | Major virulence factors for pathogenic bacteria |
| T4SSs | Secretion system protein involved in conjugation, uptake of foreign DNA from the surrounding, and transport of virulence factors into the host cell (Alvarez-Martinez and Christie 2009) |
| Ankyrin repeat domains | Interfere with host cell functions (Habyarimana et al. 2008; Al-Khodor et al. 2010) |
| Tetratricopeptide repeat domains | Mediate the translocation of virulence factors to the host cell (Cerveny et al. 2013) |
| Sel1 domains | Facilitate microbial entry through epithelial cells (Mittl and Schneider-Brachert 2007) |
| Amyloid production genes | Gene cluster responsible for curli fiber production and assembly. Mediates microbial attachment to the eukaryotic cell surface (Collinson et al. 1993) |
| Invasion-associated locus (ialB) | Virulence factor in establishing host-microbe association (Rolain et al. 2002) |
| TadE-like domain | Colonization factors (Tomich et al. 2007) |
| YadA domain | Adhesion factors (Heise and Dersch 2006) |
| tad locus | Genes essential for adherence, biofilm formation, synthesis of Flp pili, and pathogenesis (Tomich et al. 2007) |
| QS | Bacterial communication and regulation of bacterial processes such as gene expression in response to cell population density, biofilm formation, and symbioses (Ng and Bassler 2009) |
Note.—POLY-S9 involved in establishment of symbiosis with marine sponges.
Fig. 4.—

Genetic organization of T3SS and its effector molecules in Pseudovibrio sp. POLY-S9. (A) Major genes annotated for T3SS structural components are colored as red blocks. Black and brown blocks represent the hypothetical and unrelated genes, respectively. (B) Effector molecules yopJ, ipgD, and ypkA holologues responsible for the disruption of the host cell mechanism.
Fig. 5.—

Genetic organization of T6SS and T4SS gene clusters in the genome of Pseudovibrio sp. POLY-S9. (A) Two clusters representing the distribution of the genes coding for T6SS apparatus. (B) Genes coding for T6SS effector molecules (three copies of hcpl and seven copies of vgrG). (C) T4SS detected in the sequenced genome. Blocks of related genes are represented in the same color. Hypothetical and other secretion system-related protein-coding genes are colored black and gray, respectively.
Type IV Secretion System and Pathogenicity
Among the sequenced genomes of the genus Pseudovibrio, type IV SS (T4SS) protein complex was present only in Pseudovibrio sp. POLY-S9. T4SSs are involved in mediating the DNA transfer through conjugation, DNA uptake/release from the extracellular milieu, and translocation of virulence/effector proteins into the host cells (Alvarez-Martinez and Christie 2009). T4SSs loci of Pseudovibrio sp. POLY-S9 encoded the proteins homologous to several virulence genes: virB2, virB4, virB6, virB9, virB10, virB11, and virD4 (fig. 5C). These protein subunits of T4SSs form the pilus and the translocation channels spanning the bacterial cell envelop (Voth et al. 2012). Besides vir genes, the T4SSs loci also encoded the genes responsible for the conjugation (origin of transfer [oriT] and relaxase). Relaxase–oriT complex is required to initiate the lateral transfer and the incorporation of large segment of DNA (genomic island) into the bacterial chromosome (Waldor 2010).
T4SSs were reported to be overrepresented in the coral metagenome suggesting its role in establishing the successful coral–bacteria association (Carlos et al. 2014). In clinical microbes, like Haemophilus and Pseudomonas, T4SSs also mediate the horizontal gene transfer that can play a significant role in the evolution of several pathogenic characters, like antibiotic resistance and virulence (Juhas 2015). The versatile nature of the T4SSs in Pseudovibrio sp. POLY-S9 may favor genome plasticity enabling the adaptation of the bacterium to various niches in response to environmental changes.
The overrepresentation of the COG category ‘U’ for intracellular trafficking, secretion, and vesicular transport in the genome of POLY-S9 when compared with FO-BEG1 and JE062 (86, 78, and 78 genes, respectively) indicates the specific nature of interaction between Pseudovibrio sp. POLY-S9 and the sponge host (fig. 2). The genes related to the COG category ‘U’ constituted mainly virB, flagellar assembly genes, and the genes responsible for the protein export (translocase), suggesting the enhanced virulence behavior of the Pseudovibrio sp. POLY-S9.
Genomic Feature of POLY-S9 Favoring the Successful Sponge-Specific Lifestyle
Eukaryotic-like Proteins
In the genome of Pseudovibrio sp. POLY-S9, several genes were identified encoding for eukaryotic-like proteins (ELPs), which contain motifs such as ANKs, tetratrico peptide repeats (TPRs), and Sel1 repeats (table 2). These symbiosis factors (ELPs) are widely represented in the genomes of pathogenic as well as symbiotic microbes, mediating the host behavior by interfering in the eukaryotic protein–protein interactions (Mittl and Schneider-Brachert 2007; Al-Khodor et al. 2010; Cerveny et al. 2013). Sponge-associated microbes have been frequently reported to encode ELPs. For instance, metagenomic and metaproteogenomic analysis revealed the presence of ANKs and TRPs in C. concentrica (Thomas et al. 2010; Liu et al. 2012), as well as in several other marine sponge-associated microbial communities (Fan et al. 2012). Whole genome analyses of sponge symbionts, Poribacteria (Siegl et al. 2011), Deltaproteobacteria (Liu et al. 2011), and Pseudovibrio strains (Bondarev et al. 2013) also showed the presence of eukaryotic domain containing motifs emphasizing the role of ELPs pertaining to the symbiotic lifestyle.
Adhesion and Invasion Factors
Consistent with the previously sequenced Pseudovibrio strains, we detected a gene cluster responsible for amyloid (curli) production and assembly/transport components, eight homologues of the Bartonella bacilliformis ialB (invasion-associated locus B), genes coding for proteins containing YadA (Yersenia adhesin A)/TadE-like domains, and tad (tight adherence) locus (cpaABCDEF) in the genome of POLY-S9 (table 2). Detected adhesion and invasion factors play an important role in virulence due to interaction with a wide range of host protein such as extracellular matrix protein (Olsén et al. 1989; Collinson et al. 1993; Heise and Dersch 2006), bacterial colonization, biofilm formation, and pathogenesis (Schreiner et al. 2003; Tomich et al. 2007). Recurrent detection of the genes coding for adhesion and invasion in all three strains of the Pseudovibrio sp. indicate their requirement in establishing the bacterial association and increasing the colonization success with the eukaryotic sponge hosts inhabiting different environmental niches.
So far, no experimental evidences are available to demonstrate the exact role of these symbioses factors among the sponge-associated bacteria. The presence of various genes responsible for virulence, protein–protein interaction, adhesion, biofilm formation, and invasion in the genome of Pseudovibrio sp. POLY-S9 might suggests possible mechanisms involved in the symbiotic association with the marine sponge species.
Interaction among Sponge-associated Bacteria
Bacterial communication is an essential aspect in a microbial community, where competition for resource is achieved by quorum sensing (QS). QS system seems to regulate bacterial processes such as biofilm formation, virulence factor secretion, bioluminescence, motility, antibiotic production, sporulation, and DNA uptake (Ng and Bassler 2009). It is induced by diffusible signaling molecules, autoinducers (transcriptional regulators), which trigger the expression of specific genes (Fuqua et al. 1994). The genome analysis of the POLY-S9 strain revealed the presence of proteins containing DNA-binding LuxR domains, which act as transcriptional regulators of N-acyl homeserine lactones (AHL), a common proteobacterial signaling molecule. Previous genome analyses reported the presence of LuxR genes among the genus Pseudovibrio, but not the genes coding for the AHL synthase (Bondarev et al. 2013). It was hypothesized that the strains FO-BEG and JE062 respond to the autoinducers secreted by other bacterial species (Case et al. 2008). However, we could identify a luxl homologue that encodes AHL synthase in the Pseudovibrio POLY-S9 genome, suggesting the synthesis of QS molecules and the existence of intraspecific communication. The adaptive response of POLY-S9 to compete for their growth, sponge–bacteria interaction, and to survive in high cell density environment might be accomplished using QS and autoinducers.
High Mobile Genetic Elements and Genome Diversification
The comparison of previously sequenced genomes of the Pseudovibrio strains isolated from the scleractinian coral and the sponge M. laxissima (Bondarev et al. 2013) clearly shows that the Pseudovibrio sp. POLY-S9 strain has a higher number of mobile DNA genes (∼5%), a probable reason to explain its larger genome size (fig. 1). It is known that the facultative intracellular bacteria usually harbor 4-fold more mobile DNA than the obligate intracellular bacteria (Bordenstein and Reznikoff 2005), which is consistent with the prediction that the free-living bacteria contain more mobile DNA than the facultative bacteria. Bordenstein and Reznikoff (2005) estimated that the transposable elements constitute the major portion of mobile DNA among intracellular bacteria. In this study, phage-coding genes (4%) predominated among the mobile DNA in the Pseudovibrio sp. POLY-S9 strain (fig. 1B). In contrast, the sponge symbiont Pseudovibrio sp. FO-BEG1 strain and the vertically transmitted symbiont Pseudovibrio sp. JE062 strain (Bondarev et al. 2013) harbored less mobile elements (∼2.5% and ∼0.3%, respectively). The lifestyle of a bacterium might influence its genetic content, in which bacteria with small population size (obligate bacteria/FO-BEG1/JE062) might experience higher loss of mobile DNA (Toft and Andersson 2010).
Another remarkable feature of the POLY-S9 genome is the presence of several sets of GTAs (fig. 1). GTAs are phage-like entities responsible for packaging and transferring random segment of host bacterial DNA to a recipient cell of closely or distantly related lineages through horizontal gene transfer (Zhaxybayeva and Doolittle 2011). The COG analyses of the three different Pseudovibrio strains revealed that the POLY-S9 possess a higher number of COG ‘L’ category genes related to replication, recombination, and repair (fig. 3). It clearly indicates the ability of the Pseudovibrio sp. POLY-S9 strain to exchange foreign DNA between species via horizontal gene transfer, which would potentially cause genome diversification. Another study suggested the selective advantage of GTAs in enhancing the HGT process in pathogenic strains of Bartonella. The study also argues that this process facilitates the adaptive evolution of the host-adaptation systems and host-range size expansion (Guy et al. 2013). We hypothesize that the genome plasticity of the Pseudovibrio sp. POLY-S9 could explain its adaptability for the host-switching permissive lifestyle in various habitats.
Conclusions
Our genome analysis of the Pseudovibrio sp. POLY-S9 strain isolated from the intertidal marine sponge Po. penicillus shows a genomic repertoire adapted for living in close association with its eukaryotic host. Moreover, we detected the presence of several bioactive gene clusters, particularly the siderophore biosynthetic genes that warrants further evaluation. Further detailed analyses revealed that the genome plasticity of POLY-S9 is due to a large number of mobile DNA elements, which might favor the survival and the adaptation of this bacterium to various habitats and hosts through the exchange of genetic material. The bacterial genome described here represents an important resource for future investigation of microbes that live in association with intertidal marine sponges.
Supplementary Material
Acknowledgments
The authors thank two anonymous reviewers for the helpful suggestions to improve a previous version of this articlemanuscript. Anoop Alex was funded by the Fundação para a Ciência e a Tecnologia (FCT) (SFRH/BD/62356/2009 and SFRH/BPD/99251/2013). Agostinho Antunes was partially supported by the Strategic Funding UID/Multi/04423/2013 through national funds provided by FCT and European Regional Development Fund (ERDF) in the framework of the programme PT2020, and the FCT projects PTDC/AAC-AMB/121301/2010 (FCOMP-01-0124-FEDER-019490) and PTDC/AAG-GLO/6887/2014.
Literature Cited
- Alex A, Antunes A. 2015. Pyrosequencing characterization of the microbiota from Atlantic intertidal marine sponges reveals high microbial diversity and the lack of co-occurrence patterns. PLoS One 10:e0127455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alex A, Silva V, Vasconcelos V, Antunes A. 2013. Evidence of unique and generalist microbes in distantly related sympatric intertidal marine sponges (Porifera: Demospongiae). PLoS One 8:e80653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Khodor S, Price CT, Kalia A, Kwaik YA. 2010. Ankyrin-repeat containing proteins of microbes: a conserved structure with functional diversity. Trends Microbiol. 18:132–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. 1990. Basic local alignment search tool. J Mol Biol. 215:403–410. [DOI] [PubMed] [Google Scholar]
- Alvarez-Martinez CE, Christie PJ. 2009. Biological diversity of prokaryotic type IV secretion systems. Microbiol Mol Biol Rev. 73:775–808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aschtgen M-S, Bernard CS, De Bentzmann S, Lloubès R, Cascales E. 2008. SciN is an outer membrane lipoprotein required for type VI secretion in enteroaggregative Escherichia coli. J Bacteriol. 190:7523–7531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauvais C, et al. 2015. Sponging up metals: bacteria associated with the marine sponge Spongia officinalis. Mar Environ Res. 104:20–30. [DOI] [PubMed] [Google Scholar]
- Bondarev V, et al. 2013. The genus Pseudovibrio contains metabolically versatile bacteria adapted for symbiosis. Environ Microbiol. 15:2095–2113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bordenstein SR, Reznikoff WS. 2005. Mobile DNA in obligate intracellular bacteria. Nat Rev Microbiol. 3:688–699. [DOI] [PubMed] [Google Scholar]
- Carlos C, Castro DBA, Ottoboni LMM. 2014. Comparative metagenomic analysis of coral microbial communities using a reference-independent approach. PLoS One 9:e111626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Case RJ, Labbate M, Kjelleberg S. 2008. AHL-driven quorum-sensing circuits: their frequency and function among the Proteobacteria. ISME J. 2:345–349. [DOI] [PubMed] [Google Scholar]
- Cerveny L, et al. 2013. Tetratricopeptide repeat motifs in the world of bacterial pathogens: role in virulence mechanisms. Infect Immun. 81:629–635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collinson SK, et al. 1993. Thin, aggregative fimbriae mediate binding of Salmonella enteritidis to fibronectin. J Bacteriol. 175:12–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cossart P, Sansonetti PJ. 2004. Bacterial invasion: the paradigms of enteroinvasive pathogens. Science 304:242–248. [DOI] [PubMed] [Google Scholar]
- Crowley SP, O’Gara F, O’Sullivan O, Cotter PD, Dobson ADW. 2014. Marine Pseudovibrio sp. as a novel source of antimicrobials. Mar Drugs. 12:5916–5929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dale C, Moran NA. 2006. Molecular interactions between bacterial symbionts and their hosts. Cell 126:453–465. [DOI] [PubMed] [Google Scholar]
- de Caralt S, Uriz MJ, Wijffels RH. 2007. Vertical transmission and successive location of symbiotic bacteria during embryo development and larva formation in Corticium candelabrum (Porifera: Demospongiae). J Mar Biol Assoc UK. 87:1693–1699. [Google Scholar]
- Dho M, Lafont JP. 1984. Adhesive properties and iron uptake ability in Escherichia coli lethal and nonlethal for chicks. Avian Dis. 28:1016–1025. [PubMed] [Google Scholar]
- Drechsel H, Jung G. 1998. Peptide siderophores. J Pept Sci. 4:147–181. [DOI] [PubMed] [Google Scholar]
- Dupont S, et al. 2013. Diversity and biological activities of the bacterial community associated with the marine sponge Phorbas tenacior (Porifera, Demospongiae). Lett Appl Microbiol. 58:42–52. [DOI] [PubMed] [Google Scholar]
- Enticknap JJ, Kelly M, Peraud O, Hill RT. 2006. Characterization of a culturable alphaproteobacterial symbiont common to many marine sponges and evidence for vertical transmission via sponge larvae. Appl Environ Microbiol. 72:3724–3732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan L, et al. 2012. Functional equivalence and evolutionary convergence in complex communities of microbial sponge symbionts. Proc Natl Acad Sci U S A. 109:E1878–E1887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filloux A, Hachani A, Bleves S. 2008. The bacterial type VI secretion machine: yet another player for protein transport across membranes. Microbiol Read Engl. 154:1570–1583. [DOI] [PubMed] [Google Scholar]
- Fuerst JA. 2014. Diversity and biotechnological potential of microorganisms associated with marine sponges. Appl Microbiol Biotechnol. 98:7331–7347. [DOI] [PubMed] [Google Scholar]
- Fukunaga Y, et al. 2006. Pseudovibrio ascidiaceicola sp. nov., isolated from ascidians (sea squirts). Int J Syst Evol Microbiol. 56:343–347. [DOI] [PubMed] [Google Scholar]
- Fuqua WC, Winans SC, Greenberg EP. 1994. Quorum sensing in bacteria: the LuxR-LuxI family of cell density-responsive transcriptional regulators. J Bacteriol. 176:269–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao ZM, et al. 2014. Pyrosequencing reveals the microbial communities in the Red Sea sponge Carteriospongia foliascens and their impressive shifts in abnormal tissues. Microb Ecol. 68:621–632. [DOI] [PubMed] [Google Scholar]
- Gloeckner V, Lindquist N, Schmitt S, Hentschel U. 2013. Ectyoplasia ferox, an experimentally tractable model for vertical microbial transmission in marine sponges. Microb Ecol. 65:462–474. [DOI] [PubMed] [Google Scholar]
- Graça AP, et al. 2013. Antimicrobial activity of heterotrophic bacterial communities from the marine sponge Erylus discophorus (Astrophorida, Geodiidae). PLoS One 8:e78992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guy L, et al. 2013. A gene transfer agent and a dynamic repertoire of secretion systems hold the keys to the explosive radiation of the emerging pathogen Bartonella. PLoS Genet. 9:e1003393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Habyarimana F, et al. 2008. Role for the Ankyrin eukaryotic-like genes of Legionella pneumophila in parasitism of protozoan hosts and human macrophages. Environ Microbiol. 10:1460–1474. [DOI] [PubMed] [Google Scholar]
- Harjes J, et al. 2014. Draft genome sequence of the antitrypanosomally active sponge-associated bacterium Actinokineospora sp. strain EG49. Genome Announc. 2:e00160–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heise T, Dersch P. 2006. Identification of a domain in Yersinia virulence factor YadA that is crucial for extracellular matrix-specific cell adhesion and uptake. Proc Natl Acad Sci U S A. 103:3375–3380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyatt D, et al. 2010. Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics 11:119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juhas M. 2015. Type IV secretion systems and genomic islands-mediated horizontal gene transfer in Pseudomonas and Haemophilus. Microbiol Res. 170:10–17. [DOI] [PubMed] [Google Scholar]
- Kapitein N, Mogk A. 2013. Deadly syringes: type VI secretion system activities in pathogenicity and interbacterial competition. Curr. Opin. Microbiol. 16:52–58. doi: 10.1016/j.mib.2012.11.009. [DOI] [PubMed] [Google Scholar]
- Kennedy J, Codling CE, Jones BV, Dobson ADW, Marchesi JR. 2008. Diversity of microbes associated with the marine sponge, Haliclona simulans, isolated from Irish waters and identification of polyketide synthase genes from the sponge metagenome. Environ Microbiol. 10:1888–1902. [DOI] [PubMed] [Google Scholar]
- Kennedy J, et al. 2009. Isolation and analysis of bacteria with antimicrobial activities from the marine sponge Haliclona simulans collected from Irish waters. Mar Biotechnol (NY). 11:384–396. [DOI] [PubMed] [Google Scholar]
- Knöbl T, et al. 2001. Virulence properties of Escherichia coli isolated from ostriches with respiratory disease. Vet Microbiol. 83:71–80. [DOI] [PubMed] [Google Scholar]
- Kolbe DL, Eddy SR. 2011. Fast filtering for RNA homology search. Bioinformatics 27:3102–3109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lafont JP, Dho M, D’Hauteville HM, Bree A, Sansonetti PJ. 1987. Presence and expression of aerobactin genes in virulent avian strains of Escherichia coli. Infect Immun. 55:193–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagesen K, et al. 2007. RNAmmer: consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res. 35:3100–3108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laslett D, Canback B. 2004. ARAGORN, a program to detect tRNA genes and tmRNA genes in nucleotide sequences. Nucleic Acids Res. 32:11–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee OO, Chui PY, Wong YH, Pawlik JR, Qian PY. 2009. Evidence for vertical transmission of bacterial symbionts from adult to embryo in the caribbean sponge Svenzea zeai. Appl Environ Microbiol. 75:6147–6156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee OO, et al. 2011. Pyrosequencing reveals highly diverse and species-specific microbial communities in sponges from the Red Sea. ISME J. 5:650–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ling J, et al. 2013. Aerobactin synthesis genes iucA and iucC contribute to the pathogenicity of avian pathogenic Escherichia coli O2 strain E058. PLoS One. 8:e57794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu M, Fan L, Zhong L, Kjelleberg S, Thomas T. 2012. Metaproteogenomic analysis of a community of sponge symbionts. ISME J. 6:1515–1525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu MY, Kjelleberg S, Thomas T. 2011. Functional genomic analysis of an uncultured ?-proteobacterium in the sponge Cymbastela concentrica. ISME J. 5:427–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magrane M, Consortium U. 2011. UniProt Knowledgebase: a hub of integrated protein data. Database 2011:bar009–bar009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margassery LM, Kennedy J, O’Gara F, Dobson AD, Morrissey JP. 2012. Diversity and antibacterial activity of bacteria isolated from the coastal marine sponges Amphilectus fucorum and Eurypon major. Lett Appl Microbiol. 55:2–8. [DOI] [PubMed] [Google Scholar]
- Martin M. 2011. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 17:10–12. [Google Scholar]
- Mittl PRE, Schneider-Brachert W. 2007. Sel1-like repeat proteins in signal transduction. Cell Signal. 19:20–31. [DOI] [PubMed] [Google Scholar]
- Mukherjee S, et al. 2006. Yersinia YopJ acetylates and inhibits kinase activation by blocking phosphorylation. Science. 312:1211–1214. [DOI] [PubMed] [Google Scholar]
- Naim MA, et al. 2014. Host-specific microbial communities in three sympatric North Sea sponges. FEMS Microbiol Ecol. 90:390–403. [DOI] [PubMed] [Google Scholar]
- Ng WL, Bassler BL. 2009. Bacterial quorum-sensing network architectures. Annu Rev Genet. 43:197–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niebuhr K, et al. 2000. IpgD, a protein secreted by the type III secretion machinery of Shigella flexneri, is chaperoned by IpgE and implicated in entry focus formation. Mol Microbiol. 38:8–19. [DOI] [PubMed] [Google Scholar]
- Nougayrède JP, et al. 2006. Escherichia coli induces DNA double-strand breaks in eukaryotic cells. Science 313:848–851. [DOI] [PubMed] [Google Scholar]
- O’Halloran JA, et al. 2011. Diversity and antimicrobial activity of Pseudovibrio spp. from Irish marine sponges. J Appl Microbiol. 110:1495–1508. [DOI] [PubMed] [Google Scholar]
- Olsén A, Jonsson A, Normark S. 1989. Fibronectin binding mediated by a novel class of surface organelles on Escherichia coli. Nature 338:652–655. [DOI] [PubMed] [Google Scholar]
- Penesyan A, et al. 2011. Identification of the antibacterial compound produced by the marine epiphytic bacterium Pseudovibrio sp. D323 and related sponge-associated bacteria. Mar Drugs. 9:1391–1402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen TN, Brunak S, von Heijne G, Nielsen H. 2011. SignalP 4.0: discriminating signal peptides from transmembrane regions. Nat Methods. 8:785–786. [DOI] [PubMed] [Google Scholar]
- Petkau A, Stuart-Edwards M, Stothard P, Domselaar GV. 2010. Interactive microbial genome visualization with GView. Bioinformatics 26:3125–3126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Preston GM. 2007. Metropolitan microbes: type III secretion in multihost symbionts. Cell Host Microbe. 2:291–294. [DOI] [PubMed] [Google Scholar]
- Rolain J-M, et al. 2002. Bartonella quintana in human erythrocytes. Lancet 360:226–228. [DOI] [PubMed] [Google Scholar]
- Schmitt S, et al. 2012. Assessing the complex sponge microbiota: core, variable and species-specific bacterial communities in marine sponges. ISME J. 6:564–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schreiner HC, et al. 2003. Tight-adherence genes of Actinobacillus actinomycetemcomitans are required for virulence in a rat model. Proc Natl Acad Sci U S A. 100:7295–7300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarzer D, Finking R, Marahiel MA. 2003. Nonribosomal peptides: from genes to products. Nat Prod Rep. 20:275–287. [DOI] [PubMed] [Google Scholar]
- Seemann T. 2014. Prokka: rapid prokaryotic genome annotation. Bioinformatics 30:2068–2069. [DOI] [PubMed] [Google Scholar]
- Sertan-de Guzman AA, et al. 2007. Pseudovibrio denitrificans strain Z143-1, a heptylprodigiosin-producing bacterium isolated from a Philippine tunicate. FEMS Microbiol Lett. 277:188–196. [DOI] [PubMed] [Google Scholar]
- Sheng L, Lv Y, Liu Q, Wang Q, Zhang Y. 2013. Connecting type VI secretion, quorum sensing, and c-di-GMP production in fish pathogen Vibrio alginolyticus through phosphatase PppA. Vet. Microbiol. 162:652–662. [DOI] [PubMed] [Google Scholar]
- Siegl A, et al. 2011. Single-cell genomics reveals the lifestyle of Poribacteria, a candidate phylum symbiotically associated with marine sponges. ISME J. 5:61–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simister RL, Deines P, Botté ES, Webster NS, Taylor MW. 2012. Sponge-specific clusters revisited: a comprehensive phylogeny of sponge-associated microorganisms. Environ Microbiol. 14:517–524. [DOI] [PubMed] [Google Scholar]
- Staunton J, Weissman KJ. 2001. Polyketide biosynthesis: a millennium review. Nat Prod Rep. 18:380–416. [DOI] [PubMed] [Google Scholar]
- Thomas T, et al. 2010. Functional genomic signatures of sponge bacteria reveal unique and shared features of symbiosis. ISME J. 4:1557–1567. [DOI] [PubMed] [Google Scholar]
- Toft C, Andersson SGE. 2010. Evolutionary microbial genomics: insights into bacterial host adaptation. Nat Rev Genet. 11:465–475. [DOI] [PubMed] [Google Scholar]
- Tomich M, Planet PJ, Figurski DH. 2007. The tad locus: postcards from the widespread colonization island. Nat Rev Microbiol. 5:363–375. [DOI] [PubMed] [Google Scholar]
- Vizcaino MI. 2011. The chemical defense of Pseudopterogorgia americana: a focus on the antimicrobial potential of a Pseudovibrio sp [phd thesis]. [Charleston (SC)]: Medical University of South Carolina. [Google Scholar]
- Voth DE, Broederdorf LJ, Graham JG. 2012. Bacterial type IV secretion systems: versatile virulence machines. Future Microbiol. 7:241–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waldor MK. 2010. Mobilizable genomic islands: going mobile with oriT mimicry. Mol Microbiol. 78:537–540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber T, et al. 2015. antiSMASH 3.0—a comprehensive resource for the genome mining of biosynthetic gene clusters. Nucleic Acids Res. 43:W237–W243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webster NS, Taylor MW. 2012. Marine sponges and their microbial symbionts: love and other relationships. Environ Microbiol. 14:335–346. [DOI] [PubMed] [Google Scholar]
- Wiley DJ, et al. 2006. The Ser/Thr kinase activity of the Yersinia protein kinase A (YpkA) is necessary for full virulence in the mouse, mollifying phagocytes, and disrupting the eukaryotic cytoskeleton. Microb. Pathog. 40:234–243. [DOI] [PubMed] [Google Scholar]
- Wu S, Zhu Z, Fu L, Niu B, Li W. 2011. WebMGA: a customizable web server for fast metagenomic sequence analysis. BMC Genomics 12:444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zerbino DR, Birney E. 2008. Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 18:821–829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhaxybayeva O, Doolittle WF. 2011. Lateral gene transfer. Curr Biol. 21:R242–R246. [DOI] [PubMed] [Google Scholar]
- Zhou Y, Liang Y, Lynch KH, Dennis JJ, Wishart DS. 2011. PHAST: a fast phage search tool. Nucleic Acids Res. 39:W347–W352 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.