

Abstract
GC-biased gene conversion (gBGC) is a process that tends to increase the GC content of recombining DNA over evolutionary time and is thought to explain the evolution of GC content in mammals and yeasts. Evidence for gBGC outside these two groups is growing but is still limited. Here, we analyzed 36 completely sequenced genomes representing four of the five major groups in eukaryotes (Unikonts, Excavates, Chromalveolates and Plantae). gBGC was investigated by directly comparing GC content and recombination rates in species where recombination data are available, that is, half of them. To study all species of our dataset, we used chromosome size as a proxy for recombination rate and compared it with GC content. Among the 17 species showing a significant relationship between GC content and chromosome size, 15 are consistent with the predictions of the gBGC model. Importantly, the species showing a pattern consistent with gBGC are found in all the four major groups of eukaryotes studied, which suggests that gBGC may be widespread in eukaryotes.
Keywords: GC-biased gene conversion, recombination, GC content, chromosome size
During meiotic recombination, parental chromosomes undergo not only large-scale genetic exchanges by crossover but also small-scale exchanges by gene conversion. These events of gene conversion can be biased. In particular, there is evidence that in some species gene conversion affecting G/C:A/T heterozygous sites yields more frequently to G/C than to A/T alleles, a phenomenon called GC-biased gene conversion (gBGC) (Eyre-Walker 1993; Galtier et al. 2001; Marais 2003; Duret and Galtier 2009a). gBGC is expected to increase the GC content of recombining DNA over evolutionary time and is considered a major contributor to the variation in GC content within and between genomes (Eyre-Walker 1993; Galtier et al. 2001; Marais 2003; Duret and Galtier 2009a). gBGC has caught a lot of attention because it affects the probability of fixation of GC alleles and looks like selection for increasing GC, which can mislead several tests designed to detect positive selection (Galtier and Duret 2007; Berglund et al. 2009; Duret and Galtier 2009b; Galtier et al. 2009; Ratnakumar et al. 2010; Webster and Hurst 2012). It has been demonstrated that gBGC occurs during meiosis in budding yeast (Birdsell 2002; Mancera et al. 2008), and there is strong indirect evidence that this process also affects mammals, where clear-cut relationships between local GC content and recombination rates and many other observations consistent with gBGC have been reported (Galtier 2003; Montoya-Burgos et al. 2003; Spencer et al. 2006; Duret and Arndt 2008; Romiguier et al. 2010). Other studies have investigated gBGC in several organisms such as opossum, chicken, sticklebacks, Drosophila, honeybees, Caenorhabditis elegans, Arabidopsis, wheat, rice, the marine unicellular algae Ostreococcus, and the ciliate Paramecium (Marais et al. 2001, 2003; International Chicken Genome Sequencing Consortium 2004; Marais et al. 2004; Beye et al. 2006; Galtier et al. 2006; Mikkelsen et al. 2007; Duret et al. 2008; Haudry et al. 2008; Jancek et al. 2008; Escobar et al. 2010; Capra and Pollard 2011; Muyle et al. 2011; Nabholz et al. 2011). However, most of the currently available data comes from animals and plants, and we lack a global picture on gBGC in eukaryotes.
Here we wanted to investigate whether gBGC has affected genome evolution in other eukaryotic groups. One typical signature of gBGC is that, on the long term, this process leads to a positive correlation between local GC content and recombination rates (reviewed in Marais 2003; Duret and Galtier 2009a). We thus looked for such a relationship in eukaryotic species for which the genome was entirely sequenced. We focused our analyses on taxa for which the genome sequence was assembled and anchored on chromosomes. We included all species available, except for metazoans, which are clearly over-represented in genomic sequence databases, and for which we only selected a representative sample. Our dataset includes 36 species from four of the five major eukaryotic groups: Unikonts, Excavates, Stramenopiles and Plantae ([Keeling et al. 2005], see fig. 1). Recombination data are available for 17 of these species, mostly Metazoan (Unikonts) and Plantae (see table 1). Among these 17 species, 6 show a significant correlation between chromosome-averaged recombination rate and GC content (table 1). Interestingly, out of these six correlations, five are positive. Thus, when a significant correlation is detected, it is in most cases consistent with gBGC. Moreover, the mean correlation coefficient is significantly >0 (0.31, P = 0.0015), again consistent with gBGC.
Fig. 1.—
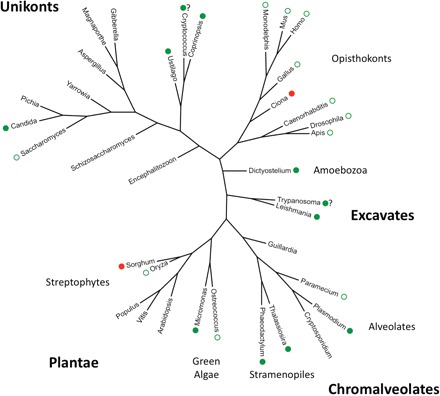
Phylogenetic tree of the 36 species studied. Major groups in eukaryotes (see Keeling et al. 2005) are indicated. Green circles indicate significant positive correlations between GC content (total GC content and/or GC3) and recombination rates (measured directly or using chromosome size as a proxy), consistent with gBGC (this work and others). Red circles indicate significant negative correlations between GC content and recombination rates, not consistent with gBGC. Filled circles indicate new observations from the present study. The “?” indicates when results using direct or indirect measures of recombination rates are not fully consistent.
Table 1.
Correlation between Recombination Rates and GC Content among Eukaryotes
| Species | Eukaryotic groupsa | Chromosome number | Genetic mapb | Total GC/rec ratesc | Chrom size/rec ratesc |
|---|---|---|---|---|---|
| Saccharomyces cerevisiae | Unikonts | 16 | 861 | 0.62* (*) | −0.6* (*) |
| Cryptococcus neoformans | Unikonts | 13 | 285 | 0.04 ns (ns) | −0.14 ns (ns) |
| Monodelphis domestica | Unikonts | 8 | 150 | 0.29 ns (ns) | −0.05 ns (ns) |
| Mus musculus | Unikonts | 19 | 10195 | 0.68* (*) | −0.5* (ns) |
| Homo sapiens | Unikonts | 22 | 28121 | 0.75*** (**) | −0.87*** (***) |
| Gallus gallus | Unikonts | 27 | 9268 | 0.89*** (***) | −0.97*** (***) |
| Ciona intestinalis | Unikonts | 13 | 276 | −0.59* (ns) | 0.21 ns (ns) |
| Caenorhabditis elegans | Unikonts | 5 | 780 | 0.5 ns (ns) | −1* (*) |
| Drosophila melanogaster | Unikonts | 4d | 67 | 0.8 ns (ns) | −0.4 ns (ns) |
| Apis mellifera | Unikonts | 16 | 2008 | 0.74* (*) | −0.35 ns (ns) |
| Trypanosoma brucei | Excavates | 11 | 119 | 0.14 ns (ns) | −0.09 ns (ns) |
| Plasmodium falciparum | Chromal | 14 | 3438 | 0.37 ns (ns) | −0.54* (ns) |
| Arabidopsis thaliana | Plantae | 5 | 676 | 0 ns (ns) | −0.2 ns (ns) |
| Populus trichocarpa | Plantae | 19 | 540 | 0.06 ns (ns) | −0.28 ns (ns) |
| Vitis vinifera | Plantae | 19 | 515 | −0.33 ns (ns) | −0.56* (*) |
| Oryza sativa | Plantae | 12 | 1202 | −0.18 ns (ns) | 0.53 ns (ns) |
| Sorghum bicolor | Plantae | 10 | 2029 | 0.5 ns (ns) | 0.21 ns (ns) |
aThe eukaryotic groups relate to those shown in figure 1. Chromal, Chromalveolates.
bNumber of markers in genetic maps.
cValues are Spearman correlation coefficients, then come P values: ns, nonsignificant, * <0.05, ** <10−3, *** <10−4 and q values (from FDR corrections for multiple tests) are indicated in parentheses.
dHere is indicated the number of chromosome arms instead of the number of chromosomes.
To investigate gBGC in a larger sample of species, including those without recombination data, we used chromosome size as a proxy for recombination rates. It has been shown that chromosome size and recombination rates are inversely correlated in many eukaryotes (e.g., Kaback 1996; Copenhaver et al. 1998; Kaback et al. 1999). This pattern reflects the fact that in many species, the proper segregation of chromosomes during meiosis requires having at least one crossover per chromosome, and that the occurrence of a crossover on a given chromosome decreases the probability of having a second one on the same chromosome (a process termed “crossover interference”). These constraints lead to a lower crossover rate (per Mb) in large chromosomes compared with small ones (Kaback 1996; Copenhaver et al. 1998; Kaback et al. 1999). Among species for which genetic maps are available, we found that in most cases (14/17) chromosome size indeed correlates negatively with recombination rates (table 1), and all significant correlations are negative (7/7). The gBGC model therefore predicts a negative correlation between chromosome size and GC content (although other explanations are possible, see Discussion below). Accordingly, this expected correlation has been found in yeast—for which there is direct evidence of gBGC—and mammals—for which there is strong indirect evidence of gBGC (Bradnam et al. 1999; Meunier and Duret 2004). Table 2 shows that among the 36 eukaryotic species studied, 13 show a significant correlation between chromosome size and chromosome-wide GC content (12 after correction for multiple testing, see table 2). Out of these 13 correlations, 12 are consistent with gBGC—that is, negative. The single exception is Trypanosoma brucei, which shows a significant positive correlation between chromosome size and GC content. Figure 2 shows three examples illustrating the different types of situations that we observed: Leishmania major (significant negative correlation), T. brucei (significant positive correlation) and Guillardia theta (no significant correlation).
Table 2.
Correlation between Chromosome Size and GC Content among Eukaryotes
| Species | Eukaryotic groupsa | Chromosome number | Mean GC content (%) | Statistical powerb (%) | GC total/chrom sizec | GC3/chrom sizec |
|---|---|---|---|---|---|---|
| Encephalitozoon cuniculi | Unikonts | 11 | 47 | 41 | 0.3 ns (ns) | 0.06 ns (ns) |
| Schizosaccharomyces pombe | Unikonts | 3 | 36 | 0 | −0.5 ns (ns) | −0.5 ns (ns) |
| Saccharomyces cerevisiae | Unikonts | 16 | 38 | 70 | −0.83*** (**) | −0.87*** (***) |
| Candida glabrata | Unikonts | 13 | 39 | 52 | −0.69* (*) | −0.71* (*) |
| Pichia stipitis | Unikonts | 8 | 41 | 25 | 0.24 ns (ns) | 0.71 ns (ns) |
| Yarrowia lipolytica | Unikonts | 6 | 49 | 14 | 0.77 ns (ns) | −0.09 ns (ns) |
| Aspergillus fumigatus | Unikonts | 8 | 50 | 25 | 0.71 ns (ns) | 0.26 ns (ns) |
| Magnaporthe grisea | Unikonts | 7 | 52 | 22 | −0.11 ns (ns) | 0.07 ns (ns) |
| Gibberella zeae | Unikonts | 4 | 48 | 0 | 0.4 ns (ns) | 0.4 ns (ns) |
| Ustilago maydis | Unikonts | 23 | 54 | 100 | −0.46* (ns) | −0.47* (*) |
| Cryptococcus neoformans | Unikonts | 14 | 49 | 58 | −0.33 ns (ns) | −0.72* (*) |
| Coprinopsis cinerea | Unikonts | 13 | 52 | 52 | −0.91*** (***) | −0.68* (*) |
| Monodelphis domestica | Unikonts | 9 | 38 | 28 | −0.1 ns (ns) | −0.07 ns (ns) |
| Mus musculus | Unikonts | 20 | 42 | 99 | −0.28 ns (ns) | −0.26 ns (ns) |
| Homo sapiens | Unikonts | 23 | 41 | 100 | −0.57* (*) | −0.54* (*) |
| Gallus gallus | Unikonts | 29 | 41 | 100 | −0.93*** (***) | −0.97*** (***) |
| Ciona intestinalis | Unikonts | 13 | 36 | 52 | 0.2 ns (ns) | 0.29 ns (ns) |
| Caenorhabditis elegans | Unikonts | 6 | 35 | 14 | −0.54 ns (ns) | −0.26 ns (ns) |
| Drosophila melanogaster | Unikonts | 5d | 42 | 7 | −0.3 ns (ns) | −0.5 ns (ns) |
| Apis mellifera | Unikonts | 16 | 35 | 70 | −0.03 ns (ns) | −0.16 ns (ns) |
| Dictyostelium discoideum | Unikonts | 6 | 22 | 14 | −0.94* (*) | −0.6 ns (ns) |
| Trypanosoma brucei | Excavates | 11 | 46 | 41 | 0.73* (*) | 0.48 ns (ns) |
| Leishmania major | Excavates | 36 | 60 | 100 | −0.85*** (***) | −0.82*** (***) |
| Guillardia theta | Chromal | 3 | 26 | 0 | −1 ns (ns) | −1 ns (ns) |
| Paramecium tetraurelia | Chromal | 114 | 28 | 100 | −0.84*** (***) | −0.89*** (***) |
| Plasmodium falciparum | Chromal | 14 | 19 | 58 | −0.8** (*) | −0.77* (*) |
| Cryptosporidium parvum | Chromal | 8 | 30 | 25 | 0.1 ns (ns) | −0.1 ns (ns) |
| Thalassiosira pseudonana | Chromal | 23 | 47 | 100 | −0.06 ns (ns) | −0.87*** (***) |
| Phaeodactylum tricornutum | Chromal | 33 | 49 | 100 | −0.18 ns (ns) | −0.46* (*) |
| Ostreococcus lucimarinus | Plantae | 19 | 60 | 94 | −0.72** (*) | −0.66* (*) |
| Micromonas pusilla | Plantae | 15 | 64 | 63 | −0.83** (**) | −0.42 ns (ns) |
| Arabidopsis thaliana | Plantae | 5 | 36 | 7 | −0.2 ns (ns) | −0.3 ns (ns) |
| Vitis vinifera | Plantae | 19 | 34 | 94 | 0.37 ns (ns) | −0.31 ns (ns) |
| Populus trichocarpa | Plantae | 19 | 33 | 94 | 0.22 ns (ns) | 0.36 ns (ns) |
| Oryza sativa | Plantae | 12 | 44 | 46 | 0.47 ns (ns) | 0.48 ns (ns) |
| Sorghum bicolor | Plantae | 10 | 44 | 36 | 0.3 ns (ns) | 0.68* (*) |
aThe eukaryotic groups relate to those shown in figure 1. Chromal, Chromalveolates.
bStatistical power for chromosome number ≥23 is set to 100%.
cValues are Spearman correlation coefficients, then come P values: ns, nonsignificant, * <0.05,** <10−3,*** <10−4 and q values (from FDR corrections for multiple tests) are indicated in parentheses.
dHere is indicated the number of chromosome arms instead of the number of chromosomes.
Fig. 2.—
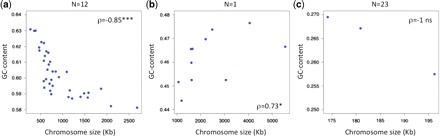
Examples of relationships between chromosome size and total GC content. (a) L. major. (b) T. brucei. (c) G. theta. The values above the plots indicate the number of similar observations that were made among the 36 species (e.g., N = 12 for [a] means 12 significant positive correlations). ρ = Spearman coefficient. Statistical significance: ns, nonsignificant, * <0.05, ** <10−3, *** <10−4.
The evolution of chromosomal GC content can be driven by various processes: point substitutions, deletions, or insertions (including repeated sequences). Interestingly, we observed similar correlations when using GC at third codon position (GC3) instead of total GC content (table 2). Given that third codon positions can only evolve by base replacement, this shows that the observed correlation is due to variation in the pattern of point substitutions, and not to variation in DNA repeat content across chromosomes (table 2). In several cases, the statistical significance of the correlation changed from the total GC content analysis to the GC3 one, but the total number of species showing data consistent with gBGC is similar (significant negative correlation: 13/36, significant positive correlation: 1/36). Both analyses gave qualitatively the same results, with—as expected—changes in statistical significance caused by slight changes of the coefficients of correlation in case of species with low chromosome number (i.e., Dictyostelium discoideum, Sorghum bicolor, T. brucei, Cryptococcus neoformans, Micromonas pusilla). Thalassiosira pseudonana and Phaeodactylum tricornutum, two diatoms with a relatively large number of chromosomes, show results consistent with gBGC only for GC3, which raises the possibility of different mutation patterns affecting coding and noncoding regions in these species.
The fact that about half of the species shows the footprint of gBGC (i.e., a significant negative correlation) may indicate gBGC is absent in the other half. It may also indicate that our approach fails to detect gBGC in many species. Indeed, the statistical significance of the correlations strongly depends on the number of chromosomes. For species with few chromosomes, our ability to detect the signature of gBGC is limited. For instance, G. theta shows a strong negative correlation between chromosome size and GC content (fig. 2c), but with only three chromosomes, the P value is obviously nonsignificant. We thus performed a statistical power analysis using human as a reference (see Materials and Methods). Table 2 shows the statistical power (from 0 to 100%) for all species of our dataset. Most species have too few chromosomes to detect any significant correlation between GC content and chromosome size. Among the 19 species for which the estimated power of our test is >50%, 14 (74%) show a significant correlation with total or third position GC content, and in all cases the correlation is consistent with gBGC. Similarly, another power analysis using a more conservative reference (yeast) revealed that 14 out of the 28 species with a power of >50% show results consistent with gBGC. Moreover, the combined analysis of all species indicated a strong significant negative correlation (for total GC content and chromosomes size: P value = 10−50, for GC3 and chromosome size: P value = 10−63). However, focusing only on the species that show individually nonsignificant correlations, the combined analysis is not significant. There is thus no clear trend emerging from this subset of species.
Given that chromosomal size is only a rough proxy for recombination rate, this result is most likely an underestimate of how widespread this pattern is in our set of species. For example, Mus musculus and Apis mellifera, which contain a high number of chromosomes, show no significant correlation between chromosome size and GC content (table 2). Yet, in both species, studies using recombination data inferred from genetic maps showed a significant positive correlation between local GC content and crossover rates (Beye et al. 2006; Khelifi et al. 2006; see table 1). In M. musculus, the absence of significant correlation between chromosome size and GC content can be explained by the lack of variance in chromosome size in that species (Meunier and Duret 2004). In A. mellifera, as in several other eukaryotes (e.g., Schizosaccharomyces pombe), chromosomes experience little or no crossover interference, and their mean recombination rate is therefore not correlated to their size, which explains that we do not observe any correlation between chromosome size and GC content in these species. Finally, it should be noted that the evolution of GC content is a slow process. If a genome has undergone recent chromosomal rearrangements, it might not show any significant correlation between chromosome size and GC content, simply because there was not enough time to establish the pattern (Duret and Arndt 2008). Given all these limitations of our test, it is remarkable that a majority of species (50–74% of all species with statistical power >50%) show correlations consistent with the predictions of the gBGC model.
Several species, however, do not fit into this general pattern: Ciona instestinalis, C. neoformans, S. bicolor and T. brucei. Cryptococcus neoformans is a species with evidence for gBGC from table 2 but not (or incompletely) from table 1. This can look surprising at first sight since we use recombination data in table 1, which is a more direct way of testing for gBGC. However, this assumption is correct if recombination data are of high quality, which might not be the case for most of the species in table 1 with a small number of markers. Too few markers will tend to shorten genetic maps, underestimating recombination rates (other important parameters are the number of meioses analyzed, the distribution of markers along chromosomes). Cryptococcus neoformans and other species in table 1 may be in this situation. It is possible that in such species, chromosome length gives a better idea of the average chromosome-wide recombination rates, which could explain why we report comparatively more species showing evidence of gBGC in table 2 than in table 1. In C. neoformans, the use of two different strains for the available genetic map and the complete genome could be an additional problem for correlating GC content and recombination rates reliably. The conflicting results in Ciona intestinalis may also come from the poor-quality map found in this species (only 276 markers, see table 1). Using two genetic maps in Plasmodium falciparum, one from 1999 with 900 markers (Su et al. 1999) and a more recent one with 3,438 markers (Jiang et al. 2011), we found very different results (GC/recombination: −0.31 nonsignificant with the 1999 version map, 0.34 nonsignificant with the 2011 version map, Chromosome size/recombination: 0.23 nonsignificant with the 1999 version map, −0.54, P < 0.05 with the 2011 version map), which confirms that the quality of recombination data is critical. Trypanosoma brucei shows a significant positive correlation between chromosome size and GC content (fig. 2b). However, it turns out that, for an unknown reason, chromosome size is not a good proxy for recombination rate in this species: the two parameters are not correlated (ρ = −0.09; P = 0.797, see table 1). Table 1 reveals that GC content correlates positively with recombination rates in T. brucei (ρ = 0.14), although not significantly (P = 0.694). It thus appears that T. brucei is not an exception to the general pattern consistent with gBGC. Again, a better map in this species would help understand more clearly the relationships between GC content, chromosome size and recombination rates (there are only 119 markers in this species, see table 1). In S. bicolor, GC3 correlates strongly with chromosome size in a positive manner (table 2). We do not have explanations for this significant correlation, which is not in agreement with gBGC. Sorghum bicolor seems therefore to represent a true exception to the general pattern.
In conclusion, we found 17 species with a significant correlation between chromosome-wide GC content and chromosome size, as a rough proxy for recombination rate. Most of them (15/17) showed a negative correlation, consistent with the gBGC model. Our results were unaltered when considering GC3, which rules out the insertion of transposable elements as a general explanation for the observed pattern. Other explanations are of course possible (mutational biases, selection on GC content). In species where these various hypotheses have been tested, gBGC has always come out as the most likely explanation (reviewed in Marais 2003; Duret and Galtier 2009a). More work will be needed, however, to test these alternative explanations and firmly establish gBGC in the species where we report data consistent with gBGC for the first time. Figure 1 shows, in our set of 36 eukaryotes, the species with a positive correlation between GC content and recombination rates (measured directly or using chromosome size as a proxy), consistent with gBGC. Remarkably, this correlation is found in all four major eukaryotic groups studied, which suggests gBGC is widespread in eukaryotes. This is in agreement with a recent study using GC content of ribosomal DNA as a proxy for gBGC, in which gBGC was inferred in several distantly related eukaryotes (Escobar et al. 2011). Firm evidence for gBGC is only available for a handful of species (yeasts and mammals) and our work suggests that gBGC should be further studied in many more species, where it could have important effects on genome evolution.
Materials and Methods
Genome Data
We selected species for which a complete genome assembly, anchored on chromosomes, was available. Animal species are clearly over-represented in public databases. As gBGC is already established in animals, we only selected a subset of species representing the main animal groups. Genome data were extracted from Hogenom version 3 (17 species [Penel et al. 2009]), the NCBI website (15 species, http://www.ncbi.nlm.nih.gov/), and the JGI website (4 species, http://www.jgi.doe.gov/). For the Paramecium genome, we selected the scaffolds that were at least chromosomal arms (Gout J-F, personal communications). The relationship between chromosome size and recombination rate only stands for recombining chromosomes and we therefore removed all the nonrecombining chromosomes (chromosomes 4 from Drosophila melanogaster, 2 and 18 from Ostreococcus lucimarinus, 1 and 17 from M. pusilla, Y and W chromosomes from mammals and chicken, respectively). For our 36 species, we thus had chromosome sizes and sequences to estimate the GC content.
Recombination Data
We got recombination data for C. elegans directly from MareyMap (Rezvoy et al. 2007), D. melanogaster from Flybase (http://flybase.org) and Saccharomyces cerevisiae from http://www.yeastgenome.org/pgMaps/pgI.shtml. Recombination data for other species was obtained from specific papers: M. musculus (Cox et al. 2009), Homo sapiens (Matise et al. 2007), Gallus gallus (Groenen et al. 2009), Monodelphis domestica (Samollow et al. 2007), A. mellifera (Beye et al. 2006), T. brucei (Cooper et al. 2008), P. falciparum (Jiang et al. 2011), Arabidopsis thaliana (Singer et al. 2006), C. intestinalis (Kano et al. 2006), S. bicolor (Mace et al. 2009), Populus trichocarpa (Yin et al. 2004), Vitis vinifera (Doligez et al. 2006), Oryza sativa (Muyle et al. 2011), and C. neoformans (Marra et al. 2004). The number of chromosomes indicated in table 1 may differ from the true chromosome number: the X and Z chromosomes were excluded from this analysis because they recombine only in one sex, and recombination patterns are thus different from those in the autosomes, and the recombination data are not available for some chromosomes (for instance, chromosome 10 for C. neoformans). The recombination rates were computed by dividing the genetic map length of each chromosome by its physical size (in bp), and are thus chromosomal-averaged estimates.
GC Content Analysis
The total GC content was computed using whole-chromosome sequences. The GC content at third codon position (GC3) was computed by collecting all the available CDS from a genome (extracting CDS from Hogenom or Ensembl, or using CDS files from JGI or Broad Institute). For both total GC content and GC3 estimates, ambiguous nucleotides were excluded. Chromosome-averaged GC values were then computed. R was used to obtain bilateral Spearman coefficients of correlation, P values, and q values (P values corrected for multiple testing using the false discovery rate method). The combined analysis was performed by first getting the P values (P) from unilateral tests on Spearman coefficients in order to test for a general trend for a negative correlation between GC content and chromosome size (null hypothesis: GC content and chromosome size are not correlated negatively). The sum of the −2 * log (P) for all species follows a chi-squared distribution with 2n degrees of freedom, n being the number of species, which gave the P value of the combined analysis (Sokal and Rohlf 2012).
Statistical Power Analysis
To estimate the power of our approach according to the number of chromosomes (N) in a given genome, we performed the following test: we took the human genome (for which there is clear evidence of gBGC and which shows a significant negative correlation between chromosome size and GC content) and we asked what would be the probability to detect a significant correlation if this genome only contained N chromosomes. We thus randomly sampled N human chromosomes, computed the Spearman coefficient between their size and GC content, repeated this for all the possible combinations (up to 50,000 samples) and measured the fraction of significant Spearman correlations in the simulated data using R. We took this fraction as the statistical power of our test for a given number of chromosomes N. The same was done using S. cerevisiae as a reference.
Acknowledgments
We thank Jean-François Gout for his help with the Paramecium genome data. This work was supported by the Centre National de la Recherche Scientifique and the Agence Nationale de la Recherche (ANR-08-GENM-036-01).
Literature Cited
- Berglund J, Pollard KS, Webster MT. Hotspots of biased nucleotide substitutions in human genes. PLoS Biol. 2009;7:e26. doi: 10.1371/journal.pbio.1000026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beye M, et al. Exceptionally high levels of recombination across the honey bee genome. Genome Res. 2006;16:1339–1344. doi: 10.1101/gr.5680406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birdsell JA. Integrating genomics, bioinformatics, and classical genetics to study the effects of recombination on genome evolution. Mol Biol Evol. 2002;19:1181–1197. doi: 10.1093/oxfordjournals.molbev.a004176. [DOI] [PubMed] [Google Scholar]
- Bradnam KR, Seoighe C, Sharp PM, Wolfe KH. G+C content variation along and among Saccharomyces cerevisiae chromosomes. Mol Biol Evol. 1999;16:666–675. doi: 10.1093/oxfordjournals.molbev.a026149. [DOI] [PubMed] [Google Scholar]
- Capra JA, Pollard KS. Substitution patterns are GC-biased in divergent sequences across the metazoans. Genome Biol Evol. 2011;3:516–527. doi: 10.1093/gbe/evr051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper A, et al. Genetic analysis of the human infective trypanosome Trypanosoma brucei gambiense: chromosomal segregation, crossing over, and the construction of a genetic map. Genome Biol. 2008;9:R103. doi: 10.1186/gb-2008-9-6-r103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Copenhaver GP, Browne WE, Preuss D. Assaying genome-wide recombination and centromere functions with Arabidopsis tetrads. Proc Natl Acad Sci U S A. 1998;95:247–252. doi: 10.1073/pnas.95.1.247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox A, et al. A new standard genetic map for the laboratory mouse. Genetics. 2009;182:1335–1344. doi: 10.1534/genetics.109.105486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doligez A, et al. An integrated SSR map of grapevine based on five mapping populations. Theor Appl Genet. 2006;113:369–382. doi: 10.1007/s00122-006-0295-1. [DOI] [PubMed] [Google Scholar]
- Duret L, Arndt PF. The impact of recombination on nucleotide substitutions in the human genome. PLoS Genet. 2008;4:e1000071. doi: 10.1371/journal.pgen.1000071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duret L, et al. Analysis of sequence variability in the macronuclear DNA of Paramecium tetraurelia: a somatic view of the germline. Genome Res. 2008;18:585–596. doi: 10.1101/gr.074534.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duret L, Galtier N. Biased gene conversion and the evolution of mammalian genomic landscapes. Annu Rev Genomics Hum Genet. 2009;10:285–311. doi: 10.1146/annurev-genom-082908-150001. [DOI] [PubMed] [Google Scholar]
- Duret L, Galtier N. Comment on “Human-specific gain of function in a developmental enhancer”. Science. 2009;323:714. doi: 10.1126/science.1165848. author reply 714. [DOI] [PubMed] [Google Scholar]
- Escobar JS, et al. An integrative test of the dead-end hypothesis of selfing evolution in Triticeae (Poaceae) Evolution. 2010;64:2855–2872. doi: 10.1111/j.1558-5646.2010.01045.x. [DOI] [PubMed] [Google Scholar]
- Escobar JS, Glemin S, Galtier N. GC-biased gene conversion impacts ribosomal DNA evolution in vertebrates, angiosperms, and other eukaryotes. Mol Biol Evol. 2011;28:2561–2575. doi: 10.1093/molbev/msr079. [DOI] [PubMed] [Google Scholar]
- Eyre-Walker A. Recombination and mammalian genome evolution. Proc Biol Sci. 1993;252:237–243. doi: 10.1098/rspb.1993.0071. [DOI] [PubMed] [Google Scholar]
- Galtier N. Gene conversion drives GC content evolution in mammalian histones. Trends Genet. 2003;19:65–68. doi: 10.1016/s0168-9525(02)00002-1. [DOI] [PubMed] [Google Scholar]
- Galtier N, Bazin E, Bierne N. GC-biased segregation of noncoding polymorphisms in Drosophila. Genetics. 2006;172:221–228. doi: 10.1534/genetics.105.046524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galtier N, Duret L. Adaptation or biased gene conversion? Extending the null hypothesis of molecular evolution. Trends Genet. 2007;23:273–277. doi: 10.1016/j.tig.2007.03.011. [DOI] [PubMed] [Google Scholar]
- Galtier N, Duret L, Glemin S, Ranwez V. GC-biased gene conversion promotes the fixation of deleterious amino acid changes in primates. Trends Genet. 2009;25:1–5. doi: 10.1016/j.tig.2008.10.011. [DOI] [PubMed] [Google Scholar]
- Galtier N, Piganeau G, Mouchiroud D, Duret L. GC-content evolution in mammalian genomes: the biased gene conversion hypothesis. Genetics. 2001;159:907–911. doi: 10.1093/genetics/159.2.907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groenen MA, et al. A high-density SNP-based linkage map of the chicken genome reveals sequence features correlated with recombination rate. Genome Res. 2009;19:510–519. doi: 10.1101/gr.086538.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haudry A, et al. Mating system and recombination affect molecular evolution in four Triticeae species. Genet Res. 2008;90:97–109. doi: 10.1017/S0016672307009032. [DOI] [PubMed] [Google Scholar]
- International Chicken Genome Sequencing Consortium. Sequence and comparative analysis of the chicken genome provide unique perspectives on vertebrate evolution. Nature. 2004;432:695–716. doi: 10.1038/nature03154. [DOI] [PubMed] [Google Scholar]
- Jancek S, Gourbiere S, Moreau H, Piganeau G. Clues about the genetic basis of adaptation emerge from comparing the proteomes of two Ostreococcus ecotypes (Chlorophyta, Prasinophyceae) Mol Biol Evol. 2008;25:2293–2300. doi: 10.1093/molbev/msn168. [DOI] [PubMed] [Google Scholar]
- Jiang H, et al. High recombination rates and hotspots in a Plasmodium falciparum genetic cross. Genome Biol. 2011;12:R33. doi: 10.1186/gb-2011-12-4-r33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaback DB. Chromosome-size dependent control of meiotic recombination in humans. Nat Genet. 1996;13:20–21. doi: 10.1038/ng0596-20. [DOI] [PubMed] [Google Scholar]
- Kaback DB, Barber D, Mahon J, Lamb J, You J. Chromosome size-dependent control of meiotic reciprocal recombination in Saccharomyces cerevisiae: the role of crossover interference. Genetics. 1999;152:1475–1486. doi: 10.1093/genetics/152.4.1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kano S, Satoh N, Sordino P. Primary genetic linkage maps of the ascidian, Ciona intestinalis. Zoolog Sci. 2006;23:31–39. doi: 10.2108/zsj.23.31. [DOI] [PubMed] [Google Scholar]
- Keeling PJ, et al. The tree of eukaryotes. Trends Ecol Evol. 2005;20:670–676. doi: 10.1016/j.tree.2005.09.005. [DOI] [PubMed] [Google Scholar]
- Khelifi A, Meunier J, Duret L, Mouchiroud D. GC content evolution of the human and mouse genomes: insights from the study of processed pseudogenes in regions of different recombination rates. J Mol Evol. 2006;62:745–752. doi: 10.1007/s00239-005-0186-0. [DOI] [PubMed] [Google Scholar]
- Mace ES, et al. A consensus genetic map of sorghum that integrates multiple component maps and high-throughput Diversity Array Technology (DArT) markers. BMC Plant Biol. 2009;9:13. doi: 10.1186/1471-2229-9-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mancera E, Bourgon R, Brozzi A, Huber W, Steinmetz LM. High-resolution mapping of meiotic crossovers and non-crossovers in yeast. Nature. 2008;454:479–485. doi: 10.1038/nature07135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marais G. Biased gene conversion: implications for genome and sex evolution. Trends Genet. 2003;19:330–338. doi: 10.1016/S0168-9525(03)00116-1. [DOI] [PubMed] [Google Scholar]
- Marais G, Charlesworth B, Wright SI. Recombination and base composition: the case of the highly self-fertilizing plant Arabidopsis thaliana. Genome Biol. 2004;5:R45. doi: 10.1186/gb-2004-5-7-r45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marais G, Mouchiroud D, Duret L. Does recombination improve selection on codon usage? Lessons from nematode and fly complete genomes. Proc Natl Acad Sci U S A. 2001;98:5688–5692. doi: 10.1073/pnas.091427698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marais G, Mouchiroud D, Duret L. Neutral effect of recombination on base composition in Drosophila. Genet Res. 2003;81:79–87. doi: 10.1017/s0016672302006079. [DOI] [PubMed] [Google Scholar]
- Marra RE, et al. A genetic linkage map of Cryptococcus neoformans variety neoformans serotype D (Filobasidiella neoformans) Genetics. 2004;167:619–631. doi: 10.1534/genetics.103.023408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matise TC, et al. A second-generation combined linkage physical map of the human genome. Genome Res. 2007;17:1783–1786. doi: 10.1101/gr.7156307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meunier J, Duret L. Recombination drives the evolution of GC-content in the human genome. Mol Biol Evol. 2004;21:984–990. doi: 10.1093/molbev/msh070. [DOI] [PubMed] [Google Scholar]
- Mikkelsen TS, et al. Genome of the marsupial Monodelphis domestica reveals innovation in non-coding sequences. Nature. 2007;447:167–177. doi: 10.1038/nature05805. [DOI] [PubMed] [Google Scholar]
- Montoya-Burgos JI, Boursot P, Galtier N. Recombination explains isochores in mammalian genomes. Trends Genet. 2003;19:128–130. doi: 10.1016/S0168-9525(03)00021-0. [DOI] [PubMed] [Google Scholar]
- Muyle A, Serres-Giardi L, Ressayre A, Escobar J, Glemin S. GC-biased gene conversion and selection affect GC content in the Oryza genus (rice) Mol Biol Evol. 2011;28:2695–2706. doi: 10.1093/molbev/msr104. [DOI] [PubMed] [Google Scholar]
- Nabholz B, Kunstner A, Wang R, Jarvis ED, Ellegren H. Dynamic evolution of base composition: causes and consequences in avian phylogenomics. Mol Biol Evol. 2011;28:2197–2210. doi: 10.1093/molbev/msr047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penel S, et al. Databases of homologous gene families for comparative genomics. BMC Bioinformatics. 2009;10(Suppl 6):S3. doi: 10.1186/1471-2105-10-S6-S3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ratnakumar A, et al. Detecting positive selection within genomes: the problem of biased gene conversion. Philos Trans R Soc Lond B Biol Sci. 2010;365:2571–2580. doi: 10.1098/rstb.2010.0007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rezvoy C, Charif D, Gueguen L, Marais GA. MareyMap: an R-based tool with graphical interface for estimating recombination rates. Bioinformatics. 2007;23:2188–2189. doi: 10.1093/bioinformatics/btm315. [DOI] [PubMed] [Google Scholar]
- Romiguier J, Ranwez V, Douzery EJ, Galtier N. Contrasting GC-content dynamics across 33 mammalian genomes: relationship with life-history traits and chromosome sizes. Genome Res. 2010;20:1001–1009. doi: 10.1101/gr.104372.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samollow PB, et al. A microsatellite-based, physically anchored linkage map for the gray, short-tailed opossum (Monodelphis domestica) Chromosome Res. 2007;15:269–281. doi: 10.1007/s10577-007-1123-4. [DOI] [PubMed] [Google Scholar]
- Singer T, et al. A high-resolution map of Arabidopsis recombinant inbred lines by whole-genome exon array hybridization. PLoS Genet. 2006;2:e144. doi: 10.1371/journal.pgen.0020144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sokal R, Rohlf F. New York: W.H. Freeman & Co Ltd; 2012. Biometry: the principles and practices of statistics in biological research. [Google Scholar]
- Spencer CC, et al. The influence of recombination on human genetic diversity. PLoS Genet. 2006;2:e148. doi: 10.1371/journal.pgen.0020148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su X, et al. A genetic map and recombination parameters of the human malaria parasite Plasmodium falciparum. Science. 1999;286:1351–1353. doi: 10.1126/science.286.5443.1351. [DOI] [PubMed] [Google Scholar]
- Webster MT, Hurst LD. Direct and indirect consequences of meiotic recombination: implications for genome evolution. Trends Genet. 2011;28:101–109. doi: 10.1016/j.tig.2011.11.002. [DOI] [PubMed] [Google Scholar]
- Yin TM, DiFazio SP, Gunter LE, Riemenschneider D, Tuskan GA. Large-scale heterospecific segregation distortion in Populus revealed by a dense genetic map. Theor Appl Genet. 2004;109:451–463. doi: 10.1007/s00122-004-1653-5. [DOI] [PubMed] [Google Scholar]