

SUMMARY
How is Polycomb (Pc), a eukaryotic negative regulator of transcription, targeted to specific mammalian genes? Our genome-wide analysis of the Pc mark H3K27me3 in murine cells that revealed Pc is preferentially associated with CpG island promoters of genes that are transcribed at a low level, and less so with promoters of genes that are either silent or more highly expressed. Studies of the CpG island promoter of the Kit gene demonstrate that Pc is largely absent when the gene is silent in myeloid cells as well as when the gene is highly expressed in mast cells. Manipulations that increase transcription in the former case, and reduce it in the latter, increase Pc occupancy. The average negative effect of Pc, we infer, is about two-fold. We suggest possible biological roles for such negative effects and propose a mechanism by which Pc might be recruited to weakly transcribed genes.
eTOC
Berrozpe et al. assayed occupancy of the polycomb mark H3K27me3 genome-wide and at a specific gene (Kit) when transcribed at different levels. They report that Polycomb is preferentially recruited to genes expressed at a low level.
INTRODUCTION
The murine Kit gene is expressed at high levels in mast cells thanks to activators functioning at the mast Kit enhancer, but is silent in myeloid cells, which do not express some or all of these activators. Previous studies suggest that spontaneous random nucleosome formation at the promoter suffices to eliminate basal transcription that would otherwise occur in myeloid cells (Berropze et al., 2013). This strategy – which does not require promoter-occluding repressors to turn genes “off” - would ensure that different enhancers can work on the same gene: if one enhancer is decommissioned by negative factors that inhibit or eliminate key activators, the promoter remains free to be re-activated by another enhancer (Berrozpe et al., 2006; Jing et al., 2008). This ‘alternative enhancer (activator)’ strategy could not hold for the typical gene in bacteria. In that case, a specific DNA binding repressor – a protein that occludes the promoter – is required to eliminate the inevitable basal transcription that would otherwise occur in the absence of an activator. Such a repressor prevents activation by other activators (Ptashne, 2014).
There are, however, negative factors in eukaryotes that apparently do not simply inhibit activators. A salient example is the Pc protein complex found in many metazoans. PRC2, one of the two major polycomb complexes, includes subunits EED, SUZ12, and the enzyme EZH2 that confers a characteristic histone modification, H3K27me3 (Margueron and Reinberg, 2011). PRC1, another component, has been suggested to bind modified nucleosomes and help inhibit transcription (Simon and Kingston, 2009). The term “Pc” is often used to describe the Drosophila polycomb complex, and PcG that found in other organisms. Here we use simply “Pc” to refer to the histone mark and/or to the Pc protein Suz12 as found in murine cells.
Polycomb is often referred to as a silencer of transcription, but its negative effects are apparently weak. Thus, polycomb- silenced genes can be activated by moderately strong activators (Simon and Kingston, 2009; Vernimmen et al., 2011; Xu et al., 2015). To take another illustration, although Pc is found at high levels associated with CpG island promoters in mammalian ES and iPS cells, removal of PRC2 has a small or undetectable effect on transcription in those cells (Chamberlain et al., 2008; Galonska et al., 2015; Leeb et al., 2010; Pasini et al., 2007; Riising et al., 2014). In flies, Pc is recruited to DNA by factors that bind PREs (Polycomb repressive elements), but mammalian genomes evidently lack such elements (Pirrotta 1997). It has been suggested that in mammals targeting of Pc is effected by binding to nascent mRNA (Davidovich et al., 2013; Kaneko et al., 2014; Rinn et al., 2007; Tsai et al., 2010; Wang and Chang, 2011) or, according to another scenario, that PRC2 binds, directly or indirectly, to CpG-rich regions such as are found in many mammalian promoters (Lynch et al., 2012; Mendenhall et al., 2010; Riising et al., 2014).
A Kit enhancer active in mast cells was first identified by deletion of a 7 Kb fragment lying some 150 Kb upstream of the gene. That deletion greatly reduced production of the Kit protein specifically in mast cells (Berrozpe 2006). We showed that this region bears sites at which DNA-bound transcription factors Gata1 and Gata2 have (separately) displaced nucleosomes, with surrounding nucleosomes at each site bearing the modifications H3K27Ac and H3K4me1. As has been found for many other enhancers, the region bears Pol II as well as Rad21, a protein believed to be important for DNA looping (Kagey et al., 2010; Guo et al., 2012; Hadjur et al., 2009; Parelho et al., 2008; Phillips et al., 2009; Rubio et al., 2008; Wendt et al., 2008). A chromosomal inversion that moves the enhancer much farther upstream (2cM) reduces Kit transcription in mast cells about 100-fold. Rad21 is present at both enhancer and promoter in wild type cells but only at the enhancer in the inversion strain. None of these proteins/modifications is present at the corresponding region in myeloid cells, consistent with the gene being inactive in those cells (Berrozpe et al., 2013).
In myeloid cells, the CpG island Kit promoter bears randomly positioned nucleosomes that, as expected from the promoter’s high GC content, occupy the promoter with high avidities (Berrozpe et al., 2013, Wang et al., 2011). In WT mast cells one such promoter nucleosome is replaced by the transcription complex, a change associated with turning the gene on. The remaining promoter nucleosomes are phased (precisely positioned) in response to the barrier erected by the bound transcription complex. In myeloid cells the region encompassing the mast-cell enhancer, and the Kit promoter, are both covered with nucleosomes that form randomly but with lower avidities than at the promoter (Berrozpe et al., 2013).
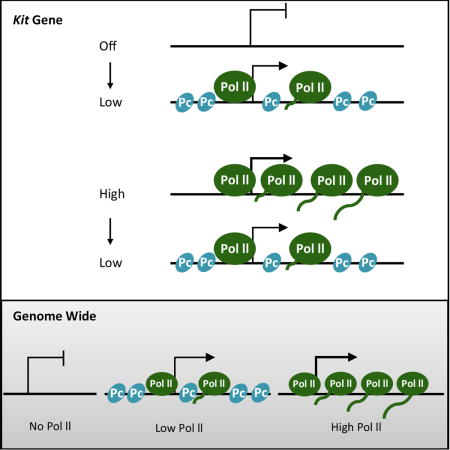
Here we investigate the relationship between levels of transcription of genes and their association with polycomb. A genome-wide analysis of murine mast cells suggests that Pc is associated predominantly (but not exclusively) with genes that are being transcribed weakly, rather than with genes that are being transcribed at higher or lower levels. These experiments also support the notion that, on average, Pc has only a small negative effect on transcription. We then focus specifically on the association of Pc with the murine Kit gene. Although Pc is not found at Kit in WT mast cells, in which transcription is high, nor in myeloid cells in which transcription is undetectable, Pc appears at this gene in the mast cell inversion strain in which Kit transcription is low. Higher levels of Pc at the Kit gene are also evoked by artificial manipulations that decrease Kit transcription in mast cells and, separately, increase Kit transcription in myeloid cells. For the latter effect we used a CRISPR-derived activator, and found (unexpectedly), that its stimulatory effect required the action of 5-AZA-C. We suggest possible roles for the small average negative effect of Pc, and a possible mechanism for targeting of Pc.
RESULTS
Genome-Wide Assay
In order to compare transcription activity and polycomb occupancy, we measured levels of Pol II (RNA polymerase II) in gene bodies, and the Pc mark (H3K27me3) in associated CpG island promoters, for some 10,000 annotated murine genes in primary murine mast cells. The polymerase measurements extended from 2 Kb downstream of the CpG island to the end of the gene, and so are likely to represent active transcription, not complicated by possible polymerase pausing in the CpG island. As shown in Figure 1A, the Pc mark was highest in promoters associated with genes bearing low to moderate levels of Pol II. i.e. genes bearing higher or lower levels of polymerase were less likely to bear the mark.
Figure 1. Levels of the Pc mark (H3K27m3) as a function of polymerase at genes and genome-wide.
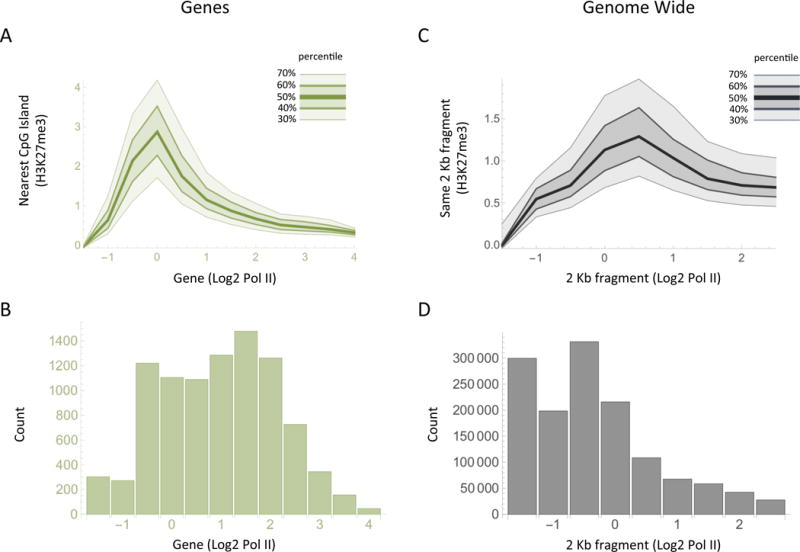
(A) The plot shows the level of the Pc mark H3K27me3, measured at CpG islands, as a function of the level of Pol II found over the nearest gene as measured from 2 Kb downstream of the promoter to the end of the gene. Each curve represents the percentile (as labeled) of the H3K27me3 measurements at each level of Pol II. (B) The bar chart shows the number of genes bearing the indicated level of Pol II. (C) As in (A), except that Pol II and H3K27me3 levels are compared for individual contiguous non-overlapping 2 Kb fragments throughout the genome. (D) The bar chart shows the number of 2 Kb fragments at each level of Pol II.
We extended the analysis by surveying the entire genome without regard to promoters or gene bodies in contiguous non-overlapping 2 Kb fragments. Figure 1C shows that, again, the Pc mark is most predominantly associated with DNA regions undergoing low to moderate levels of transcription. Figure 1B and 1D show the number of genes, or of 2 Kb fragments, at each level of Pol II. Thus, curiously, both experiments in Figure 1A and 1C indicate that at genes that have little or no polymerase, and are therefore transcribed at a very low level if at all, we see little or no H3K27me3. Rather, the results suggest that the Pc mark is most commonly associated with genes that are being transcribed at a low level.
We also assayed CpG island promoters for a mark usually associated with promoter activity (H3K4me3). We separated genes bearing a relatively high active promoter signal into two groups, one of which also bore high levels of the Pc mark, and the other low levels of this Pc mark. The distribution of the amounts of Pol II in these 2 groups of genes are plotted in Figure 2C. The figure suggests that the genes bearing relatively high levels of Pc are expressed, on average, at about a two-fold lower level than genes bearing lower levels of Pc. This finding does not show that Pc works as a repressor, but if it does, the effect is on average small, about two fold.
Figure 2. Polymerase levels in genes bearing high and low levels of Pc at associated CpG island promoters.

(A) The plot shows the distribution of Pol II levels in the gene bodies of genes with a CpG island promoter. The y-axis (density) is proportional to the fraction of genes at each level of Pol II (Log2). (B) The plot shows the distribution of Pol II levels in gene bodies of genes bearing CpG island promoters that have either a low Pc mark (<Log20.5=−1, light grey curve) or a high Pc mark (>Log23=1.58, dark grey curve) in the associated CpG island. (C) The plot shows the distribution of Pol II levels in the bodies of genes with a CpG Island promoter that have a high promoter mark H3K4me3 and either a low Pc mark H3K27me3 (light grey curve) or high Pc (dark grey curve).
Figure 2B shows that most of the genes bearing low levels of Pc (light grey) fall into two groups, one of which also bears high polymerase, the other little or no polymerase. Genes bearing high levels of Pc (darker grey) are generally associated with levels of polymerase intermediate between the extremes observed at genes with low polycomb. Figure 2A shows the distribution of polymerase at all genes bearing a CpG island promoter.
We turn now to an examination of the relationship between transcription and polycomb specifically at the murine Kit gene. We begin with a description of the Kit enhancer.
The Kit Enhancer and Kit expression
As analyzed using ChIP-Endo-Seq, the mast cell Kit enhancer extends over some 150 Kb, and is centered roughly on the previously described mast Kit enhancer (Berrozpe 2013). These experiments confirmed the presence of the enhancer ‘peak’ previously detected by ChIP-qPCR (Berrozpe et al 2013) and, in addition, revealed four additional peaks (Figure 3B). We also compared the enhancer found at its usual WT position with that in the inversion strain (mast INV) in which it is moved far upstream. As shown, the extended enhancer, like the one originally characterized, is identical (or nearly so) at its WT position and at its new position upstream. The only obvious difference is that the latter lacks the right-most peak, which lies on the proximal side of the inversion breakpoint (compare Figure 3B and 3C).
Figure 3. Increased Kit enhancer-promoter separation evokes Pc.
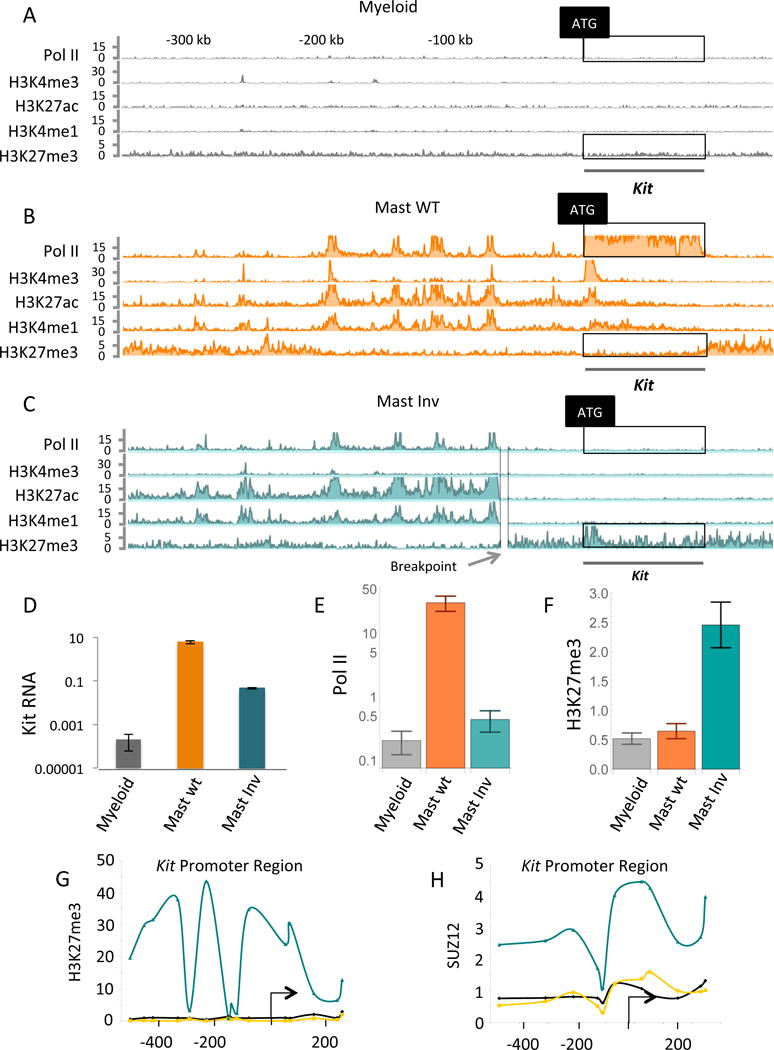
(A), (B) and (C) are ChIP-Endo-Seq experiments probing for the proteins and marks listed on the left of each line. The cells are murine primary mast cells (labeled Mast WT), and murine myeloid cell line 32D (labeled Myeloid). (C) Shows, in addition, the gene-proximal break point of the large chromosomal inversion that greatly increases enhancer-promoter separation (labeled Mast Inv). (D) Kit mRNA levels assayed by qPCR in three cell types as indicated. The significance of the increase in Kit mRNA levels of mast WT over mast Inv: p < 0.0015, of mast WT over myeloid: p < 0.028, and mast Inv over myeloid: p < 0.020. (E) Quantitation of Pol II levels in the boxed regions of the Pol II tracts of A, B and C. The significance of the increase in Pol II levels of mast WT over myeloid: p < 4.4 × 10^−22, mast WT over mast Inv: p < 2.0 × 10 ^−18, and mast Inv over myeloid: p < 0.042. P values were directly calculated by comparing negative binomial distributions (see statistical methods). (F) Quantitation of Pc mark (H3K27me3) in the boxed regions of the Pc tracts of A, B, and C. (G) And (H) show the results of ChIP-qPCR experiments probing for the Pc mark H3K27me3 and for the protein Suz12 at the Kit promoter (myeloid grey lines, mast WT orange lines, and mast Inv cyan lines). The significance of the increase in H3K27me3 levels of mast Inv over mast WT: p < 8.0 × 10 ^−11, and mast Inv over myeloid: p < 3.9 × 10^−13.
As measured by ChIP-Endo-Seq, in WT mast cells the Kit gene bears a high level of Pol II, but in WT myeloid cells there is little or no Pol II in the gene (Figure 3A and 3B). As found previously, the gene bears no indication of an active Kit enhancer in myeloid cells (Figure 3A). In the inversion strain (Figure 3C), there is a detectably higher level of Pol II relative to the myeloid cells (Figure 3C and 3E). Kit mRNA levels are some ten-fold higher in the mast inversion strain than in myeloid cells (Figure 3D).
Polycomb at the Kit Gene
In WT murine ES cells, the Kit promoter is associated with Pc (as assayed by the H3K27me3 mark). The gene is expressed at a low level (some ca. 5% of that found in mast cells). Consistent with the results of others cited in the Introduction, that level is increased about 2-fold in an Eed−/− ES cell (Figure S3). The result is consistent with the notion that Pc has only a small negative effect.
The mast inversion – that in which the Kit enhancer is moved far upstream of the gene - strain provides a local perturbation that reduces Kit transcription some 100-fold below its value in WT mast cells (Figure 3D). Two experimental methods were used to probe for Pc at the Kit gene in this strain. First, a genome-wide ChIP-Endo-Seq experiment detected a peak of the Pc mark at the promoter, with the mark extending (at a lower level) upstream and downstream of the ATG (Figure 3C). This Pc mark was not found at Kit in either WT mast nor in myeloid cells (Figure 3A, 3B). Second, ChIP-qPCR experiments detected signals for both the Pc mark and the Pc subunit Suz12 at the Kit promoter in mast cells bearing the inversion (Figure 3G and 3H, bluish lines). Pc was detected neither in WT mast nor in myeloid cells (Fig 3G and 3H, orange and grey lines). These results prompt the idea, consistent with the genome wide ChIP-Endo-Seq experiments described above, that low to moderate levels of Kit transcription evoke the appearance of Pc at the gene.
To further test this idea we examined the effects of artificially manipulating the level of Kit transcription. We decreased the level of Kit transcription in WT mast cells by ectopically expressing shRNAs directed to two different regions of Rad21 mRNA. The mRNA measurements of Figure 4C show that each of these constructs reduced Kit transcription some 10-fold, consistent with the idea that enhancer-promoter communication requires Rad21. This effect is reflected in the diminished level of Pol II at the gene (compare Figure 4B with 4A). Reduced Kit mRNA expression was accompanied by the presence of the Pc mark over the promoter as revealed in the ChIP-Endo-Seq and ChIP-qPCR experiments of Figure 4B and 4D. The bar chart of Figure 4E shows that at the segment of the Kit gene boxed in Figure 4A and 4B, the Pc track was higher in the presence of the Rad21 shRNA. We note that expression of shRad21 also decreased the heights of the Kit mast cell enhancer peaks (compare Figure 4A and 4B). We detected a total of 200 genes transcription of which decreased upon expression of the Rad21 shRNA. At those genes, the average Pc signal increased, whereas there was no increase in that signal at the many thousands of genes expression of which did not change upon expression of Rad21 shRNA (Figure 4F and 4G). Thus, the effect of Rad21 depletion on the presence of the Pc mark correlates with transcription levels.
Figure 4. Decreased Rad21 activity evokes Pc.
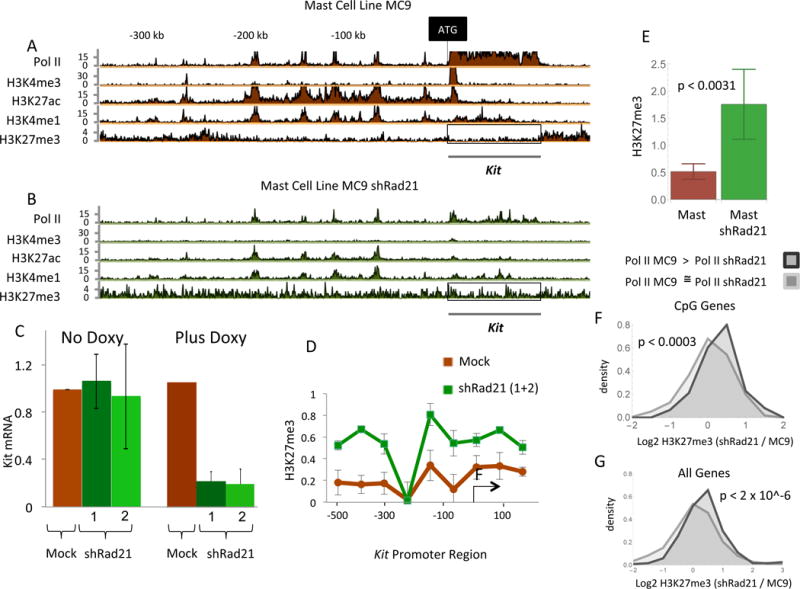
(A) And (B) are ChIP-Seq-Endo experiments as before, with the boxed regions of H3K27me3 quantitated in (E). The cells used for B were exposed to doxycycline, a treatment that induces either of two shRNAs targeted to Rad21. (C) The effect of the shRNAs in Kit expression. The significance of the decrease in Kit mRNA levels, Plus Doxy, of mock over shRad21-1: p < 0.0021, and mock over shRad21-2: p < 0.0041. (D) The H3K27me3 mark along the Kit promoter as affected by the Rad21. (E) The significances of the increase in H3K27me3 levels of mast shRad21 over mast: p < 0.0031. (F) Distribution plots of the change in H3K27me3 signal upon expression of shRad21 in MC9 cells measured at CpG island containing genes at which Pol II signal in the gene significantly decreased (FDR < 0.05, 122 genes, light curve), and at genes at which the Pol II signal did not change (Pol II MC9 / Pol II shRad21 is between 0.8 and 0.9, 660 genes, dark curve). (G) Same as (F) except that in this case all genes were used (FDR < 0.05, 234 genes, light curve), (Pol II MC9 / Pol II shRad21 is between 0.8 and 0.9, 1750 genes, dark curve).
Having studied two cases in which Kit expression was reduced in mast cells, we next investigated the effect on Pc appearance of increasing Kit expression in myeloid cells. In order to increase the level of Kit expression in myeloid cells, we used Cas9-based SAM activators kindly sent to us by Feng Zhang (Konermann et al., 2015). These activators were targeted to three regions of the Kit promoter by three different guide RNAs (Figure S1). However, the Cas-9 based activators, alone or in combination, failed to activate Kit transcription. Cognizant of the claims that DNA methylation might prevent Pc association (Meissner et al., 2008), we treated the cells with 5-AZA-C. As shown in Figure 5A, 5-AZA-C treatment alone had no effect on Kit transcription. However, in the presence of 5-AZA-C, the CRISPR activators induced low levels of Kit transcription, an effect that was accompanied by the appearance of the Pc mark (Figure 5B and 5C). As expected, 5-AZA-C significantly decreased methylation of the Kit promoter (Figure S1), and addition of the CRISPR activators had no additional demethylating effect (Figure S1). The figure also shows that the CRISPR activators, absent 5-AZA-C, had no effect on methylation at the Kit promoter. Since neither 5-AZA-C nor Cas9-activators alone had any effect on transcription or Pc association, these data support the model that the presence of the Pc mark correlates with low transcription.
Figure 5. The effect of CRISPR-derived activators on Kit mRNA expression.
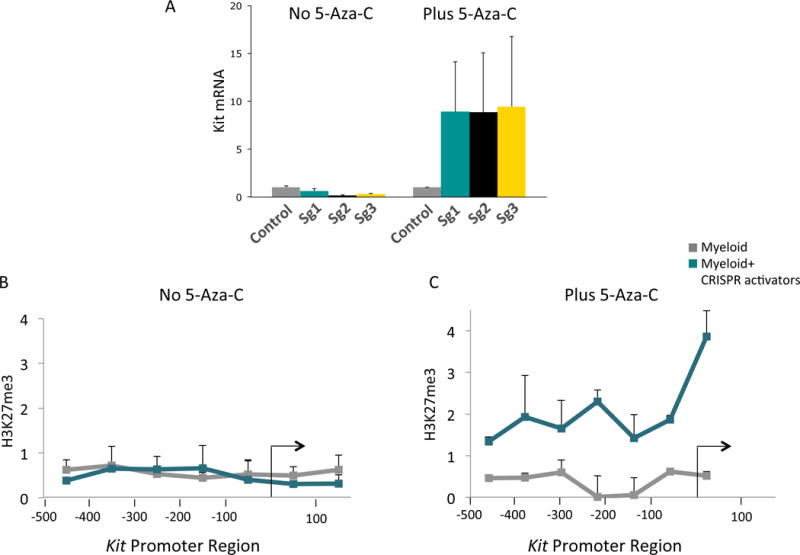
(A) Shows that no mRNA is induced in the absence of 5-AZA-C. Control stands for myeloid cells expressing the CRISPR activator protein (CAS9-SAM) but no guide RNA. Sg1, Sg2, and Sg3 refer to different guides directed to three different locations in the promoter. The significance of the increase in Kit mRNA levels, Plus 5-AZA-C, in Sg1 over control: p < 0.011, in Sg2 over control: p < 0.028, in Sg3 over control: p < 0.02. (B) And (C) are ChIP-qPCR experiments probing for H3K27me3 along the Kit promoter in the absence of any guide RNA (gray line), and the bluish line guide 3.
DISCUSSION
Our genome-wide experiments (performed with primary mast cells) reveal many genes with CpG island promoters that are “off,” but are not associated with Pc (see Figure 1). DNA methylation evidently cannot explain this inactivity, because such promoters in primary cells are largely unmethylated (Bird, 2011). Rather, the inactivity of such genes is likely explained by the absence of relevant transcriptional activators. Consistent with this surmise, the murine Kit gene is silent in myeloid cells (a cell line), and it bears no detectable Pc. It is highly methylated, but removal of those methyl groups with 5-AZA-C affects neither transcription nor the appearance of Pc. As expected from these considerations, we have been unable to detect a Kit enhancer in myeloid cells. Pc is also largely absent from genes that are highly expressed, including the Kit gene in mast cells. Pc appears at the Kit promoter under conditions in which its expression is altered to be at levels intermediate between its highest and lowest levels observed in vivo. These findings are consistent with genome-wide experiments indicating that, although not an absolute rule, Pc appears associated primarily with genes undergoing low to moderate levels of transcription. Our genome wide experiments do not assay highly repetitive DNA, and so we cannot exclude the possibility that Pc works on those sequences.
Although genes bearing Pc have been described as ‘silenced’, our genome-wide experiments, as well as those of others (see Introduction), suggest that its negative effect is small, on average a factor of two (see Figure 2C). This value might vary depending on the strengths of the activators working on any given gene, a model suggested by studies of regulation of genes at the MAT locus of budding yeast. In that case, ‘Sir’ proteins efficiently repress (some 400-fold) genes involved in specifying cell type in WT cells. But this large factor of repression holds only because the natural activators of these genes are very weak. As the strength of those activators is increased, the repressive effect rapidly diminishes (Struhl 1999; Wang et al., 2015). The low level of expression of those genes, absent Sirs, is nevertheless biologically significant. Perhaps certain genes of higher eukaryotes (e.g. Hox genes) have effects even when expressed at relatively low levels, and Pc might play an important role in dampening that expression. Another consideration might be that gene activation often requires an ensemble of activators, such as those found at typical enhancers, sometimes reinforced by activators bound outside of the enhancer. Rather than control all of these activators together, perhaps eukaryotes use Pc to (weakly) counter effects of activators unavoidably present, or present as the stronger activator ensemble is being assembled. A more general role for Pc might be to suppress unwanted basal transcription – already low – that occurs spontaneously around the genome.
How then would specificity be imposed on Pc action in mammals? As noted in the Introduction, the targeting mechanism exploited in flies apparently does not hold for mammals, nor, apparently, does the similar mechanism for imposing specificity on the Sir proteins in yeast. Rather, we would like to entertain the suggestion of others (Lynch et al., 2012; Mendenhall et al., 2010; Riising et al., 2014) that Pc is directed to CpG sequences in CpG island promoters in higher eukaryotes, keeping in mind the following. Our previous work shows that nucleosomes form with high avidity at such GC-rich sequences (Berrozpe et al., 2013; Wang et al., 2011). Those sequences would therefore be relatively sequestered when the gene is silent. When the gene is activated at least one promoter nucleosome is replaced with the transcriptional machinery, and CpG sequences, we imagine, could be at least transiently exposed, thereby allowing binding of Pc. At higher levels of transcription, we imagine, the bound Pc would tend to be evicted, replaced perhaps by transcription complexes. Such a scenario would explain the observation that Pc is associated primarily with weakly transcribed genes. The mechanism would also explain the observation that Pc appears at genes that are in the process of being turned off, but only after expression has been reduced (Riising et al., 2014; Yuan et al., 2012)
Our results are in general agreement with Riising et al., (2014), except that those authors suggest, based on genome-wide experiments, that Pc, assayed by the presence of Suz12, is primarily associated with genes that have ceased transcription rather than with weakly transcribed genes. We are not sure we can account for this difference, beyond noting that in our respective genome-wide experiments different methods were in some instances used to measure gene activity (mRNA vs. Pol II in the gene), and polycomb (Suz12 vs. the Pc histone mark). Our additional results obtained by artificially manipulating Kit expression support our surmise that transcription, not zero transcription, recruits polycomb. Our picture, moreover, allows for a coherent model for how Pc is recruited to, and then at least partially evicted from, genes.
Why the requirement for 5-AZA-C for activation of transcription by CRISPR activators in our experiments? The Kit promoter is heavily methylated in the myeloid cell line we used (Figure S1), and we suggest that, as one possible contributing factor, methylation interferes with the binding of the CRISPR activators. Such an inhibitory effect of methylation is probably small, as it has not been detected in typical CRISPR ‘cutting’ experiments. But inhibition has been observed where the CRISPR enzyme is working off target (Wu et al., 2014). Such off target interactions would be expected to be weak, and although our CRISPR activators are specifically targeted, the overall reaction – including transcriptional activation – is probably weak, and so it might be inhibited by DNA methylation. Another factor might be that the CRISPR activator could work synergistically with other activators binding near the promoter. Sp1 would be an example of such an activator, and indeed we have found that 5-AZA-C mediated demethylation increases Sp1 binding upstream of the Kit promoter (Figure S2).
EXPERIMENTAL PROCEDURES
Cell culture
Bone marrow mast cells (BMMCs) were obtained by flushing the femurs and tibias from adult mice with PBS. Mast cells, MC9, and 32D myeloid cells were grown in RPMI 1640 supplemented with 10% fetal calf serum (FCS), recombinant murine IL3 (Peprotech), nonessential amino acids, and sodium pyruvate. All procedures were approved by the MSKCC Institutional Animal Care and Use Committee protocol 12-09-015.
ChIP Experiments
ChIP experiments were performed according to a protocol provided by Upstate Biotechnology. For ChIP with BMMC, MC9, and 32D cells, 5×106 cells were used per immunoprecipitation. Briefly, the cells were cross-linked with 1% formaldehyde, collected, and washed with PBS containing protease inhibitors. The cells were resuspended in 400 μl SDS lysis buffer on ice for 10 min and then sonicated with eight sets of 12 s pulses by a Branson Sonifier 250 cell. Antibody against H3K27me3 and Rad 21, and Suz12 were purchased from Abcam. DNAs were phenol-chloroform extracted and ethanol precipitated. The immunoprecipitated DNA was analyzed by qPCR as described (Bryant and Ptashne, 2003 and Bryant et al., 2008). The exact sequences of the primers used can be given upon request.
DNA Methylation
Genomic DNA was treated with bisulfite using the EZ DNA Methylation-Direct Kit from ZYMO Research. The experiments were performed according to a protocol provided by ZYMO Research. After the treatment, the DNA was subjected to 35 cycles of PCR. The PCR products were cloned using the TOPO TA Cloning Kit according to a protocol provided by Invitrogen. We analyzed the different clones by sequencing.
RNA Isolation and Quantitative PCR Analysis
RNA was isolated using Trizol reagent, and reverse transcribed (Maxima First Strand cDNA, Thermo Scientific). cDNA was amplified by Roche Light Cycler 480 QPCR machine under the following conditions: 30 sec at 95C, 4 sec at 95, 26 sec at 59C and 4 sec at 72C for 35 cycles. The exact sequences of the primers used can be given upon request. Analyses were conducted in parallel using mouse Rpl 15 and, Rpl 17 for normalization.
Lentivirus production
293T cells were transfected with the different plasmids by calcium phosphate. Two days after the transfection, the supernatant was collected, and the lentivirus was concentrated by ultracentrifugation.
shRNA Infections
Mast cells (MC9) were transduced with lentivirus via spinfection in 6 well plates. 1×106 cells in 2 ml of media supplemented with polybrene (Sigma) were added to each well, supplemented with lentiviral supernatant and centrifuged for 1:30 hours at 1000 g. A scrambled control shRNA, and shRNA’s targeting mouse Rad21 were provided by RNAi Core Facility, Memorial Sloan Kettering Cancer Center (clones NM_009009.4_2172, NM_009009.4_2036, NM_009009.4_3153). Infected mast cells were selected using Puromycin for 2 weeks. After selection, shRad21 was induced using Doxycycline for 3 days, and then the cells were harvested as required for RNA, ChIP, and ChIP-Endo-Seq analyses.
CRISPR-SAM
sgRNA-specific Kit promoter oligo sequences were chosen to minimize the likelihood of off-target using http://tools.genome-engineering.org approach. Three different Kit oligos were cloned into the Lenti-sgRNA (MS2)_EF1A-zeo cloning vector using a Golden Gate Assembly strategy (Konermann et al., 2015). Lenti-sgRNA (MS2)_EF1A-zeo cloning backbone, Lenti_MS2-P65-HSF1_2A-Hygro, and, Lenti-dCas9_VP64_2A-Blast were kindly provided by Feng Zhang (Konermann et al., 2015). 32D myeloid cells were transduced with lentivirus via spinfection. After 2 days the cells were selected with 200 μg/ml Zeocin, 10 μg/ml Blastcidin, and 300 μg/ml Hygromycin. After selection, the cells were harvested as required for RNA, and ChIP analyses.
Statistical Methods
Error for the genomic ChIP-Endo-Seq experiments was estimated using negative binomial distribution as described in Supplemental Methods. P values for RT-qPCR experiments were calculated using students t test. P-values for significant differences comparing distribution functions are calculated using the Kolmogorov-Smirnov Test.
Supplementary Material
Highlights.
Genome-wide, Pc preferentially associated with genes transcribed at moderate levels
Modulating Kit transcription, from OFF or ON to moderately on, increased Pc occupancy
Polycomb’s repressive effect is small, on average about two-fold
Acknowledgments
We thank Xin Wang for developing a software tool to calculate p values, Antoinette Rookard for her help maintaining the mouse colony, Myles Fennell from the RNAi Facility for technical assistant, Silvana Konermann and Feng Zhang for advice and reagents for the CRISPR experiments, and Terry Magnuson for the mouse ES Eed−/− cells. This work was funded by NYSTEM (N11G-262) to MP, and the MSK Cancer Center Support Grant/Core Grant (P30 CA008748)
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
AUTHOR CONTRIBUTIONS
G.B. and K.W. performed the cell culture, the shRad21, and CRISPR experiments; G.O.B. performed and developed the genome wide experiments and analyses. D.S. assisted with qPCR. S.N. provided expertise and feedback. S.S. assisted with lentivirus production. G.B. G.O.B, and M.P. analyzed the data. G.B. G.O.B. and M.P wrote the paper.
ACCESSION NUMBERS
The accession number for the genome wide ChIP-Endo-Seq data reported in this paper is GEO:GSE94946
References
- Benjamini Y, Hochberg Y. Controlling the False Discovery Rate: a Practical and Powerful Approach to Multiple Testing. J R Statist Soc B. 1995;57:289–300. [Google Scholar]
- Berrozpe G, Agosti V, Tucker C, Blanpain C, Manova K, Besmer P. A distant upstream locus control region is critical for expression of the Kit receptor gene in mast cells. Molecular and cellular biology. 2006;26:5850–5860. doi: 10.1128/MCB.01854-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berrozpe G, Bryant GO, Warpinski K, Ptashne M. Regulation of a mammalian gene bearing a CpG island promoter and a distal enhancer. Cell reports. 2013;4:445–453. doi: 10.1016/j.celrep.2013.07.001. [DOI] [PubMed] [Google Scholar]
- Bird A. Putting the DNA back into DNA methylation. Nat Genet. 2011;43(11):1050–1051. doi: 10.1038/ng.987. [DOI] [PubMed] [Google Scholar]
- Bryant GO, Prabhu V, Floer M, Wang X, Spagna D, Schreiber D, Ptashne M. Activator control of nucleosome occupancy in activation and repression of transcription. PLoS biology. 2008;6:2928–2939. doi: 10.1371/journal.pbio.0060317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryant GO, Ptashne M. Independent recruitment in vivo by Gal4 of two complexes required for transcription. Molecular cell. 2003;11:1301–1309. doi: 10.1016/s1097-2765(03)00144-8. [DOI] [PubMed] [Google Scholar]
- Chamberlain SJ, Yee D, Magnuson T. Polycomb repressive complex 2 is dispensable for maintenance of embryonic stem cell pluripotency. Stem cells. 2008;26:1496–1505. doi: 10.1634/stemcells.2008-0102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidovich C, Zheng L, Goodrich KJ, Cech TR. Promiscuous RNA binding by Polycomb repressive complex 2. Nature structural & molecular biology. 2013;20:1250–1257. doi: 10.1038/nsmb.2679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galonska C, Ziller MJ, Karnik R, Meissner A. Ground State Conditions Induce Rapid Reorganization of Core Pluripotency Factor Binding before Global Epigenetic Reprogramming. Cell stem cell. 2015;17:462–470. doi: 10.1016/j.stem.2015.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo Y, Monahan K, Wu H, Gertz J, Varley KE, Li W, Myers RM, Maniatis T, Wu Q. CTCF/cohesin-mediated DNA looping is required for protocadherin alpha promoter choice. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:21081–21086. doi: 10.1073/pnas.1219280110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hadjur S, Williams LM, Ryan NK, Cobb BS, Sexton T, Fraser P, Fisher AG, Merkenschlager M. Cohesins form chromosomal cis-interactions at the developmentally regulated IFNG locus. Nature. 2009;460:410–413. doi: 10.1038/nature08079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jing H, Vakoc CR, Ying L, Mandat S, Wang H, Zheng X, Blobel GA. Exchange of GATA factors mediates transitions in looped chromatin organization at a developmentally regulated gene locus. Molecular cell. 2008;29:232–242. doi: 10.1016/j.molcel.2007.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kagey MH, Newman JJ, Bilodeau S, Zhan Y, Orlando DA, van Berkum NL, Ebmeier CC, Goossens J, Rahl PB, Levine SS, et al. Mediator and cohesin connect gene expression and chromatin architecture. Nature. 2010;467:430–435. doi: 10.1038/nature09380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaneko S, Bonasio R, Saldana-Meyer R, Yoshida T, Son J, Nishino K, Umezawa A, Reinberg D. Interactions between JARID2 and noncoding RNAs regulate PRC2 recruitment to chromatin. Molecular cell. 2014;53:290–300. doi: 10.1016/j.molcel.2013.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konermann S, Brigham MD, Trevino AE, Joung J, Abudayyeh OO, Barcena C, Hsu PD, Habib N, Gootenberg JS, Nishimasu H, et al. Genome-scale transcriptional activation by an engineered CRISPR-Cas9 complex. Nature. 2015;517:583–588. doi: 10.1038/nature14136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leeb M, Pasini D, Novatchkova M, Jaritz M, Helin K, Wutz A. Polycomb complexes act redundantly to repress genomic repeats and genes. Genes & development. 2010;24:265–276. doi: 10.1101/gad.544410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch MD, Smith AJ, De Gobbi M, Flenley M, Hughes JR, Vernimmen D, Ayyub H, Sharpe JA, Sloane-Stanley JA, Sutherland L, et al. An interspecies analysis reveals a key role for unmethylated CpG dinucleotides in vertebrate Polycomb complex recruitment. The EMBO journal. 2012;31:317–329. doi: 10.1038/emboj.2011.399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margueron R, Reinberg D. The Polycomb complex PRC2 and its mark in life. Nature. 2011;469:343–349. doi: 10.1038/nature09784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meissner A, Mikkelsen TS, Gu H, Wernig M, Hanna J, Sivachenko A, Zhang X, Bernstein BE, Nusbaum, et al. Genome-scale DNA methylation maps of pluripotent and differentiated cells. Nature. 2008;454:766–770. doi: 10.1038/nature07107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendenhall EM, Koche RP, Truong T, Zhou VW, Issac B, Chi AS, Ku M, Bernstein BE. GC-rich sequence elements recruit PRC2 in mammalian ES cells. PLoS genetics. 2010;6:e1001244. doi: 10.1371/journal.pgen.1001244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parelho V, Hadjur S, Spivakov M, Leleu M, Sauer S, Gregson HC, Jarmuz A, Canzonetta C, Webster Z, Nesterova T, et al. Cohesins functionally associate with CTCF on mammalian chromosome arms. Cell. 2008;132:422–433. doi: 10.1016/j.cell.2008.01.011. [DOI] [PubMed] [Google Scholar]
- Pasini D, Bracken AP, Hansen JB, Capillo M, Helin K. The polycomb group protein Suz12 is required for embryonic stem cell differentiation. Molecular and cellular biology. 2007;27:3769–3779. doi: 10.1128/MCB.01432-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips JE, Corces VG. CTCF: master weaver of the genome. Cell. 2009;137:1194–1211. doi: 10.1016/j.cell.2009.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pirrotta V. PcG complexes and chromatin silencing. Current opinion in genetics & development. 1997;7:249–258. doi: 10.1016/s0959-437x(97)80135-9. [DOI] [PubMed] [Google Scholar]
- Ptashne M. The chemistry of regulation of genes and other things. The Journal of biological chemistry. 2014;289:5417–5435. doi: 10.1074/jbc.X114.547323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riising EM, Comet I, Leblanc B, Wu X, Johansen JV, Helin K. Gene silencing triggers polycomb repressive complex 2 recruitment to CpG islands genome wide. Molecular cell. 2014;55:347–360. doi: 10.1016/j.molcel.2014.06.005. [DOI] [PubMed] [Google Scholar]
- Rinn JL, Kertesz M, Wang JK, Squazzo SL, Xu X, Brugmann SA, Goodnough LH, Helms JA, Farnham PJ, Segal E, et al. Functional demarcation of active and silent chromatin domains in human HOX loci by noncoding RNAs. Cell. 2007;129:1311–1323. doi: 10.1016/j.cell.2007.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubio ED, Reiss DJ, Welcsh PL, Disteche CM, Filippova GN, Baliga NS, Aebersold R, Ranish JA, Krumm A. CTCF physically links cohesin to chromatin. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:8309–8314. doi: 10.1073/pnas.0801273105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon JA, Kingston RE. Mechanisms of polycomb gene silencing: knowns and unknowns. Nature reviews Molecular cell biology. 2009;10:697–708. doi: 10.1038/nrm2763. [DOI] [PubMed] [Google Scholar]
- Struhl K. Fundamentally different logic of gene regulation in eukaryotes and prokaryotes. Cell. 1999;98:1–4. doi: 10.1016/S0092-8674(00)80599-1. [DOI] [PubMed] [Google Scholar]
- Tsai MC, Manor O, Wan Y, Mosammaparast N, Wang JK, Lan F, Shi Y, Segal E, Chang HY. Long noncoding RNA as modular scaffold of histone modification complexes. Science. 2010;329:689–693. doi: 10.1126/science.1192002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vernimmen D, Lynch MD, De Gobbi M, Garrick D, Sharpe JA, Sloane-Stanley JA, Smith AJ, Higgs DR. Polycomb eviction as a new distant enhancer function. Genes & development. 2011;25:1583–1588. doi: 10.1101/gad.16985411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang KC, Chang HY. Molecular mechanisms of long noncoding RNAs. Molecular cell. 2011;43:904–914. doi: 10.1016/j.molcel.2011.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Bai L, Bryant GO, Ptashne M. Nucleosomes and the accessibility problem. Trends in genetics : TIG. 2011;27:487–492. doi: 10.1016/j.tig.2011.09.001. [DOI] [PubMed] [Google Scholar]
- Wang X, Bryant G, Zhao A, Ptashne M. Nucleosome avidities and transcriptional silencing in yeast. Current biology : CB. 2015;25:1215–1220. doi: 10.1016/j.cub.2015.03.004. [DOI] [PubMed] [Google Scholar]
- Wendt KS, Yoshida K, Itoh T, Bando M, Koch B, Schirghuber E, Tsutsumi S, Nagae G, Ishihara K, Mishiro T, et al. Cohesin mediates transcriptional insulation by CCCTC-binding factor. Nature. 2008;451:796–801. doi: 10.1038/nature06634. [DOI] [PubMed] [Google Scholar]
- Wu X, Scott DA, Kriz AJ, Chiu AC, Hsu PD, Dadon DB, Cheng AW, Trevino AE, Konermann S, Chen S, et al. Genome-wide binding of the CRISPR endonuclease Cas9 in mammalian cells. Nature biotechnology. 2014;32:670–676. doi: 10.1038/nbt.2889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J, Shao Z, Li D, Xie H, Kim W, Huang J, Taylor JE, Pinello L, Glass K, Jaffe JD, et al. Developmental control of polycomb subunit composition by GATA factors mediates a switch to non-canonical functions. Molecular cell. 2015;57:304–316. doi: 10.1016/j.molcel.2014.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan W, Wu T, Fu H, Dai C, Wu H, Liu N, Li X, Xu M, Zhang Z, Niu T, et al. Dense chromatin activates Polycomb repressive complex 2 to regulate H3 lysine 27 methylation. Science. 2012;337:971–975. doi: 10.1126/science.1225237. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.