

Abstract
Photosynthetic diatom plastids have long been suggested to have originated by the secondary endosymbiosis of a red alga. However, recent phylogenomic studies report a high number of diatom nuclear genes phylogenetically related to green algal and green plant genes. These were interpreted as endosymbiotic gene transfers (EGT) from a cryptic green algal endosymbiosis. We reanalyzed this issue using a larger set of red algal genomic data. We show that previous studies suffer from a taxonomic sampling bias and point out that a majority of gene phylogenies are either poorly resolved or do not describe EGT events. We finally show that genes having a complete descent from cyanobacteria to diatoms through primary and secondary EGTs have been mostly transferred via a red alga. We conclude that, even if some diatom genes still support a putative green algal origin, these are not sufficient to argue for a cryptic green algal secondary endosymbiosis.
Keywords: diatoms, phylogenomics, plastid, endosymbiotic gene transfers
Diatoms (Bacillariophyceae) are well-known microbial eukaryotes, most of them photosynthetic and important members of phytoplanktonic communities (Armbrust 2009). They belong to the Stramenopiles (or Heterokonta), a group that also includes various other photosynthetic phyla, such as the brown, yellow–green, and golden algae (Phaeophyceae, Xanthophyceae, and Chrysophyceae, respectively), but also a variety of heterotrophic phyla (e.g., Bicosoecida, Oomycetes, and Labyrinthulomycetes). Photosynthetic diatoms carry four-membrane plastids which, based on their pigment content and on the analysis of their plastid genomes, have for long been suggested to have originated by the secondary endosymbiosis of a red alga (Cavalier-Smith 2002). However, as for the majority of secondary endosymbioses at the origin of different eukaryotic photosynthetic lineages, the number of symbiotic events and the nature of the partners involved remain uncertain (Elias and Archibald 2009; Baurain et al. 2010; Keeling 2010). These questions can be addressed using phylogenomic approaches since, during the endosymbiotic process, a high number of genes are usually transferred from the symbiont to the host genome (Martin et al. 1998). These endosymbiotic gene transfers (EGT) provide numerous marker candidates that may carry enough phylogenetic signals to retrace their symbiotic origin.
Phylogenomics of EGT in Diatoms
Recently, some studies have reported the existence of genes of green algal origin in diatom nuclear genomes. Some of them were limited to the study of the origin of genes involved in specific pathways such as carotenoid biosynthesis or sugar phosphate metabolism (Frommolt et al. 2008; Allen et al. 2012), whereas some others scanned entire genomes and retrieved thousands of green algal-related genes (Moustafa et al. 2009, reviewed in Prihoda et al. 2012). In particular, the phylogenomic survey led by Moustafa et al. (2009) detected 4956 genes putatively acquired by EGT in the completely sequenced genomes of two diatom species (2533 in Thalassiosira pseudonana and 2423 in Phaeodactylum tricornutum). The analysis of the phylogenies of the corresponding proteins showed that 3619 (>70%) of these putative diatom EGT genes were more closely related to green algae and plants (Viridiplantae) than to red algae (fig. 1A). This was a surprising result because this amount of EGT seemed huge and also because if a red alga was the precursor of diatom plastids, as commonly accepted, the vast majority of EGT genes should be related to red algal homologs. Considering this result, Moustafa et al. concluded that diatoms, and perhaps other related phyla, acquired their plastid through an endosymbiotic event involving a cell related to green algae before replacing it by the red algal-like plastid found today.
Fig. 1.—
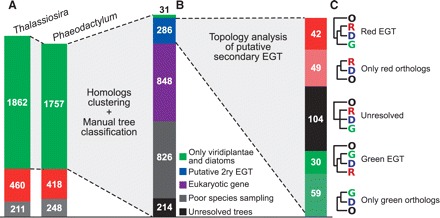
Phylogenetic reanalysis of putative EGT genes of green algal and plant origin in diatom genomes. (A) Summary of the results reported by Moustafa et al. (2009) and used as a reference for this work. (B) Classification of the protein-based phylogenetic tree topologies in five classes: Black—Trees not sufficiently resolved to allow any inference about gene evolutionary origin; Gray—Similarity searches retrieved a poor set of species, leading to inconclusive trees; Purple—Trees supporting vertical transmission in all eukaryotes. Blue—Trees compatible with a secondary EGT scenario; Green—Only Viridiplantae and diatom homologous sequences were retrieved by similarity searches. (C) Identification of the donor group (red or green alga) for the remaining putative EGT candidates.
Such massive phylogeny-based approaches aimed at pinpointing EGT genes left by endosymbiotic events are not trivial. Indeed, they require efficiently retrieving the subtle phylogenetic signal distinguishing genes of different origins. When used to detect genes of cyanobacterial origin in a eukaryotic genome (i.e., genes transferred by EGT during primary endosymbiosis of a cyanobacterium within a heterotrophic eukaryote), the task is rather easy because of the large phylogenetic distance between the two partners (Martin et al. 1998). However, when addressing transfers of eukaryotic genes into another eukaryotic genome, the evolutionary distance between genes in phylogenetic trees can be much shorter and the inference of gene evolutionary origin becomes more difficult (Stiller 2011). This may have occurred in the work described above when the authors tried to infer the origin of each diatom gene as either transferred from red or green algae by EGT or just derived from an ancestral eukaryotic homolog. In fact, this work may have suffered from two major issues: (1) A severe taxonomic sampling bias, because only one genome sequence of red algae, the highly reduced species Cyanidioschyzon merolae (Matsuzaki et al. 2004), was incorporated in the study in comparison with 13 genomes of Viridiplantae; and (2) The use of an automatic phylogenetic tree topology analysis software which may have inferred wrong relationships when facing trees that were poorly resolved or contained hidden paralogs. Unfortunately, many diatom gene phylogenies probably fall into one or both of these categories. Indeed, eukaryotic genomes usually present a high frequency of gene duplication, increasing the risk of observing hidden paralogy. Furthermore, the short evolutionary distance between Viridiplantae and Rhodophyta requires a strong phylogenetic signal to identify a specific relationship of a particular gene with one of these two sister groups (especially when using automated procedures). Finally, known difficulties to reconstruct robust phylogenetic trees when using single eukaryotic proteins can lead to weakly supported trees that sometimes can artificially resemble EGT events. Therefore, there is a real need for testing the robustness of the phylogenetic trees as well as the accuracy of the automatic tree topology analysis that led to the groundbreaking conclusions proposed by Moustafa et al. This test can be done by improving the taxonomic sampling. If the putative green-EGT proteins identified were indeed of green algal origin, their phylogenetic relationship with Viridiplantae should not be affected by the incorporation of additional red algal sequences to the analysis.
Reevaluation of EGT Traces in Diatom Genomes
To test this idea, we have searched the homologs of all the diatom proteins of putative EGT origin in the genome sequence of the red alga Galdieria sulphuraria (http://genomics.msu.edu/galdieria/), as well as in various red algal EST resources: Chondrus crispus, Furcellaria lumbricalis, Porphyra yezoensis, Porphyra haitanensis, Gracilaria changii, Eucheuma denticulatum, Calliarthron tuberculosum, and Porphyridium cruentum (Nikaido et al. 2000; Aspilla et al. 2010; Chan et al. 2011; and unpublished data available in GenBank) and incorporated them into our own local genome database (refer to Materials and Methods). We directly excluded from the analysis 66 putative EGT proteins that did not retrieve more than two similar proteins in the database. For each putative EGT protein left, we reconstructed a maximum likelihood phylogenetic tree. Using these trees, we first clustered highly similar diatom genes that were counted twice or more by Moustafa et al. as independent EGTs although they actually represent a single case of EGT followed by diversification linked to the separation of the diatom species or to gene duplication(s) in a single diatom species (see Materials and Methods). In this way, the initial 4956 proteins (minus 66, see above) could be grouped in a list of 2205 unique putative green-EGT proteins (fig. 1A). We then manually inspected the phylogenetic trees corresponding to this list to assess the robustness of the tree reconstruction and to identify the closest relatives of diatom sequences. All trees were sorted into five groups based on simple and strict rules: (1) Poor sampling: the number of species having proteins similar to the putative green-EGT diatom protein was too restricted (either for the whole tree, or for the subtree containing the diatom sequences) to infer properly its evolutionary history (fig. 2C); (2) Unresolved trees: major accepted monophyletic groups (e.g., animals, fungi, or land plants) could not be retrieved or a probable hidden paralogy lowered the tree resolution (fig. 2E); (3) Eukaryotic gene: trees where most of the major monophyletic phyla were recovered with good support and where eukaryotic sequences were grouped apart from prokaryotic ones. We interpreted these trees as cases of vertical gene transmission (fig. 2A); (4) Only greens and diatoms: proteins similar to the EGT candidate were found only in diatoms and in Viridiplantae, thus being poorly informative with respect to their putative EGT origin (fig. 2B); (5) Putative secondary EGT: trees compatible with a secondary EGT transmission scenario. This included trees where a primary EGT event from prokaryotic source was observed as well as its transmission through a secondary EGT event (fig. 2D). This also included trees showing no transmission from prokaryotic sources but where eukaryotic phyla branched in such way that a putative secondary EGT transmission between two groups of eukaryotes was more plausible than a vertical descent relationship. An evaluation of the quality of the sequence alignments corresponding to each of these classes of trees did not show any particular bias (see Supplementary Material online). Figure 1B summarizes the number of trees that were assigned to each class. In addition, the complete list of diatom genes assigned to each class is available in the Supplementary Material online.
Fig. 2.—
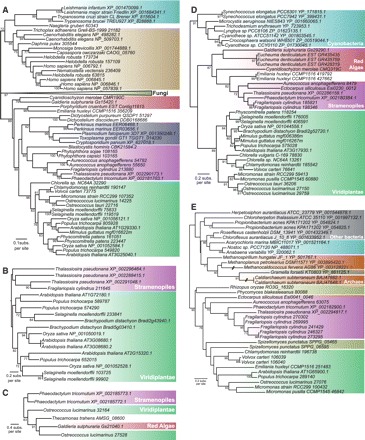
Examples of ML phylogenetic trees recovered in each of the five classes. Node supports lower than 70% are hidden. (A) Eukaryotic gene; (B) only Viridiplantae and diatoms; (C) poor species sampling; (D) putative secondary EGT; (E) unresolved tree. Sequence accession numbers are from GenBank or JGI.
Quantitatively, the two major classes of trees that we retrieved corresponded to either a poor species sampling (826 trees) or to proteins just having an ancient eukaryotic origin without any evidence of EGT (848 trees). This suggested that the automated tree topology analysis carried out by Moustafa et al. was not sufficiently accurate. Indeed, most of the trees in the “Eukaryotic gene” class were not ambiguous. Even if we found a considerable amount of “Eukaryotic” phylogenies where Viridiplantae were closer to diatoms than expected, these phylogenies were not actually in favor of an EGT scenario because, most often, they corresponded to trees with complex patterns of paralogy or to trees with a very small number of red algal homologs. Moreover, we noticed that in many trees containing an insufficient species sampling (included in the “Poor sampling” class), a single diatom protein had a single green algal protein as closest relative and both were isolated in the tree, for example, within a large group of bacterial sequences. Such a topology is not compatible with a true green-EGT scenario where diatoms should emerge nested within a large set of green algal and plant sequences. The automated analysis used by Moustafa et al., probably designed to detect paired sequences with no consideration about their phylogenetic environment, was inappropriate to identify and exclude those problematic cases.
In the end, after manual inspection of all phylogenies, we drastically reduced the number of tree topologies compatible with secondary EGTs to only 286. A deeper analysis of these trees allowed to determine if the corresponding genes were transmitted from red or from green algae to diatoms during secondary endosymbiosis. We encountered three situations (fig. 1C): (1) The corresponding subpart of the tree was not sufficiently resolved to determine the origin of the EGT; (2) The subtree was resolved and we could determine the donor; and (3) The subtree was resolved but only contained green algae plus diatoms, or red algae plus diatoms. We consider the latter situation as partly inconclusive because one cannot reject the possibility that red or green algal species having the corresponding homologous protein in their genome are missing from our database, which is likely the case for many red algae given the relatively small amount of sequence data for this group. For the well-resolved topologies, the number of inferred red algal EGTs and green algal EGTs was comparable (89 and 91, respectively). This new data, in addition to the 253 cases already detected as red algal EGTs by Moustafa et al., switch the overall perspective toward a much higher amount of red algal EGTs than of putative green algal EGTs in diatom nuclear genomes.
Issues Regarding Secondary EGT Candidates
Among all the putative EGT cases that we detected, many should be considered with precaution. These are the ones showing a sister relationship between Archaeplastida (Viridiplantae plus Rhodophyta plus Glaucophyta) and diatoms. In fact, the only difference between a vertically inherited gene phylogeny and a secondary EGT phylogeny is that, in the second case, diatoms should branch nested within the Archaeplastida instead of being one of its related groups. Only a slight difference of a few nodes can switch a moderately supported tree from one topology to the other. Moreover, some trees may appear to support an EGT but actually be the result of hidden paralogy or sampling bias. In our opinion, the most robust and unambiguous cases regarding the question of secondary EGT are those for which we can phylogenetically trace the complete gene pedigree from cyanobacteria, through Archaeplastida (primary EGT), and finally to diatoms (secondary EGT). We thus selected from the complete set of EGT candidates detected by Moustafa et al. (4956 proteins) the ones that had homologs in cyanobacteria, Archaeplastida, and diatoms. We reconstructed and manually inspected all the corresponding protein phylogenetic trees to keep only those that presented a strong support for the monophyly of Archaeplastida plus diatoms (and other secondary photosynthetic eukaryotes) with cyanobacteria as closest outgroup. We collected 209 phylogenetic trees that suited these conditions and manually checked in every tree if the diatom group was emerging from red or from green algae (table 1). The majority of these trees (126) displayed a topology compatible with a scenario where red algae were the EGT donors. Green algae were the sensu stricto EGT source in 16 trees, and were apparent donors (by absence of red algal homologs) in 12 trees.
Table 1.
Sources of EGT Inferred from Phylogenetic Trees Showing a Complete History of Gene Transmission from Primary EGT to Secondary EGT
| EGT Source | Topology | Compatible Trees |
|---|---|---|
| Red algae | (Cyanobacteria, (Viridiplantae, (Rhodophyta, Diatoms))) | 126 |
| (Cyanobacteria, (Rhodophyta, Diatoms)) | 13 | |
| Green algae | (Cyanobacteria, (Rhodophyta, (Viridiplantae, Diatoms))) | 16 |
| (Cyanobacteria, (Viridiplantae, Diatoms)) | 12 | |
| Unresolved | (Cyanobacteria, (Viridiplantae, Rhodophyta, Diatoms)) | 42 |
| Total | 209 | |
Materials and Methods
Genome Database and Similarity Searches
A local genome database was constructed for this study to host protein sequences of 494 full genomes as well as six-frame translated EST sequences for nine species (refer to Supplementary Material online for a complete list of species). Every proteins from P. tricornutum or T. pseudonana reported as of putative green-EGT origin by Moustafa et al. was tagged in the database and used as query for similarity searches. BlastP searches (Altschul et al. 1997) were carried out against the local database and used to collect a set of similar proteins (up to 300, with an e-value threshold of 1e-5) for each query protein.
Phylogenetic Reconstruction
Sets of similar protein sequences were aligned using MAFFT with default parameters (Katoh and Toh 2008) and conserved regions of the resulting multiple sequence alignments were identified with BMGE (Criscuolo and Gribaldo 2010) allowing a maximum of 50% gaps per position. The quality of the resulting alignments was estimated using GUIDANCE (Penn et al. 2010). Maximum likelihood (ML) tree reconstruction and likelihood calculation were done with the program TREEFINDER (Jobb et al. 2004) using the WAG + Γ model.
Gene Clustering and Phylogenetic Tree Classification
A homemade python script was used to cluster homologous proteins (orthologs in P. tricornutum and T. pseudonana or paralogs in a single diatom species) into “single EGT event” groups using phylogenetic trees. Two diatom proteins were considered as “homologous” if they were separated by less than five nodes in the corresponding tree. As a control, we ran a similar clustering procedure based on BlastP results by grouping proteins sharing more than 60% of sequence identity. Both methods gave a similar amount and an almost identical list of clusters (2205 and 2179, respectively). Trees corresponding to every cluster were visually checked to classify them in various topology groups, as described in the main text.
Discussion
In this study, we have tried to reevaluate the contribution of green algal genes to diatom genomes. For this purpose, we have recovered a set of phylogenetic trees comparable with the one produced by Moustafa et al., but with an extended red algal sequence sampling. We manually analyzed these trees using rigorous criteria to try to find topologies compatible with ancient secondary EGT events. This manual method was preferred to the automated tree sorting procedure used by Moustafa et al. to avoid misinterpretations due to classical phylogenetic problems. Indeed, hidden or visible paralogies, as well as the lack of support due to insufficient conservation of the phylogenetic signal, can often be easily detected by a human eye, but are very difficult to translate into flexible patterns that could be sorted by an algorithm. In fact, our detection of a large number of phylogenies that actually support vertical descent but were considered as cases of EGT by Moustafa et al., provides a clear example of the inaccuracy of automated tree sorting to address complex phylogenetic questions.
By trying to balance the number of red algal genome or transcriptome sequences compared with those of Viridiplantae in our database, we wanted to test if a considerable number of EGT events were falsely attributed to green algae simply because of the absence of a red algal counterpart. The addition of a limited amount of genomic data for red algae has been sufficient to change the topology of many trees, either toward a probable EGT of red algal origin or to significantly lower the support of an EGT scenario toward a classical gene transmission by vertical descent from a eukaryotic ancestor. Without completely excluding the possibility of a cryptic green algal endosymbiosis in diatoms, our reanalysis significantly lowers its credibility. Nevertheless, even after including additional red algal data, our database remains overwhelmingly dominated by green algae and land plant sequences, maintaining a high probability of erroneous phylogenetic inference because of unbalanced taxonomic sampling. Thus, as hypothesized by Dagan and Martin (2009), including red algal species with bigger genomes should again increase the number of EGT toggling from Viridiplantae to red algal origin. This is even more plausible for EGT phylogenies spanning from cyanobacteria to diatoms, where the few remaining cases of green EGTs may definitely switch to the red side. We cannot exclude the possibility that a limited residual amount of genes will be confirmed as of real green algal origin, but there would be so few that an alternative scenario of conventional gene transfers (e.g., isolated horizontal gene transfers) should be considered (Keeling and Palmer 2008). A similar situation has been found in another group of eukaryotic microalgae, the chromerids. Though a first analysis of ESTs in the species Chromera velia suggested that 513 of them being of EGT origin from both green and red algae (with a 1 : 1 ratio, see Woehle et al. 2011), a recent analysis of the draft genome sequence suggested that only 51 of the chromera genes evolved by EGT, with a majority of red algal origin (Burki et al. 2012). Available genomic data are very restricted and unevenly distributed among the tree of eukaryotes, providing an incomplete view of the real evolutionary history of genes and species. This is particularly problematic for automated massive phylogenomic analyses. Unfortunately, the time-consuming visual inspection of phylogenetic trees still remains far more accurate.
Supplementary Material
Acknowledgments
We thank the French CNRS for support and the two anonymous reviewers of this article for their useful comments. This work was funded by the Université Paris-Sud (programme attractivité).
Literature Cited
- Allen AE, et al. Evolution and functional diversification of fructose bisphosphate aldolase genes in photosynthetic marine diatoms. Mol Biol Evol. 2012;29:367–379. doi: 10.1093/molbev/msr223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altschul SF, et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armbrust EV. The life of diatoms in the world’s oceans. Nature. 2009;459:185–192. doi: 10.1038/nature08057. [DOI] [PubMed] [Google Scholar]
- Aspilla PS, Antonio AACB, Zuccarello GC, Rojas NRL. A partial expressed sequence tag (EST) library of the economically important red alga Eucheuma denticulatum. Philipp Sci Lett. 2010;3:109–120. [Google Scholar]
- Baurain D, et al. Phylogenomic evidence for separate acquisition of plastids in cryptophytes, haptophytes, and stramenopiles. Mol Biol Evol. 2010;27:1698–1709. doi: 10.1093/molbev/msq059. [DOI] [PubMed] [Google Scholar]
- Burki F, et al. Re-evaluating the green versus red signal in eukaryotes with secondary plastid of red algal origin. Genome Biol Evol. 2012;4:626–635. doi: 10.1093/gbe/evs049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavalier-Smith T. Chloroplast evolution: secondary symbiogenesis and multiple losses. Curr Biol. 2002;12:R62–R64. doi: 10.1016/s0960-9822(01)00675-3. [DOI] [PubMed] [Google Scholar]
- Chan CX, et al. Red and green algal monophyly and extensive gene sharing found in a rich repertoire of red algal genes. Curr Biol. 2011;21:328–333. doi: 10.1016/j.cub.2011.01.037. [DOI] [PubMed] [Google Scholar]
- Criscuolo A, Gribaldo S. BMGE (Block Mapping and Gathering with Entropy): a new software for selection of phylogenetic informative regions from multiple sequence alignments. BMC Evol Biol. 2010;10:210. doi: 10.1186/1471-2148-10-210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dagan T, Martin W. Microbiology. Seeing green and red in diatom genomes. Science. 2009;324:1651–1652. doi: 10.1126/science.1175765. [DOI] [PubMed] [Google Scholar]
- Elias M, Archibald JM. Sizing up the genomic footprint of endosymbiosis. Bioessays. 2009;31:1273–1279. doi: 10.1002/bies.200900117. [DOI] [PubMed] [Google Scholar]
- Frommolt R, et al. Ancient recruitment by chromists of green algal genes encoding enzymes for carotenoid biosynthesis. Mol Biol Evol. 2008;25:2653–2667. doi: 10.1093/molbev/msn206. [DOI] [PubMed] [Google Scholar]
- Jobb G, von Haeseler A, Strimmer K. TREEFINDER: a powerful graphical analysis environment for molecular phylogenetics. BMC Evol Biol. 2004;4:18. doi: 10.1186/1471-2148-4-18. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Katoh K, Toh H. Recent developments in the MAFFT multiple sequence alignment program. Brief Bioinform. 2008;9:286–298. doi: 10.1093/bib/bbn013. [DOI] [PubMed] [Google Scholar]
- Keeling PJ. The endosymbiotic origin, diversification and fate of plastids. Philos Trans R Soc Lond B Biol Sci. 2010;365:729–748. doi: 10.1098/rstb.2009.0103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keeling PJ, Palmer JD. Horizontal gene transfer in eukaryotic evolution. Nat Rev Genet. 2008;9:605–618. doi: 10.1038/nrg2386. [DOI] [PubMed] [Google Scholar]
- Martin W, et al. Gene transfer to the nucleus and the evolution of chloroplasts. Nature. 1998;393:162–165. doi: 10.1038/30234. [DOI] [PubMed] [Google Scholar]
- Matsuzaki M, et al. Genome sequence of the ultrasmall unicellular red alga Cyanidioschyzon merolae 10D. Nature. 2004;428:653–657. doi: 10.1038/nature02398. [DOI] [PubMed] [Google Scholar]
- Moustafa A, et al. Genomic footprints of a cryptic plastid endosymbiosis in diatoms. Science. 2009;324:1724–1726. doi: 10.1126/science.1172983. [DOI] [PubMed] [Google Scholar]
- Nikaido I, et al. Generation of 10,154 expressed sequence tags from a leafy gametophyte of a marine red alga, Porphyra yezoensis. DNA Res. 2000;7:223–227. doi: 10.1093/dnares/7.3.223. [DOI] [PubMed] [Google Scholar]
- Penn O, Privman E, Landan G, Graur D, Pupko T. An alignment confidence score capturing robustness to guide-tree uncertainty. Mol Biol Evol. 2010;27:1759–1767. doi: 10.1093/molbev/msq066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prihoda J, et al. Chloroplast-mitochondria cross-talk in diatoms. J Exp Bot. 2012;63:1543–1557. doi: 10.1093/jxb/err441. [DOI] [PubMed] [Google Scholar]
- Stiller JW. Experimental design and statistical rigor in phylogenomics of horizontal and endosymbiotic gene transfer. BMC Evol Biol. 2011;11:259. doi: 10.1186/1471-2148-11-259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woehle C, Dagan T, Martin WF, Gould SB. Red and problematic green phylogenetic signals among thousands of nuclear genes from the photosynthetic and apicomplexa-related Chromera velia. Genome Biol Evol. 2011;3:1220–1230. doi: 10.1093/gbe/evr100. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.