

Subgenome differences in levels of transposon methylation mirror increases in subgenome-specific expression bias observed over generations following hybridization and allopolyploidization in Mimulus.
Abstract
Recent studies have shown that one of the parental subgenomes in ancient polyploids is generally more dominant, having retained more genes and being more highly expressed, a phenomenon termed subgenome dominance. The genomic features that determine how quickly and which subgenome dominates within a newly formed polyploid remain poorly understood. To investigate the rate of emergence of subgenome dominance, we examined gene expression, gene methylation, and transposable element (TE) methylation in a natural, <140-year-old allopolyploid (Mimulus peregrinus), a resynthesized interspecies triploid hybrid (M. robertsii), a resynthesized allopolyploid (M. peregrinus), and progenitor species (M. guttatus and M. luteus). We show that subgenome expression dominance occurs instantly following the hybridization of divergent genomes and significantly increases over generations. Additionally, CHH methylation levels are reduced in regions near genes and within TEs in the first-generation hybrid, intermediate in the resynthesized allopolyploid, and are repatterned differently between the dominant and recessive subgenomes in the natural allopolyploid. Subgenome differences in levels of TE methylation mirror the increase in expression bias observed over the generations following hybridization. These findings provide important insights into genomic and epigenomic shock that occurs following hybridization and polyploid events and may also contribute to uncovering the mechanistic basis of heterosis and subgenome dominance.
INTRODUCTION
Whole-genome duplications (WGDs) have been an important recurrent process throughout the evolutionary history of eukaryotes (McLysaght et al., 2002; Dehal and Boore, 2005; Otto, 2007), including having contributed to the origin of novel traits and shifts in net diversification rates (Levin, 1983; Wright et al., 1998; Crow and Wagner, 2006; Chao et al., 2013; Edger et al., 2015; Tank et al., 2015). WGDs are especially widespread across flowering plants (Cui et al., 2006; Vanneste et al., 2014; Soltis and Soltis, 2016), with both deep WGD events (all extant angiosperms share at least two events; Jiao et al., 2011) and a plethora of more recent events including those unique to our model system, Mimulus (Vallejo-Marín et al., 2015). Polyploids, species that have three or more complete sets of genomes, are grouped into two main categories: autopolyploids (WGD that occurred within a species) and allopolyploids (WGD coupled with an interspecific hybridization) (Ramsey and Schemske, 1998). Previous studies indicate that allopolyploids are more likely to persist and become ecologically established, a fact that has partially been attributed to heterosis due to transgressive gene expression and fixed heterozygosity (McLysaght et al., 2002; Crow and Wagner, 2006; Rapp et al., 2009; Barker et al., 2016). It is worth noting that this finding may be affected by the fact that it is often easier to identify allopolyploids than autopolyploids.
Newly formed allopolyploids face the unique challenge of organizing two genomes (i.e., subgenomes), each contributed by different parental species, that have independently evolved in separate contexts, but which now exist within a single nucleus (Comai, 2005). Homoploid hybridization and allopolyploidization may disrupt both genetic and epigenomic processes resulting in altered DNA methylation patterns (Shaked et al., 2001; Mittelsten Scheid et al., 2003; Salmon et al., 2005; Lukens et al., 2006; Chen, 2007; Song and Chen, 2015; Rigal et al., 2016), changes in gene expression (Adams et al., 2003; Adams and Wendel, 2005; Buggs et al., 2010; Chelaifa et al., 2010; Coate et al., 2014; Renny-Byfield et al., 2015, 2017) and transposable element (TE) reactivation (Dion-Côté et al., 2014), commonly referred to as genomic shock (McClintock, 1984). These genome-wide changes are associated with novel phenotypic variation in newly formed allopolyploids (Madlung et al., 2002; Gaeta et al., 2007), which likely contributed to the survival and ultimate success of polyploids (Kagale et al., 2014; Vanneste et al., 2014).
One observation that may be linked to the long-term success of allopolyploids is that homoeologous genes (orthologous genes encoded on different parental subgenomes) are often expressed at nonequal levels, with genome-wide expression abundance patterns being highly skewed toward one of the subgenomes. Examples of plants with evidence for subgenome-specific expression include maize (Zea mays; Schnable et al., 2011), Brassica (Cheng et al., 2016), cotton (Gossypium spp; Renny-Byfield et al., 2015), wheat (Triticum aestivum; Li et al., 2014), Tragopogon (Buggs et al., 2010), Spartina (Chelaifa et al., 2010), and Arabidopsis thaliana (Wang et al., 2004). Additionally, it has been shown that the less expressed subgenome tends to be more highly fractionated (i.e., accumulate more deletions), a pattern thought to be due to relaxed selective constraints. Collectively, these phenomena are referred to as “subgenome dominance” (Woodhouse et al., 2010). For example, in Brassica rapa, a three-way battle ensued following a whole-genome triplication event that occurred over ten million years ago resulting in a single dominant subgenome emerging and two highly fractionated subgenomes (Tang et al., 2012). The most highly fractionated subgenome lost more than double the total number of genes than the least fractionated, dominant subgenome.
It remains largely unknown how one subgenome becomes more highly expressed, with respect to either whole genome patterns or specific genes. Another unanswered question is, on what time scale (i.e., how quickly) does subgenome dominance become established? Subgenome dominance in newly formed hybrids and allopolyploids could have substantial implications for our understanding of plant hybridization in both ecological and agricultural contexts. In addition, a mechanistic understanding of these phenomena is fundamental to better understanding the long-term evolutionary advantages of WGDs. One hint at a mechanism may be that gene expression can be impacted by the proximity to and methylation status of nearby TEs (Hollister and Gaut, 2009). Prompted by the finding that the density of methylated TEs is negatively correlated with gene expression magnitude, Freeling et al. (2012) hypothesized that the relationship between TE repression and the expression of neighboring genes might explain patterns of observed subgenome dominance. The degree of methylation repatterning and reestablishment genome-wide, specifically nearby genes, following hybridization and/or WGD is largely unknown. Here, we tested this hypothesis by assessing (1) the overall rate that subgenome expression dominance is established following hybridization and WGD, (2) genome-wide methylation repatterning following hybridization and WGD, and (3) the influence of methylation repatterning on biased expression of parental subgenomes.
Research in most polyploid systems is hindered by at least one of two major difficulties: (1) lack of genomic resources for extant parental progenitors (if parents are known) or (2) the inability to confidently partition the polyploid genome to each of the parental subgenomes. Here, we used the recently and recurrently formed natural allopolyploid, Mimulus peregrinus, to overcome these hurdles. M. peregrinus (6x) is derived from the hybridization of M. luteus (4x) and M. guttatus (2x), which produced a sterile triploid intermediate M. x robertsii (3x) that underwent a subsequent WGD to regain fertility (Vallejo-Marín, 2012; Vallejo-Marín et al., 2015) (Figure 1). Importantly, M. luteus (native to Chile) and M. guttatus (native to Western North America) only recently came into contact following a documented introduction into the UK in the early 1800s (Vallejo-Marín et al., 2015). Thus, we have a narrow time window for the formation of M. peregrinus. Moreover, the natural allopolyploid (M. peregrinus) still exists with its introduced parents in the UK, which allows us to recreate hybrids and synthetic allopolyploids in the lab. Furthermore, the M. guttatus genome was recently published (Hellsten et al., 2013), and we complement this with a new genome assembly for M. luteus. M. luteus is an allopolyploid formed from two unknown diploid progenitors, which may be long extinct due to the uncertain age of the polyploid event. These resources and the unique natural history of M. peregrinus have provided an unprecedented opportunity to properly investigate patterns of homoeolog expression bias (measured by the ratio of expression of the homoeologs) across subgenomes in a neo-allopolyploid and its relation to DNA methylation and TE density differences.
Figure 1.

Whole-Genome Duplications in Mimulus and Related Species.
(A) Tree showing locations of whole-genome duplications (stars) on Lamiales phylogeny. Coalescence-based phylogeny of 96 single copy loci estimated in ASTRAL. Node labels represent bootstrap values for 100 replicates. Nodes with bootstrap values less than 80 were collapsed. Green stars are published WGD events not identified in this study. Red stars indicate events identified in this study. Blue stars represent uncertainty in the nature of either a single event with varying support for the timing of paralog coalescence or two individual events.
(B) to (E) Mimulus species used in this study. M. guttatus (2x) hybridized with M. luteus (4x) (C) to produce a sterile triploid M. robertsii (3x) (D), which underwent a subsequent whole-genome duplication giving rise to fertile natural allopolyploid M. peregrinus (6x) (E).
(F) Graphic showing chromosome complement of individuals ([B] to [E]) in the middle panel. The allotetraploid M. luteus has two distinct subgenomes; La and Lb represent the two distinct subgenomes.
RESULTS
M. luteus Genome Assembly
Here, we present the draft genome of M. luteus with a total genome size estimate of 640 to 680 Mb based on flow cytometry and kmer spectrum analysis (Vallejo-Marín, 2012). We sequenced the genome of an inbred line (EY7). The assembly contains 6439 scaffolds spanning 410 Mb with an N50 of 283 kb, representing roughly 60% of the genome, with gene content analyses supporting the recovery of nearly the entire gene space. A total of 46,855 protein-coding genes were annotated in M. luteus genome, which is nearly double the number of protein coding genes (26,718) previously annotated in the M. guttatus genome (430 Mb estimated; 300 Mb assembled) (Hellsten et al., 2013). This difference in gene content supports a tetraploid event unique to M. luteus, further supported by a 2:1 syntenic block ratio when compared with the M. guttatus genome, plus base chromosome number differences between these species (Vallejo-Marin, 2012). The vast majority of BUSCO (Benchmarking Universal Single-Copy Orthologs) (Simão et al., 2015) groups, 931 of 956 (97.4%), were identified in the M. luteus genome assembly with 837 duplicates. The high number of identified duplicates supports both the tetraploid event and assembly completeness of the genic regions from both subgenomes. We mapped all mate pair reads back to the M. luteus assembly to screen for possible chimeric contigs or scaffolds between the two subgenomes (La and Lb). No instance of chimeric fusions between subgenomes was identified in the assembly (Supplemental Figures 1 and 2), which was largely enabled by the overall sequence divergence of the two subgenomes (genome-wide average = 89.33% sequence similarity across genes) and leveraging overlapping paired-end sequences as single long reads. We reannotated the M. guttatus genome with identical methods used for M. luteus, reducing the total number to 25,465 protein coding genes. The reannotation of this genome permits us to make proper genomic and transcriptomic comparisons by removing artifacts that arise due to differences in genome annotation pipelines. A total of 319,944 and 451,448 TEs or TE fragments were annotated in the M. guttatus and M. luteus genomes, respectively. To annotate TEs, the TE exemplar library (see Methods) was used to identify TEs in the M. guttatus and M. luteus genomes using RepeatMasker. We combined these two genomes to represent M. peregrinus.
History of WGD in Mimulus
A shared ancient whole-genome duplication was detected in both genomes, termed Mimulus-alpha, with a mean Ks of 0.92 and phylogenetically placed at the most recent common ancestor of Mimulus (Phrymaceae) and nearly all other Lamiales families (Figure 1; Supplemental Figures 3 to 5). Support was found at the base of the species tree for two possible WGD events: one prior to the diversification of all sampled Lamiales species and one after the divergence of Oleaceae. The PUG algorithm estimates the timing of WGD by identifying when coalescence of paralogs occurs in the context of a species tree. It is possible that the putative event placed after the divergence of Oleaceae is the result of artifactual gene tree reconciliation to the species tree. Another possibility is the event after the divergence of Oleaceae is the result of hybridization between the Oleaceae and the rest of Lamiales as suggested by Julca et al. (2017). Follow-up studies are needed to verify these findings with additional species sampling. A recent mean-date estimate for that phylogenetic node is 71 million years before present (Magallón et al., 2015), while other studies have obtained earlier and later date estimates (Tank et al., 2015; Wikström et al., 2015). Our taxon sampling did not include the earliest diverging lineage (family Plocospermataceae) in Lamiales (Refulio-Rodriguez and Olmstead, 2014; Magallón et al., 2015; Stull et al., 2015). The Mimulus-alpha event is shared by all other surveyed Lamiales families. A total of 757 unique shared duplications from Mimulus-alpha were identified in all surveyed taxa. Two additional duplication events detected across Lamiales were not shared with Phrymaceae. The first detected event was shared by Orobanche (Orobanchaceae) and Striga (Oleaceae) and was supported by 1738 shared duplicate pairs. The second WGD event was shared by Olea and Fraxinus and supported by 3312 shared duplicate pairs.
M. luteus experienced an additional whole genome duplication event not shared with M. guttatus. As a result, for every M. guttatus gene, M. luteus typically has two corresponding homoeologs encoded on subgenomes (La and Lb). The mean per base divergence at neutral sites between the two M. luteus subgenomes is ∼0.11 Ks (synonymous substitutions per synonymous site). This level of divergence supports previous claim that this species is an allopolyploid (Mukherjee and Vickery, 1962) and by our previous genomic analysis, which suggested an allopolyploid origin for this taxon (Vallejo-Marín et al., 2015, 2016). Over 50 years ago, Mukherjee and Vickery (1962) hypothesized that M. luteus may be an allopolyploid formed by the hybridization of two distinct species with one more closely related to M. guttatus than the other species (Mukherjee and Vickery, 1962). We sought to test this hypothesis with phylogenetic analyses of syntenic orthologs between M. guttatus and from both M. luteus subgenomes. We determine that in the majority of cases (1853 of 2200 gene trees), homoeologs encoded on subgenome A were more closely related to M. guttatus than it was to other subgenome supporting their hypothesis. However, it is important to note the M. luteus subgenomes are both quite distinct from M. guttatus at the sequence level (genome-wide average = 91.52% sequence similarity across genes). This suggests that the diploid progenitor species of either subgenome is likely not a close relative of M. guttatus, but rather a more distantly related species in the past. Thus, the newly formed allopolyploid M. peregrinus consists of three unique subgenomes; two from M. luteus (La and Lb) and one from M. guttatus.
Investigating the Establishment of Subgenome Dominance
The goal of this study was to determine whether homoeologs from a given subgenome are expressed at higher levels than homoeologs from another subgenome. This goal guided our sampling strategy and analysis approach. Specifically, we sought to determine whether one homoeolog was the dominantly expressed form over multiple tissue types (our goal is not to determine tissue-specific expression bias).
Toward this end, for each individual represented in this study (parents, hybrid, and natural allopolyploids), three separate RNA-seq data sets derived from three different tissues were generated. RNA-seq data sets for calyx, stem, and petals were generated for the F1 hybrid M. x robertsii (M. guttatus x M. luteus), the resynthesized allopolyploid M. peregrinus, and the naturally derived allopolyploid M. peregrinus and parental taxa, M. luteus and M. guttatus. For each tissue type, we calculated the expression ratios of homoeologs encoded on separate subgenomes. Thus, our three biological replicates were the expression ratio of homoeologs across three tissues. These data were used to measure homoeolog-specific gene expression following the M. luteus WGD event as well as in a contemporary hybrid and neo-allopolyploids (M. peregrinus). All RNA samples were collected within a narrow time range to control for major diurnal rhythmic expression differences. In hybrids and allopolyploids, subgenome-specific (parental) single-nucleotide polymorphisms were identified and used to measure homoeolog-specific gene expression. For the analysis of our RNA-seq data, we developed a likelihood ratio test (LRT) involving three nested hypotheses to identify cases of homoeolog expression bias that do not involve tissue-specific expression differences. The null hypothesis is that both homoeologs are expressed at equal levels (ratio of homoeolog-1 to homoeolog-2 equals 1 for all three tissues). The first alternate hypothesis is that homoeologs are expressed at different levels, but similar ratios, across all three tissue types. The second alternate hypothesis is that homoeologs are expressed at different levels and at different ratios across all three tissues.
Using the expression data and the nested hypotheses we test for (1) expression bias and subgenome dominance following the M. luteus-specific WGD, comparing La and Lb homoeolog expression, in M. luteus, hybrid M. guttatus x M. luteus, and M. peregrinus (both natural and resynthesized allopolyploid) and (2) expression bias and subgenome dominance of the M. guttatus or M. luteus homoeologs in the hybrid and allopolyploid M. peregrinus. Importantly, the first comparison will allow us to understand long-term patterns of expression bias in Mimulus (the M. luteus WGD is a relatively ancient event), whereas the second comparison will test for expression bias and subgenome dominance in a newly formed hybrid and allopolyploid Mimulus.
Expression Bias of Homoeologs from M. luteus-Specific WGD Event
We sought to compare expression of homoeologs within M. luteus to each other, thereby addressing the question of whether La homoeologs or Lb homoeologs are more highly expressed within M. luteus, M. x robertsii (F1 hybrid), or M. peregrinus (both a resynthesized and natural allopolyploid). Using the likelihood ratio test mentioned above, we identified cases of homoeolog expression bias that did not involve tissue-specific expression differences. In each case, over 1100 homoeolog pairs were tested; the test was only applied when both homoeologs were expressed in all three tissues. For each homoeolog pair, we quantified expression bias as
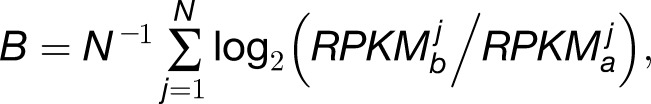 |
where the subscripts a and b denote the two distinct subgenomes (La and Lb), and j is an index over the N tissues. An expression bias of 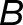 = −2 indicates a 4x expression bias toward the “a” homoeolog, while an expression bias of
= −2 indicates a 4x expression bias toward the “a” homoeolog, while an expression bias of 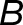 = 3 is 8x toward the “b” homoeolog. The histograms in Figure 2 summarize the measured expression bias of homoeologs resulting from the M. luteus-specific WGD event in M. luteus, F1 hybrid, synthetic allopolyploid, and natural allopolyploid. For M. luteus (top panel), the gray histogram shows the distribution of expression bias for all testable homoeolog pairs indicating a slight average bias toward the La subgenome (
= 3 is 8x toward the “b” homoeolog. The histograms in Figure 2 summarize the measured expression bias of homoeologs resulting from the M. luteus-specific WGD event in M. luteus, F1 hybrid, synthetic allopolyploid, and natural allopolyploid. For M. luteus (top panel), the gray histogram shows the distribution of expression bias for all testable homoeolog pairs indicating a slight average bias toward the La subgenome (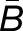 = −0.09, NLa = 388 > 336 = NLb) (Figure 2). Using the likelihood ratio test developed in this article, we found that over half of the homoeolog pairs in M. luteus (NLa + NLb = 724, ∼53% of the total N) were biased toward one of the subgenomes with no tissue-specific expression differences (hypothesis one; see Methods). Of these, a small majority of homoeologs (NLa = 388, ∼54% of biased homoeologs) were dominantly expressed from the La subgenome. In contrast to M. luteus, in the hybrid, synthetic allopolyploid, and natural allopolyploid, the Lb subgenome is slightly dominant in either number or average (see Figure 2, where ∼52, 51, and 53% of biased homoeologs are dominantly expressed from the Lb subgenome and
= −0.09, NLa = 388 > 336 = NLb) (Figure 2). Using the likelihood ratio test developed in this article, we found that over half of the homoeolog pairs in M. luteus (NLa + NLb = 724, ∼53% of the total N) were biased toward one of the subgenomes with no tissue-specific expression differences (hypothesis one; see Methods). Of these, a small majority of homoeologs (NLa = 388, ∼54% of biased homoeologs) were dominantly expressed from the La subgenome. In contrast to M. luteus, in the hybrid, synthetic allopolyploid, and natural allopolyploid, the Lb subgenome is slightly dominant in either number or average (see Figure 2, where ∼52, 51, and 53% of biased homoeologs are dominantly expressed from the Lb subgenome and 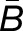 = 0.05, 0.01, and 0.02, respectively).
= 0.05, 0.01, and 0.02, respectively).
Figure 2.

Expression Bias of Homoeologs Resulting from M. luteus-Specific WGD Event in M. luteus, F1 Hybrid, Synthetic Allopolyploid, and Natural Allopolyploid.
Gray histograms show distribution of expression bias (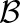 ) for all testable homoeolog pairs. Testable homoeolog pairs (N) are those that could clearly be identified as homologous and had at least 1 read in each tissue sampled. Homoeolog pairs significantly biased toward the M. guttatus-like homoeolog are crosshatched, while pairs significantly biased toward the “other” homoeolog are shown in solid blue. Across all three hybrid individuals (F1, synthetic, and natural allopolyploid) the “other” subgenome dominates the M. guttatus-like subgenome either by the number of homoeologs biased toward it (NLb > NLa) or on average,
) for all testable homoeolog pairs. Testable homoeolog pairs (N) are those that could clearly be identified as homologous and had at least 1 read in each tissue sampled. Homoeolog pairs significantly biased toward the M. guttatus-like homoeolog are crosshatched, while pairs significantly biased toward the “other” homoeolog are shown in solid blue. Across all three hybrid individuals (F1, synthetic, and natural allopolyploid) the “other” subgenome dominates the M. guttatus-like subgenome either by the number of homoeologs biased toward it (NLb > NLa) or on average, 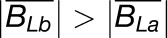 , where
, where  and
and 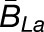 are averages over all homoeolog pairs that were biased toward Lb or La, respectively.
are averages over all homoeolog pairs that were biased toward Lb or La, respectively.
Expression Bias of M. guttatus and M. luteus Homoeologs in the Hybrid, Resynthesized Allopolyploid, and Naturally Occurring Neo-Allopolyploid Mimulus
To test for expression bias that arises instantaneously following the merger of two genomes, we compared homoeologs in the hybrid, resynthesized allopolyploid, and natural allopolyploid, which contain both a M. guttatus and M. luteus subgenome. We asked two questions. First, when considering M. luteus weighted average of its homoeologs compared with M. guttatus, do we see expression dominance toward either species (Figure 3)? Second, when we consider the M. luteus homoeologs separately (Lb or La) and compare these to their M. guttatus homoeolog (G), do we see expression dominance toward either species or a particular subgenome? The weighted average of expression of the two M. luteus homoeologs was calculated by dividing the sum of read count of the two M. luteus homoeologs by the sum of their individual gene lengths. The answer to the first question may indicate which parental species overall is more expressed, while the second question will shed light whether one single subgenome is most dominant. For each homoeolog pair, we quantified expression bias as
Figure 3.

Homoeolog Expression Bias in Hybrid and Allopolyploids, Comparing the M. guttatus Homoeolog to the Weighted Average Expression of Its Pair of M. luteus Homoeologs.
The weighted average of expression of the two M. luteus homoeologs was calculated by dividing the sum of read count of the two M. luteus homoeologs by the sum of their individual gene lengths. Gray histograms show distribution of expression bias (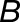 ) for all testable homoeolog pairs. Only genes that had a clear 2 to 1 (M. luteus to M. guttatus) homology were considered. Homoeolog pairs significantly biased toward the M. guttatus homoeolog are shown in yellow, while pairs significantly biased toward the M. luteus homoeolog are shown in blue. Across all three hybrid individuals (F1, synthetic, and natural allopolyploid) the pair of M. luteus homoeologs, when added together, dominates the M. guttatus homoeolog (i.e.,
) for all testable homoeolog pairs. Only genes that had a clear 2 to 1 (M. luteus to M. guttatus) homology were considered. Homoeolog pairs significantly biased toward the M. guttatus homoeolog are shown in yellow, while pairs significantly biased toward the M. luteus homoeolog are shown in blue. Across all three hybrid individuals (F1, synthetic, and natural allopolyploid) the pair of M. luteus homoeologs, when added together, dominates the M. guttatus homoeolog (i.e., 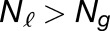 and
and 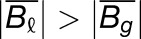 , where
, where 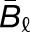 and
and 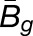 are averages over all homoeolog pairs).
are averages over all homoeolog pairs).
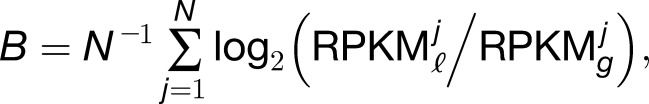 |
where the subscripts 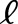 and
and 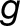 denote the M. luteus and M. guttatus subgenome, respectively, and j is an index over the N tissues. Considering the M. luteus homoeologs separately allows us to control for additive expression levels.
denote the M. luteus and M. guttatus subgenome, respectively, and j is an index over the N tissues. Considering the M. luteus homoeologs separately allows us to control for additive expression levels.
The histograms in Figure 3 summarize the measured expression bias in hybrid and allopolyploids, comparing the expression of the M. guttatus homoeolog to the weighted average expression of its pair of M. luteus homoeologs. This weighted average of expression and is a proxy for the average expression activity of homoeolog pairs from the M. luteus genome. On average, there is considerable bias toward the M. luteus subgenome with 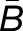 = 0.57, 0.51, and 1.05 in the first generation hybrid, synthetic allopolyploid, and natural allopolyploid, respectively. Using the LRT (at significance level α = 0.01), a total of 1893 (57%), 2072 (66%), and 2096 (70%) homoeolog pairs were found to be significantly biased. Of these biased pairs, the M. luteus homoeolog was dominant in the vast majority of cases (
= 0.57, 0.51, and 1.05 in the first generation hybrid, synthetic allopolyploid, and natural allopolyploid, respectively. Using the LRT (at significance level α = 0.01), a total of 1893 (57%), 2072 (66%), and 2096 (70%) homoeolog pairs were found to be significantly biased. Of these biased pairs, the M. luteus homoeolog was dominant in the vast majority of cases (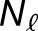 = 1330 (70%), 1397 (67%), and 1573 (75%), respectively).
= 1330 (70%), 1397 (67%), and 1573 (75%), respectively).
The histograms in Figure 4 summarize the measured expression bias in hybrid and allopolyploids, comparing the expression of the M. guttatus homoeolog to expression of each of its M. luteus homoeologs separately. That is, for each M. guttatus gene (G), there are two M. luteus homoeologs, La and Lb. Figure 4 includes the comparisons: G versus La and G versus Lb. In other words, each G is used in two different comparisons. When considering the M. luteus homoeologs separately, expression is still considerably biased on average toward M. luteus with 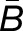 = 0.51, 0.48, and 1.00, in the first generation hybrid, resynthesized allopolyploid, and natural allopolyploid, respectively (Figure 4). Next, using the LRT, across all comparisons, 55, 64, and 69% homoeologs were significantly biased. Of these biased homoeolog pairs, the M. luteus homoeolog was the dominantly expressed homoeolog in 69, 65, and 74% of the comparisons (Figure 4). Additionally, among biased homoeologs, the average bias toward the M. luteus subgenome is greater than the average bias toward the M. guttatus subgenome
= 0.51, 0.48, and 1.00, in the first generation hybrid, resynthesized allopolyploid, and natural allopolyploid, respectively (Figure 4). Next, using the LRT, across all comparisons, 55, 64, and 69% homoeologs were significantly biased. Of these biased homoeolog pairs, the M. luteus homoeolog was the dominantly expressed homoeolog in 69, 65, and 74% of the comparisons (Figure 4). Additionally, among biased homoeologs, the average bias toward the M. luteus subgenome is greater than the average bias toward the M. guttatus subgenome 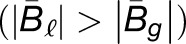 in all cases (Figure 4). It is also worth noting that the degree of bias (as measured by the fraction of biased homoeologs and B of biased homoeologs) increased from the first generation hybrid, to the resynthesized allopolyploid, and to the natural allopolyploid.
in all cases (Figure 4). It is also worth noting that the degree of bias (as measured by the fraction of biased homoeologs and B of biased homoeologs) increased from the first generation hybrid, to the resynthesized allopolyploid, and to the natural allopolyploid.
Figure 4.

Homoeolog Expression Bias in Hybrid and Allopolyploids, Comparing the M. guttatus Homoeolog to Each of Its M. luteus Homoeologs Separately.
Gray histograms show distribution of expression bias (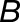 ) for all testable homoeolog pairs. Homoeolog pairs significantly biased toward the M. guttatus homoeolog are shown in yellow, while pairs significantly biased toward the M. luteus homoeolog are shown in blue. Across all three hybrid individuals (F1, synthetic, and natural allopolyploid) the M. luteus homoeolog dominates the M. guttatus homoeolog (i.e.,
) for all testable homoeolog pairs. Homoeolog pairs significantly biased toward the M. guttatus homoeolog are shown in yellow, while pairs significantly biased toward the M. luteus homoeolog are shown in blue. Across all three hybrid individuals (F1, synthetic, and natural allopolyploid) the M. luteus homoeolog dominates the M. guttatus homoeolog (i.e., 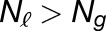 and
and 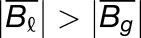 , where
, where 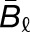 and
and 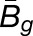 are averages over all homoeolog pairs).
are averages over all homoeolog pairs).
Expression Bias in Three Separate Hybrid Lineages
While it is clear that the M. luteus homoeologs are dominantly expressed in the hybrid and allopolyploid lineages, we sought to determine whether the same homoeologs were repeatedly biased across independent hybrid and allopolyploids. A Venn diagram reveals that homoeologs biased in one individual are far more likely to be biased in the other two lineages than would be expected by random chance (Figure 5). Moreover, measured levels of individual homoeolog expression bias are correlated across all three lineages (Figure 5). Interestingly, levels of expression bias B in the first generation hybrid and resynthesized allopolyploid are much more correlated with each other (r2 = 0.70) than either sample is with the natural allopolyploid (r2 = 0.35 and 0.33; Figure 5).
Figure 5.

Expression Bias in Three Separate Hybrid Lineages.
(A) Venn diagram of the number of biased homoeolog pairs across hybrid lineages (1532 homoeolog pairs were biased in all three lineages).
(B) to (D) Scatterplots of expression bias (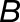 ) for these 1532 homoeolog pairs comparing hybrid to synthetic allopolyploid, hybrid to natural allopolyploid, and synthetic to natural allopolyploid (red line is linear regression; thin blue line is identity).
) for these 1532 homoeolog pairs comparing hybrid to synthetic allopolyploid, hybrid to natural allopolyploid, and synthetic to natural allopolyploid (red line is linear regression; thin blue line is identity).
Transposon Density Linked to Gene Expression
One possibility we considered was whether proximal transposon (TE) loads were related to homoeolog expression bias. In order to test this, it was necessary to annotate TEs in M. guttatus and M. luteus genome assemblies. Using a homology and structured based annotation, as well as de novo annotation, we identified the transposons in the M. guttatus and M. luteus genomes. Our survey revealed that 50% of the M. guttatus genome assembly is composed of TE sequences that are classified into 863 families. We compiled a TE exemplar library with 1439 sequences representing the TE composition of the genome. After annotating TEs in 10-kb windows (10 kb upstream and downstream of genes as well as within genic region), we calculated the total number of TEs and the number of TE bases. On average, M. luteus homoeologs and M. guttatus homoeologs have TE densities of 0.31 and 0.34 (fraction of bases that occur within a transposon), respectively. In the parents, hybrid, and allopolyploid individuals, this measure of proximal TE density is negatively correlated with gene expression (Figure 6; Supplemental Figures 6 and 7).
Figure 6.

TE Density in a Window Spanning 10 kb Upstream to 10 kb Downstream of a Gene Is Negatively Related to Gene Expression.
The vertical axis is gene expression in RPKM. The horizontal axis is transposon density, binned into 10 windows with width proportional to the number of data points it contains. Horizontal gray line indicates the mean of the response, log(RPKM). TE density is negatively related to gene expression in M. guttatus (A) and M. luteus (B).
Altered DNA Methylation Patterns in Parents, Hybrid, and Allopolyploids
Building on the finding of expression bias in the hybrid and neo-allopolyploids, we asked whether DNA methylation changes mirror the observed expression bias. First, whole-genome bisulfite sequencing was used to determine the methylation status and patterns of methylation change in hybrid and neo-allopolyploid lineages as well as in each parent at CHH (where H = C, A, T), CHG, and CG sites. Next, we tested the hypothesis that changes in TE methylation between parents and hybrid or allopolyploids may explain patterns of subgenome dominance.
Methylation patterns in TE and genes and their upstream and downstream regions were compared. CG and CHG methylation patterns in genes and TE are unchanged in the hybrid and allopolyploid lineages (Figure 7). CHG methylation levels are marginally lower in upstream and downstream regions of genes in the hybrid and slightly higher in the synthetic and natural allopolyploid. CHH methylation levels are decreased significantly in upstream and downstream genic regions in the first generation hybrid, decreased slightly in the resynthesized allopolyploid, and returned to parental levels in the natural allopolyploid. Similar to findings in genes, transposon bodies and up- and downstream regions of transposons are depleted in CHH methylation in the hybrid and synthetic allopolyploid (Figure 7; Supplemental Figure 8). Transposon CHH methylation levels are lowest in the first generation hybrid and remained at a reduced level in the resynthesized allopolyploid compared with parental levels. In the natural allopolyploid, CHH methylation of TEs across the M. guttatus subgenome returned to near parental levels while M. luteus subgenome TE methylation remained lower compared with parental levels. Methylation repatterning across the subgenomes closely reflects the pattern of homoeolog expression bias observed between the two subgenomes in the hybrid and allopolyploids.
Figure 7.

Subgenome-Specific Methylation Repatterning in Hybrid and Allopolyploid Mimulus.
M. guttatus and M. luteus subgenome-specific patterns of gene (top two rows) and transposon (bottom two rows) methylation. The y axis is the weighted methylation level. The x axis shows the gene body (TSS = transcription start site and TTS = transcription termination site) or TE body and 1 kb upstream and downstream. CG, CHG, and CHH methylation levels are shown in the first, second, and third column, respectively. Methylation levels of each individual are shown in unique colors (parents = red; F1 hybrid = light blue; synthetic allopolyploid = dark green; natural allopolyploid = yellow).
DISCUSSION
Investigating the aftermath of WGDs across both deep and recent time scales provides clearer insight into the collective evolutionary processes that occur in a polyploid nucleus (Mayfield-Jones et al., 2013), including the emergence and establishment of subgenome dominance. Subgenome dominance has largely been investigated in ancient polyploids, including Arabidopsis (Thomas et al., 2006), maize (Schnable et al., 2011), and Brassica (Cheng et al., 2016), which revealed the presence of a dominant subgenome with significantly greater gene content and which contributes more to the global transcriptome than the other subgenome(s).
Gene expression bias toward one of the subgenomes has also been observed in more recent allopolyploids including those formed as a product of domestication over the past 10 thousand years, e.g., wheat (Triticum aestivum; Li et al., 2014) and within recently formed natural allopolyploids, namely, Tragopogon mirus (Buggs et al., 2010). Due to the recent time scale of these WGD events, gene fractionation bias toward the recessive subgenome is not observed. It remains largely unknown how quickly subgenome dominance is established following an allopolyploid event. Subgenome dominance can also manifest itself in ways other than homoeolog expression bias. For instance, in Nicotiana, repeat abundances and rDNA levels exhibit subgenome-specific changes (Kovarik et al., 2008; Renny-Byfield et al., 2011), and in Brassica, expression bias of rRNA genes from a single parent in a hybrid or allopolyploid has been observed (Chen and Pikaard, 1997). In the future, exploring these questions in Mimulus may lead to a more comprehensive understanding of subgenome bias.
Here, we report that subgenome dominance becomes established in the first generation hybrid of two Mimulus species. Our analyses show that homoeologs from M. luteus, compared with M. guttatus, are significantly more expressed in the interspecific F1 hybrid and that this expression bias increases over subsequent generations, with the greatest bias observed in the natural (∼140 years old) neo-allopolyploid M. peregrinus. Using the LRT, we determined that the number of biased homoeolog pairs also increases with additional generations.
Genome-wide methylation analyses uncovered that CHH methylation levels are greatly reduced in the F1 hybrid. The greatest changes in CHH methylation are observed near gene bodies, near TE bodies, and within TE bodies. The methylation status of many of these CHH sites are regained in the second-generation resynthesized allopolyploid, indicating the onset of repatterning of DNA methylation. The methylation status of CHH sites near genes returned to parental levels across both subgenomes in the natural allopolyploid. Similarly, the CHH methylation status of TEs across M. guttatus subgenome returned to near parental levels in the natural allopolyploid. However, this pattern for CHH methylation is not observed across the dominant M. luteus subgenomes in the natural allopolyploid, with methylation levels within and near TEs remaining noticeably below parental levels. The methylation of CHG sites near TE and gene bodies was also impacted upon hybridization, but to a lesser degree than CHH methylation, and quickly returned to either at or above parental levels in the resynthesized allopolyploid. It is important to note that although M. luteus has 84% percent more genes than M. guttatus, it only has 41% percent more TEs. This means that the M. luteus genome has evolved to have fewer TEs at a genome-wide level. These observations, a dominantly expressed subgenome with lower TE abundance and lower CHH methylation levels near genes compared with the recessive subgenome, support the predictions made by Freeling et al. (2012) to explain subgenome dominance. However, given these data alone, we are unable at this time to distinguish between causation and correlation.
Our analyses confirm that the density of TEs negatively impacts the expression of nearby genes in Mimulus, similar to observations made in Arabidopsis (Hollister and Gaut, 2009). Furthermore, here, we show that the repatterning of TE methylation levels is different for the two subgenomes in M. peregrinus, mirroring the expression bias observed over the generations following the hybridization. Our results suggest that subgenome dominance may be at least partially due to subgenome-specific differences in the epigenetic silencing of TEs, which was established long ago in the ancestors of the diploid progenitors. The strong correlation between homoeolog expression bias (Β) in the F1 hybrid, resynthesized allopolyploid, and independently established natural allopolyploid indicate that the observed subgenome expression dominance is biologically real and likely heritable. The fact that homoeolog expression bias (B) in Mimulus hybrids mirrors methylation repatterning and subgenome-specific TE densities supports the original hypothesis for the mechanistic basis of subgenome dominance.
The observed methylation differences between homoeologs present on the different subgenomes may represent early earmarks for the ultimate loss (i.e., fractionation) of a duplicate gene copy. Fractionation is the loss of genes, regulatory elements, or other genomic features from a subgenome over time. Recessive subgenomes in ancient polyploids may be more highly fractionated and contribute less to the overall transcriptome compared with the dominant subgenome(s) (Tang et al., 2012). Although duplicate genes on either subgenome are not physically lost yet in M. peregrinus, many homoeologs on the recessive subgenome are already functionally absent (low to no expression). Due to selection acting on maintaining proper stoichiometry in dosage-sensitive macromolecular complexes and gene-interaction networks (Birchler et al., 2007; Edger and Pires, 2009), stoichiometric balance is likely best maintained by retaining the more highly expressed copy of interacting genes. One of the biggest opportunities arising from any gene duplication is the possibility of sub- or neofunctionalization. The finding of strong and immediate homoeolog expression bias in a hybrid and neo-allopolyploid may have important implications for our understanding of these processes.
Additionally, our analyses revealed that the two subgenomes in M. luteus are each dominant over the M. guttatus subgenome in the F1 hybrid and allopolyploid M. peregrinus. This observed pattern of dominance between the three subgenomes in Mimulus is opposite of what was reported for B. rapa (Tang et al., 2012), which similarly underwent a whole-genome triplication. Tang et al. (2012) hypothesized that the subgenome which joined the “battle” last, in a two-step process toward hexaploidy, emerged as the dominantly expressed and gene rich subgenome. In resynthesized and natural M. peregrinus, the most dominantly expressed subgenomes joined the “battle” first in M. luteus, which are both dominant over the M. guttatus subgenome. This suggests that the dominant subgenome(s) in higher polyploids (e.g., hexa-, octo-, and deca-) is likely not determined by the order by which subgenomes are merged into a single nucleus. Instead, our results collectively suggest that subgenome dominance is at least partially due to subgenome-specific epigenetic differences.
In conclusion, there appears to be clear trade-off between the benefits of epigenetic silencing of TEs (this inhibits their proliferation across the genome) and the effects of TE methylation on neighboring gene expression (Hollister and Gaut, 2009). Our results support the idea that subgenome dominance may be the result of lineage-specific genomic evolution shaping TE densities and methylation levels. In addition, subgenome expression dominance should not be unique to interspecific hybrids, but should also occur in intraspecies crosses between lines with different TE loads. These results have major implications to a number of research fields ranging from ecological studies to crop breeding efforts.
METHODS
Genome Assembly and Annotation
Genome assembly of Mimulus luteus was performed using the ALLPATHS-LG assembler v45395 (Gnerre et al., 2011) with ∼33x Illumina paired-end (2 × 100 bp) reads and a TruSeq mate-pair (5-kb insert) library. Additional gap closing was undertaken using GapCloser (v. 1.12, BGI). The assembly totaled 409 Mb in 6349 scaffolds with a scaffold N50/L50 of 283/439 kb, a contig N50 of 52 kb, and 96% of bases called (IUPAC ambiguous bases converted to N). This is 60% of the total genome size (680 Mb) anticipated by the assembler from the Kmer depth distribution. Genome completeness in terms of gene content was assessed using BUSCO (Simão et al., 2015) with default settings and a set of universal single-copy orthologs. A read coverage depth analysis was also run to verify that homoeologous regions in the M. luteus genome were not collapsed during the assembly. TEs were annotated using a combination of sequence and structure homology as well as de novo approaches (see “Methods for Transposon Annotation”).
The MAKER genome annotation pipeline was used to annotate the M. luteus and M. guttatus genome assemblies using SNAP, AUGUSTUS, and Fgenesh gene prediction programs (Salamov and Solovyev, 2000; Stanke and Waack, 2003; Korf, 2004; Cantarel et al., 2008). Species-specific transcript assemblies as well as TAIR Arabidopsis thaliana and plant-specific SwissProt protein sequences were aligned to the repeat-masked genome assemblies and used as evidence to aid the gene prediction programs (Berardini et al., 2015). Gene predictions with transcript, protein, or protein domain support were retained as final high-quality gene predictions (Campbell et al., 2014). The assembled genome sequence and annotation files (GFFs), including gene models, are deposited in CoGe (https://genomevolution.org/coge; genome IDs 22656 and 22665) and Dryad (http://dx.doi.org/10.5061/dryad.d4vr0).
Whole-Genome Alignments and Phylogenetic Analyses
Pairwise genomic alignments between the M. luteus and M. guttatus genomes were made using SynMap (Lyons et al., 2008) and QUOTA-ALIGN (Tang et al., 2011) and then filtered to identify syntenic orthologs between the two genomes and retained homoeologs in M. luteus. Data for various members of the order Lamiales were downloaded from NCBI-SRA and combined with newly generated transcriptome data for M. peregrinus and genomic data from M. luteus, M. guttatus (v.1.2) (Hellsten et al., 2013), and Solanum lycopersicum (ITAG2.3) (Tomato Genome Consortium, 2012) for phylogenetic analysis of retained duplicates.
Transcriptomes were assembled using Trinity v.2.1.1 (Haas et al., 2013). Reads were filtered for quality and length with adapter trimming (ILLUMINACLIP:TruSeq2-PE.fa:2:30:10), a sliding window of 10 bases with an average phred score of 20 (SLIDINGWINDOW:10:20), and a minimum length of 40 bp (MINLEN:40). Reads were normalized using Trinity’s in silico read normalization option with default parameters and a max coverage of 50 and assembled as paired-end with no assumed directionality. Assemblies were FPKM (per kilobase of exon per million fragments mapped) filtered by aligning reads to assembled contigs using bowtie v.0.12.7 (Langmead and Salzberg, 2012), and abundance was estimated using RSEM v.1.2.17 (Li and Dewey, 2011) and the “align_and_estimate_abundance.pl” script available with Trinity. Isoforms with a minimum of 1% of total mapped fragments to a component were kept as well-supported assemblies.
Transcripts were translated using the same method as McKain et al. (2016). For translation, the coding sequence (CDS) from M. guttatus was used as a reference for the RefTrans pipeline (https://github.com/mrmckain/RefTrans) and GeneWise v.2.2.0 (Birney et al., 2004). Assembled contigs were BLASTed (BLASTX) against amino acid sequences from a reference data set with a minimum e-value cutoff of 1e-10. Reference sequences were identified by filtering all BLASTXhits with a bidirectional overlap of 85% for each contig-reference pair.
Orthogroups were identified using OrthoFinder v.0.4 (Emms and Kelly, 2015) from translated transcriptome and full genome data sets with default parameters. Sequences were sorted into individual orthogroups, and amino acids were aligned using PASTA v.1.6.3 (Mirarab et al., 2014) under default parameters. CDS was overlaid to amino acid alignments using PAL2NAL v.14 (Suyama et al., 2006). Codon alignments from CDS were used to reconstruct gene trees using RAxML v.7.3.0 (Stamatakis, 2006) under a GTR+gamma model and 500 bootstrap replicates.
Single-copy gene trees were identified using the clone_reducer method outlined by Estep et al. (2014) and McKain et al. (2016). In short, for each gene family tree, clades with bootstrap values of 50 or greater that consisted of only a single taxon were reduced to a single, longest representative sequence. A total of 96 single copy orthogroups were identified. Reduced orthogroup phylogenies were then estimated in RAxML under a GTR+gamma model with 500 bootstrap replicates. A coalescence-based tree was estimated using Astral v.4.10.2 (Mirarab and Warnow, 2015) with 100 bootstrap replicates using the 96 single copy orthogroup trees and their associated bootstrap trees. A concatenation-based tree was estimated by first concatenating alignments for all 96 orthogroups and then estimating the phylogeny using RAxML under a GTR+gamma model with 500 bootstrap replicates.
Both the concatenation and coalescence species trees were used as guide trees to estimate the phylogenetic placement of putative whole-genome duplication events with the PUG software (https://github.com/mrmckain/PUG; McKain et al., 2016). PUG was used to identify all putative paralog pairs for all sampled taxa across non-single-copy gene families. A total of 1,731,351 putative paralogs were estimated from 10,888 gene trees. For each putative paralog pair, PUG identifies the node of coalescence in a gene tree and queries the subtree against the species tree. PUG then verifies that the taxa present in the sister lineage to the minimal species subtree are also present in the lineage sister to the paralog subtree. If this is found to be true, the paralog subtree is counted as a gene duplication event. The bootstrap value of the paralog coalescence node is recorded and all coalescence subtrees are filtered for minimal bootstrap values of 50 and 80. All data presented in this manuscript are for nodes with a bootstrap value of at least 80.
Mimulus Individuals Used in This Study
Two outbred individuals of M. guttatus and M. luteus s.l. from two populations in the UK (DBL and COL, respectively), derived from manual cross-fertilization of field-collected plants, were hybridized to produce triploid seed as described by Vallejo-Marín et al. (2016). Triploid seeds from a single hybrid cross (H003b; product of crossing accessions CG-1-1 and CS-4-3) were treated in 2013 with an aqueous solution of 0.1% colchicine to induce somatic polyploidization. Seeds were then planted, grown to flower, and screened with flow cytometry. A single mixoploid individual (3x and 6x nuclei; SYN-1) producing fertile pollen was self-pollinated to generate viable seeds. These seeds were then germinated and screened using flow cytometry to confirm that they were stable hexaploids (13-SYN-seed). For details on localities and taxonomy of the accessions used in the hybridization experiment, see Vallejo-Marín et al. (2016). The natural allohexaploid used in this study was accession 11-LED-seed-1 from Leadhills, Scotland.
RNA Sequencing and Quantification of Gene Expression
RNA-seq libraries were constructed using the TruSeq RNA kit (Illumina) from total RNA isolated from calyx, stem, and petals and then sequenced with single-end 100-bp reads on an Illumina HiSeq 2000 at the University of Missouri Sequencing Core for three tissue types for both M. guttatus and M. luteus. Illumina reads were quality filtered using NextGENe v2.3.3.1 (SoftGenetics), removing adapter sequences, reads with a median quality score of less than 22, trimmed reads at positions that had three consecutive bases with a quality score of <20, and any trimmed reads with a total length <40 bp. This resulted in 87.9% of the reads passing the quality-score filter. Expression levels, in FPKM (fragments per kilobase per million reads), were determined for all genes in the M. guttatus and M. luteus genomes. Quality-filtered reads for each library were aligned to the respective genomes using NextGENe v2.3.3.1 and counting only uniquely mapped reads, using parameters (A, matching requirement: >40 bases and >99%; B, allow ambiguous mapping: FALSE; and C, rigorous alignment: TRUE), resulting in the alignment of over 189.6 million reads to the M. guttatus and M. luteus genomes. The raw read counts and normalized RPKM values for each gene and every library used in this study are also deposited on Dryad (http://dx.doi.org/10.5061/dryad.d4vr0). Using these data, we compared expression of each individual homoeolog (Lb, La, or G) to each other. We also compared the expression of M. luteus to M. guttatus by comparing the weighted average of two homoeologs of M. luteus to M. guttatus. To do this, the expression of the two M. luteus homoeologs was calculated by dividing the sum of read count of the two M. luteus homoeologs by the sum of their individual gene lengths (weighted average). The Venn diagram of biased genes (Figure 5A) was drawn using eulerAPE (Micallef and Rodgers, 2014).
Whole-Genome Methylation Sequencing and Analysis
MethylC-seq libraries were prepared according to the following protocol (Urich et al., 2015), which involves sodium bisulfite treatment and the deamination of unmethylated cytosines to uracil, while methylated cytosines remain the same. Libraries were Illumina HiSeq 2500 platform with 150-bp reads at the University of Georgia. Cutadapt v1.9 (Martin, 2011) was used to trim adapter sequences, and then Bowtie 1.1.0 (Langmead and Salzberg, 2012) aligned reads to both a converted forward strand (cytosines to thymines) and converted reverse strand (guanines to adenines) reference genomes as previously described (Schmitz et al., 2013). Residue substitution of the reference genome adjusts for sodium bisulfite conversion and strand of the reads. Only uniquely aligned reads were retained. Mitochondrial sequence of M. guttatus (which is fully unmethylated) was used as control to calculate the sodium bisulfite reaction nonconversion rate of unmodified cytosines. For metaplots, both upstream and downstream regions were divided into 20 bins each of 50 bp in length for a total 1 kb in each direction. Gene and TE bodies were separated every 5%, for a total of 20 bins. Weighted methylation levels were computed for each bin as described previously (Schultz et al., 2012). These data are deposited in the Gene Expression Omnibus (GSE95799).
LRT to Detect Homoeolog Expression Bias
We defined homoeolog expression bias (B) as
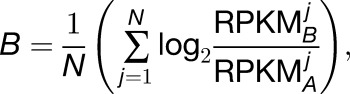 |
where A and B represent M. luteus and M. guttatus. In this formula, the observed expression levels of the homoeologs are normalized by gene length and the total number of mapped reads and j is an index over the N tissues. This measure of homoeolog expression bias can be computed for any homoeolog pair with non-zero read counts (testable homoeolog pairs).
For the analysis of our RNA-seq data, we developed a likelihood ratio test involving three nested hypotheses to identify cases of homoeolog expression bias that do not involve tissue-specific expression differences. The null hypothesis (H0) is that homoeologs are expressed at equal levels (ratio of homoeolog-1 to homoeolog-2 equals 1 for all three tissues). The first alternative hypothesis (H1) is that homoeologs are expressed at different levels, but similar ratios, across all three tissue types. The second alternative hypothesis (H2) is that homoeologs are expressed at different levels and at different ratios across all three tissues.
A brief description of our likelihood ratio test for homoeolog expression bias that does not involve tissue-specific expression differences follows (Smith et al., 2017). Denote the true but unknown expression levels (per kilobase of coding DNA sequence) of homoeologs 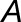 and
and 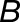 as
as 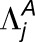 and
and 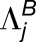 . The expected numbers of reads in tissue j (λAj and λBj) are
. The expected numbers of reads in tissue j (λAj and λBj) are
where 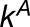 and
and 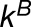 and are the transcript lengths of homoeologs
and are the transcript lengths of homoeologs 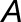 and
and 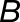 , respectively, and d is the total number of reads generated. Define
, respectively, and d is the total number of reads generated. Define 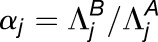 to be the “expression scaling factor” of
to be the “expression scaling factor” of 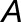 compared with
compared with 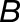 in tissue j, and
in tissue j, and 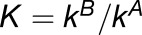 to be the ratio of the homoeologs’ transcript lengths. In that case, the expected number of reads,
to be the ratio of the homoeologs’ transcript lengths. In that case, the expected number of reads, 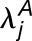 and
and 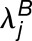 , are related by
, are related by
where 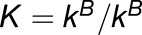 is a known quantity.
is a known quantity.
Because we are working with single replicates (one observation for each homoeolog pair in each tissue), we model the observed read counts (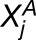 and
and 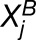 ) as Poisson distributed random variables with parameters
) as Poisson distributed random variables with parameters 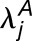 and
and 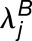 , respectively. The parameter sets associated with the three nested hypotheses for our likelihood ratio test are as follows:
, respectively. The parameter sets associated with the three nested hypotheses for our likelihood ratio test are as follows:
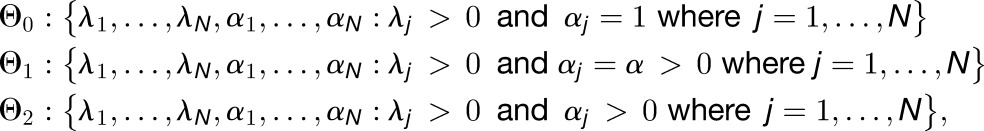 |
where Θ0⊂Θ1⊂Θ2 . The parameter set Θ0 corresponds to the null hypothesis (H0) that, after accounting for differences in transcript length, there are no tissue- or subgenome-specific differences in expression levels. The parameter set Θ1 corresponds the first alternative hypothesis (H1) that there are subgenome- but not tissue-specific differences in expression levels. The parameter set Θ2 corresponds to the second alternative hypothesis (H2) that there are tissue-specific differences in expression levels.
Assuming the probability models described above, we analytically derived the likelihood functions for H0, H1, and H2. Using the observed counts for each homoeolog pair (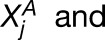
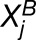 ), maximum likelihood estimation of the free parameters was performed using MATLAB’s built-in nonlinear system solver (fsolve) for H1, while H0 and H2 admit analytic solutions. Subsequently, two likelihood ratio tests were performed (using significance levels of 0.01). The first test was to determine whether we could reject H0 in favor of H1. If so, a second test determined whether we could reject H1 in favor of H2. Rejecting H0, but not rejecting H1, is interpreted as statistically significant homoeolog expression bias that does not involve tissue-specific expression differences. The Benjamini-Hochberg correction (Benjamini and Hochberg, 1995) was made to correct for multiple testing error for the first test, but not the second, since we were asking whether we were unable to reject H1 in favor of H2.
), maximum likelihood estimation of the free parameters was performed using MATLAB’s built-in nonlinear system solver (fsolve) for H1, while H0 and H2 admit analytic solutions. Subsequently, two likelihood ratio tests were performed (using significance levels of 0.01). The first test was to determine whether we could reject H0 in favor of H1. If so, a second test determined whether we could reject H1 in favor of H2. Rejecting H0, but not rejecting H1, is interpreted as statistically significant homoeolog expression bias that does not involve tissue-specific expression differences. The Benjamini-Hochberg correction (Benjamini and Hochberg, 1995) was made to correct for multiple testing error for the first test, but not the second, since we were asking whether we were unable to reject H1 in favor of H2.
Methods for Transposon Annotation
Autonomous Cut-and-Paste TEs
There are five superfamilies of cut-and-paste DNA TEs in angiosperm genomes (i.e., Tc1/mariner, PIF/Harbinger, Mutator-like elements [MULEs], hAT, and CACTA; Yuan and Wessler, 2011). They can be readily distinguished by the size of their target-site duplications (TSDs) and sequence similarity of the encoded transposase (TPase). The DDE/D domain alignment profile for each of the five superfamilies (obtained from Yuan and Wessler, 2011) was used as query to search against the M. guttatus genome assembly by TBLASTN (Altschul et al., 1997), as implemented in the TARGeT pipeline (Han et al., 2009). TARGeT outputs the DNA sequences with 10 kb upstream and downstream of the matched regions and a guide tree depicting the phylogenetic relationships of these putative elements. The ends of each putative element were then determined by aligning two closely related elements with their 20-kb flanking sequences, using NCBI-BLAST 2 Sequences on the NCBI server. Usually the breakpoint of a pairwise alignment is the boundary of a full-length element, which can be subsequently refined by identifying the terminal inverted repeats and TSDs around the breakpoint. Full-length elements were then classified into individual families following the guidelines proposed by Wicker et al. (2007).
Nonautonomous elements do not have coding capacity and thus cannot be identified effectively using sequence homology-based approaches. However, virtually all plant nonautonomous cut-and-paste TEs have terminal inverted repeats (10–400 bp) on both ends, are flanked by TSDs (2–10 bp), and are <3 kb in length. These structure features have been incorporated into an annotation pipeline MITE-Hunter (Han and Wessler, 2010), which was primarily designed to search miniature inverted repeat transposable elements (MITEs), but also can be used to search other non-autonomous cut-and-paste TEs. MITE-Hunter was employed to search the M. guttatus genome assembly, and the output consensus sequence for each putative nonautonomous family was used as query to retrieve all family members with 200-bp flanking sequences from the genome, using BLASTN implemented in TARGeT. Inspection of the multiple alignment profile of all family members leads to verification or refinement of the element boundary.
Helitrons
The program HelSearch (Yang and Bennetzen, 2009) was used to identify putative Helitron 3′ end, characterized by a CTRR terminus that is followed by a target site T residue and is preceded 5 to 10 bp by a 18-bp hairpin structure. HelSearch assigns candidate 3′ ends into groups based on the hairpin stem sequences and generates multiple alignment of flanking sequences for each group. These alignments were manually inspected to define authentic 3′ end and false positives were discarded. The last 100 nucleotides of each remaining alignment of authentic 3′ end were then used as query to retrieve the 15-kb upstream sequences using TARGeT. The 5′ ends were determined by inspection of the breakpoints of pairwise alignments of these upstream sequences using NCBI-BLAST 2 Sequences on the NCBI server. A typical Helitron 5′ prime end starts with TC, following a target site A residue, and is conspicuously AT-rich in the first 18-bp region. Once the 5′ ends were identified, 70 nucleotides from both 5′ and 3′ ends for each group were joined together to form a pseudo-element, which was used as a query to retrieve all related Helitrons from the genome using TARGet, allowing the sequence between the two ends to be up to 15 kb. Sequence similarity of the 70-bp region at the 5′ end was the basis for family assignment.
LTR Retrotransposons
LTR retrotransposons were mined using LTR retrotransposons were mined using LTR_STRUC (McCarthy and McDonald, 2003), a structural data-searching program that identifies LTR retrotransposons based on the presence of the LTR pairs, the 4- to 6-bp TSDs flanking the LTRs, the primer binding site and polypurine tract, as well as the canonical TG/CA dinucleotides at the 5′ and 3′ end of each LTR. A copia- and gypsy-specific reference sequence from the most conserved region of the reverse transcriptase (RT) domain (corresponding to Pfam family PF07727 and PF00078, respectively) was used to BLAST the full-length retrotransposon sequences found by LTR_STRUC. A phylogenetic tree was generated for all the matched copia-like and gypsy-like elements, respectively. Elements that have the RT domain were then classified into families based on the RT phylogeny. The remaining elements that do not have the RT domain (i.e., nonautonomous) were classified into families based on an all-to-all BLAST, following the 80-80 rule suggested by Wicker et al. (2007).
LINEs
The protocol of LINE identification is very similar to that of autonomous cut-and-paste TEs. There are two major clades/superfamilies of LINEs in plants, L1-like and RTE-like (Kapitonov et al., 2009). The RT amino acid sequence alignments of representative elements of these two clades were retrieved from Kapitonov et al. (2009) and were used as query to search all putative LINEs with their flanking sequences using TARGeT. Pairwise alignments were then used to determine the element boundary. The 3′ end is usually characterized by a poly(A) tail followed by a 7- to 21-bp TSD sequence. The TSD, in turn, can be used to demarcate the 5′ end. The RT phylogeny was used to guide family assignments.
SINEs
The M. guttatus genome assembly was subjected to RepeatModeler open-1.0 (Smit et al., 1996), a de novo repeat detection package that searches putative interspersed repeats and builds consensus models of identified repeat families. A TE exemplar set (see below) was built from all other elements identified by the homology-based and structure-based methods, as described above, and was used to mask the de novo repeat library output from RepeatModeler. The unmasked consensus sequences were then subjected to manual examination individually, to identify SINEs and additional TEs that were missed by structure- and homology-based methods. Sequences that are short (<500 bp) and have two conserved motifs corresponding to the Pol III internal promoter A and B boxes within the first 100-bp region are candidate SINEs (Kumar and Bennetzen, 1999; Schmidt, 1999). Each candidate consensus sequence was used as query to retrieve all related sequences through TARGeT. A 7- to 21-bp TSD flanking most of these sequences would verify bono fide SINEs.
TE Examplar Sequences
One to a few exemplar sequences were chosen to represent each family for building an exemplar library that can represent the entire TE content of the genome with a minimum number of sequences. For autonomous elements, these exemplar sequences were chosen based on two criteria: (1) sequence similarity between the exemplar and all other family members is at least 80%; (2) encoded open reading frames should be the most intact. The first criterion ensures that the exemplar library can be used to mask almost all TEs in the genome prior to gene model predictions and other kinds of genome annotations. The second criterion renders convenience to use the exemplars for comparative analysis between different genomes because elements satisfying the second criterion are often among the youngest and most abundant members of a family and the encoded protein products can be translated with minimal extent of interruption. For nonautonomous elements, only the first criterion was applied. Helitrons are much more heterogeneous in sequence between family members than other TE types, so usually many more exemplars need to be included for each family to capture the diversity. The complete M. guttatus TE exemplar library is available at http://monkeyflower.uconn.edu/resources/. The TE exemplar library was used to identify TEs in the M. guttatus and M. luteus genomes using RepeatMasker (http://www.repeatmasker.org).
Accession Numbers
Sequence data from this article can be found under the following accession numbers. M. luteus genome sequence, including transcript assembly, and GFF genome annotation files are available on CoGe. The new version of M. guttatus gene annotation is available on COGE. COGE genome IDs are 22656 and 22665. Illumina fastq reads (mate-pair and paired-end reads) used to assemble the M. luteus genome are available on SRA (PRJNA377704). Gene expression data (counts and RPKM data) are available on Dryad (http://dx.doi.org/10.5061/dryad.d4vr0). All called orthogroups are available Dryad (http://dx.doi.org/10.5061/dryad.d4vr0). Syntenic gene sets are available Dryad (http://dx.doi.org/10.5061/dryad.d4vr0). The M. guttatus gene annotation file is on Dryad (http://dx.doi.org/10.5061/dryad.d4vr0). The M. luteus gene annotation file is on Dryad (http://dx.doi.org/10.5061/dryad.d4vr0). The M. luteus genome assembly file is on Dryad (http://dx.doi.org/10.5061/dryad.d4vr0). Methylation sequencing data are available under accession number GSE95799. The alignments for generating the trees used in this study are available in the DRYAD repository (http://dx.doi.org/10.5061/dryad.d4vr0.2).
Supplemental Data
Supplemental Figure 1. Chimera analysis.
Supplemental Figure 2. Substitution mean.
Supplemental Figure 3. Concatenation tree.
Supplemental Figure 4. Astral tree coalescence-based phylogeny.
Supplemental Figure 5. Results of PUG analysis using the coalescence tree.
Supplemental Figure 6. Relationship of TE density and gene expression for M. luteus subgenomic regions.
Supplemental Figure 7. Relationship of TE density and gene expression for M. guttatus subgenomic regions.
Supplemental Figure 8. Patterns of methylation in Mimulus parents and hybrids.
Acknowledgments
This work was supported by The College of William and Mary Research Award to J.R.P., by USDA-NIFA HATCH 1009804 to P.P.E., and by Murdock Life Sciences Grant 2013265 to A.M.C.
AUTHOR CONTRIBUTIONS
J.R.P. and P.P.E. designed the research. J.R.P., P.P.E., J.D.W., and J.C.P. collected samples and performed RNA sequencing. R.S., G.D.S., and J.R.P. developed the likelihood ratio test for expression bias. J.R.P., P.P.E., and R.S. performed homoeolog expression analyses. M.R.M. and P.P.E. conducted phylogenetic analyses. J.R.P., A.M.C., P.P.E., A.E.P., M.J.B., and K.L.C. performed genome sequencing, assembly, and annotation. R.J.S., A.J.B., and L.J. performed methylation analyses. M.V.-M. created synthetic polyploids and identified naturally occurring polyploid plants. Y.Y. and J.R.P. conducted transposon related analyses. J.R.P., P.P.E., and R.S. wrote the article.
Glossary
- WGD
whole-genome duplication
- TE
transposable element
- LRT
likelihood ratio test
- CDS
coding sequence
- TSD
target-site duplication
Footnotes
Articles can be viewed without a subscription.
References
- Adams K.L., Wendel J.F. (2005). Novel patterns of gene expression in polyploid plants. Trends Genet. 21: 539–543. [DOI] [PubMed] [Google Scholar]
- Adams K.L., Cronn R., Percifield R., Wendel J.F. (2003). Genes duplicated by polyploidy show unequal contributions to the transcriptome and organ-specific reciprocal silencing. Proc. Natl. Acad. Sci. USA 100: 4649–4654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altschul S.F., Madden T.L., Schäffer A.A., Zhang J., Zhang Z., Miller W., Lipman D.J. (1997). Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25: 3389–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barker M.S., Arrigo N., Baniaga A.E., Li Z., Levin D.A. (2015). On the relative abundance of autopolyploids and allopolyploids. New Phytol. 210: 391–398. [DOI] [PubMed] [Google Scholar]
- Benjamini Y., Hochberg Y. (1995). Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Stat. Soc. B 57: 289–300. [Google Scholar]
- Berardini T.Z., Reiser L., Li D., Mezheritsky Y., Muller R., Strait E., Huala E. (2015). The Arabidopsis information resource: Making and mining the “gold standard” annotated reference plant genome. Genesis 53: 474–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birchler J.A., Yao H., Chudalayandi S. (2007). Biological consequences of dosage dependent gene regulatory systems. Biochim. Biophys. Acta 1769: 422–428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birney E., Clamp M., Durbin R. (2004). GeneWise and Genomewise. Genome Res. 14: 988–995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buggs R.J., Elliott N.M., Zhang L., Koh J., Viccini L.F., Soltis D.E., Soltis P.S. (2010). Tissue-specific silencing of homoeologs in natural populations of the recent allopolyploid Tragopogon mirus. New Phytol. 186: 175–183. [DOI] [PubMed] [Google Scholar]
- Campbell M.S., Holt C., Moore B., Yandell M. (2014). Genome annotation and curation using MAKER and MAKER-P. Curr. Protoc. Bioinformatics 48: 1–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cantarel B.L., Korf I., Robb S.M., Parra G., Ross E., Moore B., Holt C., Sánchez Alvarado A., Yandell M. (2008). MAKER: an easy-to-use annotation pipeline designed for emerging model organism genomes. Genome Res. 18: 188–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chao D.-Y., Dilkes B., Luo H., Douglas A., Yakubova E., Lahner B., Salt D.E. (2013). Polyploids exhibit higher potassium uptake and salinity tolerance in Arabidopsis. Science 341: 658–659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chelaifa H., Monnier A., Ainouche M. (2010). Transcriptomic changes following recent natural hybridization and allopolyploidy in the salt marsh species Spartina x townsendii and Spartina anglica (Poaceae). New Phytol. 186: 161–174. [DOI] [PubMed] [Google Scholar]
- Chen Z.J. (2007). Genetic and epigenetic mechanisms for gene expression and phenotypic variation in plant polyploids. Annu. Rev. Plant Biol. 58: 377–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z.J., Pikaard C.S. (1997). Epigenetic silencing of RNA polymerase I transcription: a role for DNA methylation and histone modification in nucleolar dominance. Genes Dev. 11: 2124–2136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng F., Sun C., Wu J., Schnable J., Woodhouse M.R., Liang J., Cai C., Freeling M., Wang X. (2016). Epigenetic regulation of subgenome dominance following whole genome triplication in Brassica rapa. New Phytol. 211: 288–299. [DOI] [PubMed] [Google Scholar]
- Coate J.E., Bar H., Doyle J.J. (2014). Extensive translational regulation of gene expression in an allopolyploid (Glycine dolichocarpa). Plant Cell 26: 136–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Comai L. (2005). The advantages and disadvantages of being polyploid. Nat. Rev. Genet. 6: 836–846. [DOI] [PubMed] [Google Scholar]
- Crow K.D., Wagner G.P.; SMBE Tri-National Young Investigators (2006). Proceedings of the SMBE Tri-National Young Investigators’ Workshop 2005. What is the role of genome duplication in the evolution of complexity and diversity? Mol. Biol. Evol. 23: 887–892. [DOI] [PubMed] [Google Scholar]
- Cui L., et al. (2006). Widespread genome duplications throughout the history of flowering plants. Genome Res. 16: 738–749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dehal P., Boore J.L. (2005). Two rounds of whole genome duplication in the ancestral vertebrate. PLoS Biol. 3: e314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dion-Côté A.-M., Renaut S., Normandeau E., Bernatchez L. (2014). RNA-seq reveals transcriptomic shock involving transposable elements reactivation in hybrids of young lake whitefish species. Mol. Biol. Evol. 31: 1188–1199. [DOI] [PubMed] [Google Scholar]
- Edger P.P., Pires J.C. (2009). Gene and genome duplications: the impact of dosage-sensitivity on the fate of nuclear genes. Chromosome Res. 17: 699–717. [DOI] [PubMed] [Google Scholar]
- Edger P.P., et al. (2015). The butterfly plant arms-race escalated by gene and genome duplications. Proc. Natl. Acad. Sci. USA 112: 8362–8366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emms D.M., Kelly S. (2015). OrthoFinder: solving fundamental biases in whole genome comparisons dramatically improves orthogroup inference accuracy. Genome Biol. 16: 157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Estep M.C., McKain M.R., Vela Diaz D., Zhong J., Hodge J.G., Hodkinson T.R., Layton D.J., Malcomber S.T., Pasquet R., Kellogg E.A. (2014). Allopolyploidy, diversification, and the Miocene grassland expansion. Proc. Natl. Acad. Sci. USA 111: 15149–15154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freeling M., Woodhouse M.R., Subramaniam S., Turco G., Lisch D., Schnable J.C. (2012). Fractionation mutagenesis and similar consequences of mechanisms removing dispensable or less-expressed DNA in plants. Curr. Opin. Plant Biol. 15: 131–139. [DOI] [PubMed] [Google Scholar]
- Gaeta R.T., Pires J.C., Iniguez-Luy F., Leon E., Osborn T.C. (2007). Genomic changes in resynthesized Brassica napus and their effect on gene expression and phenotype. Plant Cell 19: 3403–3417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gnerre S., et al. (2011). High-quality draft assemblies of mammalian genomes from massively parallel sequence data. Proc. Natl. Acad. Sci. USA 108: 1513–1518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haas B.J., et al. (2013). De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat. Protoc. 8: 1494–1512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han Y., Wessler S.R. (2010). MITE-Hunter: a program for discovering miniature inverted-repeat transposable elements from genomic sequences. Nucleic Acids Res. 38: e199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han Y., Burnette J.M. III, Wessler S.R. (2009). TARGeT: a web-based pipeline for retrieving and characterizing gene and transposable element families from genomic sequences. Nucleic Acids Res. 37: e78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hellsten U., Wright K.M., Jenkins J., Shu S., Yuan Y., Wessler S.R., Schmutz J., Willis J.H., Rokhsar D.S. (2013). Fine-scale variation in meiotic recombination in Mimulus inferred from population shotgun sequencing. Proc. Natl. Acad. Sci. USA 110: 19478–19482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollister J.D., Gaut B.S. (2009). Epigenetic silencing of transposable elements: a trade-off between reduced transposition and deleterious effects on neighboring gene expression. Genome Res. 19: 1419–1428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiao Y., et al. (2011). Ancestral polyploidy in seed plants and angiosperms. Nature 473: 97–100. [DOI] [PubMed] [Google Scholar]
- Julca I., Marcet-Houben M., Vargas P., Gabaldon T. (2017). Phylogenomics of the olive tree (Olea europaea) disentangles ancient allo-and autopolyploidizations in Lamiales. bioRxiv doi/10.1101/163063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kagale S., Robinson S.J., Nixon J., Xiao R., Huebert T., Condie J., Kessler D., Clarke W.E., Edger P.P., Links M.G., Sharpe A.G., Parkin I.A. (2014). Polyploid evolution of the Brassicaceae during the Cenozoic era. Plant Cell 26: 2777–2791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapitonov V.V., Tempel S., Jurka J. (2009). Simple and fast classification of non-LTR retrotransposons based on phylogeny of their RT domain protein sequences. Gene 448: 207–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korf I. (2004). Gene finding in novel genomes. BMC Bioinformatics 5: 59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovarik A., Dadejova M., Lim Y.K., Chase M.W., Clarkson J.J., Knapp S., Leitch A.R. (2008). Evolution of rDNA in Nicotiana allopolyploids: a potential link between rDNA homogenization and epigenetics. Ann. Bot. (Lond.) 101: 815–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar A., Bennetzen J.L. (1999). Plant retrotransposons. Annu. Rev. Genet. 33: 479–532. [DOI] [PubMed] [Google Scholar]
- Langmead B., Salzberg S.L. (2012). Fast gapped-read alignment with Bowtie 2. Nat. Methods 9: 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levin D.A. (1983). Polyploidy and novelty in flowering plants. Am. Nat. 112: 1–25. [Google Scholar]
- Li A., et al. (2014). mRNA and small RNA transcriptomes reveal insights into dynamic homoeolog regulation of allopolyploid heterosis in nascent hexaploid wheat. Plant Cell 26: 1878–1900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B., Dewey C.N. (2011). RSEM: accurate transcript quantification from RNA-seq data with or without a reference genome. BMC Bioinformatics 12: 323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukens L.N., Pires J.C., Leon E., Vogelzang R., Oslach L., Osborn T. (2006). Patterns of sequence loss and cytosine methylation within a population of newly resynthesized Brassica napus allopolyploids. Plant Physiol. 140: 336–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyons E., Pedersen B., Kane J., Freeling M. (2008). The value of nonmodel genomes and an example using SynMap within CoGe to dissect the hexaploidy that predates the rosids. Trop. Plant Biol. 1: 181–190. [Google Scholar]
- Madlung A., Masuelli R.W., Watson B., Reynolds S.H., Davison J., Comai L. (2002). Remodeling of DNA methylation and phenotypic and transcriptional changes in synthetic Arabidopsis allotetraploids. Plant Physiol. 129: 733–746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magallón S., Gómez-Acevedo S., Sánchez-Reyes L.L., Hernández-Hernández T. (2015). A metacalibrated time-tree documents the early rise of flowering plant phylogenetic diversity. New Phytol. 207: 437–453. [DOI] [PubMed] [Google Scholar]
- Martin M. (2011). Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J.17: 10. [Google Scholar]
- Mayfield-Jones D., Washburn J.D., Arias T., Edger P.P., Pires J.C., Conant G.C. (2013). Watching the grin fade: tracing the effects of polyploidy on different evolutionary time scales. Semin. Cell Dev. Biol. 24: 320–331. [DOI] [PubMed] [Google Scholar]
- McCarthy E.M., McDonald J.F. (2003). LTR_STRUC: a novel search and identification program for LTR retrotransposons. Bioinformatics 19: 362–367. [DOI] [PubMed] [Google Scholar]
- McClintock B. (1984). The significance of responses of the genome to challenge. Science 226: 792–801. [DOI] [PubMed] [Google Scholar]
- McKain M.R., Tang H., McNeal J.R., Ayyampalayam S., Davis J.I., dePamphilis C.W., Givnish T.J., Pires J.C., Stevenson D.W., Leebens-Mack J.H. (2016). A phylogenomic assessment of ancient polyploidy and genome evolution across the Poales. Genome Biol. Evol. 8: 1150–1164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLysaght A., Hokamp K., Wolfe K.H. (2002). Extensive genomic duplication during early chordate evolution. Nat. Genet. 31: 200–204. [DOI] [PubMed] [Google Scholar]
- Micallef L., Rodgers P. (2014). eulerAPE: drawing area-proportional 3-Venn diagrams using ellipses. PLoS One 9: e101717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirarab S., Warnow T. (2015). ASTRAL-II: coalescent-based species tree estimation with many hundreds of taxa and thousands of genes. Bioinformatics 31: i44–i52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirarab S., Nguyen N., Warnow T. (2014). PASTA: ultra-large multiple sequence alignment. In Research in Computational Molecular Biology: 18th Annual International Conference, RECOMB 2014, Pittsburgh, PA, April 2–5, 2014, R. Sharan, ed (Springer), pp. 177–191. [Google Scholar]
- Mittelsten Scheid O., Afsar K., Paszkowski J. (2003). Formation of stable epialleles and their paramutation-like interaction in tetraploid Arabidopsis thaliana. Nat. Genet. 34: 450–454. [DOI] [PubMed] [Google Scholar]
- Mukherjee B.B., Vickery R.K. (1962). Chromosome counts in the section Simiolus of the genus Mimulus (Scrophulariaceae). V. The chromosomal homologies of M. guttatus and its allied species and varieties. Madroño 16: 141–155. [Google Scholar]
- Otto S.P. (2007). The evolutionary consequences of polyploidy. Cell 131: 452–462. [DOI] [PubMed] [Google Scholar]
- Ramsey J., Schemske D.W. (1998). Pathways, mechanisms, and rates of polyploid formation in flowering plants. Annu. Rev. Ecol. Syst. 29: 467–501. [Google Scholar]
- Rapp R.A., Udall J.A., Wendel J.F. (2009). Genomic expression dominance in allopolyploids. BMC Biol. 7: 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Refulio-Rodriguez N.F., Olmstead R.G. (2014). Phylogeny of Lamiidae. Am. J. Bot. 101: 287–299. [DOI] [PubMed] [Google Scholar]
- Renny-Byfield S., Rodgers-Melnick E., Ross-Ibarra J. (2017). Gene fractionation and function in the ancient subgenomes of maize. Mol. Biol. Evol. 34: 1825–1832. [DOI] [PubMed] [Google Scholar]
- Renny-Byfield S., Gong L., Gallagher J.P., Wendel J.F. (2015). Persistence of subgenomes in paleopolyploid cotton after 60 my of evolution. Mol. Biol. Evol. 32: 1063–1071. [DOI] [PubMed] [Google Scholar]
- Renny-Byfield S., Chester M., Kovařík A., Le Comber S.C., Grandbastien M.-A., Deloger M., Nichols R.A., Macas J., Novák P., Chase M.W., Leitch A.R. (2011). Next generation sequencing reveals genome downsizing in allotetraploid Nicotiana tabacum, predominantly through the elimination of paternally derived repetitive DNAs. Mol. Biol. Evol. 28: 2843–2854. [DOI] [PubMed] [Google Scholar]
- Rigal M., Becker C., Pélissier T., Pogorelcnik R., Devos J., Ikeda Y., Weigel D., Mathieu O. (2016). Epigenome confrontation triggers immediate reprogramming of DNA methylation and transposon silencing in Arabidopsis thaliana F1 epihybrids. Proc. Natl. Acad. Sci. USA 113: E2083–E2092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salamov A.A., Solovyev V.V. (2000). Ab initio gene finding in Drosophila genomic DNA. Genome Res. 10: 516–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salmon A., Ainouche M.L., Wendel J.F. (2005). Genetic and epigenetic consequences of recent hybridization and polyploidy in Spartina (Poaceae). Mol. Ecol. 14: 1163–1175. [DOI] [PubMed] [Google Scholar]
- Schmidt T. (1999). LINEs, SINEs and repetitive DNA: non-LTR retrotransposons in plant genomes. Plant Mol. Biol. 40: 903–910. [DOI] [PubMed] [Google Scholar]
- Schmitz R.J., Schultz M.D., Urich M.A., Nery J.R., Pelizzola M., Libiger O., Alix A., McCosh R.B., Chen H., Schork N.J., Ecker J.R. (2013). Patterns of population epigenomic diversity. Nature 495: 193–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnable J.C., Springer N.M., Freeling M. (2011). Differentiation of the maize subgenomes by genome dominance and both ancient and ongoing gene loss. Proc. Natl. Acad. Sci. USA 108: 4069–4074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schultz M.D., Schmitz R.J., Ecker J.R. (2012). ‘Leveling’ the playing field for analyses of single-base resolution DNA methylomes. Trends Genet. 28: 583–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaked H., Kashkush K., Ozkan H., Feldman M., Levy A.A. (2001). Sequence elimination and cytosine methylation are rapid and reproducible responses of the genome to wide hybridization and allopolyploidy in wheat. Plant Cell 13: 1749–1759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simão F.A., Waterhouse R.M., Ioannidis P., Kriventseva E.V., Zdobnov E.M. (2015). BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31: 3210–3212. [DOI] [PubMed] [Google Scholar]
- Smit A.F., Hubley R., Green P. (1996). RepeatMasker Open-3.0, http://www.repeatmasker.org.
- Smith R.D., Kinser T.J., Smith G.D., Puzey J.R. (2017). A likelihood ratio test for changes in homeolog expression bias. bioRxiv doi/10.1101/119438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soltis P.S., Soltis D.E. (2016). Ancient WGD events as drivers of key innovations in angiosperms. Curr. Opin. Plant Biol. 30: 159–165. [DOI] [PubMed] [Google Scholar]
- Song Q., Chen Z.J. (2015). Epigenetic and developmental regulation in plant polyploids. Curr. Opin. Plant Biol. 24: 101–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stamatakis A. (2006). RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics 22: 2688–2690. [DOI] [PubMed] [Google Scholar]
- Stanke M., Waack S. (2003). Gene prediction with a hidden Markov model and a new intron submodel. Bioinformatics 19 (suppl. 2): ii215–ii225. [DOI] [PubMed] [Google Scholar]
- Stull G.W., Duno de Stefano R., Soltis D.E., Soltis P.S. (2015). Resolving basal lamiid phylogeny and the circumscription of Icacinaceae with a plastome-scale data set. Am. J. Bot. 102: 1794–1813. [DOI] [PubMed] [Google Scholar]
- Suyama M., Torrents D., Bork P. (2006). PAL2NAL: robust conversion of protein sequence alignments into the corresponding codon alignments. Nucleic Acids Res. 34: W609–W612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang H., Lyons E., Pedersen B., Schnable J.C., Paterson A.H., Freeling M. (2011). Screening synteny blocks in pairwise genome comparisons through integer programming. BMC Bioinformatics 12: 102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang H., Woodhouse M.R., Cheng F., Schnable J.C., Pedersen B.S., Conant G., Wang X., Freeling M., Pires J.C. (2012). Altered patterns of fractionation and exon deletions in Brassica rapa support a two-step model of paleohexaploidy. Genetics 190: 1563–1574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tank D.C., Eastman J.M., Pennell M.W., Soltis P.S., Soltis D.E., Hinchliff C.E., Brown J.W., Sessa E.B., Harmon L.J. (2015). Nested radiations and the pulse of angiosperm diversification: increased diversification rates often follow whole genome duplications. New Phytol. 207: 454–467. [DOI] [PubMed] [Google Scholar]
- Thomas B.C., Pedersen B., Freeling M. (2006). Following tetraploidy in an Arabidopsis ancestor, genes were removed preferentially from one homeolog leaving clusters enriched in dose-sensitive genes. Genome Res. 16: 934–946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomato Genome Consortium (2012). The tomato genome sequence provides insights into fleshy fruit evolution. Nature 485: 635–641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urich M.A., Nery J.R., Lister R., Schmitz R.J., Ecker J.R. (2015). MethylC-seq library preparation for base-resolution whole-genome bisulfite sequencing. Nat. Protoc. 10: 475–483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vallejo-Marín M. (2012). Mimulus peregrinus (Phrymaceae): A new British allopolyploid species. PhytoKeys 14: 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vallejo-Marín M., Cooley A.M., Lee M.Y., Folmer M., McKain M.R., Puzey J.R. (2016). Strongly asymmetric hybridization barriers shape the origin of a new polyploid species and its hybrid ancestor. Am. J. Bot. 103: 1272–1288. [DOI] [PubMed] [Google Scholar]
- Vallejo-Marín M., Buggs R.J., Cooley A.M., Puzey J.R. (2015). Speciation by genome duplication: Repeated origins and genomic composition of the recently formed allopolyploid species Mimulus peregrinus. Evolution 69: 1487–1500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanneste K., Baele G., Maere S., Van de Peer Y. (2014). Analysis of 41 plant genomes supports a wave of successful genome duplications in association with the Cretaceous-Paleogene boundary. Genome Res. 24: 1334–1347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J., Tian L., Madlung A., Lee H.-S., Chen M., Lee J.J., Watson B., Kagochi T., Comai L., Chen Z.J. (2004). Stochastic and epigenetic changes of gene expression in Arabidopsis polyploids. Genetics 167: 1961–1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wicker T., et al. (2007). A unified classification system for eukaryotic transposable elements. Nat. Rev. Genet. 8: 973–982. [DOI] [PubMed] [Google Scholar]
- Wikström N., Kainulainen K., Razafimandimbison S.G., Smedmark J.E., Bremer B. (2015). A revised time tree of the asterids: establishing a temporal framework for evolutionary studies of the coffee family (Rubiaceae). PLoS One 10: e0126690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodhouse M.R., Schnable J.C., Pedersen B.S., Lyons E., Lisch D., Subramaniam S., Freeling M. (2010). Following tetraploidy in maize, a short deletion mechanism removed genes preferentially from one of the two homologs. PLoS Biol. 8: e1000409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright R.J., Thaxton P.M., El-Zik K.M., Paterson A.H. (1998). D-subgenome bias of Xcm resistance genes in tetraploid Gossypium (cotton) suggests that polyploid formation has created novel avenues for evolution. Genetics 149: 1987–1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L., Bennetzen J.L. (2009). Structure-based discovery and description of plant and animal Helitrons. Proc. Natl. Acad. Sci. USA 106: 12832–12837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan Y.-W., Wessler S.R. (2011). The catalytic domain of all eukaryotic cut-and-paste transposase superfamilies. Proc. Natl. Acad. Sci. USA 108: 7884–7889. [DOI] [PMC free article] [PubMed] [Google Scholar]