

Abstract
Background
Prefrontal global brain connectivity with global signal regression (GBCr) was proposed as a robust biomarker of depression, and was associated with ketamine’s mechanism of action. Here, we investigated prefrontal GBCr in treatment-resistant depression (TRD) at baseline and following treatment. Then, we conducted a set of pharmacological challenges in healthy subjects to investigate the glutamate neurotransmission correlates of GBCr.
Methods
In study A, we used functional magnetic resonance imaging (fMRI) to compare GBCr between 22 TRD and 29 healthy control. Then, we examined the effects of ketamine and midazolam on GBCr in TRD patients 24h post-treatment. In study B, we acquired repeated fMRI in 18 healthy subjects to determine the effects of lamotrigine (a glutamate release inhibitor), ketamine, and lamotrigine-by-ketamine interaction.
Results
In study A, TRD patients showed significant reduction in dorsomedial and dorsolateral prefrontal GBCr compared to healthy control. In TRD patients, GBCr in the altered clusters significantly increased 24h following ketamine (effect size = 1.0 [0.3 1.8]), but not midazolam (effect size = 0.5 [−0.6 1.3]). In study B, oral lamotrigine reduced GBCr 2h post-administration, while ketamine increased medial prefrontal GBCr during infusion. Lamotrigine significantly reduced the ketamine-induced GBCr surge. Exploratory analyses showed elevated ventral prefrontal GBCr in TRD and significant reduction of ventral prefrontal GBCr during ketamine infusion in healthy subjects.
Conclusions
This study provides first replication of the ability of ketamine to normalize depression-related prefrontal dysconnectivity. It also provides indirect evidence that these effects may be triggered by the capacity of ketamine to enhance glutamate neurotransmission.
Keywords: ketamine, glutamate, functional MRI, global brain connectivity, rapid acting antidepressants, Treatment resistant depression
Introduction
Major depressive disorder (MDD) is a disabling mental illness with poorly understood pathophysiology and high rates of inadequate treatment response (1, 2). Accumulating evidence over the past 2 decades strongly implicated glutamate neurotransmission alterations in the pathophysiology and treatment of MDD (3, 4), particularly the exciting discovery of rapid acting antidepressant effects induced by subanesthetic doses of ketamine, a glutamate modulator (5–8). However, with the exception of ketamine, translating the glutamate findings into novel antidepressants has been difficult, with various agents showing no antidepressant effects or exiting development pipelines by pharmaceutical industry due to failure reaching primary targets (9–11). A major challenge in the field is to establish robust in vivo in human biomarkers that could serve as measures of target engagement and target validation in the development of rapid acting antidepressants (12–14). These biomarkers would advance our understanding of the neurobiology of depression and could play a critical role in early phases of drug development. In this report, we used a promising biomarker of functional network connectivity, termed global brain connectivity with global signal regression (GBCr) (15, 16), to establish the effects of ketamine on prefrontal connectivity in MDD and healthy subjects, and to demonstrate a direct link between large scale connectivity networks and underlying glutamate neurotransmission.
Ketamine, an N-methyl-D-aspartate (NMDA) receptor antagonist, is a rapid acting antidepressant that is believed to exert its therapeutic effect by inducing a glutamate neurotransmission surge leading to increased synaptogenesis and reversal of depression-related glutamate synaptic deficits (17, 18). To date, two non-mutually exclusive models have been proposed for the mechanisms through which ketamine induces glutamate neurotransmission (19, 20). The NMDA model proposes that subanesthetic doses of ketamine preferentially block NMDA receptors on a subpopulation of interneurons precipitating disinhibition of glutamate neurotransmission (21, 22). A more recent model suggested the presence of ketamine metabolites (Z-6-Hydroxynorketamine) that activate synaptic glutamate neurotransmission independent of inhibiting NMDA receptors, through a yet unidentified mechanism (20). Both models converge on the role of AMPA-dependent activation of glutamate neurotransmission as a common pathway to induce downstream synaptogenesis and rapid acting antidepressant effects (23). Preclinical work showed a robust glutamate neurotransmission surge after administering ketamine or other rapid acting antidepressants (24). In humans, proton magnetic resonance spectroscopy (1H MRS) has been used to provide indirect evidence of the ability of ketamine to stimulate glutamate (25, 26), although one study failed to support this conclusion (27). In depressed individuals, preliminary evidence in 8 subjects showed increased mPFC Glx during ketamine infusion (28). Others failed to show occipital cortical glutamate changes 3h and 24h post-ketamine (29). Together, these data provided mechanistic evidence of a ketamine-induced mPFC glutamate surge during infusion in healthy and depressed subjects. However, the inconsistencies affect the potential utility of these methods as a robust biomarker for drug development. These inconsistencies are potentially influenced by the limited spatial and temporal resolutions of the approach and by the fact that 1H MRS measures total levels of glutamate (i.e., intra- and extra-cellular) that may not sufficiently capture glutamate activation and transmission.
GBCr is a robust well-validated graph theory measure of large scale functional connectivity networks. Using functional magnetic resonance imaging (fMRI), GBCr is measured as the average correlation between each voxel and all other voxels in the brain gray matter. These global brain connectivity values, a.k.a. functional connectivity strength, have been used to identify major brain networks (30), were found to predict cognitive functioning and intelligence (31), and were shown to significantly correlate with regional cerebral blood flow (32, 33). Accumulating evidence has consistently shown reduced PFC GBCr across various psychiatric disorders marked by chronic stress (15, 34–39). Thus, it was proposed that these PFC GBCr abnormalities may reflect depression and stress-induced glutamate alterations (16, 17, 34). Depressive features and chronic stress in animal models reduce prefrontal glutamate synaptic density and strength, abnormalities that are reversed by ketamine treatment (40). Consistent with this model, we have recently demonstrated widespread GBCr reduction in the dorsomedial, frontolateral, and ventral PFC in patients with MDD (35). In treatment-resistant depression (TRD), we found comparable GBCr reduction in dorsomedial and frontolateral, but not ventral (Limbic network; vPFC), PFC in patients at baseline (16). These GBCr abnormalities were primarily located in the Ventral Attention and Frontoparietal networks within the PFC (41). At 24h following ketamine administration, we found a pattern of normalization of pre-treatment PFC GBCr alterations and a significant increase in frontolateral GBCr that positively correlated with improvement of depression (16).
Considering preclinical evidence of glutamate synaptic transmission normalization 24h post-ketamine administration in models of depression (40), our previous ketamine findings in TRD suggested a crucial role for PFC connectivity in TRD and raised an essential translational question, whether the ketamine-induced increases in synaptic glutamate neurotransmission is causally related to fMRI measure of GBCr. In the current report, we first replicated, in a new independent cohort of TRD patients, the presence of PFC GBCr abnormalities in TRD and the ability of ketamine to reverse these GBCr alterations 24h post-treatment. Then, we employed experimental pharmacoimaging challenges in healthy subjects to demonstrate a correlational relationship, as well as to probe for a putative causal link, between glutamate neurotransmission and functional connectivity as measured by GBCr. The demonstration of a direct link between synaptic transmission and GBCr would provide insight into the neurobiology of depression and the mechanism of action of rapid acting antidepressants. It will also present GBCr as a potential biomarker of target validation. Moreover, considering the robust ketamine induction of glutamate neurotransmission during infusion, GBCr could serve as a biomarker of target engagement if a direct link between GBCr and underlying synaptic transmission is demonstrated.
To accomplish the study aims, we first compared PFC GBCr between TRD and healthy controls (HC), predicting reduced GBCr in prefrontal regions within the Ventral Attention and Frontoparietal, but not Limbic networks. We hypothesized that ketamine would reverse these PFC GBCr alterations 24h following treatment. Then, to investigate the relationship between glutamate neurotransmission and GBCr, we examined whether lamotrigine, an inhibitor of glutamate neurotransmission, would induce a reduction in PFC GBCr. We also examined the effects of ketamine on PFC GBCr during infusion, hypothesizing that ketamine would increase dorsomedial and frontolateral GBCr but reduce vPFC GBCr –comparable to previous report of reduced vPFC activity during ketamine infusion (42). Finally, to provide experimental evidence of a direct link between ketamine induction of glutamate neurotransmission and GBCr, we hypothesized that lamotrigine would significantly reduce the ketamine-induced PFC GBCr changes.
Methods
Participants
The studies were approved by institutional review boards. All subjects completed an informed consent process prior to participation (ClinicalTrials.gov identifier: NCT00768430). All imaging data presented in this report are new and have not been previously published. Cohort A study was conducted at one site (Houston, TX), as an add-on to a ketamine clinical trial that was previously published (6). Cohort B study was conducted at one site (New Haven, CT), aimed to investigate the modulation of ketamine effects by lamotrigine.
Cohort A included 22 TRD (54% males, mean age ±SEM = 44 ±2.3 years) and 29 HC (55% males, mean age ±SEM = 44 ±1.8 years). All participants completed fMRI scans at baseline. Of the 22 TRD, 20 subjects were randomized to ketamine (n = 13; 0.5mg/kg over 40 minutes) or to active placebo midazolam (n = 7; 0.045 mg/kg over 40 minutes). Seventeen TRD subjects (10 ketamine; 7 midazolam) completed fMRI scans at 24h following treatment. Study criteria for the TRD group included current MDD diagnosis, treatment resistant to at least 3 antidepressants according to the Antidepressant Treatment History Form (ATHF), no psychotropic medication for at least 1 week (4 weeks for fluoxetine), no substance use disorder for at least 24 months, no MR contraindications, no unstable medical condition, and no history of bipolar or psychotic disorders. HC had no psychiatric disorders and no MR contraindications. Primary outcome of depression severity was the Montgomery-Åsberg Depression Rating Scale (MADRS) at baseline and 24h post-treatment. Secondary measure included the Inventory of Depressive Symptomatology (IDS).
Cohort B included 18 males between the ages of 22 and 36 (mean ±SEM = 28 ±0.9 years). Participants were excluded if they had an unstable medical illness, a psychiatric disorder, or an MR contraindication. The study used a double-blind crossover design in which participants were randomized to three study sessions: I- oral placebo followed by normal saline infusion, then intravenous placebo (saline) (Plc-Plc); II- oral placebo followed by normal saline infusion, then intravenous ketamine (Plc-Ket); III- oral lamotrigine (300 mg), followed by normal saline infusion, then intravenous ketamine (Lamo-Ket). Subjects, but not investigators, were blind to the fact that the first infusion was always saline. The oral drug (placebo or lamotrigine) was administered 2 hours prior to normal saline. This lamotrigine dose was selected as it was the largest dose that had been safely administered to lamotrigine-naïve subjects for research purposes (43). Study sessions were separated by at least 1 week. Ketamine was administered as bolus (0.23 mg/kg in 2 min) followed by 0.58 mg/kg over approximately 70 min, a well-validated infusion regimen to maintain a steady-state level of ketamine during fMRI acquisition and subsequent clinical assessments. This ketamine dose is also comparable to doses that have been shown to produce a prefrontal glutamate surge during infusion in healthy subjects (26). The Brief Psychiatric Rating Scale (BPRS) and Clinician-Administered Dissociative States Scale (CADSS) were used to assess the psychotomimetic effects of ketamine during infusion.
Neuroimaging
In Cohort A, resting-state fMRI scans (voxel size = 3.4×3.4×4 mm, TR = 2000 ms, 150 frames) were completed at baseline for all subjects (TRD and HC) and repeated 24h following ketamine and midazolam infusions in TRD participants. In Cohort B, fMRI scans were acquired while subjects performed a visual oddball task (voxel size = 3.4×3.4×4.5 mm; TR = 2000 ms, 210 frames) and were repeated during each session. Functional connectivity measures were computed on the visual oddball task fMRI data comprising three 7 min. runs during normal saline infusion and three 7 min. runs during study drug infusion. The results of the visual oddball task will be reported elsewhere. Participants in both cohorts completed structural MRI scans for coregistration. GBCr values were computed as the average of the correlations of the BOLD time series between each voxel and all other voxel in the brain gray matter. GBCr methods have been detailed in previous reports (16, 34) and summarized in the Supplemental Information (SI).
Statistical Analyses
For additional details, please see SI. Briefly, independent t-tests were used to compare TRD and HC. Paired t-tests were used to examine the effects of lamotrigine and ketamine on GBCr. Correlational analyses examined the relationships between GBCr and behavioral measures. To examine the effects of lamotrigine, we conducted a voxel-wise paired t-test comparing PFC GBCr post lamotrigine and post placebo (fMRI runs prior to the infusion of the study drug). To examine the effects of ketamine on PFC GBCr, we compared delta GBCr (drug – saline) between the placebo-ketamine and placebo-placebo sessions using voxel-wise paired t-tests. To examine the effects of lamotrigine on the ketamine-induced GBCr changes, we extracted the average delta GBCr in the clusters that showed significant increase during ketamine infusion and conducted a pairwise paired t-test comparing delta GBCr between each of the study arms. All voxel-wise analyses were limited to the PFC and only voxels surviving correction for multiple comparisons are reported in the main text and figures. Type I error correction for voxel-wise analyses was based on permutation methods (44). Significance was set at p ≤ 0.05.
Results
TRD and HC were well matched for age and sex. Demographics and clinical characteristics are reported in Table 1. On average, TRD participants had moderate to severe depression with considerable history of treatment failures. Comparable to the parent clinical trial (6), there was a treatment by time interaction in the 20 randomized subjects (F(1,18) = 5.8; p = 0.03), with significant reduction in MADRS severity in the ketamine group compared to placebo (Fig. S1). The full description of the behavioral measures and treatment effects in TRD subjects were previously reported (6).
Table 1.
Demographics and Clinical Characteristics
| TRD (n = 22) |
HC (n = 29) |
pa | |
|---|---|---|---|
|
| |||
| Mean ± SEM | Mean ± SEM | ||
| Age (y) | 44.8 ± 2.3 | 44.3 ± 1.8 | 0.88 |
| Female (N; %) | 10 (46%) | 13 (45%) | 0.97 |
| Failed antidepressant trials | 5.1 ± 0.4 | ||
| Age of onset (y) | 23.4 ± 2.2 | ||
| MADRS | 32 ± 1.2 | ||
Independent t-test or Chi square test (significance set at p ≤ .05);
Abbreviations: MDD: Major Depressive Disorder; y: years; MADRS: Montgomery-Åsberg Depression Rating Scale; Number of treatment failures was determined by the Antidepressants Treatment History Form (ATHF);
GBCr in TRD & Post-ketamine
Following correction for multiple comparisons, a voxel-wise analysis comparing TRD and HC in Cohort A showed significantly lower GBCr in TRD in three PFC clusters (Fig. 1A; Table S1). These clusters were located in the bilateral dorsomedial and right dorsolateral PFC. GBCr abnormalities overlapped with the Ventral Attention and Frontoparietal networks (Fig. 1A).
Figure 1. Functional Dysconnectivity in TRD.
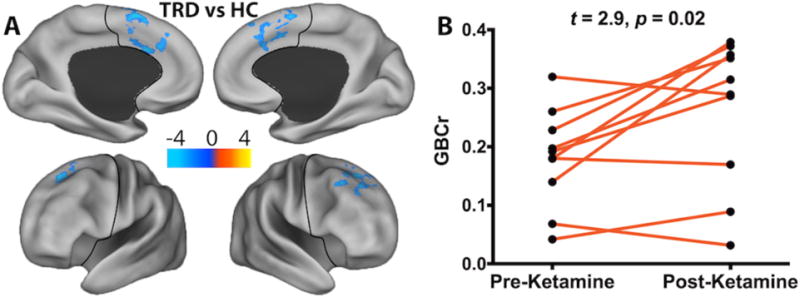
(A) Voxel-wise comparisons of PFC GBCr showed significant reductions of GBCr in TRD, relative to HC, in three clusters (blue). (B) Ketamine increased GBCr 24h post-treatment in these clusters. The color bar depicts the z values of reduced (TRD < HC; blue) and increased (TRD > HC; yellow-red) GBCr. The black line delineates the PFC region. Abbreviations: TRD = Treatment-Resistant Depression; HC = Healthy Control; PFC = prefrontal cortex; GBCr = Global Brain Connectivity with global signal regression.
As shown in Fig. 1B, the average GBCr in the altered clusters significantly increased following ketamine treatment (mean ±SEM pre-treatment = 0.18 ±0.03; post-treatment = 0.26 ±0.04; t = 2.9; p = 0.017). However, we found no significant changes in the placebo group (t = 1.2; p = 0.29). Considering the small samples, the effects of ketamine and placebo were confirmed using non-parametric tests and using a bootstrapping approach with 10,000 iterations. These follow-up analyses similarly showed significant increase in GBCr following ketamine (p = 0.03, effect size = 1.0, CI = 0.3 to 1.8), but no significant changes were found following placebo (p = 0.03, effect size = 0.5, CI = −0.6 to 1.3). In the full TRD group, we found no correlation between baseline GBCr in the altered clusters and MADRS (r = 0.1, p = 0.7) or IDS severity (r = −0.1, p = 0.8).
GBCr & Glutamate Modulators
In Cohort B, lamotrigine significantly reduced GBCr in a cluster containing part of the bilateral rostral anterior cingulate and medial PFC (Fig. 2A; Table S1). This cluster primarily overlapped with the Default Mode network within the PFC (Fig. 2A). Ketamine significantly increased GBCr in two clusters located in the bilateral dorsomedial and left frontolateral PFC (Fig. 2B; Table S1). These clusters primarily overlapped with the Frontoparietal and Default Mode networks (Fig. 2B). Lamotrigine pretreatment significantly reduced the ketamine-induced GBCr increases in these two clusters (Fig. 3).
Figure 2. Effects of Lamotrigine and Ketamine on Prefrontal Functional Connectivity.
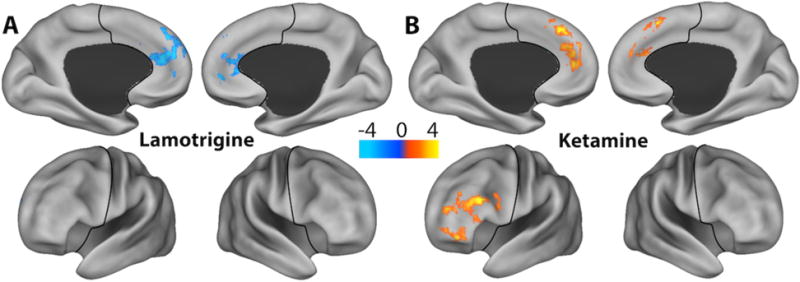
(A) Voxel-wise paired t-tests with family-wise-error correction revealed significant reductions of GBCr 2h post lamotrigine (blue) and (B) increased GBCr during ketamine infusion (red). The color bar depicts the z values of reduced (blue) and increased (yellow-red) GBCr. The black line delineates the prefrontal cortex region. Abbreviations: GBCr = Global Brain Connectivity with global signal regression;
Figure 3. Interaction between lamotrigine and ketamine.
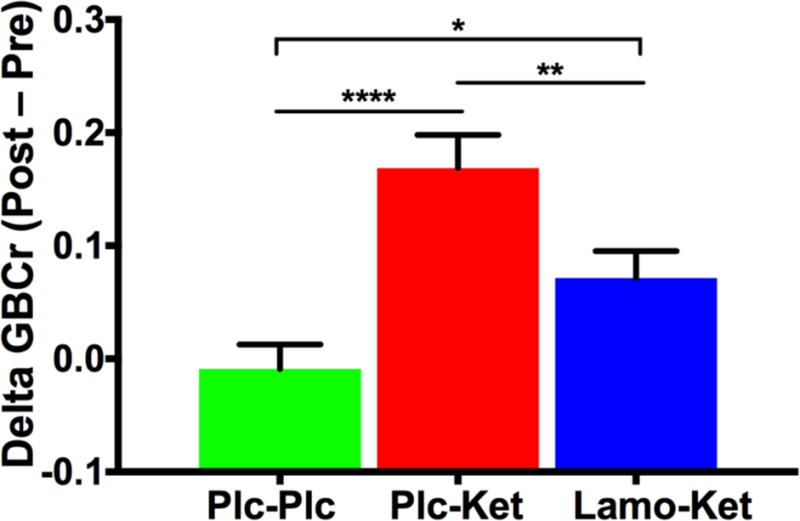
Lamotrigine significantly inhibited the ketamine-induced GBCr surge. Abbreviations: GBCr = Global Brain Connectivity with global signal regression; Plc = Placebo; Ket = ketamine; Lamo = lamotrigine; * p < 0.05; * p < 0.01; * p < 0.0001
Ketamine significantly increased BPRS (z = 3.5, p < 0.001) and CADSS scores (z = 3.7, p < 0.001). Pretreatment with lamotrigine had no significant effects on the ketamine-induced increases in BPRS (z = 1, p = 0.29) and CADSS scores (z = 0, p = 1). Similarly, follow-up analysis showed ketamine-induced increases in BPRS Positive (z = 2.8, p = 0.005) and Negative symptoms (z = 3.2, p = 0.001), but there were no effects of lamotrigine pretreatment (all p > 0.1).
PFC Limbic Network
The PFC limbic area plays a critical role in the pathophysiology of depression. In addition, previous reports associated this brain area with TRD and ketamine infusion (16, 35, 42). Therefore, we conducted a follow-up region of interest (ROI) analysis examining the average GBCr in the vPFC (Fig. 4A). We found significantly higher vPFC GBCr in TRD compared to HC (t = 2.4, p = 0.02; Fig. 4B) and the number of previous treatment failures as measured by ATHF positively correlated with vPFC GBCr (r = 0.47, p = 0.03; Fig. 5A). However, ketamine treatment did not significantly reduce vPFC GBCr 24h post-treatment in TRD subjects (t = −1.3, p = 0.2; Fig. 4C). In healthy subjects, ketamine produced a robust reduction in vPFC GBCr during infusion (t = −4.1, p = 0.001; Fig. 4D). Following pretreatment with lamotrigine, ketamine showed no significant effects on vPFC GBCr (t = −1.4, p = 0.17).
Figure 4. Prefrontal Limbic Connectivity.
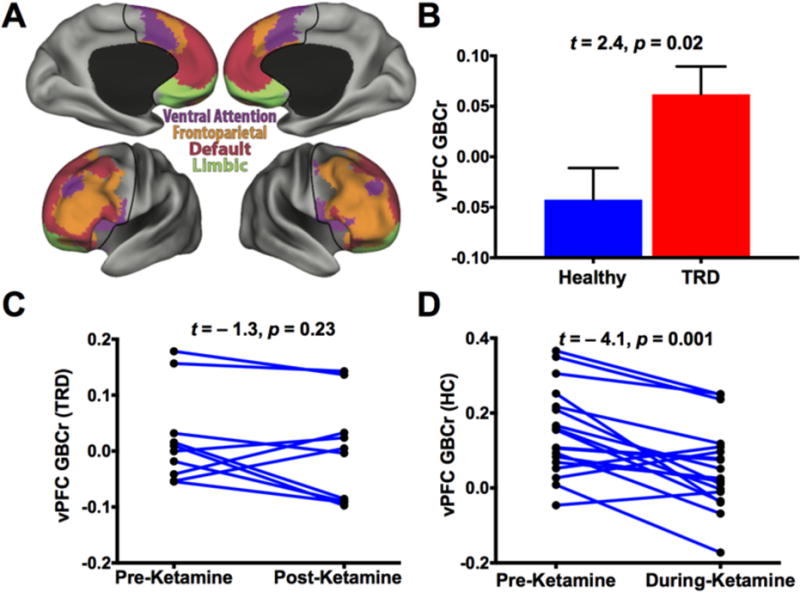
(A) Labels of the 4 major subnetworks within the prefrontal cortex (PFC). The ventral PFC (vPFC) area is colored with green. (B) TRD group had higher GBCr in the vPFC than the control group. (C) GBCr was numerically reduced in 7 of 10 TRD patients 24h post-ketamine treatment, but this reduction was not statistically significant. (D) In healthy subjects, ketamine significantly reduced vPFC GBCr during infusion. Abbreviations: GBCr = Global Brain Connectivity with global signal regression; TRD = Treatment-Resistant Depression;
Figure 5. Association Between Depression Treatment and Function Connectivity.
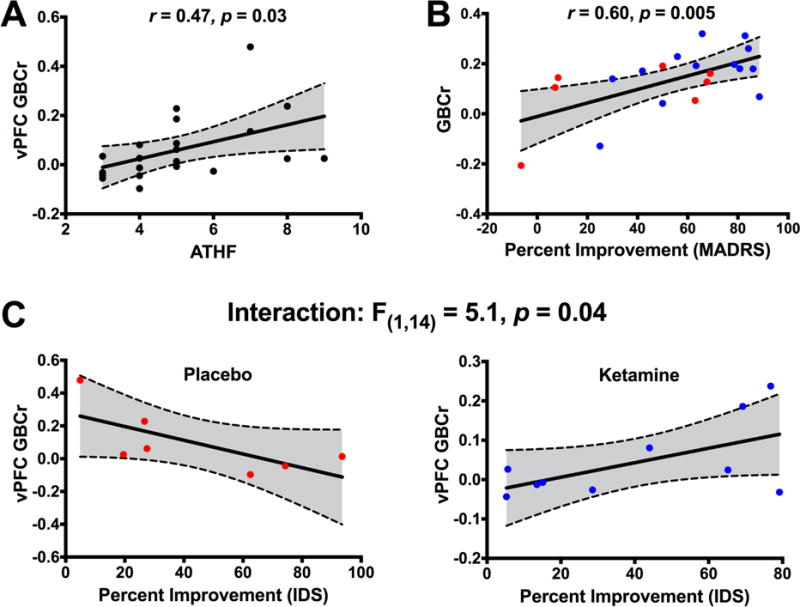
(A) Correlation between ventral prefrontal cortex (vPFC) GBCr and the number of treatment failures as measured by the Antidepressant Treatment History Form (ATHF). (B) Correlation between improvement in depression severity and average baseline GBCr in the altered clusters. (C) The relationship between improvement in depression severity and baseline vPFC GBCr. Abbreviations: GBCr = Global Brain Connectivity with global signal regression; MADRS = Montgomery-Åsberg Depression Rating Scale; IDS = Inventory of Depressive Symptomatology.
GBCr & Treatment Response
Exploratory analyses were conducted to examine whether baseline GBCr predicted the antidepressant effects of ketamine or placebo. Regardless of treatment, baseline GBCr in the altered clusters significantly predicted improvement in MADRS (F(1,17) = 7.1; p = 0.02; Fig. 5B), but not improvement in IDS (F(1,14) = 2.5; p = 0.14). We found no significant interaction between GBCr and treatment (all p > 0.1). There were no significant correlations between GBCr changes and improvement (all p > 0.1). Using baseline vPFC GBCr, we found a significant vPFC GBCr by treatment interaction, which differentially predicted improvement in MADRS (F(1,17) = 4.2; p = 0.05) and IDS (F(1,14) = 5.1; p = 0.04; Fig. 5C).
Discussion
This study affirmed its primary objectives. We have confirmed the presence of reduced PFC GBCr in treatment resistant MDD patients. Comparable to our previous report in TRD, the GBCr reduction was evident in the Ventral Attention and Frontoparietal, but not the Limbic networks within the PFC. Using a set of pharmacological challenges, we have demonstrated that the inhibition of glutamate transmission reduces mPFC GBCr. Similarly, the previously shown ketamine-induced glutamate increases in the mPFC (25, 26, 28) were paralleled by robust ketamine-induced increases in mPFC GBCr, which we interpret as putative evidence of an association between increased cortical glutamate transmission and GBCr. Further supporting a direct link between glutamate neurotransmission and GBCr, we have shown that the glutamate release inhibitor lamotrigine significantly reduces GBCr as well as the ketamine-induced GBCr surge. Yet, it is noticeable that ketamine continued to induce a significant GBCr surge even following lamotrigine pretreatment, which could potentially explain the inability of lamotrigine 300 mg to block the antidepressant effects of ketamine in a previous trial (45). Future studies should investigate whether the PFC GBCr alterations at 24h post-treatment in TRD could serve as a biomarker of target validation, while the ketamine-induced PFC GBCr increases during infusion may serve as biomarker of target engagement.
The ROI analysis showed higher vPFC GBCr in TRD compared to HC. In addition, patients with a history of more severe treatment failures had higher levels of vPFC GBCr. Along with previous reports (16, 35), the current study findings associate vPFC GBCr increases with TRD as defined by failure to respond to adequate trials of monoaminergic drugs and traditional antidepressants. Intriguingly, subjects with more prominent baseline vPFC GBCr abnormalities showed an enhanced response to ketamine, but poor response to placebo. While ketamine did not significantly reduce GBCr 24h post-treatment in the TRD group, ketamine significantly reduced vPFC GBCr during infusion in the healthy group. The latter finding is consistent with a previous report in healthy subjects receiving a comparable dose of ketamine, which showed ketamine-induced reduction in vPFC activity during infusion (42). Therefore, we interpret this vPFC finding as evidence of an association between ketamine-induced reduction in neurotransmission and GBCr in the vPFC. The vPFC findings in TRD and during ketamine infusion are of great interest to the field reflecting the critical role of this region in the pathophysiology of depression (12).
Comparable to our previous reports, baseline severity of depression was not associated with the level of GBCr abnormalities (16, 35). Our current working model of depression (46, 47), which accounts for the latter finding, is that MDD patients could be stratified into two groups: Glutamate-Based Depression (GBD) and non-GBD. Thus, while both GBD and non-GBD patients may report severe symptoms, we speculate that certain GBCr abnormalities would be most pronounced in the GBD subgroup. If this hypothesis is confirmed in future studies, this MDD pathophysiological heterogeneity could explain the lack of correlation between connectivity alterations and behavioral reports of severity.
The GBD model is based on the synaptic hypothesis of depression which proposes that depression and chronic stress precipitate glutamate dysregulation leading to excitotoxicity and neuronal atrophy in the PFC and hippocampus (48, 49). The predictions of this preclinical hypothesis are that MDD patients will have reduced gray matter integrity and altered amino acid neurotransmitters in the PFC. While a plethora of data support these predictions, it is overwhelmingly clear that not all MDD patients have amino acid and gray matter abnormalities. In fact, accumulating evidence shows that these abnormalities are most prominent in a subgroup of patients who often present with anhedonia and/or treatment refractory symptoms (50–60). Using cortical levels of amino acid neurotransmitters, we have recently shown that, compared to non-GBD, the GBD group has abnormally low levels of mPFC glutamate and gamma-aminobutyric acid (GABA), and considerable reduction in hippocampal volume (46). We have also shown the limitation of stratifying patients into GBD based on their TRD status, compared to using brain biomarkers (46). Preliminary findings suggested that glutamate-based antidepressants may be particularly effective in patients with GBD (61, 62). More recently, we found that the non-GBD group has increased nucleus accumbens volume (47). In addition, ketamine treatment appeared to normalize gray matter abnormalities by increasing hippocampal, but reducing nucleus accumbens, volumes (47).
Finally, comparable to our previous study in depressed subjects (45), lamotrigine did not block the ketamine-induced psychotomimetic effects. This finding is in contrast to previous reports by our group (43) and others (42). We speculate that the larger dose of ketamine used in our previous study (43) may have afforded us the ability to detect the modulating effects of lamotrigine on the ketamine-induced psychotomimetic symptoms. Nonetheless, this inconsistency of the behavioral outcomes across studies further highlight the urgent need for robust biomarkers with high sensitivity to use as targets for drug development and optimization, rather than solely relying on behavioral outcomes in early exploratory phases of clinical trials.
The main limitation of the current study is the small number of TRD subjects who received ketamine and post-treatment scans. This may have affected our statistical power to detect a relationship between GBCr increases and improvement of depression, a finding that was evident in our previous cohort (16). In addition, the longitudinal GBCr assessment in Cohort A was constrained by the study design of the parent clinical trial, which used 2:1 randomization and the psychotropically active midazolam. Thus, our neuroimaging data lacked both an appropriate non-active placebo control (i.e., saline) and statistical power (i.e., only 7 midazolam subjects) to determine the specificity of the findings to ketamine using a time-by-treatment interaction analysis. Future studies randomizing patients to ketamine vs. saline in a 1:1 design would be required to discern the pharmacological, course of illness, and other non-specific effects. Another limitation is the lack of human evidence directly showing blockade of ketamine-induced glutamate neurotransmission by lamotrigine. Thus, our interpretation is based on extensive in vitro and preclinical evidence supporting the inhibition of glutamate release by lamotrigine. The glutamate release inhibition is believed to be the result of blocking sodium channels. However, other off-site effects have been related to lamotrigine, including weak blockade of other channels (e.g., calcium) and modulation of the monoaminergic system [for further details see (63, 64)]. Therefore, it is plausible that the observed GBCr modulation by lamotrigine could be affected by mechanisms other than glutamate neurotransmission.
Conclusion
The current report builds on the exciting discovery of ketamine-induced rapid acting antidepressant effects and the subsequent wealth of preclinical and clinical studies over the past decade dissecting the neurobiology of depression and the mechanisms of ketamine (1, 3, 4, 8, 11, 65). Capitalizing on the rapid and robust effects of ketamine, state-of-the-art network analyses and a pharmacoimaging design were employed to further delineate the role of prefrontal global dysconnectivity in the pathophysiology and treatment of depression. Finally, using interventional methods often implemented in basic science, the study attempted to provide evidence of a direct link between large scale networks and underlying molecular alterations, an approach that perhaps goes beyond circumstantial associations and begins to probe for causality.
Supplementary Material
Acknowledgments
The authors would like to thank the individuals who participated in these studies for their invaluable contribution. Funding support was provided by NIMH (K23MH101498; R01MH081870), the VA National Center for PTSD and a VA Research Career Development Award (DHM), Brain & Behavior Foundation (NARSAD), Patterson Trust Award, GlaxoSmithKline for an investigator initiated study (DHM), the Johnson Family Chair for Research, and the use of resources and facilities at the Michael E. Debakey VA Medical Center, Houston, Texas (VHA 5I01CX000994). The content is solely the responsibility of the authors and does not necessarily represent the official views of the sponsors, the Department of Veterans Affairs, NIH, or the U.S. Government.
Footnotes
Conflict of Interests
CGA has served as a consultant and/or on advisory boards for Genentech and Janssen, and on editorial for Sage Publications, Inc. JHK is a consultant for AbbVie, Inc., Amgen, Astellas Pharma Global Development, Inc., AstraZeneca Pharmaceuticals, Biomedisyn Corporation, Bristol-Myers Squibb, Eli Lilly and Company, Euthymics Bioscience, Inc., Neurovance, Inc., FORUM Pharmaceuticals, Janssen Research & Development, Lundbeck Research USA, Novartis Pharma AG, Otsuka America Pharmaceutical, Inc., Sage Therapeutics, Inc., Sunovion Pharmaceuticals, Inc., and Takeda Industries; is on the Scientific Advisory Board for Lohocla Research Corporation, Mnemosyne Pharmaceuticals, Inc., Naurex, Inc., and Pfizer; is a stockholder in Biohaven Medical Sciences; holds stock options in Mnemosyne Pharmaceuticals, Inc.; holds patents for Dopamine and Noradrenergic Reuptake Inhibitors in Treatment of Schizophrenia, U.S. Patent No. 5,447,948 (issued Sep 5, 1995), and Glutamate Modulating Agents in the Treatment of Mental Disorders, U.S. Patent No. 8,778,979 (issued Jul 15, 2014); and filed a patent for Intranasal Administration of Ketamine to Treat Depression. U.S. Application No. 14/197,767 (filed on Mar 5, 2014); U.S. application or Patent Cooperation Treaty international application No. 14/306,382 (filed on Jun 17, 2014). SJM: In the past 12 months, Dr. Mathew has received consulting fees from Acadia, Alkermes, Allergan, Cerecor, Otsuka, and Valeant, and serves on an Advisory Board for VistaGen Therapeutics. He has received research support from Janssen Research & Development. DHM is a consultant for Boehringer Ingelheim, Takeda, Alkermes, and Upsher-Smith. All other authors report no competing interests.
References
- 1.Ionescu DF, Rosenbaum JF, Alpert JE. Pharmacological approaches to the challenge of treatment-resistant depression. Dialogues Clin Neurosci. 2015;17:111–126. doi: 10.31887/DCNS.2015.17.2/dionescu. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Coplan JD, Gopinath S, Abdallah CG, Berry BR. A neurobiological hypothesis of treatment-resistant depression - mechanisms for selective serotonin reuptake inhibitor non-efficacy. Frontiers in behavioral neuroscience. 2014;8:189. doi: 10.3389/fnbeh.2014.00189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Niciu MJ, Ionescu DF, Richards EM, Zarate CA., Jr Glutamate and its receptors in the pathophysiology and treatment of major depressive disorder. J Neural Transm (Vienna) 2014;121:907–924. doi: 10.1007/s00702-013-1130-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Murrough JW, Abdallah CG, Mathew S. Targeting glutamate signaling in depression: progress and prospects. Nature Reviews: Drug Discovery. 2017 doi: 10.1038/nrd.2017.16. In Press. [DOI] [PubMed] [Google Scholar]
- 5.Zarate CA, Jr, Singh JB, Carlson PJ, Brutsche NE, Ameli R, Luckenbaugh DA, et al. A randomized trial of an N-methyl-D-aspartate antagonist in treatment-resistant major depression. Arch Gen Psychiatry. 2006;63:856–864. doi: 10.1001/archpsyc.63.8.856. [DOI] [PubMed] [Google Scholar]
- 6.Murrough JW, Iosifescu DV, Chang LC, Al Jurdi RK, Green CE, Perez AM, et al. Antidepressant efficacy of ketamine in treatment-resistant major depression: a two-site randomized controlled trial. Am J Psychiatry. 2013;170:1134–1142. doi: 10.1176/appi.ajp.2013.13030392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Abdallah CG, Averill LA, Krystal JH. Ketamine as a promising prototype for a new generation of rapid-acting antidepressants. Ann N Y Acad Sci. 2015;1344:66–77. doi: 10.1111/nyas.12718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bobo WV, Voort JL, Croarkin PE, Leung JG, Tye SJ, Frye MA. Ketamine for Treatment-Resistant Unipolar and Bipolar Major Depression: Critical Review and Implications for Clinical Practice. Depress Anxiety. 2016 doi: 10.1002/da.22505. [DOI] [PubMed] [Google Scholar]
- 9.Zarate CA, Jr, Singh JB, Quiroz JA, De Jesus G, Denicoff KK, Luckenbaugh DA, et al. A double-blind, placebo-controlled study of memantine in the treatment of major depression. Am J Psychiatry. 2006;163:153–155. doi: 10.1176/appi.ajp.163.1.153. [DOI] [PubMed] [Google Scholar]
- 10.Sanacora G, Johnson MR, Khan A, Atkinson SD, Riesenberg RR, Schronen JP, et al. Adjunctive Lanicemine (AZD6765) in Patients with Major Depressive Disorder and History of Inadequate Response to Antidepressants: A Randomized, Placebo-Controlled Study. Neuropsychopharmacology. 2016 doi: 10.1038/npp.2016.224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Newport DJ. Carpenter LL, McDonald WM, Potash JB, Tohen M, Nemeroff CB, et al. Ketamine and Other NMDA Antagonists: Early Clinical Trials and Possible Mechanisms in Depression. Am J Psychiatry. 2015;172:950–966. doi: 10.1176/appi.ajp.2015.15040465. [DOI] [PubMed] [Google Scholar]
- 12.Dunlop BW, Mayberg HS. Neuroimaging-based biomarkers for treatment selection in major depressive disorder. Dialogues Clin Neurosci. 2014;16:479–490. doi: 10.31887/DCNS.2014.16.4/bdunlop. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Insel TR, Sahakian BJ, Voon V, Nye J, Brown VJ, Altevogt BM, et al. Drug research: a plan for mental illness. Nature. 2012;483:269–269. doi: 10.1038/483269a. [DOI] [PubMed] [Google Scholar]
- 14.Smart OL, Tiruvadi VR, Mayberg HS. Multimodal approaches to define network oscillations in depression. Biol Psychiatry. 2015;77:1061–1070. doi: 10.1016/j.biopsych.2015.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cole MW, Anticevic A, Repovs G, Barch D. Variable global dysconnectivity and individual differences in schizophrenia. Biol Psychiatry. 2011;70:43–50. doi: 10.1016/j.biopsych.2011.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Abdallah CG, Averill LA, Collins KA, Geha P, Schwartz J, Averill C, et al. Ketamine Treatment and Global Brain Connectivity in Major Depression. Neuropsychopharmacology. 2016 doi: 10.1038/npp.2016.186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Abdallah CG, Sanacora G, Duman RS, Krystal JH. Ketamine and rapid-acting antidepressants: a window into a new neurobiology for mood disorder therapeutics. Annu Rev Med. 2015;66:509–523. doi: 10.1146/annurev-med-053013-062946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Niciu MJ, Henter ID, Luckenbaugh DA, Zarate CA, Jr, Charney DS. Glutamate receptor antagonists as fast-acting therapeutic alternatives for the treatment of depression: ketamine and other compounds. Annu Rev Pharmacol Toxicol. 2014;54:119–139. doi: 10.1146/annurev-pharmtox-011613-135950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li N, Lee B, Liu RJ, Banasr M, Dwyer JM, Iwata M, et al. mTOR-dependent synapse formation underlies the rapid antidepressant effects of NMDA antagonists. Science. 2010;329:959–964. doi: 10.1126/science.1190287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zanos P, Moaddel R, Morris PJ, Georgiou P, Fischell J, Elmer GI, et al. NMDAR inhibition-independent antidepressant actions of ketamine metabolites. Nature. 2016;533:481–486. doi: 10.1038/nature17998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Homayoun H, Moghaddam B. NMDA receptor hypofunction produces opposite effects on prefrontal cortex interneurons and pyramidal neurons. J Neurosci. 2007;27:11496–11500. doi: 10.1523/JNEUROSCI.2213-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Neymotin SA, Lazarewicz MT, Sherif M, Contreras D, Finkel LH, Lytton WW. Ketamine disrupts theta modulation of gamma in a computer model of hippocampus. J Neurosci. 2011;31:11733–11743. doi: 10.1523/JNEUROSCI.0501-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Abdallah CG. What’s the Buzz About Hydroxynorketamine? Is It the History, the Story, the Debate, or the Promise? Biol Psychiatry. 2017;81:e61–e63. doi: 10.1016/j.biopsych.2017.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chowdhury GM, Zhang J, Thomas M, Banasr M, Ma X, Pittman B, et al. Transiently increased glutamate cycling in rat PFC is associated with rapid onset of antidepressant-like effects. Mol Psychiatry. 2016 doi: 10.1038/mp.2016.34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rowland LM, Bustillo JR, Mullins PG, Jung RE, Lenroot R, Landgraf E, et al. Effects of ketamine on anterior cingulate glutamate metabolism in healthy humans: a 4-T proton MRS study. Am J Psychiatry. 2005;162:394–396. doi: 10.1176/appi.ajp.162.2.394. [DOI] [PubMed] [Google Scholar]
- 26.Stone JM, Dietrich C, Edden R, Mehta MA, De Simoni S, Reed LJ, et al. Ketamine effects on brain GABA and glutamate levels with lH-MRS: relationship to ketamine-induced psychopathology. Mol Psychiatry. 2012;17:664–665. doi: 10.1038/mp.2011.171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Taylor MJ, Tiangga ER, Mhuircheartaigh RN, Cowen PJ. Lack of effect of ketamine on cortical glutamate and glutamine in healthy volunteers: a proton magnetic resonance spectroscopy study. J Psychopharmacol. 2012;26:733–737. doi: 10.1177/0269881111405359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Milak MS, Proper CJ, Mulhern ST, Parter AL, Kegeles LS, Ogden RT, et al. A pilot in vivo proton magnetic resonance spectroscopy study of amino acid neurotransmitter response to ketamine treatment of major depressive disorder. Mol Psychiatry. 2016;21:320–327. doi: 10.1038/mp.2015.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Valentine GW, Mason GF, Gomez R, Fasula M, Watzl J, Pittman B, et al. The antidepressant effect of ketamine is not associated with changes in occipital amino acid neurotransmitter content as measured by [(l)H]-MRS. Psychiatry Res. 2011;191:122–127. doi: 10.1016/j.pscychresns.2010.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cole MW, Pathak S, Schneider W. Identifying the brain’s most globally connected regions. NeuroImage. 2010;49:3132–3148. doi: 10.1016/j.neuroimage.2009.11.001. [DOI] [PubMed] [Google Scholar]
- 31.Cole MW, Yarkoni T, Repovs G, Anticevic A, Braver TS. Global connectivity of prefrontal cortex predicts cognitive control and intelligence. J Neurosci. 2012;32:8988–8999. doi: 10.1523/JNEUROSCI.0536-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Liang X, Connelly A, Calamante F. Graph analysis of resting-state ASL perfusion MRI data: nonlinear correlations among CBF and network metrics. Neuroimage. 2014;87:265–275. doi: 10.1016/j.neuroimage.2013.11.013. [DOI] [PubMed] [Google Scholar]
- 33.Liang X, Zou Q, He Y, Yang Y. Coupling of functional connectivity and regional cerebral blood flow reveals a physiological basis for network hubs of the human brain. Proc Natl Acad Sci U S A. 2013;110:1929–1934. doi: 10.1073/pnas.1214900110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Abdallah CG, Wrocklage KM, Averill CL, Akiki T, Schweinsburg B, Roy A, et al. Anterior hippocampal dysconnectivity in posttraumatic stress disorder: a dimensional and multimodal approach. Translational psychiatry. 2017;7:el045. doi: 10.1038/tp.2017.12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Murrough JW, Abdallah CG, Anticevic A, Collins KA, Geha P, Averill LA, et al. Reduced global functional connectivity of the medial prefrontal cortex in major depressive disorder. Hum Brain Mapp. 2016 doi: 10.1002/hbm.23235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Anticevic A, Brumbaugh MS, Winkler AM, Lombardo LE, Barrett J, Corlett PR, et al. Global prefrontal and fronto-amygdala dysconnectivity in bipolar I disorder with psychosis history. Biol Psychiatry. 2013;73:565–573. doi: 10.1016/j.biopsych.2012.07.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Anticevic A, Hu S, Zhang S, Savic A, Billingslea E, Wasylink S, et al. Global resting-state functional magnetic resonance imaging analysis identifies frontal cortex, striatal, and cerebellar dysconnectivity in obsessive-compulsive disorder. Biol Psychiatry. 2014;75:595–605. doi: 10.1016/j.biopsych.2013.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Anticevic A, Corlett PR, Cole MW, Savic A, Gancsos M, Tang Y, et al. N-methyl-D-aspartate receptor antagonist effects on prefrontal cortical connectivity better model early than chronic schizophrenia. Biol Psychiatry. 2015;77:569–580. doi: 10.1016/j.biopsych.2014.07.022. [DOI] [PubMed] [Google Scholar]
- 39.Wang L, Dai Z, Peng H, Tan L, Ding Y, He Z, et al. Overlapping and segregated resting-state functional connectivity in patients with major depressive disorder with and without childhood neglect. Hum Brain Mapp. 2014;35:1154–1166. doi: 10.1002/hbm.22241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Li N, Liu RJ, Dwyer JM, Banasr M, Lee B, Son H, et al. Glutamate N-methyl-D-aspartate receptor antagonists rapidly reverse behavioral and synaptic deficits caused by chronic stress exposure. Biol Psychiatry. 2011;69:754–761. doi: 10.1016/j.biopsych.2010.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yeo BT, Krienen FM, Sepulcre J, Sabuncu MR, Lashkari D, Hollinshead M, et al. The organization of the human cerebral cortex estimated by intrinsic functional connectivity. J Neurophysiol. 2011;106:1125–1165. doi: 10.1152/jn.00338.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Deakin JF, Lees J, McKie S, Hallak JE, Williams SR, Dursun SM. Glutamate and the neural basis of the subjective effects of ketamine: a pharmaco-magnetic resonance imaging study. Arch Gen Psychiatry. 2008;65:154–164. doi: 10.1001/archgenpsychiatry.2007.37. [DOI] [PubMed] [Google Scholar]
- 43.Anand A, Charney DS, Oren DA, Berman RM, Hu XS, Cappiello A, et al. Attenuation of the neuropsychiatrie effects of ketamine with lamotrigine: support for hyperglutamatergic effects of N-methyl-D-aspartate receptor antagonists. Arch Gen Psychiatry. 2000;57:270–276. doi: 10.1001/archpsyc.57.3.270. [DOI] [PubMed] [Google Scholar]
- 44.Winkler AM, Ridgway GR, Webster MA, Smith SM, Nichols TE. Permutation inference for the general linear model. Neuroimage. 2014;92:381–397. doi: 10.1016/j.neuroimage.2014.01.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mathew SJ, Murrough JW, aan het Rot M, Collins KA, Reich DL, Charney DS. Riluzole for relapse prevention following intravenous ketamine in treatment-resistant depression: a pilot randomized, placebo-controlled continuation trial. Int J Neuropsychopharmacol. 2010;13:71–82. doi: 10.1017/S1461145709000169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Abdallah CG, Jaekowski A, Sato JR, Mao X, Kang G, Cheema R, et al. Prefrontal cortical GABA abnormalities are associated with reduced hippocampal volume in major depressive disorder. Eur Neuropsychopharmacol. 2015;25:1082–1090. doi: 10.1016/j.euroneuro.2015.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Abdallah CG, Jackowski A, Salas R, Gupta S, Sato JR, Mao X, et al. The Nucleus Accumbens and Ketamine Treatment in Major Depressive Disorder. Neuropsychopharmacology. 2017 doi: 10.1038/npp.2017.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.McEwen BS, Bowles NP, Gray JD, Hill MN, Hunter RG, Karatsoreos IN, et al. Mechanisms of stress in the brain. Nat Neurosci. 2015;18:1353–1363. doi: 10.1038/nn.4086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Popoli M, Yan Z, McEwen BS, Sanacora G. The stressed synapse: the impact of stress and glucocorticoids on glutamate transmission. Nat Rev Neurosci. 2012;13:22–37. doi: 10.1038/nrn3138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gabbay V, Mao X, Klein RG, Ely BA, Babb JS, Panzer AM, et al. Anterior cingulate cortex gamma-aminobutyric acid in depressed adolescents: relationship to anhedonia. Arch Gen Psychiatry. 2012;69:139–149. doi: 10.1001/archgenpsychiatry.2011.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Petty F, Kramer GL, Gullion CM, Rush AJ. Low plasma gamma-aminobutyric acid levels in male patients with depression. Biol Psychiatry. 1992;32:354–363. doi: 10.1016/0006-3223(92)90039-3. [DOI] [PubMed] [Google Scholar]
- 52.Roy A, Dejong J, Ferraro T. CSF GABA in depressed patients and normal controls. Psychol Med. 1991;21:613–618. doi: 10.1017/s0033291700022248. [DOI] [PubMed] [Google Scholar]
- 53.Sanacora G, Gueorguieva R, Epperson CN, Wu YT, Appel M, Rothman DL, et al. Subtype-specific alterations of gamma-aminobutyric acid and glutamate in patients with major depression. Arch Gen Psychiatry. 2004;61:705–713. doi: 10.1001/archpsyc.61.7.705. [DOI] [PubMed] [Google Scholar]
- 54.Levinson AJ, Fitzgerald PB, Favalli G, Blumberger DM, Daigle M, Daskalakis ZJ. Evidence of cortical inhibitory deficits in major depressive disorder. Biol Psychiatry. 2010;67:458–464. doi: 10.1016/j.biopsych.2009.09.025. [DOI] [PubMed] [Google Scholar]
- 55.Zhang C, Li Z, Wu Z, Chen J, Wang Z, Peng D, et al. A study of N-methyl-D-aspartate receptor gene (GRIN2B) variants as predictors of treatment-resistant major depression. Psychopharmacology (Berl) 2013 doi: 10.1007/s00213-013-3297-0. [DOI] [PubMed] [Google Scholar]
- 56.Merkl A, Schubert F, Quante A, Luborzewski A, Brakemeier EL, Grimm S, et al. Abnormal cingulate and prefrontal cortical neurochemistry in major depression after electroconvulsive therapy. Biol Psychiatry. 2011;69:772–779. doi: 10.1016/j.biopsych.2010.08.009. [DOI] [PubMed] [Google Scholar]
- 57.Grimm S, Luborzewski A, Schubert F, Merkl A, Kronenberg G, Colla M, et al. Region-specific glutamate changes in patients with unipolar depression. J Psychiatr Res. 2012;46:1059–1065. doi: 10.1016/j.jpsychires.2012.04.018. [DOI] [PubMed] [Google Scholar]
- 58.Portella MJ, de Diego-Adelino J, Gomez-Anson B, Morgan-Ferrando R, Vives Y, Puigdemont D, et al. Ventromedial prefrontal spectroscopic abnormalities over the course of depression: a comparison among first episode, remitted recurrent and chronic patients. J Psychiatr Res. 2011;45:427–434. doi: 10.1016/j.jpsychires.2010.08.010. [DOI] [PubMed] [Google Scholar]
- 59.de Diego-Adelino J, Portella MJ, Gomez-Anson B, Lopez-Moruelo O, Serra-Blasco M, Vives Y, et al. Hippocampal abnormalities of glutamate/glutamine, N-acetylaspartate and choline in patients with depression are related to past illness burden. Journal of psychiatry & neuroscience : JPN. 2013;38:107–116. doi: 10.1503/jpn.110185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zhang J, Narr KL, Woods RP, Phillips OR, Alger JR, Espinoza RT. Glutamate normalization with ECT treatment response in major depression. Mol Psychiatry. 2013;18:268–270. doi: 10.1038/mp.2012.46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Abdallah CG, Coplan JD, Jackowski A, Sato JR, Mao X, Shungu DC, et al. A pilot study of hippocampal volume and N-acetylaspartate (NAA) as response biomarkers in riluzole-treated patients with GAD. Eur Neuropsychopharmacol. 2013;23:276–284. doi: 10.1016/j.euroneuro.2012.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Abdallah CG, Salas R, Jackowski A, Baldwin P, Sato JR, Mathew SJ. Hippocampal volume and the rapid antidepressant effect of ketamine. J Psychopharmacol. 2015;29:591–595. doi: 10.1177/0269881114544776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ketter TA, Manji HK, Post RM. Potential mechanisms of action of lamotrigine in the treatment of bipolar disorders. J Clin Psychopharmacol. 2003;23:484–495. doi: 10.1097/01.jcp.0000088915.02635.e8. [DOI] [PubMed] [Google Scholar]
- 64.Cunningham MO, Jones RS. The anticonvulsant, lamotrigine decreases spontaneous glutamate release but increases spontaneous GABA release in the rat entorhinal cortex in vitro. Neuropharmacology. 2000;39:2139–2146. doi: 10.1016/s0028-3908(00)00051-4. [DOI] [PubMed] [Google Scholar]
- 65.Abdallah CG, Adams TG, Kelmendi B, Esterlis I, Sanacora G, Krystal JH. Ketamine’s Mechanism of Action: A Path to Rapid-Acting Antidepressants. Depress Anxiety. 2016 doi: 10.1002/da.22501. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.