

Abstract
Coronary reactive hyperemia (CRH) protects the heart against ischemia. Adenosine A2AAR– deficient (A2AAR−/−) mice have increased expression of soluble epoxide hydrolase (sEH); the enzyme responsible for breaking down the cardioprotective epoxyeicosatrienoic acids (EETs) to dihydroxyeicosatrienoic acids (DHETs). she–inhibition enhances CRH, increases EETs, and modulates oxylipin profiles. We investigated the changes of oxylipins and their impact on CRH in A2AAR−/− and wild type (WT) mice. We hypothesized that the attenuated CRH in A2AAR−/− mice is mediated by changes in oxylipin profiles, and that it can be reversed by either sEH– or ω -hydroxylases–inhibition. Compared to WT mice, A2AAR−/− mice had attenuated CRH and changed oxylipin profiles, which were consistent between plasma and heart perfusate samples, including decreased EET/DHET ratios, and increased hydroxyeicosatetraenoic acids (HETEs).
Plasma oxylipns in A2AAR−/− mice indicated an increased proinflammatory state including increased ω-terminal HETEs, decreased epoxyoctadecaenoic / dihydroxyoctadecaenoic acids (EpOMEs/DiHOMEs) ratios, increased 9-hydroxyoctadecadienoic acid, and increased prostanoids. Inhibition of either sEH or ω-hydroxylases reversed the reduced CRH in A2AAR−/− mice. In WT and sEH −/− mice, blocking A2AAR decreased CRH. These data demonstrate that A2AAR–deletion was associated with changes in oxylipin profiles, which may contribute to the attenuated CRH. Also, inhibition of sEH and ω-hydroxylases reversed the reduction in CRH.
Keywords: Adenosine A2A receptor, coronary reactive hyperemia, ω-hydroxylases, soluble epoxid, hydrolase, heart perfusate oxylipins, plasma oxylipins
Introduction
Reactive hyperemia (RH) is a physiologic phenomenon whose function is to rapidly restore blood perfusion by increasing blood flow to an ischemic organ or tissue [1]. The myocardium is perpetually active and responds to coronary occlusion through the same mechanism: coronary reactive hyperemia (CRH) [2]. Ischemia is inherently harmful, and the increased blood flow associated with CRH seems to serve a protective role by increasing blood supply to the deprived cardiac tissue [1, 3]. The importance of CRH is reiterated by the observation that several cardiovascular pathologic conditions affecting the coronary circulation, such as the metabolic syndrome [4], cardiac hypertrophy [5], coronary artery disease, and congestive heart failure [6] have documented abnormalities in CRH. In addition to a number of mediators, including hydrogen peroxide (H2O2) [5], nitric oxide (NO) [5], and Katp channels [5], adenosine mediates CRH through its receptor subtype A2AAR [5, 7, 8]. Our lab has recently reported that several arachidonic- and linoleic-acid derived oxylipins, such as EETs, DHETs, EpOMEs, DiHOMEs, mid-chain HETEs, and hydroxyoctadecadienoic acids (HODEs), could impact CRH [3, 9-11]. Arachidonic and linoleic-acids are two important ω-6 PUFAs (polyunsaturated fatty acids), which can be oxidized to oxylipins known to mediate myriad of physiologic effects [12]. Cytochrome P450 epoxygenases generate epoxyeicosatrienoic acids (EETs) from arachidonic acid (AA) by epoxidation, whereas Cytochrome P450 ω-hydroxylases hydroxylate AA to form ω-terminal HETEs [3]. EETs have protective functions in the cardiovascular system including vasodilation through hyperpolarization [13, 14]. A cardioprotective effect against ischemia/reperfusion injury was observed by the increased generation of EETs in cardiomyocytes in mice [15]. Soluble epoxide hydrolase (sEH) breaks EETs down to form DHETs, which are less active. To investigate the effects of EETs in experimental studies, sEH is often utilized either by pharmacologic inhibition [9, 16], genetic deletion [9, 15, 17], or over-expression [3, 18]. In the isolated mouse heart model, sEH inhibition was associated with enhanced CRH and increased EET/DHET ratio [9], whereas endothelial over-expression of sEH attenuated CRH [3], decreased EET/DHET ratio, and impaired endothelium-dependent vasodilation in mouse cerebral circulation [18]. Of the ω-terminal HETEs, 20-HET, which is a potent vasoconstrictor, is produced by CYP4A from arachidonic acid [19]. 20-HETE promotes hypertension, vasoconstriction, and vascular dysfunction [20, 21]. Like arachidonic acid, linoleic acid is converted to vasoactive oxylipins, such as EpOMEs and HODEs. At physiological levels, EpOMEs were protective against hypoxia injury [22, 23]. One of the two isomers, 12,13-EpOME, was protective for rabbits' renal proximal tubular cell cultures against hypoxia/reoxygenation injury [24]. sEH metabolizes EpOMEs to DiHOMEs; the latter have deleterious effects in the heart and are cytotoxic to cells in tissue culture at high concentrations [25].
Adenosine is generally cytoprotective; it is an endogenous nucleoside produced in response to stressful conditions, such as inflammation and ischemia [26]. Adenosine's cardioprotective effects are mediated by its receptor subtypes A2AAR and A2BAR [27]; A2AAR is important in mediating the vasodilatory effect of adenosine [28], including coronary reactive hyperemia in response to ischemia in dogs [8]. Adenosine inhibits platelet aggregation through its A2AAR, which is expressed on murine and human platelets [29]. A genetic variant in adenosine A2AAR in humans, ADORA2A TT variant, predisposes to increased changes in mean and peak systolic blood pressure in response to caffeine [30]. Moreover, a single-nucleotide polymorphism variant of A2AAR, rs4822489, was linked to the severity of chronic heart failure in a group of Chinese people [27]. EETs mediate A2AAR–induced vasodilation in rat arcuate arteries [31]. The A2AAR–EET pathway is further supported by the finding that A2AAR–activation results in increased EETs' release in isolated perfused kidney [32]. Our lab demonstrated that sEH expression was increased in A2AAR−/− (A2AAR-null) mice compared to A2AAR+/+ (wild type) mice [33]. Increased sEH expression was associated with attenuated CRH [3], and decreased EET/DHET ratio [18].
The effect of sEH– and ω-hydroxylases–inhibition on CRH in A2AAR−/− mice in response to ischemia, and the changes in oxylipin profiles in plasma and heart perfusate associated with genetic deletion of A2AAR in A2AAR−/− mice have not been investigated. We hypothesized that the attenuated CRH in A2AAR−/− mice is partially mediated by changes in oxylipin profiles, and that it can be reversed either by soluble epoxide hydrolase– or ω-hydroxylases–inhibition.
Methods
Animals
The mice used in this paper, sEH −/−, A2AAR−/−, and wild type (WT), were of the C57BL/6 genetic background. A2AAR−/− mice (initially from C. Ledent, Universite Libre de Bruxelles, Brussels, Belgium) were obtained from Dr. Stephen Tilley, UNC Chapel Hill. A2AAR−/− mice were backcrossed 12 generations to the C57BL/6 background as previously reported [34]. The breeder pairs of A2AAR −/− mice were generously provided by Dr. Jamal Mustafa (WVU). The generation of sEH −/− mice was described by Sinal et al. [35]. sEH −/− mice were provided by Dr. Darryl Zeldin, National Institute of Environmental Health Sciences/National Institutes of Health (NIH). Animal care and experimentation protocols were approved and performed in conformity with the West Virginia University Institutional Animal Care and Use Committee and were in compliance with the guidelines of the NIH's Guide for the Care and Use of Laboratory Animals. Equal ratios of male and female mice, age 16 –18 weeks, were used in this study. Mice were cared for in cages with a 12:12 h light-dark cycle and allowed access to standard chow (Cat #2018, Envigo, Indianapolis, IN) and water. Diet 2018 was used and contained 6.2% fat by weight, including 1.2% oleic %, 0.3% linolenic, 0.2% stearic, 0.7% palmitic, and 3.1% linoleic acids.
Langendorff-Perfused Heart Preparation
A constant pressure mode of 80 mmHg was used in the Langendorff isolated heart perfusion as previously described [3, 9-11]. Briefly, sEH−/−, A2AAR−/−, and wild-type (WT) mice (16–18 wks.) were euthanized using 100 mg/kg body weight sodium pentobarbital intra-peritoneally. Hearts were excised and instantly placed into ice-cold Krebs-Henseleit buffer containing (in mM) 1.2 KH2PO4, 11.0 glucose, 4.7 KCl, 119.0 NaCl, 1.2 MgSO4, 2.5 CaCl2, 2.0 pyruvate, 22.0 NaHCO3, 0.5 EDTA, and heparin (5 U/mL). The lungs and surrounding tissues around the heart were then removed, the aorta cannulated, and the heart continuously perfused with warm (37°C) buffer bubbled with 95% O2. Measurement of left ventricular developed pressure (LVDP) and heart rate (HR) was provided by a water-filled balloon placed into the left ventricle and connected to a pressure transducer. Coronary flow (CF) was measured by a flow transducer with an ultrasonic flow probe (Transonic Systems, Ithaca, NY). Data acquisition and recording was done by a Power–Lab Chart (AD Instruments, Colorado Springs, CO). Hearts were left to stabilize for 30–40 min before experimentation. Hearts with CF increase by less than two-fold in response to a 15-second total occlusion, as well as hearts with LVDP <80 mmHg or persistent arrhythmias were excluded from analysis.
Coronary Reactive Hyperemic Response
Upon stabilization, the valve above the cannulated heart was closed for 15 seconds to produce CRH. Pre- and post-CRH repayment volume (RV), CF, repayment duration (RD), LVDP, and HR were analyzed for all hearts. A microinjection pump (Harvard Apparatus, Holliston, MA) was used to infuse the investigational drugs for 15 minutes. A second CRH was produced and the same data collected again for the same heart. Drugs were dissolved in dimethyl sulfoxide (DMSO) and brought to the target concentration by adding PBS (Phosphate buffered saline). DMSO's final concentration was < 0.1%. Drugs' infusion rate was 1% of CF. The final concentrations, after standardization of dose (0.01, 0.1, 1, & 10 μM) response for the used drugs were 0.1 μM SCH-58261, selective A2AAR-antagonist, and 1 μM DDMS (dibromo-dodecenyl-methylsulfimide, CYP4A-blocker), and 10 μM t-AUCB (trans-4-[4-(3-adamantan-1-yl-ureido)-cyclohexyloxy]-benzoic acid (a selective sEH-inhibitor, University of California, Davis) [36]. These concentrations were less than or equal to concentrations used in earlier studies: t-AUCB, 10 μM; [3, 9, 10, 37], DDMS, 1 μM [38].
Effect of SCH-58261 (A2AAR-antagonist) on CRH Response in WT and sEH−/− Mice
The hearts from WT and sEH −/− mice were exposed to 15 seconds of occlusion. CRH parameters (RV, RD, baseline CF, HR, and LVDP) were recorded and averaged. SCH-58261's final concentration was 0.1 μM and was injected for 15 min. Then, another CRH followed and the same recordings taken again.
Effect of the sEH-inhibitor, t-AUCB, on CRH Response in A2AAR−/− Mice
Similar to the previous section, but using A2AAR−/− mice, the sEH-inhibitor, t-AUCB, was infused at 10 μM for 15 min. CRHs before and after t-AUCB infusion were analyzed and compared.
Effect of DDMS (ω-hydroxylases-inhibitor) on CRH Response in A2AAR−/− Mice
As described above, CRH in A2AAR−/− mice before and after the ω-hydroxylases-inhibitor (DDMS) treatment (at a 1 μM for 15 min) were compared.
LC–MS/MS Oxylipin Analysis
Oxylipins (EETs and their metabolites DHETs, mid-chain and ω-terminal HETEs, EpOMEs and their metabolites DiHOMEs, HODEs, and prostanoids) were measured in plasma and heart perfusate samples of A2AAR−/− mice by liquid chromatography, tandem mass spectroscopy (LC-MS/MS) as described previously [9, 39]. Briefly, we collected the heart perfusate at baseline and after occlusion. For the plasma samples, submandibular blood collection in the amount of 0.5 -0.7 mL was carried out by a trained lab staff. Each mouse was restrained and a puncture in its cheek was made with a 5.0 mm lancet. The blood was then collected in 1.5 mL–Eppendorf tubes placed on ice, gently shaken, and allowed to settle for 1 hour. After the desired amount was collected, gauze was placed on the mouse's cheek to ensure that the bleeding had stopped. Each blood sample was then centrifuged at 10,000 g for 10 min, and the supernatant was collected. Both heart perfusate and plasma samples were stored at −80°C until processing. The reader is referred to our published paper [9] for details on the quantification procedure.
Statistical and Data Analyses
The reader is referred to our paper [9] for definitions and calculations of flow debt and repayment volume. Values are presented as means ± standard error; n denotes number of animals used in experiments and analysis. Data from two independent groups were analyzed by Student t-test, whereas data from more than two groups were analyzed using two-way ANOVA. Differences were considered statistically significant when P < 0.05.
Results
CRH Response
Effect of SCH-58261 (A2AAR–antagonist) on CRH Response in WT and sEH−/− Mice
The selective A2AAR–antagonist, SCH-58261, decreased CRH in both WT and sEH−/− mice. Repayment volume (RV) was decreased in both WT mice (from 5.0 ± 0.6 to 2.3 ± 0.3 mL/g; P < 0.05) and sEH−/− mice (from 7.4 ± 0.7 to 3.4 ± 0.4 mL/g; P < 0.05, Fig 1A). Repayment/debt (R/D) ratio was also decreased in WT (from 1.3 ± 0.2 to 0.6 ± 0.1; P < 0.05) and sEH−/− mice (from 1.8 ± 0.2 to 1.3 ± 0.1; P < 0.05, Fig 1B). Repayment duration (RD) was decreased in WT (from 1.4 ± 0.3 to 0.4 ± 0.1 min; P < 0.05) and sEH−/− mice (from 2.1 ± 0.3 to 0.9 ± 0.2 min; P < 0.05, Fig 1C). Blocking A2AAR by SCH-58261 decreased baseline CF in WT (from 15.9 ± 0.7 to 13.0 ± 0.6 mL/min/g) and sEH−/− mice (from 15.0 ± 0.7 to 10.8 ± 0.8 mL/min/g; P < 0.05, Fig 1D). Comparing the untreated groups (WT vs. sEH−/− mice) demonstrated significantly increased RV, R/D ratio, and RD in sEH−/− compared to WT mice (Fig 1A-C). There was no significant difference between SCH-58261–treated WT and SCH-58261–treated sEH−/− mice in the above– mentioned parameters based on the 2-way ANOVA Analyses of Interaction between the 2 variables: mouse genotype and drug (SCH-58261) treatment. LVPD and HR were not different between and within the two groups (P > 0.05; Fig 1E-F). Due to the lack of sex difference in CRH response, we used equal ratios of male and female mice from each strain for the remainder of the study.
Fig 1. Effect of the selective A2AAR–antagonist (SCH-58261, 0.1 μM) on coronary reactive hyperemia (CRH) in WT and sEH−/− mice.
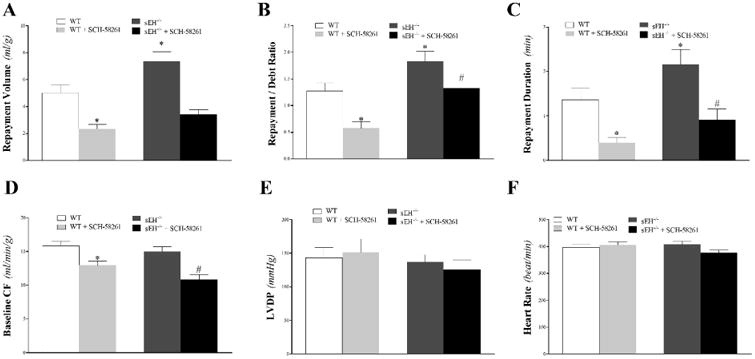
The selective A2AAR–antagonist, SCH-58261, decreased CRH in both WT and sEH−/− mice. Repayment volume (A), repayment/debt ratio (B), and repayment duration (C) were more increased in sEH−/− compared to WT mice. They, and baseline CF (D), were decreased by SCH-58261 in both WT and sEH−/− mice. No significant difference between SCH-58261–treated WT and SCH-58261–treated sEH−/− mice in the above–mentioned parameters was observed. Baseline CF (D), LVPD (E), and HR (F) were not different between and within the two groups. * P < 0.05 versus untreated WT. # P < 0.05 versus SCH-58261–treated WT. n = 8 per group.
Effect of A2AAR–deletion on CRH Response
Mice with deleted A2AAR (A2AAR−/−) had attenuated CRH compared to WT mice. Compared to WT mice, A2AAR−/− mice had decreased repayment volume by 25% (6.1 ± 0.5 and 4.9 ± 0.2 mL/g, respectively; P < 0.05, Fig 2A), decreased repayment/debt ratio (2.5 ± 0.2 and 1.9 ± 0.2, respectively; P < 0.05; Fig 2B), and decreased repayment duration (2.5 ± 0.3 and 1.2 ± 0.1 min, respectively; P < 0.05; Fig 2C). There was no statistically significant difference in LVPD, baseline CF, and HR between the two groups (P > 0.05; Fig 2D-F). Time-matched control experiments with WT mouse hearts, employing three consecutive inductions of CRH, showed no change in the CRH response and no difference in baseline heart functions, including LVDP, baseline CF, and HR (data not shown).
Fig 2. Comparison of coronary reactive hyperemia (CRH) between WT and A2AAR−/− mice.
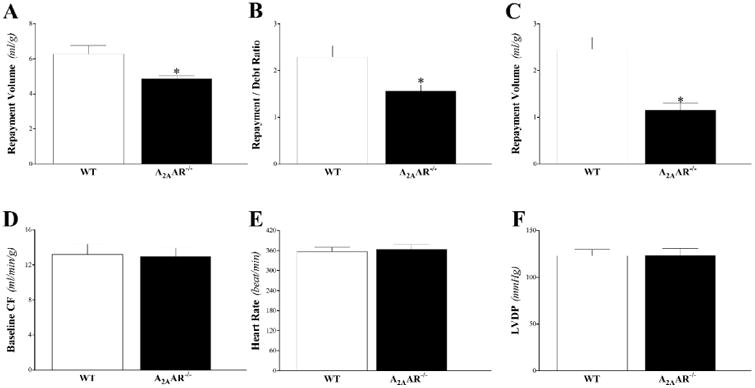
Repayment volume (A), repayment/debt ratio (B), and repayment duration (C), were decreased in A2AAR−/− compared to WT mice (P < 0.05). Baseline CF (D), LVPD (E), and HR (F) were not different between the two groups. * P < 0.05 versus A2AAR−/−. n = 8 per group.
Effect of t-AUCB (sEH-inhibitor) on CRH Response in A2AAR−/− Mice
The sEH-inhibitor, t-AUCB, enhanced CRH in A2AAR−/− mice. Repayment volume was increased by 22% (from 5.8 ± 0.5 to 7.1 ± 0.8 mL/g; P < 0.05, Fig 3A), repayment/debt ratio from 1.6 ± 0.1 to 2.0 ± 0.2 (P < 0.05, Fig 3B), and repayment duration from 1.6 ± 0.2 to 2.3 ± 0.3 min (P < 0.05, Fig 3C). There was no significant difference between t-AUCB–treated and untreated A2AAR−/− mice in baseline CF, LVPD, and HR (P > 0.05; Fig 3D-F).
Fig 3. Effect of the selective sEH-inhibitor (t-AUCB, 10 μM) on coronary reactive hyperemia (CRH) in A2AAR−/− mice.
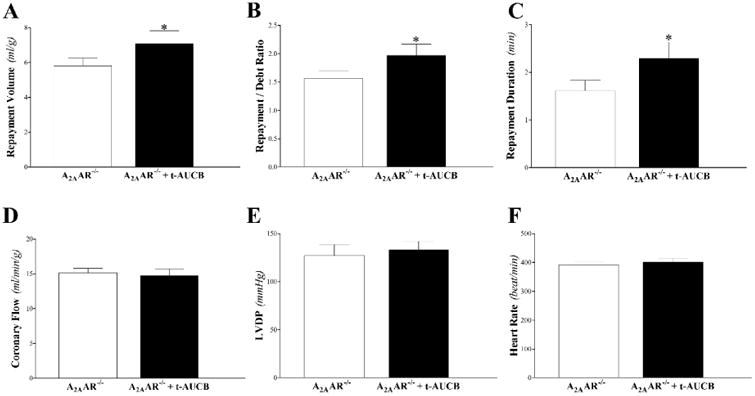
Repayment volume (A), repayment/debt ratio (B), and repayment duration (C), were enhanced in A2AAR−/− mice by t-AUCB (P < 0.05). Baseline CF (D), LVPD (E), and HR (F) were not different between the two groups. * P < 0.05 versus A2AAR−/−. n = 10 per group.
Effect of DDMS (ω-hydroxylases-inhibitor) on CRH Response
DDMS enhanced CRH in A2AAR−/− mice. Repayment volume was increased from 7.8 ± 0.7 to 11.8 ± 0.7 mL/g (P < 0.05, Fig 4A), repayment/debt ratio from 2.0 ± 0.2 to 3.0 ± 0.4 min (P < 0.05, Fig 4B), and repayment duration from 2.8 ± 0.6 to 4.6 ± 0.7 min (P < 0.05, Fig 4C). Baseline CF, LVPD, and HR were not different between the two groups (P > 0.05; Fig 4D-F).
Fig 4. Effect of the ω-hydroxylases-inhibitor (DDMS, 1 μM) on coronary reactive hyperemia (CRH) in A2AAR−/− mice.
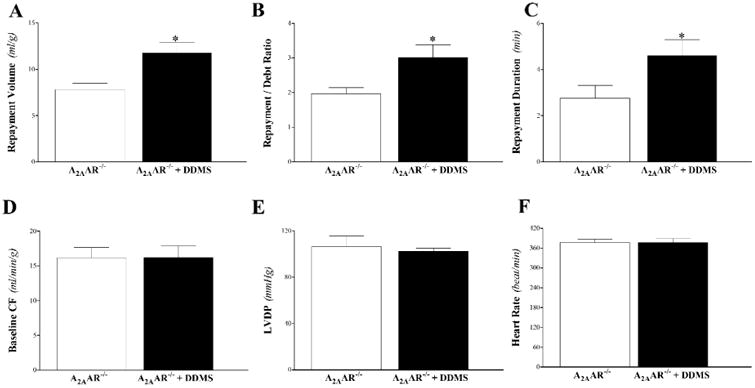
Repayment volume (A), repayment/debt ratio (B), and repayment duration (C), were enhanced in A2AAR−/− mice by DDMS (P < 0.05). Baseline CF (D), LVPD (E), and HR (F) were not different between the two groups. * P < 0.05 versus A2AAR−/−. n = 6 per group.
Oxylipin Analysis of Heart Perfusate and Plasma in WT and A2AAR−/− Mice
LC–MS/MS was used to determine oxylipin levels in heart perfusate and plasma samples.
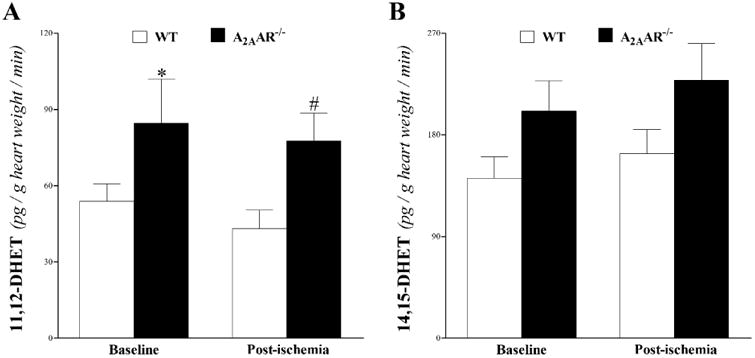
EETs and DHETs: we were able to detect 11,12-DHET and 14,15-DHET in the heart perfusate, which were increased in A2AAR−/− vs. WT mice at baseline and post-ischemia (Fig 5). However, only 11,12-DHET's increase vs. WT was statistically significant (P < 0.05; Fig 5A), whereas 14,15-DHET was not (P > 0.05; Fig 5B).
Fig 5. LC–MS/MS analysis for DHETs (11, 12–, and 14, 15–DHETs) levels in WT and A2AAR−/− mouse heart perfusate at baseline and post-ischemia.
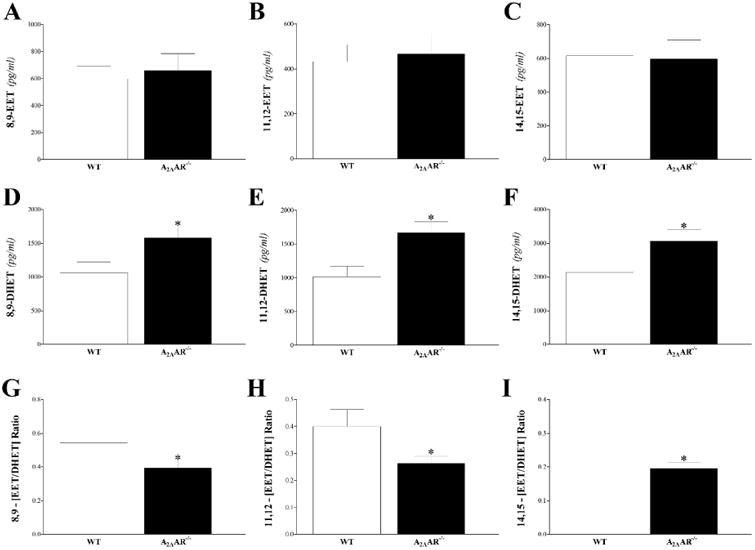
Both 11,12-, and 14,15-DHETs were increased in A2AAR−/− vs. WT mice at baseline and post-ischemia. Only 11,12-DHET's increase (A) was statistically significant. * P < 0.05 versus WT. n = 6 per group.
In plasma samples, 8,9-, 11,12-, and 14,15-EETs and their corresponding metabolites (8,9-, 11,12-, and 14,15-DHETs) were detected. The measured EETs (8,9-, 11,12-, and 14,15-EETs) were not significantly different between WT and A2AAR−/− mice, (P > 0.05; Fig 6A-C). All measured DHETs were increased in A2AAR−/− compared to WT mice (P < 0.05; Fig 6D-F). As a result, 8,9-, 11,12-, and 14,15-EET/DHET ratios decreased in A2AAR−/− compared to WT mice (P < 0.05; Fig 6G-I).
Fig 6. LC–MS/MS analysis for EETs (8, 9–, 11, 12–, and 14, 15–) and DHETs (8, 9–, 11, 12–, and 14, 15–) levels in WT and A2AAR−/− mouse plasma.
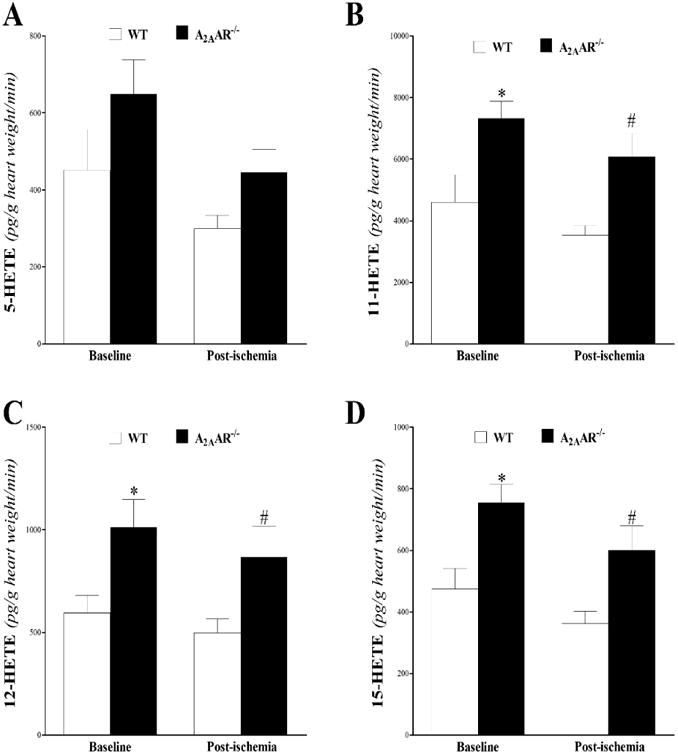
Plasma 8,9-EET (A), 11,12-EET (B), and 14,15-EET (C) were not different, whereas, 8,9-DHET (D), 11,12- DHET (E), and 14,15- DHET (F) were increased in A2AAR −/− compared to WT mice. As a result, 8,9- (G), 11,12- (H), and 14,15- (I) EET/DHET ratios decreased in A2AAR−/− compared to WT mice. * P < 0.05 versus WT. n = 10 per group.
Mid-chain HETEs: in heart perfusate, 5-, 11-, 12-, and 15-HETEs were detected in WT and A2AAR−/− mice. Baseline and post-ischemic levels of 5-, 11-, 12-, and 15-HETE were increased in A2AAR−/− compared to WT mice, and was significant for 11-, 12-, and 15-HETEs (P < 0.05; Fig 7B-D), but not for 5-HETE (P > 0.05; Fig 7A). In plasma, the same mid-chain HETEs were detected. However, only 5-, 11-, and 15-HETEs were increased in A2AAR−/− compared to WT mice (P < 0.05; Fig 8A, B and D), whereas 12-HETE was not different between the two groups (P > 0.05; Fig 8C).
Fig 7. LC–MS/MS analysis for mid-chain HETE (5-, 11-, 12-, and 15-HETE) levels in WT and A2AAR−/− mouse heart perfusate at baseline and post-ischemia.
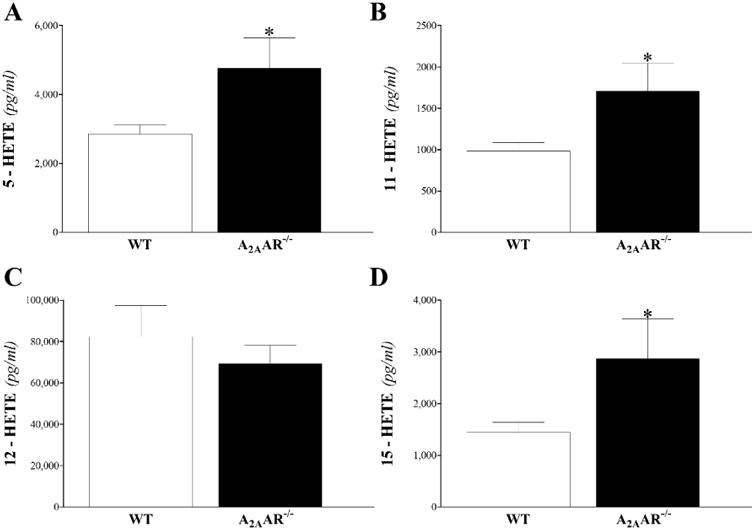
Baseline and post-ischemic levels of 5- (A), 11- (B), 12- (C), and 15- (D) HETE were increased in A2AAR−/− compared to WT mice, and was significant for 11-, 12-, and 15-HETEs. * P < 0.05 versus WT. n = 6 per group.
Fig 8. LC–MS/MS analysis for mid-chain HETE (5-, 11-, 12-, and 15-HETE) levels in WT and A2AAR−/− mouse plasma.
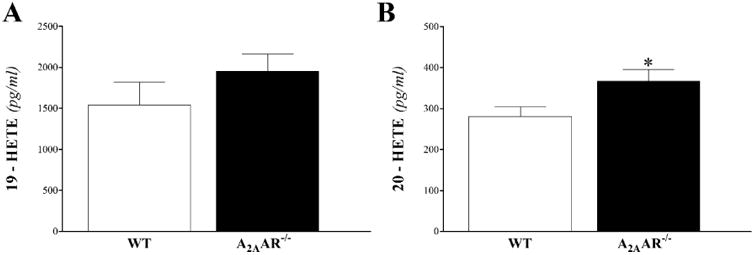
Plasma 5- (A), 11- (B), and 15- (D) HETEs, but not 12- (C) HETE, were significantly increased in A2AAR−/− compared to WT mice. * P < 0.05 versus WT. n = 9 per group.
ω–Terminal HETEs were detected only in plasma samples of WT and A2AAR−/− mice. Both 19-and 20-HETE were increased in A2AAR−/− compared to WT mice, but it was statistically significant in 20-HETE (P < 0.05; Fig 9B), not in 19-HETE (P > 0.05; Fig 9A)
Fig 9. LC–MS/MS analysis for ω–Terminal HETEs (19- and 20-HETE) levels in WT and A2AAR−/− mouse plasma.
Plasma 19-HETE (A) and 20-HETE (B) were increased in A2AAR−/− compared to WT mice, but only 20-HETE was statistically significant. * P < 0.05 versus WT. n = 9 per group.
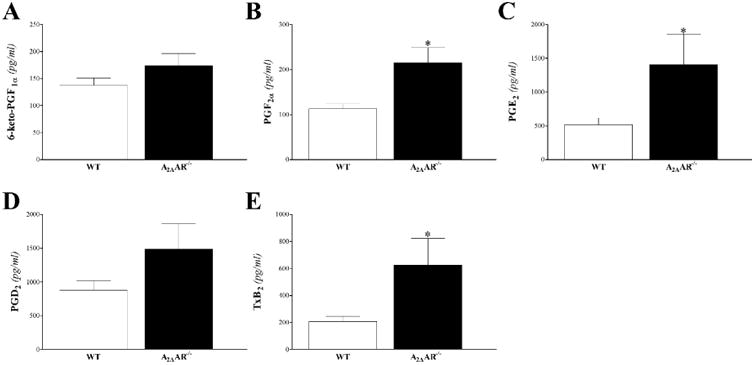
Prostanoids: 6-keto-prostaglandin (PG)-F1α, PG-F2α, PG-D2, PG-E2, thromboxane B2 (T×B2) were detected in the plasma samples (Fig 10). These prostanoids were increased in A2AAR-/compared to WT mice, but were statistically significant only for PG-F2α, PG-E2, and T×B2 (P < 0 05; Fig 10B, C and E)
Fig 10. LC-MS/MS analysis for prostanoids (6-keto-PG-F1α, PG-F2α, PG-D2, PG-E2, and T×B2) levels in WT and A2AAR−/− mouse plasma.
Plasma 6-keto-PG-F1α (A), PG-F2α (B), PG-E2 (C), PG-D2 (D), and T×B2 (E) were increased in A2AAR−/− compared to WT mice, but were significant for PG-F2α (A), PG-E2 (C), and T×B2 (E). * P < 0.05 versus WT. n = 9 per group.
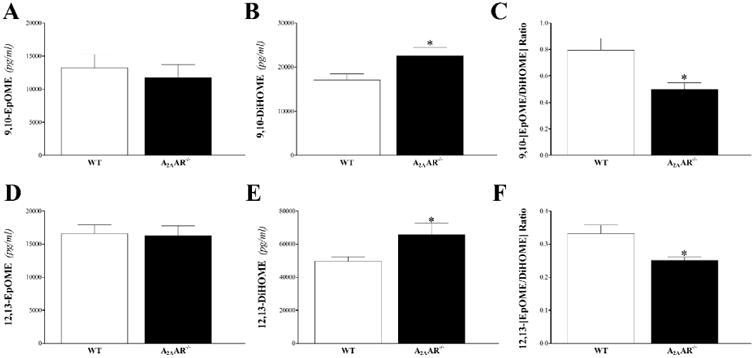
EpOMEs and DiHOMEs: these epoxides of linoleic acid were detected only in plasma samples. Compared to WT, A2AAR−/− mice had similar levels of 9,10- and 12,13-EpOMEs (P > 0.05; Fig 11A and D), increased 9,10- and 12,13-DiHOMEs (P < 0.05; Fig 11B and E), and as a result, decreased 9,10- and 12,13- EpOME/DiHOME ratios (P < 0.05; Fig 11C and F).
Fig 11. LC–MS/MS analysis for EpOME and DiHOME levels in WT and A2AAR−/− mouse plasma.
Compared to WT, A2AAR−/− mice had similar levels of plasma 9,10- (A) and 12,13-EpOMEs (D), increased 9,10- (B) and 12,13-DiHOMEs (E), and as a result, decreased 9,10- (C) and 12,13- EpOME/DiHOME ratios (F). * P < 0.05 versus WT. n = 9 per group.
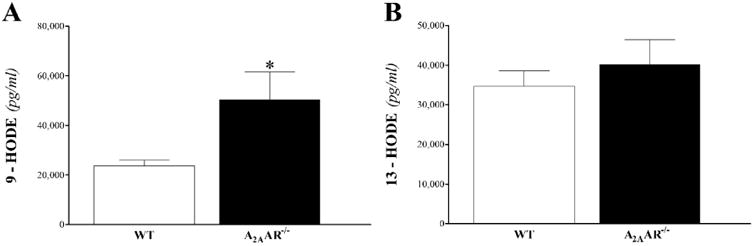
HODEs: the two hydroxylated metabolites of LA, 9- and 13-HODEs (Fig 12), were detected only in plasma samples. 9-HODE, but not 13-HODE, was increased in A2AAR −/− compared to WT mice (P < 0.05; Fig 12B).
Fig 12. LC–MS/MS analysis for HODE levels in WT and A2AAR−/− mouse plasma.
Plasma 9-HODE (A), but not 13-HODE (B), was increased in A2AAR−/− compared to WT. * P < 0.05 versus WT. n = 9 per group.
The observed results and their possible impact on CRH were summarized into a proposed schematic diagram (Fig. 15).
Table 1 lists the statistical mean values and the standard error of the mean (SEM) for the measured plasma oxylipins for the two groups of mice. Also, the observed results and their possible impact on CRH were summarized into a proposed schematic diagram (Fig. 15).
Table 1.
Oxylipin concentration in plasma samples (pg / mL) determined by LC-MS/MS in wild type (WT) and A2AAR-null (A2AAR−/−) mice.
| Oxylipin | WT | A2AAR-/- | ||||
|---|---|---|---|---|---|---|
| Arachidonic Acid-derived Oxylipins | ||||||
| 8,9-EET (pg/mL) | 598 | ± | 94 | 660 | ± | 125 |
| 11,12-EET (pg/mL) | 432 | ± | 76 | 467 | ± | 91 |
| 14,15-EET (pg/mL) | 615 | ± | 88 | 598 | ± | 110 |
| 8,9-DHET (pg/mL) | 1,062 | ± | 162 | 1,585 | ± | 150 * |
| 11,12-DHET (pg/mL) | 1,014 | ± | 155 | 1,666 | ± | 165 * |
| 14,15-DHET (pg/mL) | 2,136 | ± | 306 | 3,062 | ± | 343 * |
| 8,9- (EET/DHET) ratio | 0.56 | ± | 0.05 | 0.38 | ± | 0.03 * |
| 11,12- (EET/DHET) ratio | 0.40 | ± | 0.05 | 0.32 | ± | 0.05 * |
| 14,15- (EET/DHET) ratio | 0.30 | ± | 0.03 | 0.27 | ± | 0.05 * |
| 5-HETE (pg/mL) | 2,854 | ± | 262 | 4,772 | ± | 878 * |
| 11-HETE (pg/mL) | 981 | ± | 106 | 1,710 | ± | 334 * |
| 12-HETE (pg/mL) | 82,335 | ± | 15,143 | 69,265 | ± | 8,986 |
| 15-HETE (pg/mL) | 1,441 | ± | 197 | 2,870 | ± | 772 * |
| 19-HETE (pg/mL) | 1,542 | ± | 277 | 1,958 | ± | 204 |
| 20-HETE (pg/mL) | 281 | ± | 23 | 367 | ± | 28 * |
| 6-keto-PGF1α (pg/ml) | 138 | ± | 13 | 174 | ± | 22 |
| PGF2α (pg/ml) | 113 | ± | 11 | 215 | ± | 34 * |
| PGD2 (pg/ml) | 879 | ± | 139 | 1,488 | ± | 376 |
| PGE2 (pg/ml) | 514 | ± | 105 | 1,410 | ± | 451 * |
| T×B2 (pg/ml) | 209 | ± | 38 | 624 | ± | 198 * |
| Linoleic Acid-derived Oxylipins | ||||||
| 9,10-EpOME (pg/mL) | 13,198 | ± | 2,068 | 11,782 | ± | 1,913 |
| 12,13-EpOME (pg/mL) | 16,553 | ± | 1,375 | 16,270 | ± | 1,482 |
| 9,10-DiHOME (pg/mL) | 17,089 | ± | 1,340 | 22,560 | ± | 1,939 * |
| 12,13-DiHOME (pg/mL) | 49,477 | ± | 2,978 | 65,670 | ± | 6,909 * |
| 9,10-(EpOME/DiHOME) ratio | 0.80 | ± | 0.09 | 0.50 | ± | 0.05 * |
| 12,13-(EpOME/DiHOME) ratio | 0.33 | ± | 0.03 | 0.25 | ± | 0.01 * |
| 9-HODE (pg/mL) | 23,725 | ± | 2,327 | 50,297 | ± | 11279 * |
| 13-HODE (pg/mL) | 34,629 | ± | 3,942 | 40,159 | ± | 6,274 |
P < 0.05 versus WT. Values are means ± standard error. n = 10 per group.
Discussion
This study demonstrated that deletion of sEH (sEH −/−) is associated with increased EET levels, increased A2AAR expression, and enhanced CRH. Mice which are A2AAR–deficient (A2AAR−/−) had decreased CRH and increased expression of she, which breaks down the protective EETs to DHETs. It was shown previously that EETs mediated A2AAR–induced vasodilation. Also, change in sEH activity is associated with corresponding changes in arachidonic and linoleic acids-derived oxylipin profiles, which have been demonstrated to have vasoactive effects. One important group of the evaluated oxylipins is HETEs; both mid-chain and ω-terminal HETEs have vasoconstrictive effects and decrease CRH. In isolated mouse hearts, the relationship among A2AAR–deletion, subsequent oxylipin changes, sEH, ω-hydroxylases, and modulation of CRH is not known. Therefore, this study was designed to investigate the changes in oxylipin profiles associated with A2AAR–deficiency (A2AAR −/− mice) and the impact of sEH and ω -hydroxylases on the decreased CRH in the absence A2AAR in mice. Our data demonstrated that: 1) Blocking A2AAR (by SCH 58261) decreased CRH in both wild type and sEH –/– mice; 2) Inhibition of sEH (by t-AUCB) reversed the decrease in CRH associated with A2AAR–deletion in A2AAR−/− mice; 3) Inhibition of ω-hydroxylases (by DDMS) reversed the decrease in CRH associated with A2AAR–deletion in A2AAR−/− mice; 4) A2AAR–deletion was associated with changes in plasma oxylipin profiles, including decrease in EET/DHET ratios, increase in mid-chain and ω-terminal HETEs, decrease in EpOME/DiHOME ratios, increase in 9-HODE, and increase in prostanoids; 5) A2AAR–deletion was also associated with changes in oxylipin profiles in heart perfusate, with increase in DHET, and increase in mid-chain HETEs.
A2AAR–antagonist (SCH 58261) intensely decreased CRH in both wild type and sEH –/– mice. A2AAR mediated CRH in mice [40] and dogs [8]. SCH 58261 reduced CRH in response to 15-second ischemia in dogs [8]. Still, the effect of SCH 58261 on CRH in sEH–/– mice has not been reported. CRH is the cardiac response to brief ischemia [2]; its goal is to restore cardiac perfusion [1], and thus may protect the heart against ischemia's potential damage [9]. The importance of CRH is reiterated by several investigations which linked reduced CRH to cardiovascular pathologies affecting the coronary arteries [4-6]. We aimed to investigate the interaction between A2AAR and sEH in regards to CRH. Adenosine's effects are mediated by four distinct receptor subtypes: A1, A2A, A2B and A3 [33]. Both A2AAR and A2BAR mediate the cardioprotective effects of adenosine [27]. The relationship between adenosine A2AAR and CYP-epoxygenase–EET pathway is multifaceted. A2AAR–activation resulted in increased EETs' release [32], and A2AAR−/− (A2A-null) mice had increased expression of EETs'–metabolizing enzyme sEH [33]. Additionally, our lab has reported that sEH−/− mice had increased aortic expression of A2AAR [41] and increased EET levels [10]. At the functional level, sEH−/− mice demonstrated more aortic relaxation in response to an A2AAR–agonist, which is consistent with the increased expression of A2AAR [41], and enhanced CRH, which is in line with the increase in EET levels [10]. In this study, blocking A2AAR attenuated CRH in sEH−/− mice such that it became comparable to that of wild type. In other words, the molecular changes associated with sEH–deletion (increased EET levels and increased expression of A2AAR), which promote vasorelaxation, were defused by blocking A2AAR. Therefore, these data suggest that blocking A2AAR attenuates CRH partly through inhibiting A2AAR–induced increase in EETs' release; an observation that supports previous findings of a link between A2AAR activation and EETs' release [32, 33].
In A2AAR−/− mice, CRH was decreased in response to brief ischemia compared to WT mice. As mentioned earlier, A2AAR–activation resulted in increased EETs' release [32], and A2AAR−/− mice had increased expression of EETs'–metabolizing enzyme sEH [33]. EETs are hydrated to their corresponding metabolites, DHETs, by sEH [42]. Increased EETs or EET/DHET ratios partly mediate the enhanced CRH in sEH–deleted [10] and in sEH–inhibited mouse heart in response to brief ischemia [9]. At the same time, endothelial over-expression of human sEH decreased CRH in isolated mouse heart [3], increased conversion of EETs to DHETs, and impaired endothelium-dependent cerebral vasodilation in mice [18]. These and other reports reconfirm the beneficial cardiovascular effects of EETs [25, 42-44], including vasodilation in renal preglomerular vasculature [45], intestinal vasculature [46], and brain vessels [47], and protection against ischemia/reperfusion injury [15]. The A2AAR–EET pathway was demonstrated by several studies; EETs mediated A2AAR–induced vasodilation in rat arcuate arteries [31]. Also, A2AAR–activation was followed by increased EETs' release in isolated perfused kidney [32]. Our lab revealed yet another facet of the interaction between CYP-epoxygenase–EET pathway and adenosine A2AAR, where A2AAR−/− (A2A-null) mice had increased sEH expression compared to A2AAR+/+ (wild type) mice [33]. The increased sEH expression is a common finding between this mouse genotype, A2AAR−/−, and Tie2-sEH Tr mice, which had endothelial-specific over-expression of sEH [3]; they both had decreased CRH compared to their controls [3]. The increased expression of sEH was also associated with increased conversion of EETs to DHETs [18], which would decrease EET/DHET ratios. In this study, pharmacologic inhibition of sEH, by t-AUCB, reversed the decreased CRH observed in A2AAR−/− mice and decreased EET/DHET ratios (as mentioned below). sEH role in CRH has been repeatedly demonstrated [3, 9, 10]. Therefore, the ability of an sEH–inhibitor to reverse the decreased CRH in A2AAR−/− mice, suggests that the absence of A2A receptors in A2AAR−/− mice could be associated with increased activity of sEH; a possibility which was supported by the measured EETs and DHETs in heart perfusate and plasma as demonstrated below.
Oxylipins were analyzed in plasma and heart perfusate of A2AAR−/− and A2AAR+/+ mice. The heart perfusate data have the advantage of demonstrating the impact of ischemia on oxylipin levels as they are assessed before and after the ischemic interruption of perfusion. However, collecting the heart perfusate poses a number of challenges including subjecting the functioning heart to the air while moving it from its warm water-jacketed buffer bath to the collection beaker, which could affect its overall functions. Besides, the collection beaker has to contain warmed buffer to simulate the warm bath it is usually placed in before collection; this entails that the collected perfusate is further diluted, and probably partially accounts for the high variability we noticed in oxylipin levels in previous experiments [3, 9-11]. Additionally, the diluted heart perfusate samples lower the available oxylipins to undetectable ranges as noticed for some of the oxylipins we tried to measure in this study. Therefore, we sought to measure the oxylipin levels in the plasma. This approach allowed us to have more accurate readings of oxylipins with less variability, detected oxylipins which were undetectable in the heart perfusate, and offered a unique chance to compare the oxylipin data between the two sources: plasma and heart perfusate. In the current study, deletion of A2AAR (A2AAR−/− mice) was associated with increased levels of DHETs compared to WT mice in the heart perfusate. However, we could not detect EETs in the same samples. We experienced similar difficulty in detecting all EETs in the heart perfusate samples in previously published work [9] and this could be attributed to the short half-life of EETs [48]. DHETs are the sEH–catalyzed metabolic breakdown products of EETs. The plasma levels of both EETs and DHETs supported the findings in the heart perfusate samples; EET/DHET ratio was decreased by the elevated DHET levels. We earlier reported that global deletion (sEH–/– mice) [9] and pharmacologic inhibition of sEH [9] decreased DHETs in heart perfusate. The increased expression of sEH reported in A2AAR−/− mice [33] is probably what caused the observed increase in DHETs' generation in our study and drove the decrease in EET/DHET ratios in this mouse genotype. EETs have anti-inflammatory properties [49]. In mouse carotid arteries, EETs prevented leukocyte adhesion and decreased the number of adherent mononuclear cells to the vascular wall by inhibiting the transcription of NF-κB following TNF-α injection; an independent effect of their membrane-hyperpolarizing effects [50]. This observation in A2AAR−/− mice, along with the other plasma oxylipin data below, is interesting since it suggests a possible decrease in the anti-inflammatory role of EETs. Therefore, the attenuated CRH in A2AAR−/− mice could be partially due to decreased EET/DHET ratio and increased DHETs, which could be attributed to the increased expression of EETs' metabolizing enzyme (sEH) and may indicate an overall reduction in EETs vasodilatory and anti-inflammatory activity in A2AAR−/− mice.
Mid-chain (5-, 8-, 11-, 12- and 15-) HETE levels were increased in A2AAR−/− compared to WT mice. Lipoxygenase produces mid-chain HETEs by allylic oxidation from AA [24]. CYP-epoxygenase 1B1 also produces mid-chain HETEs by bis-allylic oxidation [51]. Mid-chain HETEs have pro-inflammatory and vasoconstrictive effects [51, 52]. Also, cardiovascular dysfunction was associated with increased formation of mid-chain HETEs [53-56]. Both of our samples, heart perfusate and plasma, demonstrated increase in mid-chain HETEs in A2AAR−/− compared to WT mice. Based on that, the increase in mid-chain HETEs in A2AAR−/− mice, along with the decreased EET/DHET ratios, indicates an increase in the proinflammatory state associated with A2AAR–deletion, and may have contributed to the decreased CRH in A2AAR−/− mice.
Like mid-chain HETEs, ω-terminal HETEs are generated from AA, but through CYP ω-hydroxylases, primarily CYP4A and CYP4F subfamilies [19]. The primary metabolite of ω-terminal HETEs is 20-HETE [19], which promotes hypertension, vasoconstriction, and vascular dysfunction [20, 21]. Waldman et al. demonstrated that blocking 20-HETE synthesis reduced mean arterial pressure in old spontaneously hypertensive female rats [57]. We have recently reported that inhibiting ω-hydroxylases by DDMS enhanced CRH in sEH-overexpressed (Tie2-sEH Tr) and control mice. Similarly, targeting the ω-terminal-HETEs pathway in this study reversed the decreased CRH in A2AAR−/− mice. Interestingly, our lab reported that higher levels of the 20-HETE-generating enzyme, CYP4A, were expressed in the aorta of A2AAR −/− compared to A2AAR+/+ mice [58]. Therefore, ω-hydroxylases–inhibition reversed the decreased CRH in A2AAR−/− mice, which is consistent with the reported increased expression of CYP4A in the same mice [58]. We then went further to investigate the impact of CYP4A's increased expression on the levels of ω-terminal HETEs (19- and 20-HETEs), but they were too low to be detected in the heart perfusate by our technique, which we encountered and reported earlier [3]. Fortunately, we were able to detect these ω-terminal HETEs in plasma samples, and observed increased levels of ω-terminal HETEs, particularly 20-HETE. This observation may account for ω-hydroxylase–inhibitor's efficacy to reverse the decreased CRH in A2AAR–deficient mice. Therefore, the increased 20-HETE in A2AAR−/− mice, along with the other changes in oxylipins, suggests an increase in the proinflammatory state associated with A2AAR–deficiency and may have contributed to the attenuated CRH in A2AAR−/− mice.
Deletion of A2AAR in A2AAR−/− mice was associated with increased plasma prostanoid levels, including PG-F2α, PG-E2, PG-D2, and T×B2. Prostanoids include two groups of metabolites: thromboxanes (T×A2 and T×B2) and the four main bioactive prostaglandins (PG-F2α, PG-E2, PG-I2, PG-D2) [59, 60]. Prostaglandins are generally pro-inflammatory [60]. For instance, microvascular permeability was increased by PG-E2 [60]. 6-keto-PG-F1α, the inactive metabolite of prostacyclin (PG-I2), was not different between A2AAR−/− and WT mice [60]. T×B2 is the inactive degradation product of T×A2, which mediates platelet aggregation, smooth muscle contraction, and endothelial inflammation [60]. Since sEH-expression was higher in A2AAR−/− mice [58], the observed increase in prostanoids is in agreement with our, as well as others', reported data that disrupting sEH activity, through pharmacologic inhibition or genetic deletion, was associated with decreased prostanoids [9, 61, 62]. Still, neither could we infer a relationship between prostanoid levels and CRH [3, 9, 10] nor did Hellmann et al., who suggested that PGs were not involved in post-occlusive reactive hyperemia in the skin [63]. Therefore, the increased prostanoid levels in A2AAR−/− mice may have contributed to an increase in the proinflammatory state in A2AAR−/− mice, but its impact on the attenuated CRH is yet to be determined.
The role of sEH in cardiovascular biology extends beyond its role in EET hydrolysis; it plays a central role in the metabolism of other arachidonic-, linoleic- and omega-3-derived oxylipins [24]. For example, EpOMEs, whose parental fatty acid is linoleic acid, are hydrolyzed to DiHOMEs by sEH. We reported earlier that global deletion of sEH (sEH−/− mice) [9] and pharmacologic inhibition of sEH by t-AUCB [9] increased EpOMEs, decreased DiHOMEs, and increased EpOME/DiHOME ratio in heart perfusate. In these two published studies, the increased EpOME/DiHOME ratio was believed to contribute to the enhancement of CRH [9], which suggested that increased EpOME/DiHOME ratio may have had a positive role in mediating vasodilation. A notion supported by other reports demonstrating a protective effect against hypoxia/reoxygenation injury by EpOMEs in primary cultures of rabbit renal proximal tubular cells; an effect which was unattainable with DiHOMEs [23]. On the other hand, endothelium- dependent vasodilation in the cerebral circulation was impaired by decreased EpOME/DiHOME ratio in Tie2-sEH Tr mice [18]. Likewise, in this study, A2AAR−/− mice had decreased EpOME/DiHOME ratio driven by increased DiHOMEs in the plasma. DiHOMEs were reported to have deleterious properties, including cytotoxic, cardiodepressive and vasoconstrictive [17, 25]. Therefore, the decreased EpOME/DiHOME ratio and increased DiHOMEs in A2AAR−/− mice may have contributed to the attenuated CRH in A2AAR−/− mice.
The other oxylipins derived from linoleic acid (LA), in addition to EpOMEs, are HODEs, which are generated through hydroxylation of LA by CYP epoxygenases or lipoxygenases [24]. 9-HODE, but not 13-HODE, was increased in A2AAR−/− compared to A2AAR+/+ mice. Although the physiologic functions of HODEs are not widely investigated [24], 9-HODE is thought to be pro-inflammatory [64, 65], whereas13-HODE could be anti-inflammatory [66-70]. The opposite effects of these two isomers makes this observation consistent with earlier reported finding by our lab of increased 13–HODE level in sEH–deleted (sEH−/−) mice [9]. The increased 9-HODE in A2AAR−/− mice may be linked to the reported increase in sEH-expression in A2AAR−/− mice [33]. Thus, the increased 9-HODE in A2AAR−/− mice possibly have contributed to the attenuated CRH in A2AAR−/− mice.
Conclusion
The findings of this study suggest that the previously established link between A2AAR– activation and EETs' release in the aortic tissue is also valid in the coronary arteries. Also, our data demonstrated that the decreased CRH associated with A2AAR–deficiency can be reversed by augmenting the CYP-epoxygenase–EETs pathway (through sEH–inhibition) and by blocking the ω-hydroxylase pathway (through CYP4A–inhibition). To the best of our knowledge, this is the first time oxylipins are evaluated in A2AAR−/− mice from the perspective of coronary reactive hyperemia. The changes in heart perfusate and plasma oxylipins were consistent and may partly be responsible for the reduced CRH, including decreased EET/DHET ratio, increased mid-chain and ω-terminal HETEs, decreased EpOME/DiHOME ratio, and increased 9-HODE. Also, these changes in oxylipins, along with the increase in prostanoids, suggest an increased proinflammatory state in A2AAR −/− mice.
Fig 13. A schematic diagram comparing the changes in heart perfusate and plasma oxylipin profiles observed in response to A2AAR-deletion as well as their possible effect on coronary reactive hyperemia (CRH) in WT and A2AAR−/− mice.
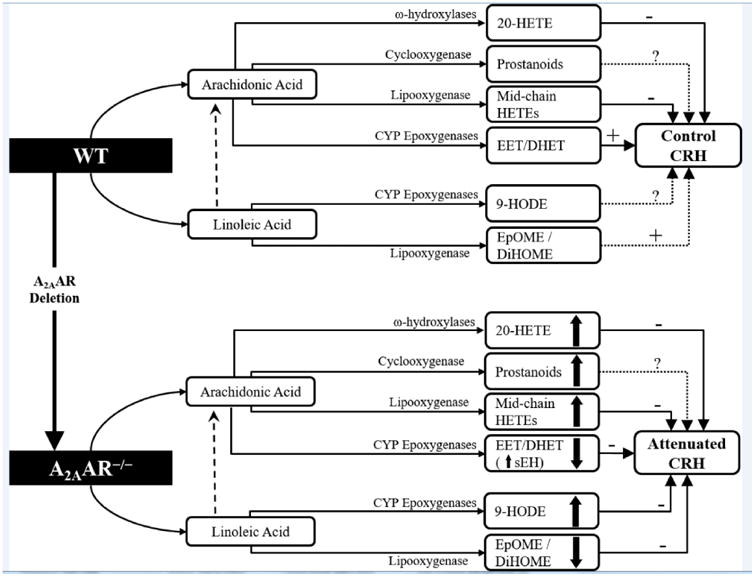
The observed changes in the measured heart perfusate and plasma oxylipin profiles collectively resulted in attenuated CRH in A2AAR−/− compared to WT mice. These changes included increased plasma 20-HETE, increased heart perfusate and plasma mid-chain HETEs, decreased heart perfusate and plasma EET/DHET ratio, increased plasma 9-HODE, and decreased plasma EpOME/DiHOME ratio. It is not clear what impact the increased prostanoid levels has on CRH, but it may, along with the other changes in oxylipins, contribute to the increased proinflammatory state in A2AAR−/− mice.
Highlights.
Adenosine A2AAR is involved in the cardiprotective coronary reactive hyperemia (CRH) in response to ischemia.
Soluble epoxide hydrolase (sEH) breaks down epoxyeicosatrienoic acids (EETs), which are cardioprotective metabolites.
A2AAR deletion is associated with changed oxylipin profiles, which were consistent between plasma and heart perfusate samples, and indicate an increased proinflammatory state including increased ω-terminal HETEs, decreased epoxyoctadecaenoic / dihydroxyoctadecaenoic acids ratios, increased 9-hydroxyoctadecadienoic acid, and increased prostanoids.
Inhibition of either sEH or ω-hydroxylases reversed the reduced CRH in A2AAR −/− mice.
Acknowledgments
This work was supported by National Institutes of Health's grant HL-114559 to M. A. Nayeem, and the Intramural Research Program of the NIH, National Institute of Environmental Health Sciences grant Z01 ES025034 to D. C. Zeldin.
Footnotes
The authors report no conflicts of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Monsuez JJ. [Mediators of reactive hyperemia] Archives des maladies du coeur et des vaisseaux. 2001;94:591–599. [PubMed] [Google Scholar]
- 2.Coffman JD, Gregg DE. Reactive hyperemia characteristics of the myocardium. Am J Physiol. 1960;199:1143–1149. doi: 10.1152/ajplegacy.1960.199.6.1143. [DOI] [PubMed] [Google Scholar]
- 3.Hanif A, Edin ML, Zeldin DC, Morisseau C, Falck JR, Nayeem MA. Vascular Endothelial Over-Expression of Human Soluble Epoxide Hydrolase (Tie2-sEH Tr) Attenuates Coronary Reactive Hyperemia in Mice: Role of Oxylipins and omega-Hydroxylases. PLoS One. 2017;12 doi: 10.1371/journal.pone.0169584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Borbouse L, Dick GM, Payne GA, Berwick ZC, Neeb ZP, Alloosh M, Bratz IN, Sturek M, Tune JD. Metabolic syndrome reduces the contribution of K+ channels to ischemic coronary vasodilation. Am J Physiol Heart Circ Physiol. 2010;298:H1182–H1189. doi: 10.1152/ajpheart.00888.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kingsbury MP, Turner MA, Flores NA, Bovill E, Sheridan DJ. Endogenous and exogenous coronary vasodilatation are attenuated in cardiac hypertrophy: a morphological defect? J Mol Cell Cardiol. 2000;32:527–538. doi: 10.1006/jmcc.1999.1097. [DOI] [PubMed] [Google Scholar]
- 6.Huang AL, Silver AE, Shvenke E, Schopfer DW, Jahangir E, Titas MA, Shpilman A, Menzoian JO, Watkins MT, Raffetto JD, Gibbons G, Woodson J, Shaw PM, Dhadly M, Eberhardt RT, Keaney JF, Jr, Gokce N, Vita JA. Predictive value of reactive hyperemia for cardiovascular events in patients with peripheral arterial disease undergoing vascular surgery. Arterioscler Thromb Vasc Biol. 2007;27:2113–2119. doi: 10.1161/ATVBAHA.107.147322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kitakaze M, Hori M, Takashima S, Iwai K, Sato H, Inoue M, Kitabatake A, Kamada T. Superoxide dismutase enhances ischemia-induced reactive hyperemic flow and adenosine release in dogs. A role of 5′-nucleotidase activity. Circ Res. 1992;71:558–566. doi: 10.1161/01.res.71.3.558. [DOI] [PubMed] [Google Scholar]
- 8.Berwick ZC, Payne GA, Lynch B, Dick GM, Sturek M, Tune JD. Contribution of adenosine A(2A) and A(2B) receptors to ischemic coronary dilation: role of K(V) and K(ATP) channels. Microcirculation. 2010;17:600–607. doi: 10.1111/j.1549-8719.2010.00054.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hanif A, Edin ML, Zeldin DC, Morisseau C, Nayeem MA. Effect of Soluble Epoxide Hydrolase on the Modulation of Coronary Reactive Hyperemia: Role of Oxylipins and PPARgamma. PLoS One. 2016;11 doi: 10.1371/journal.pone.0162147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hanif AM, Edin ML, Zeldin DC, Morisseau C, Nayeem MA. Deletion of Soluble Epoxide Hydrolase Enhances Coronary Reactive Hyperemia in Isolated Mouse Heart: Role of Oxylipins and PPARgamma. Am J Physiol Regul Integr Comp Physiol. 2016;3:1–22. doi: 10.1152/ajpregu.00237.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hanif A, Edin ML, Zeldin DC, Morisseau C, Falck JR, Nayeem MA. Vascular endothelial overexpression of human CYP2J2 (Tie2-CYP2J2 Tr) modulates cardiac oxylipin profiles and enhances coronary reactive hyperemia in mice. PLoS One. 2017;12:e0174137. doi: 10.1371/journal.pone.0174137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gabbs M, Leng S, Devassy JG, Monirujjaman M, Aukema HM. Advances in Our Understanding of Oxylipins Derived from Dietary PUFAs. Adv Nutr. 2015;6:513–540. doi: 10.3945/an.114.007732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Campbell WB, Gebremedhin D, Pratt PF, Harder DR. Identification of epoxyeicosatrienoic acids as endothelium-derived hyperpolarizing factors. Circ Res. 1996;78:415–423. doi: 10.1161/01.res.78.3.415. [DOI] [PubMed] [Google Scholar]
- 14.Ellinsworth DC, Shukla N, Fleming I, Jeremy JY. Interactions between thromboxane A(2), thromboxane/prostaglandin (TP) receptors, and endothelium-derived hyperpolarization. Cardiovasc Res. 2014;102:9–16. doi: 10.1093/cvr/cvu015. [DOI] [PubMed] [Google Scholar]
- 15.Seubert J, Yang B, Bradbury JA, Graves J, Degraff LM, Gabel S, Gooch R, Foley J, Newman J, Mao L, Rockman HA, Hammock BD, Murphy E, Zeldin DC. Enhanced postischemic functional recovery in CYP2J2 transgenic hearts involves mitochondrial ATP-sensitive K+ channels and p42/p44 MAPK pathway. Circ Res. 2004;95:506–514. doi: 10.1161/01.RES.0000139436.89654.c8. [DOI] [PubMed] [Google Scholar]
- 16.Nayeem MA, Zeldin DC, Boegehold MA, Falck JR. Salt modulates vascular response through adenosine A(2A) receptor in eNOS-null mice: role of CYP450 epoxygenase and soluble epoxide hydrolase. Mol Cell Biochem. 2011;350:101–111. doi: 10.1007/s11010-010-0686-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Edin ML, Wang Z, Bradbury JA, Graves JP, Lih FB, DeGraff LM, Foley JF, Torphy R, Ronnekleiv OK, Tomer KB, Lee CR, Zeldin DC. Endothelial expression of human cytochrome P450 epoxygenase CYP2C8 increases susceptibility to ischemia-reperfusion injury in isolated mouse heart. Faseb J. 2011;25:3436–3447. doi: 10.1096/fj.11-188300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang W, Davis CM, Edin ML, Lee CR, Zeldin DC, Alkayed NJ. Role of endothelial soluble epoxide hydrolase in cerebrovascular function and ischemic injury. PLoS One. 2013;8 doi: 10.1371/journal.pone.0061244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hoopes SL, Garcia V, Edin ML, Schwartzman ML, Zeldin DC. Vascular actions of 20-HETE. Prostaglandins Other Lipid Mediat. 2015;120:9–16. doi: 10.1016/j.prostaglandins.2015.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dunn KM, Renic M, Flasch AK, Harder DR, Falck J, Roman RJ. Elevated production of 20-HETE in the cerebral vasculature contributes to severity of ischemic stroke and oxidative stress in spontaneously hypertensive rats. Am J Physiol Heart Circ Physiol. 2008;295:24. doi: 10.1152/ajpheart.00512.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cheng J, Ou JS, Singh H, Falck JR, Narsimhaswamy D, Pritchard KA, Jr, Schwartzman ML. 20-hydroxyeicosatetraenoic acid causes endothelial dysfunction via eNOS uncoupling. Am J Physiol Heart Circ Physiol. 2008;294:H1018–H1026. doi: 10.1152/ajpheart.01172.2007. [DOI] [PubMed] [Google Scholar]
- 22.Hou HH, Hammock BD, Su KH, Morisseau C, Kou YR, Imaoka S, Oguro A, Shyue SK, Zhao JF, Lee TS. N-terminal domain of soluble epoxide hydrolase negatively regulates the VEGF-mediated activation of endothelial nitric oxide synthase. Cardiovasc Res. 2012;93:120–129. doi: 10.1093/cvr/cvr267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nowak G, Grant DF, Moran JH. Linoleic acid epoxide promotes the maintenance of mitochondrial function and active Na+ transport following hypoxia. Toxicol Lett. 2004;147:161–175. doi: 10.1016/j.toxlet.2003.11.002. [DOI] [PubMed] [Google Scholar]
- 24.Konkel A, Schunck WH. Role of cytochrome P450 enzymes in the bioactivation of polyunsaturated fatty acids. Biochim Biophys Acta. 2011;1:210–222. doi: 10.1016/j.bbapap.2010.09.009. [DOI] [PubMed] [Google Scholar]
- 25.Moghaddam MF, Grant DF, Cheek JM, Greene JF, Williamson KC, Hammock BD. Bioactivation of leukotoxins to their toxic diols by epoxide hydrolase. Nat Med. 1997;3:562–566. doi: 10.1038/nm0597-562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Linden J. Adenosine in tissue protection and tissue regeneration. Molecular pharmacology. 2005;67:1385–1387. doi: 10.1124/mol.105.011783. [DOI] [PubMed] [Google Scholar]
- 27.Zhai YJ, Liu P, He HR, Zheng XW, Wang Y, Yang QT, Dong YL, Lu J. The association of ADORA2A and ADORA2B polymorphisms with the risk and severity of chronic heart failure: a case-control study of a northern Chinese population. International journal of molecular sciences. 2015;16:2732–2746. doi: 10.3390/ijms16022732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.de Lera Ruiz M, Lim YH, Zheng J. Adenosine A2A receptor as a drug discovery target. J Med Chem. 2014;57:3623–3650. doi: 10.1021/jm4011669. [DOI] [PubMed] [Google Scholar]
- 29.Johnston-Cox HA, Koupenova M, Ravid K. A2 adenosine receptors and vascular pathologies. Arterioscler Thromb Vasc Biol. 2012;32:870–878. doi: 10.1161/ATVBAHA.112.246181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Renda G, Zimarino M, Antonucci I, Tatasciore A, Ruggieri B, Bucciarelli T, Prontera T, Stuppia L, De Caterina R. Genetic determinants of blood pressure responses to caffeine drinking. The American journal of clinical nutrition. 2012;95:241–248. doi: 10.3945/ajcn.111.018267. [DOI] [PubMed] [Google Scholar]
- 31.Cheng MK, Doumad AB, Jiang H, Falck JR, McGiff JC, Carroll MA. Epoxyeicosatrienoic acids mediate adenosine-induced vasodilation in rat preglomerular microvessels (PGMV) via A2A receptors. Br J Pharmacol. 2004;141:441–448. doi: 10.1038/sj.bjp.0705640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Carroll MA. Role of the adenosine(2A) receptor-epoxyeicosatrienoic acid pathway in the development of salt-sensitive hypertension. Prostaglandins Other Lipid Mediat. 2012;98:39–47. doi: 10.1016/j.prostaglandins.2011.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pradhan I, Ledent C, Mustafa SJ, Morisseau C, Nayeem MA. High salt diet modulates vascular response in AAR and A AR mice: role of sEH, PPARgamma, and K channels. Mol Cell Biochem. 2015;5:87–96. doi: 10.1007/s11010-015-2368-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ledent C, Vaugeois JM, Schiffmann SN, Pedrazzini T, El Yacoubi M, Vanderhaeghen JJ, Costentin J, Heath JK, Vassart G, Parmentier M. Aggressiveness, hypoalgesia and high blood pressure in mice lacking the adenosine A2a receptor. Nature. 1997;388:674–678. doi: 10.1038/41771. [DOI] [PubMed] [Google Scholar]
- 35.Sinal CJ, Miyata M, Tohkin M, Nagata K, Bend JR, Gonzalez FJ. Targeted disruption of soluble epoxide hydrolase reveals a role in blood pressure regulation. J Biol Chem. 2000;275:40504–40510. doi: 10.1074/jbc.M008106200. [DOI] [PubMed] [Google Scholar]
- 36.Hwang SH, Tsai HJ, Liu JY, Morisseau C, Hammock BD. Orally bioavailable potent soluble epoxide hydrolase inhibitors. J Med Chem. 2007;50:3825–3840. doi: 10.1021/jm070270t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cai Z, Zhao G, Yan J, Liu W, Feng W, Ma B, Yang L, Wang JA, Tu L, Wang DW. CYP2J2 overexpression increases EETs and protects against angiotensin II-induced abdominal aortic aneurysm in mice. J Lipid Res. 2013;54:1448–1456. doi: 10.1194/jlr.M036533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lukaszewicz KM, Paudyal MP, Falck JR, Lombard JH. Role of vascular reactive oxygen species in regulating cytochrome P450-4A enzyme expression in dahl salt-sensitive rats. Microcirculation. 2016;18:12304. doi: 10.1111/micc.12304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lee CR, Imig JD, Edin ML, Foley J, DeGraff LM, Bradbury JA, Graves JP, Lih FB, Clark J, Myers P, Perrow AL, Lepp AN, Kannon MA, Ronnekleiv OK, Alkayed NJ, Falck JR, Tomer KB, Zeldin DC. Endothelial expression of human cytochrome P450 epoxygenases lowers blood pressure and attenuates hypertension-induced renal injury in mice. Faseb J. 2010;24:3770–3781. doi: 10.1096/fj.10-160119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zatta AJ, Headrick JP. Mediators of coronary reactive hyperaemia in isolated mouse heart. Br J Pharmacol. 2005;144:576–587. doi: 10.1038/sj.bjp.0706099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nayeem MA, Pradhan I, Mustafa SJ, Morisseau C, Falck JR, Zeldin DC. Adenosine A2A receptor modulates vascular response in soluble epoxide hydrolase-null mice through CYP-epoxygenases and PPARgamma. Am J Physiol Regul Integr Comp Physiol. 2013;304:R23–32. doi: 10.1152/ajpregu.00213.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fang X, Weintraub NL, McCaw RB, Hu S, Harmon SD, Rice JB, Hammock BD, Spector AA. Effect of soluble epoxide hydrolase inhibition on epoxyeicosatrienoic acid metabolism in human blood vessels. Am J Physiol Heart Circ Physiol. 2004;287:H2412–H2420. doi: 10.1152/ajpheart.00527.2004. [DOI] [PubMed] [Google Scholar]
- 43.Node K, Ruan XL, Dai J, Yang SX, Graham L, Zeldin DC, Liao JK. Activation of Galpha s mediates induction of tissue-type plasminogen activator gene transcription by epoxyeicosatrienoic acids. J Biol Chem. 2001;276:15983–15989. doi: 10.1074/jbc.M100439200. [DOI] [PubMed] [Google Scholar]
- 44.Fang X, Kaduce TL, Weintraub NL, Harmon S, Teesch LM, Morisseau C, Thompson DA, Hammock BD, Spector AA. Pathways of epoxyeicosatrienoic acid metabolism in endothelial cells. Implications for the vascular effects of soluble epoxide hydrolase inhibition. J Biol Chem. 2001;276:14867–14874. doi: 10.1074/jbc.M011761200. [DOI] [PubMed] [Google Scholar]
- 45.Imig JD, Navar LG, Roman RJ, Reddy KK, Falck JR. Actions of epoxygenase metabolites on the preglomerular vasculature. J Am Soc Nephrol. 1996;7:2364–2370. doi: 10.1681/ASN.V7112364. [DOI] [PubMed] [Google Scholar]
- 46.Proctor KG, Falck JR, Capdevila J. Intestinal vasodilation by epoxyeicosatrienoic acids: arachidonic acid metabolites produced by a cytochrome P450 monooxygenase. Circ Res. 1987;60:50–59. doi: 10.1161/01.res.60.1.50. [DOI] [PubMed] [Google Scholar]
- 47.Gebremedhin D, Ma YH, Falck JR, Roman RJ, VanRollins M, Harder DR. Mechanism of action of cerebral epoxyeicosatrienoic acids on cerebral arterial smooth muscle. Am J Physiol. 1992;263:H519–525. doi: 10.1152/ajpheart.1992.263.2.H519. [DOI] [PubMed] [Google Scholar]
- 48.Catella F, Lawson JA, Fitzgerald DJ, Fitz Gerald GA. Endogenous biosynthesis of arachidonic acid epoxides in humans: increased formation in pregnancy-induced hypertension. Proc Natl Acad Sci U S A. 1990;87:5893–5897. doi: 10.1073/pnas.87.15.5893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Imig JD. Epoxides and soluble epoxide hydrolase in cardiovascular physiology. Physiol Rev. 2012;92:101–130. doi: 10.1152/physrev.00021.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Node K, Huo Y, Ruan X, Yang B, Spiecker M, Ley K, Zeldin DC, Liao JK. Anti-inflammatory properties of cytochrome P450 epoxygenase-derived eicosanoids. Science (New York, NY) 1999;285:1276–1279. doi: 10.1126/science.285.5431.1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Maayah ZH, El-Kadi AO. The role of mid-chain hydroxyeicosatetraenoic acids in the pathogenesis of hypertension and cardiac hypertrophy. Arch Toxicol. 2016;90:119–136. doi: 10.1007/s00204-015-1620-8. [DOI] [PubMed] [Google Scholar]
- 52.Burhop KE, Selig WM, Malik AB. Monohydroxyeicosatetraenoic acids (5-HETE and 15-HETE) induce pulmonary vasoconstriction and edema. Circ Res. 1988;62:687–698. doi: 10.1161/01.res.62.4.687. [DOI] [PubMed] [Google Scholar]
- 53.Conrad DJ, Kuhn H, Mulkins M, Highland E, Sigal E. Specific inflammatory cytokines regulate the expression of human monocyte 15-lipoxygenase. Proc Natl Acad Sci U S A. 1992;89:217–221. doi: 10.1073/pnas.89.1.217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Stern N, Yanagawa N, Saito F, Hori M, Natarajan R, Nadler J, Tuck M. Potential role of 12 hydroxyeicosatetraenoic acid in angiotensin II-induced calcium signal in rat glomerulosa cells. Endocrinology. 1993;133:843–847. doi: 10.1210/endo.133.2.8344221. [DOI] [PubMed] [Google Scholar]
- 55.Wen Y, Nadler JL, Gonzales N, Scott S, Clauser E, Natarajan R. Mechanisms of ANG II-induced mitogenic responses: role of 12-lipoxygenase and biphasic MAP kinase. Am J Physiol. 1996;271:C1212–1220. doi: 10.1152/ajpcell.1996.271.4.C1212. [DOI] [PubMed] [Google Scholar]
- 56.Patricia MK, Kim JA, Harper CM, Shih PT, Berliner JA, Natarajan R, Nadler JL, Hedrick CC. Lipoxygenase products increase monocyte adhesion to human aortic endothelial cells. Arterioscler Thromb Vasc Biol. 1999;19:2615–2622. doi: 10.1161/01.atv.19.11.2615. [DOI] [PubMed] [Google Scholar]
- 57.Waldman M, Peterson SJ, Arad M, Hochhauser E. The role of 20-HETE in cardiovascular diseases and its risk factors. Prostaglandins Other Lipid Mediat. 2016;7:30033–30038. doi: 10.1016/j.prostaglandins.2016.05.007. [DOI] [PubMed] [Google Scholar]
- 58.Pradhan I, Zeldin DC, Ledent C, Mustafa JS, Falck JR, Nayeem MA. High salt diet exacerbates vascular contraction in the absence of adenosine A(2)A receptor. J Cardiovasc Pharmacol. 2014;63:385–394. doi: 10.1097/FJC.0000000000000058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ricciotti E, Fitz Gerald GA. Prostaglandins and inflammation. Arterioscler Thromb Vasc Biol. 2011;31:986–1000. doi: 10.1161/ATVBAHA.110.207449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Tam VC. Lipidomic profiling of bioactive lipids by mass spectrometry during microbial infections. Semin Immunol. 2013;25:240–248. doi: 10.1016/j.smim.2013.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Schmelzer KR, Inceoglu B, Kubala L, Kim IH, Jinks SL, Eiserich JP, Hammock BD. Enhancement of antinociception by coadministration of nonsteroidal anti-inflammatory drugs and soluble epoxide hydrolase inhibitors. Proc Natl Acad Sci U S A. 2006;103:13646–13651. doi: 10.1073/pnas.0605908103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Liu Y, Zhang Y, Schmelzer K, Lee TS, Fang X, Zhu Y, Spector AA, Gill S, Morisseau C, Hammock BD, Shyy JY. The antiinflammatory effect of laminar flow: the role of PPARgamma, epoxyeicosatrienoic acids, and soluble epoxide hydrolase. Proc Natl Acad Sci U S A. 2005;102:16747–16752. doi: 10.1073/pnas.0508081102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hellmann M, Gaillard-Bigot F, Roustit M, Cracowski JL. Prostanoids are not involved in postocclusive reactive hyperaemia in human skin. Fundam Clin Pharmacol. 2015;29:510–516. doi: 10.1111/fcp.12135. [DOI] [PubMed] [Google Scholar]
- 64.Obinata H, Izumi T. G2A as a receptor for oxidized free fatty acids. Prostaglandins Other Lipid Mediat. 2009;89:66–72. doi: 10.1016/j.prostaglandins.2008.11.002. [DOI] [PubMed] [Google Scholar]
- 65.Hattori T, Obinata H, Ogawa A, Kishi M, Tatei K, Ishikawa O, Izumi T. G2A plays proinflammatory roles in human keratinocytes under oxidative stress as a receptor for 9-hydroxyoctadecadienoic acid. J Invest Dermatol. 2008;128:1123–1133. doi: 10.1038/sj.jid.5701172. [DOI] [PubMed] [Google Scholar]
- 66.Emerson MR, LeVine SM. Experimental allergic encephalomyelitis is exacerbated in mice deficient for 12/15-lipoxygenase or 5-lipoxygenase. Brain Res. 2004;17:140–145. doi: 10.1016/j.brainres.2004.06.045. [DOI] [PubMed] [Google Scholar]
- 67.Belvisi MG, Mitchell JA. Targeting PPAR receptors in the airway for the treatment of inflammatory lung disease. Br J Pharmacol. 2009;158:994–1003. doi: 10.1111/j.1476-5381.2009.00373.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Altmann R, Hausmann M, Spottl T, Gruber M, Bull AW, Menzel K, Vogl D, Herfarth H, Scholmerich J, Falk W, Rogler G. 13-Oxo-ODE is an endogenous ligand for PPARgamma in human colonic epithelial cells. Biochem Pharmacol. 2007;74:612–622. doi: 10.1016/j.bcp.2007.05.027. [DOI] [PubMed] [Google Scholar]
- 69.Stoll LL, Morland MR, Spector AA. 13-HODE increases intracellular calcium in vascular smooth muscle cells. Am J Physiol. 1994;266:C990–996. doi: 10.1152/ajpcell.1994.266.4.C990. [DOI] [PubMed] [Google Scholar]
- 70.Fritsche KL. Too much linoleic acid promotes inflammation-doesn't it? Prostaglandins Leukot Essent Fatty Acids. 2008;79:173–175. doi: 10.1016/j.plefa.2008.09.019. [DOI] [PubMed] [Google Scholar]