

Abstract
KRAS is frequently mutated in lung cancers and is associated with aggressive biology and chemotherapy resistance. Therefore, innovative approaches are needed to treat these lung cancers. Prior work implicated the interferon-stimulated gene 15 (ISG15) deubiquitinase (DUB) USP18 as having anti-neoplastic activity by regulating lung cancer growth and oncoprotein stability. This study demonstrates that USP18 affects the stability of the KRAS oncoprotein. Interestingly, loss of USP18 reduced KRAS expression and engineered gain of USP18 expression increased KRAS protein levels in lung cancer cells. Using the protein synthesis inhibitor cycloheximide (CHX), USP18 knockdown significantly reduced the half-life of KRAS, but gain of USP18 expression significantly increased its stability. Intriguingly, loss of USP18 altered KRAS subcellular localization by mislocalizing KRAS from the plasma membrane. To explore the biological consequences, immunohistochemical (IHC) expression profiles of USP18 were compared in lung cancers of KrasLA2/+ versus cyclin E engineered mouse models. USP18 expression was higher in Kras-driven murine lung cancers, indicating a link between KRAS and USP18 expression in vivo. To solidify this association, loss of Usp18 in KrasLA2/+/Usp18−/− mice was found to significantly reduce lung cancers as compared to parental KrasLA2/+ mice. Lastly, translational relevance was confirmed in a human lung cancer panel by showing USP18 IHC expression was significantly higher in KRAS mutant versus wild-type lung adenocarcinomas.
Keywords: KRAS, USP18, deubiquitinase, protein stability and lung cancer
Introduction
Kirsten rat sarcoma viral oncogene homolog (KRAS) is the most commonly mutated species of the RAS oncogenes (1). It functions as a molecular switch that controls cell growth, but when mutated, it is constitutively active resulting in uncontrolled cell growth and carcinogenesis (1). KRAS mutations occur in human lung cancers (1,2). Innovative strategies are needed to combat KRAS mutant lung cancers because these tumors are often resistant to therapy (3). Inhibiting activated KRAS is challenging, prompting investigators to examine other ways to inactivate KRAS (4). One potential approach is to reduce the stability of KRAS protein. Indeed, engaging protein destabilization pathways is a promising strategy for reduction of oncogenic proteins levels (5).
We previously found that specific oncogenic proteins including PML/RARα and cyclin D1 were substrates of the ubiquitin-like interferon-stimulated gene 15 (ISG15) modification (ISGylation) pathway (6–8). This pathway engages the E1 activating enzyme UBE1L, the E2 conjugating enzyme UBCH8 and the E3 ligase that enables ISG15 to bind protein substrates and regulate their stability (9–11). The deubiquitinating (DUB) enzyme ubiquitin-specific protease 18 (USP18) is the major enzyme involved in ISG15 removal from substrate proteins (12). USP18 activity reduces ISG15 protein conjugation and promotes tumorigenesis if an oncogenic substrate is stabilized by USP18 (6–8). USP18 also functions as a negative regulator of interferon (IFN) signaling independent of its ISG15 deconjugase activity (13). DUB activity can affect carcinogenesis and these enzymes are potential antineoplastic targets (14).
We reported that altering USP18 levels affects stability of cyclin D1 protein in lung cancer (8). This study extends that prior work by showing loss of USP18 expression destabilizes KRAS protein in murine and human lung cancer cell lines. After observing this association between USP18 and KRAS proteins in lung cancer cells, it was hypothesized that reducing USP18 expression coordinately repressed KRAS protein. Repression of USP18 not only decreased KRAS protein stability, but also conferred KRAS mislocalization from the plasma membrane to the endomembrane compartment. In depth analyses of murine and human lung cancers confirmed an elevated level of USP18 occurred in murine and human lung cancers with mutant KRAS as compared to those with wild-type KRAS expression. Notably, in engineered compound mice having Kras activation and loss of USP18 expression there was a significant (P < 0.05) reduction in lung cancers when compared to mice with basal USP18 levels. These and other findings presented here provide evidence that inhibiting the DUB USP18 is a way to combat lung cancers that express the KRAS oncoprotein.
Materials and Methods
Cell Culture
Murine (ED1, 344P, 393P and LKR13) lung cancer cell lines, human (H522, H2122 and HOP62) lung cancer cell lines and the immortalized murine pulmonary epithelial cell line C10 were cultured in RPMI 1640 medium (Invitrogen) supplemented with 10% fetal bovine serum (FBS) (Thermo Scientific). 293T human embryonic kidney cells and Platinum-E retroviral packaging cells (Plat-E) were cultured in DMEM medium (Invitrogen) supplemented with 10% FBS. Medium for Plat-E cells was supplemented with 10 μg/ml blasticidin (Invitrogen), 1 μg/ml puromycin (Invitrogen) and 1% penicillin-streptomycin (Invitrogen). Cells were cultured at 37°C in a humidified incubator with 5% CO2. Cell lines were obtained from American Type Culture Collection except for the murine ED1 lung cancer cell line (15), Plat-E cell line (Cell Biolabs), and 344P, 393P and LKR13 lung cancer cell lines (16). All cell lines were authenticated as previously described (15,16) and screened for mycoplasma using the MycoAlert PLUS Mycoplasma Detection Kit (Lonza). KRAS mutations were confirmed in studied cell lines, as previously reported (17).
Plasmids, siRNAs and Transient Transfection
Plasmids pCMV-HA-ISG15, pSG5-UBE1L, pCMV2-UBCH8, pcDNA4-USP18, pCGN-KRAS(G12V), pPUR and pRetroX-IRES-ZsGreen1-USP18 were described before (6,8,18). Lentiviral pCMV-dR8.2 dvpr and pMD2.G plasmids were purchased (Addgene) along with the pCMV-GFP-USP18 plasmid (GeneCopoeia). Candidate TRC pLKO.1 lentiviral shRNAs repressing USP18 were purchased from GE Dharmacon. Respective vector controls were purchased. QuikChange II Site-Directed Mutagenesis Kits (Agilent) rendered pcDNA4-USP18 enzymatically-inactive, as described (8). DNA sequence analysis confirmed desired engineered species. RISC-free control siRNA and two siRNAs independently targeting USP18 were purchased (GE Dharmacon). These sequences were: murine USP18 siRNA 1 (5′-CGTTGTTTGTCCAGCACGA-3′), murine siRNA 2 (5′-AGGAACTCGAGGACGGAAA-3′), human USP18 siRNA 1 (5′-CTGCATATCTTCTGGTTTA-3′), and human USP18 siRNA 2 (5′GGAAGAAGACAGCAACATG-3′). Plasmids and siRNAs were transfected into desired cells using Lipofectamine 2000 reagent (Invitrogen) and Opti-MEM medium (Gibco Thermo Scientific).
Immunoblot Assays
Cells were washed with phosphate-buffered saline (PBS, Corning) and lysed with ice-cold Pierce RIPA Lysis and Extraction Buffer (Thermo Scientific) supplemented with Halt Protease and Phosphatase Inhibitor Cocktail (Thermo Scientific). Proteins were resolved by SDS-PAGE before transfer to nitrocellulose membranes (Whatman). Membranes were blocked in 5% nonfat milk in Tris-buffered saline with 0.1% Tween 20 (TBS-T) for at least 1 hour before overnight incubation at 4°C with primary antibody diluted in 5% nonfat milk or 5% bovine serum albumin (BSA) in TBS-T. This was followed by three, 10 minute washes in TBS-T and a 40-minute incubation with secondary antibody diluted in 5% nonfat milk. After three additional, 10-minute TBS-T washes, antibody binding was visualized by Luminata Forte (EMD Millipore) and quantified by ImageJ software (imagej.nih.gov/ij). Antibodies independently used were: anti-USP18 (Cell Signaling #4813), anti-KRAS (Abcam #ab84573), anti-KRAS (Abcam #ab55391), anti-ISG15 (Cell Signaling #2743), anti-AKT (Cell Signaling #9272), anti-Phospho-AKT (Ser473) (Cell Signaling #9271), anti-p44/42 MAPK (Erk1/2) (Cell Signaling #9102), anti-Phospho-p44/42 MAPK (Erk1/2) (Thr202/Tyr204) (Cell Signaling #9101) and anti-ß-Actin (Cell Signaling #3700). Antibodies that recognize USP18 were previously described (6). Secondary anti-mouse and anti-rabbit antibodies were purchased (Amersham Biosciences). Immunoblots were stripped using Restore PLUS Western Blot Stripping Buffer (Thermo Scientific). To assess KRAS protein stability, cells were treated with cycloheximide (CHX, 60 μg/ml) (Sigma-Aldrich) for 12 to 15 hours.
Quantitative Real-Time PCR Assays
For qRT-PCR assays, Kras forward primer (5′-AACAGGCTCAGGACTTAGCAA-3′) and reverse primer (5′-AAGGCATCATCAACACCCTGT-3′); murine Usp18 forward primer (5′-TTGGGCTCCTGAGGAAACC-3′) and reverse primer (5′-CGATGTTGTGTAAACCAACCAGA-3′); murine Gapdh forward primer (5′-AGGTCGGTGTGAACGGATTTG-3′) and reverse primer (5′-TGTAGACCATGTAGTTGAGGTCA-3′); human USP18 forward primer (5′-TGTGCCATGGAGAGTAGCAG-3′) and reverse primer (5′-AGGTGGATTGTCAGGGTCTG-3′); human GAPDH forward primer (5′-TGCACCACCAACTGCTTA-3′) and reverse primer (5′-GGCATGGACTGTGGTCATGAG-3′) were used. Assays were performed, as before (6).
Stable Transfectants and Fluorescence Microscopy
Generation of stable KRAS transfectants was performed, as before (18). Retroviruses for stable gain of USP18 expression were generated in Plat-E cells and shRNA lentiviruses for USP18 knockdown were generated in 293T cells, as described (8). KRAS expressing lentivirus to generate stable transfectants for immunofluorescence studies was described previously (19–21). Briefly, USP18 shRNA transfectants were infected with lentivirus co-expressing mGFP-KRAS wild-type or KRAS G12V and mCherry-CAAX, an endomembrane marker (22). Colocalization of mGFP-KRAS with mCherry-CAAX was calculated using Manders coefficients (19).
Proliferation Assays
Cell proliferation was measured using the CellTiter-Glo Assay Kit (Promega). To assess for differences in interferon (IFN) response, cells were treated with or without Universal Type I IFNα (1000U/ml) (PBL Assay Science) for 3 days.
Immunohistochemistry
Formalin-fixed, paraffin-embedded non-small cell lung cancer (NSCLC) cases with adjacent histopathologically normal lung tissues were obtained from the Pathology Department of Dartmouth-Hitchcock Medical Center. In total, 82 adenocarcinomas with known KRAS mutational status were studied. USP18 immunohistochemistry (IHC) was with a Leica BOND-MAX™ automated stainer and Leica Bond Polymer Refine Detection reagents (Leica Microsystems Inc.) (6). Antibody specificity was confirmed using a blocking peptide (8). USP18 immunostaining was scored as follows: negative USP18 expression (0% of cells stained positive for USP18), low USP18 expression (+1 or 0–50% of cells stained for USP18) and high USP18 expression (+2 to +3 or 50–100% of cells stained for USP18). Cyclin D1 immunostaining of lung tumors from KrasLA2/+Usp18+/+ and KrasLA2/+Usp18−/− mice was described previously (23) with negative cyclin D1 expression (0% of cells stained positive for cyclin D1), low cyclin D1 expression (+1 or 0–50% of cells stained for cyclin D1) and high cyclin D1 expression (+2 to +3 or 50–100% of cells stained for cyclin D1). Average staining intensity and percentages of immunoreactive cells were recorded. USP18 IHC was independently performed in paired normal-malignant lung tissues from cyclin E as well as Kras-driven lung cancers in engineered mouse models (24,25). Digital pathology image analysis (Aperio Image Toolbox, Leica Biosystems) determined H-score and measured percentages of stained cells (0–100%) and staining intensity (+0–3). This determined an IHC score ranging from 0 and 300. DAB (3,3-diaminobenzidine) HRP substrate was used for USP18 immunostaining and hematoxylin (Leica Biosystems) was used for counterstaining.
Mouse Models
FVB-Usp18 heterozygous mice were purchased (Jackson Laboratory). Mice were bred to generate FVB-Usp18 knockout mice, as before (26,27). FVB-KrasLA2/+ mice were provided by Dr. A. Balmain, University of California (25). Genotyping for Usp18 and Kras was with PCR assays and an established protocol (Jackson Laboratory) (Usp18 #007225 and Kras #008185). FVB-KrasLA2/+ and FVB-Usp18−/− mice were crossed to generate KrasLA2/+Usp18+/+, KrasLA2/+Usp18+/− and KrasLA2/+Usp18−/− mice. Mice were housed in a temperature- and humidity-controlled and pathogen-free environment with a 12 hour light/dark cycle and ad libitum access to water and standard laboratory chow. Male mice that were KrasLA2/+Usp18+/+ (n = 25), KrasLA2/+Usp18+/− (n = 19) and KrasLA2/+Usp18−/− (n = 20) were sacrificed at four months and lung tissues were harvested, formalin-fixed and paraffin-embedded.
Study Approvals
All animal studies were reviewed and approved by the Institutional Animal Care and Use Committee (IACUC) at the Geisel School of Medicine at Dartmouth. The Dartmouth Committee for Protection of Human Subjects reviewed and approved studies with these human lung tissues that were unlinked to clinical identifiers. Studies were performed in accordance with the Institutional Review Board (IRB) oversight.
Statistics
Data are presented as mean with standard deviation bars, unless otherwise stated. Two-tailed Student’s t tests compared differences between groups. One-way ANOVA with Bonferroni correction was used for multiple comparisons. Results of independent experiments were pooled to assess statistical significance. Experiments were completed in triplicate with at least two replicates. Statistical significance was defined as P < 0.05.
Results
KRAS is Associated with USP18 in Lung Cancer Cell Lines
USP18 is a DUB that regulates stability of specific oncoproteins (6,8). Prior work reveals that repression of USP18 protein reduced growth of lung cancer cell lines (8). As expected, introduction of enzymatically-active USP18 (8) significantly (P < 0.01) overcame growth inhibition by USP18 knockdown (Supplementary Fig.S1). This indicates that loss of ISG15 deconjugase activity is responsible for growth inhibition caused by engineered USP18 repression in lung cancer cells. While investigating the effect of USP18 gain versus loss on cell growth in a broader panel of murine and human lung cancer cell lines, it was observed that proliferation of lung cancer cell lines expressing activated KRAS was affected by USP18 expression. Gain of USP18 expression enhanced (Fig.1A) and loss of USP18 significantly (P < 0.05) reduced (Fig.1B and Supplementary Fig.S2) growth of KRAS mutant lung cancer cell lines. Prior work found these changes were accompanied by augmented apoptosis (8). Knockdown of USP18 increased growth inhibitory in response to IFNα treatment (8) in diverse lung cancer lines, including those with activated KRAS expression (Supplementary Fig.S3A and S3B).
Figure 1. KRAS is associated with USP18 expression in murine lung cancer cells.
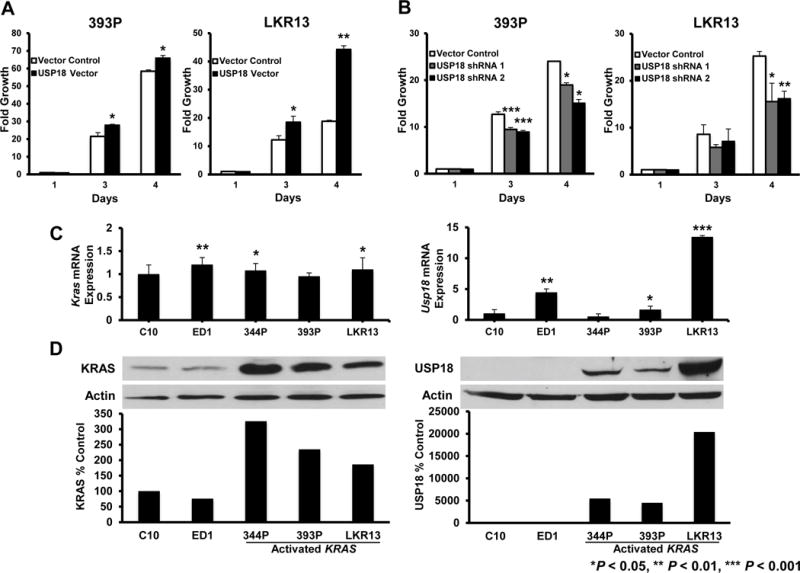
(A) Engineered stable gain of USP18 expression increased lung cancer cell growth and (B) Constitutive loss of USP18 expression by shRNAs decreased cell growth in 393P and LKR13 murine lung cancer cell lines. Proliferation was monitored over 4 days. Growth of these cell lines was normalized to their respective proliferation on day 1. Representative immunoblots confirmed engineered expression and knockdown respectively of USP18 protein in Figure 2A and B. (C) There is no association between Kras and Usp18 mRNA levels in murine lung cancer cell lines. Expression of Kras and Usp18 mRNAs were analyzed by qRT-PCR assays. Kras and Usp18 mRNA expression profiles were quantified relative to respective Gapdh mRNA and normalized to expression in C10 murine lung epithelial cells. (D) There is an association between KRAS and USP18 protein levels in murine lung cancer cell lines. Basal KRAS and USP18 protein expression profiles were quantified relative to respective actin expression and normalized to levels in C10 cells. The representative USP18 immunoblot was exposed for a brief time to avoid overexposure in lanes with abundant USP18 expression.
These findings showed that reduced USP18 levels can antagonize growth of KRAS mutant lung cancers, Yet, destabilization of ISGylated proteins downstream of KRAS (8) might also be responsible for the growth inhibition observed after USP18 loss. To investigate whether there was a relationship between KRAS and USP18, basal expression levels of KRAS and USP18 were determined in immortalized lung epithelial and lung cancer cell lines. Quantitative real-time PCR (qRT-PCR) assays revealed differences in Kras and Usp18 mRNA expression between the murine immortalized pulmonary epithelial line (C10), the cyclin E-driven murine transgenic lung cancer cell line (ED1) and murine lung cancer cell lines expressing activated Kras (Fig.1C). Immunoblot analyses revealed Kras mutant lung cancer cell lines that expressed high levels of KRAS protein also had abundant levels of USP18 protein (Fig.1D). Given this, we hypothesized that manipulating USP18 levels altered KRAS protein expression.
USP18 Regulates KRAS Protein Stability
Because there is an association between endogenous KRAS and USP18 protein levels in lung cancer cell lines, we sought to learn if engineered loss or gain of USP18 expression influenced KRAS levels in these lung cancer cell lines. Independent engineered loss (Fig.2A, Supplementary Fig.S4) and gain of USP18 expression (Fig.2B) respectively decreased and increased KRAS protein levels relative to controls, despite the presence of KRAS mutations. Changes in KRAS mRNA levels were not observed with loss of USP18 expression (Supplementary Fig.S5) indicating USP18 regulates KRAS post-transcriptionally. Restoration of USP18 levels in USP18-repressed cells augmented KRAS expression versus vector controls (Fig.2C). Lung cancer cell lines with repressed USP18 displayed decreased phosphorylation of AKT and MAPK, major downstream signaling pathways of KRAS (Supplementary Fig.S6A (28). This was also observed in Usp18 null mouse lung tissues (Supplementary Fig.S6B) that showed significant (P < 0.001) and a trend towards significant (P < 0.1) declines in expression of phosphorylated-AKT and –MAPK, respectively, relative to Usp18 wild-type mouse lung tissues.
Figure 2. Modifying USP18 expression alters KRAS protein levels.
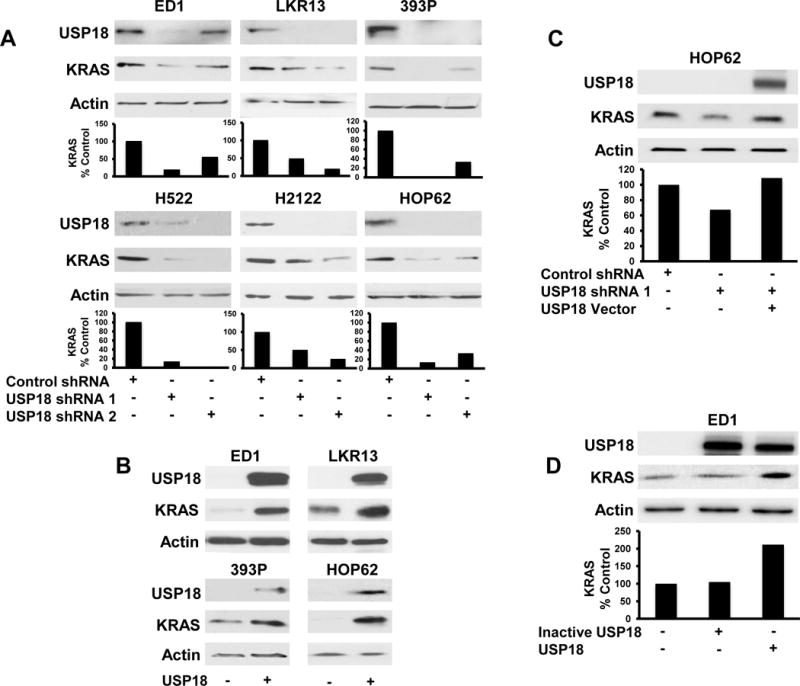
(A) Stable reduction of endogenous USP18 expression by shRNAs independently introduced into murine and human lung cancer cells decreased endogenous KRAS protein. Murine and human USP18 proteins were detected with mouse and human-specific antibodies, respectively. KRAS expression was quantified relative to actin expression and normalized to vector-transfected lung cancer cell lines. (B) Stable gain of expression of human USP18 by retrovirus introduced independently into murine and human lung cancer cells increased endogenous KRAS protein levels. Human USP18 protein was detected using a human-specific USP18 antibody. Brief exposure times avoided overexposure of exogenous USP18 protein. (C) Decreased KRAS protein conferred by reduced USP18 expression was restored by transiently overexpressing human USP18 in HOP62 lung cancer cells. Immunoblots were individually probed with KRAS and human-specific USP18 antibodies. KRAS was normalized to actin expression and compared between vector control and USP18 shRNA- as well as USP18 shRNA/USP18 expression vector-transfected lung cancer cell lines. Reduced USP18 expression in HOP62 lung cancer cells after introduction of USP18-shRNA appears in panel A. (D) Expression of human enzymatically-inactive USP18 species did not change KRAS expression in ED1 lung cancer cells. Immunoblots were individually probed with KRAS and USP18 specific antibodies. KRAS levels were normalized to actin expression and compared between vector control, enzymatically-inactive USP18 and enzymatically-active (wild-type) USP18 transfected lung cancer cell lines.
Enzymatic activity of USP18 enables this DUB to remove the post-translational ISG15 modification from protein substrates (8,12,29). To determine that the ISG15-removing enzymatic activity of USP18 and not its IFN receptor inhibitory activity (13) conferred changes in KRAS protein levels, enzymatically-inactive USP18 was transfected into ED1 lung cancer cells and KRAS protein expression was analyzed. Enzymatically-inactive USP18 did not affect KRAS protein relative to vector control (Fig.2D). Increased KRAS protein levels were only observed after transfection of enzymatically-active (wild-type) USP18. Recent work revealed that ISG15 and KRAS can regulate each other in breast cancer (30). This prior work was extended to show that the ISG15 pathway modifies KRAS in lung cancer cell lines (Supplementary Fig.S7A). This modification is decreased by gain of USP18 expression (Supplementary Fig.S7B). This indicates that USP18 likely regulates KRAS through its post-translational modification of ISGylation.
To explore consequences of USP18 repression on KRAS protein stability, KRAS expression in vector control transfectants were compared to those with gain versus loss of USP18 expression in lung cancer cell lines. These cells were also studied following treatment with the protein synthesis inhibitor, CHX. USP18 levels are reduced in ED1 murine lung cancer cell lines, KRAS was substantially destabilized by ~50% after 8 hours of CHX treatment as compared to control cells that retained basal KRAS expression at the same time point (Fig.3A). Expression of KRAS in USP18-repressed lung cancer cell lines was significantly reduced by 12 (P < 0.01) and 15 (P < 0.05) hours after CHX treatment. When overexpressed in HOP62 human lung cancer cell lines, USP18 stabilized KRAS after 12 hours of CHX treatment versus control cells where KRAS protein was destabilized after only 6 hours of treatment (Fig.3B). KRAS stability in USP18 expressed lung cancer cell lines significantly increased at 10 (P < 0.01) and 12 (P < 0.001) hours after CHX treatment. Since KRAS protein is able to undergo degradation when mislocalized from the plasma membrane (31), it was determined whether loss of USP18 affected KRAS membrane localization.
Figure 3. USP18 regulates endogenous KRAS protein stability.
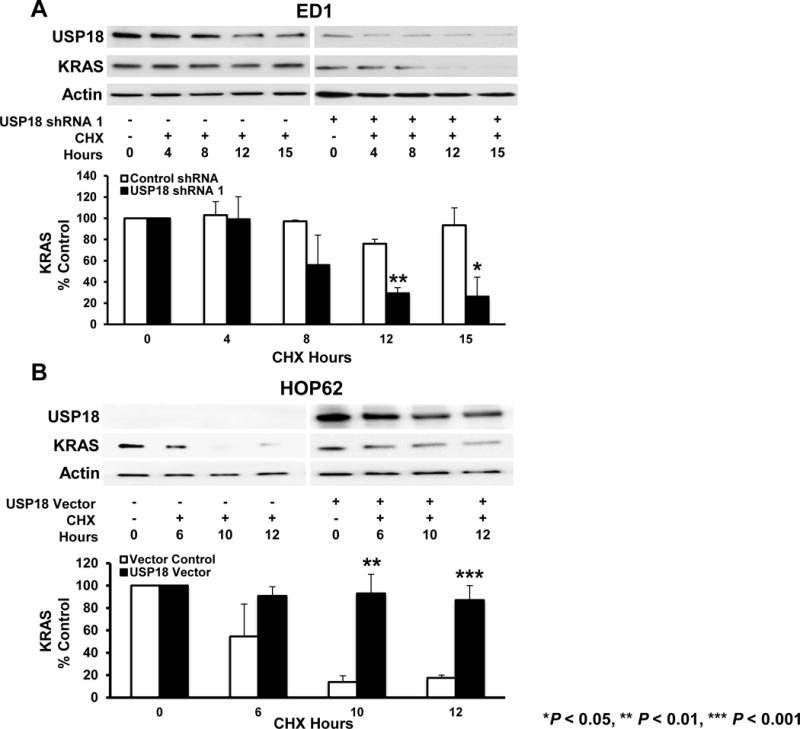
(A) Endogenous KRAS protein stability decreased with USP18 knockdown. ED1 murine lung cancer cells stably expressing shRNA against USP18 were treated with cycloheximide (CHX) for 15 hours and results were compared to vector-transfected (control) cells. KRAS expression was quantified relative to actin expression at indicated times and normalized to the 0 hour time point (before CHX treatment). Murine USP18 protein expression was detected with a mouse-specific USP18 antibody. (B) Endogenous KRAS protein was stabilized by gain of USP18 expression. HOP62 lung cancer cells transiently overexpressing human GFP-tagged USP18 were treated with CHX for 12 hours and results were compared to vector transfected (control) cells. KRAS protein was quantified relative to actin expression at indicated times and normalized to the 0 hour time point (before CHX treatment). Human USP18 protein expression was detected using a human-specific USP18 antibody.
Reduced USP18 Expression Mislocalizes KRAS Protein from the Plasma Membrane
KRAS protein localized to the plasma membrane can mediate downstream growth-regulatory signals (32). Aberrant post-translational processing of KRAS interferes with KRAS plasma membrane localization and affects its oncogenic signaling (19,33). Whether reduced USP18 expression affected KRAS localization was investigated. Individual lung cancer cell lines engineered with reduced USP18 expression were co-transfected with wild-type or mutant KRAS species along with the endomembrane marker CAAX. Subcellular localization and co-localization of each species with an endomembrane marker were analyzed by immunofluorescence. A greater proportion of wild-type KRAS (Fig.4A and Supplementary Fig.S8A) and mutant KRAS protein (Fig.4B and Supplementary Fig.S8B) was mislocalized from the plasma membrane when USP18 was repressed. USP18 knockdown increased localization of both wild-type and activated KRAS protein to endomembrane compartments, providing a basis for the observed destabilization of these proteins.
Figure 4. Knockdown of USP18 protein redistributes KRAS protein from the plasma membrane to the endomembrane compartment.
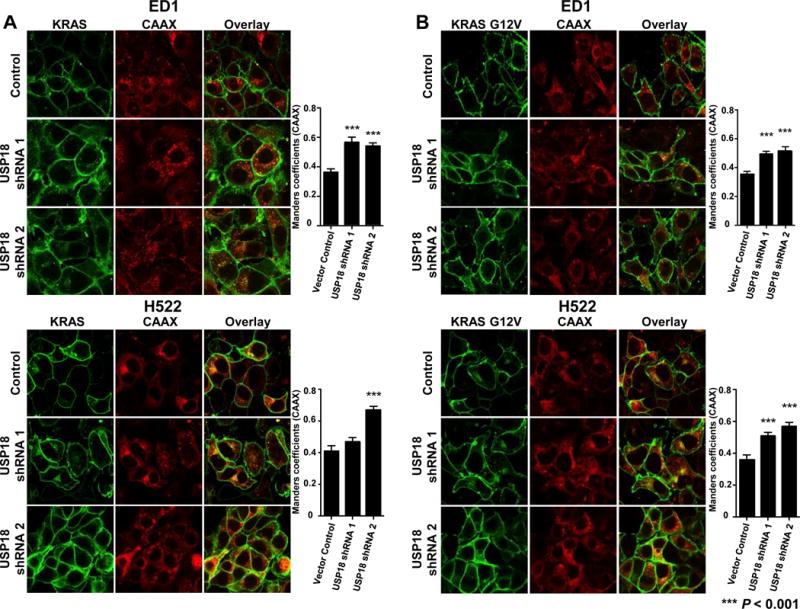
(A) ED1 murine lung cancer cells and H522 human lung cancer cells with constitutive knockdown of USP18 (Figure 2A) were stably transfected with mCherry-CAAX and mGFP-KRAS (wild-type) or (B) mGFP-KRAS G12V (mutant) species. Cells were fixed and imaged by confocal microscopy. Manders coefficients were used to quantify the relative amount of KRAS associated with the endomembrane marker CAAX. Repression of USP18 significantly (P < 0.001) mislocalized KRAS from the plasma membrane to endomembrane compartment. All magnifications are 60X.
Loss of USP18 Reduces Tumorigenicity of Kras-driven Lung Cancers in Mice
Studies that explored the relationship between KRAS and USP18 expression were extended to genetically-engineered mouse models of lung cancer. Activation of the Kras oncogene in KrasLA2/+ mice formed lung cancers that express high levels of KRAS protein (34). It was hypothesized that lung cancers in mice with somatic activation of Kras exhibited elevated USP18 protein levels. Normal and malignant lung tissues from FVB-KrasLA2/+ mice expressing activated Kras and also from a FVB-cyclin E-driven transgenic mice that do not express activated Kras species were each immunostained for USP18 expression. USP18 expression levels were quantified and compared between malignant lung tissues from these respective mouse models. USP18 expression was significantly higher (P < 0.001) in lung tumors driven by activated Kras as compared to those driven by aberrant cyclin E expression (Fig.5A). These in vivo findings extend and support in vitro lung cancer cell line data shown in Fig.1C.
Figure 5. USP18 expression is higher in Kras mutant murine lung cancers and USP18 loss reduces tumorigenicity of Kras-driven murine lung cancers.
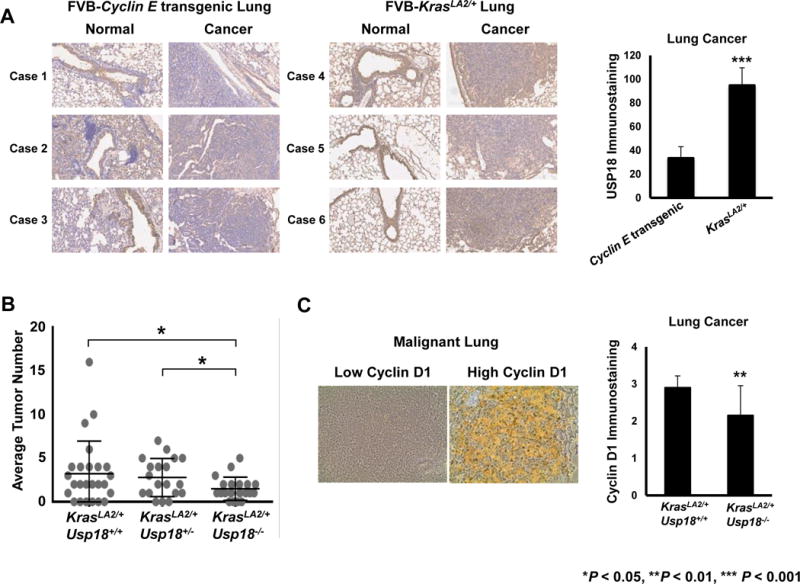
(A) USP18 expression is significantly (P < 0.001) elevated in Kras-driven murine lung cancers relative to cyclin E-driven murine lung cancers. Representative USP18 immunostaining of normal versus malignant lung tissues harvested independently from cyclin E-transgenic mice (n = 3) and KrasLA2/+ mice (n = 3). All magnifications are 20X. Average USP18 immunostaining in malignant lung was determined. (B) Loss of Usp18 significantly (P < 0.05) decreased the number of Kras-driven murine lung cancers. Average lung tumor numbers of KrasLA2/+Usp18+/+ (n = 25), KrasLA2/+Usp18+/− (n = 19) and KrasLA2/+Usp18−/− (n = 20) mice were determined for each respective group. (C) Loss of Usp18 significantly (P < 0.01) decreased cyclin D1 expression in Kras-driven murine lung cancers. Representative cyclin D1 immunostaining of malignant lung with low versus high cyclin D1 expression are shown. All magnifications are 40X. Average cyclin D1 immunostaining of lung cancer was determined in KrasLA2/+Usp18+/+ (n = 16) and KrasLA2/+Usp18−/− (n = 15) mice, respectively.
Since in vitro data revealed USP18 knockdown decreased KRAS protein stability it was sought to learn if Usp18 loss (Supplementary Fig.S9) affected lung cancer formation in the Kras-driven mouse model. Interestingly, KrasLA2/+Usp18−/− compound mice exhibited a significantly (P < 0.05) reduced average lung tumor number as compared to KrasLA2/+Usp18+/+ and KrasLA2/+Usp18+/− mice (Fig.5B). We next examined expression in these tissues of the growth-regulator cyclin D1 to determine if reduced USP18 levels affected this oncoprotein downstream of KRAS (8,35). Histological analyses of normal and neoplastic lung tissues revealed that KrasLA2/+Usp18−/− compound mice had significantly (P < 0.01) lower expression of cyclin D1 as compared to KrasLA2/+Usp18+/+ compound mice (Fig.5C). No significant differences were apparent in atypical adenomatous hyperplasia (AAH) or adenocarcinoma in-situ (AIS) lesions when comparing KrasLA2/+Usp18+/+ and KrasLA2/+Usp18−/− lung tissues (data not shown). The translational relevance of the relationship between KRAS and USP18 expression was next examined in human lung adenocarcinomas.
USP18 is Augmented by Activated KRAS Expression in Human Lung Cancers
Activated KRAS is associated with increased KRAS mRNA (Supplementary Fig.S10A) and KRAS protein (36) expression in human lung cancers. It was proposed that human lung cancers with activated KRAS expression would have higher USP18 levels than wild-type KRAS lung cancer cases. This hypothesis was based on in vitro data showing that USP18 protein was enhanced in lung cancer cell lines with engineered expression of activated KRAS (Supplementary Fig.S10B). USP18 expression profiles were then examined in 82 lung adenocarcinoma cases and results were compared to adjacent histopathologically normal human lung tissues. Representative images are displayed for these NSCLC cases having low or high USP18 immunostaining (Fig.6A). Representative expression profiles of USP18 immunostaining in normal versus malignant lung tissues were shown in cases with wild-type or activating KRAS mutations (Fig.6B). As expected from prior work (8), quantitative analysis of USP18 immunostaining revealed that USP18 expression was significantly (P < 0.001) increased in malignant versus normal lung tissues (Fig.6C). Confirming translational relevance, KRAS mutant lung cancers had a significantly (P < 0.05) larger proportion of cases with increased USP18 expression as compared to wild-type KRAS lung cancer cases (Fig.6D).
Figure 6. USP18 immunostaining is augmented in human lung adenocarcinomas with activating KRAS mutations.
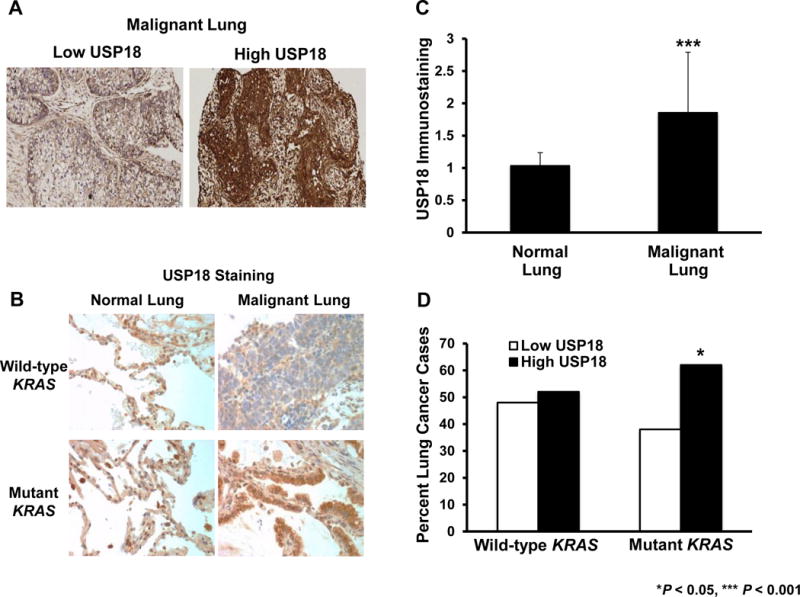
Representative immunostaining of human lung adenocarcinomas with (A) low versus high USP18 expression. (B) Representative USP18 expression profiles are shown for KRAS wild-type versus mutant lung cancers relative to adjacent normal lung tissues. All magnifications are 40X. (C) USP18 immunostaining in normal (n = 76) versus malignant (n = 81) lung was compared and significantly higher (P < 0.001) USP18 levels were detected in the malignant lung. (D) USP18 immunostaining was compared in KRAS wild-type (n = 42) versus mutant (n = 39) lung cancer cases and significantly (P < 0.05) higher USP18 expression was evident in cases where mutant KRAS was detected.
Discussion
Despite being one of the most frequently mutated oncogenes in cancer, there are currently no FDA-approved therapies for lung cancer cases with activated KRAS expression (1–3). This study adds to prior literature by reporting that the ISG15-specific DUB USP18 can regulate KRAS protein stability. This finding raised the prospect that inhibition of USP18 is useful in the treatment of KRAS mutant lung cancers. Of the many DUBs encoded in the human genome, the majority act in removal of ubiquitin from protein substrates (37). It is thus challenging to determine which DUB for ubiquitin optimally serves as an antineoplastic target. Conversely, USP18 is the major DUB for ISG15 and its loss substantially increases levels of ISG15-conjugated oncoproteins (8,12).
Prior work found that a regulatory loop exists between ISG15 and KRAS in breast cancer where ISG15 stabilizes KRAS by inhibiting its lysosomal degradation (30). This finding underscored that effects of KRAS ISGylation likely depend on tissue and cancer contexts. Indeed, this was observed in the ubiquitination of p53 (38). The data presented here demonstrate that ISG15 and KRAS can complex. Likewise, the ISG15-specific DUB USP18 regulates KRAS protein stability. Preliminary mass spectrometry and site-directed mutational evidence points to potential ISG15 binding lysines within the C-terminal of KRAS that affect stability of this complexed protein (data not shown). Interestingly, these residues reside in the polybasic HVR region of KRAS that is involved in anchorage to the plasma membrane (32). Bulky post-translational modifications could interfere with the ability of KRAS to associate with the plasma membrane (33,39). ISG15 complex formation with the HVR domain of KRAS provides a mechanistic basis for mislocalizing KRAS from the plasma membrane. Future studies will uncover whether mutations in the HVR domain can directly abrogate ISGylation and alter KRAS localization and stability. That work will determine the precise pathways involved in regulating KRAS stability once USP18 is repressed in lung cancer cells.
Both wild-type and mutant KRAS proteins are regulated by levels of USP18 expression. Yet, murine lung tumors and human lung cancer array data revealed that USP18 levels were significantly higher in cases with KRAS activation. Activated KRAS protein is known to be degraded more rapidly than wild-type KRAS protein (40). It is also possible that activated KRAS protein upregulates USP18 expression through IFN-ß-dependent signaling mechanisms (41). This would limit destabilization of proteins that undergo complex formation with ISG15. Thus, inhibition of USP18 would likely have a greater differential effect on mutant KRAS protein.
Findings revealed engineered KrasLA2/+/Usp18−/− mice developed significantly (P < 0.05) fewer lung cancers as compared to KrasLA2/+ mice. Previous work showed that enhanced immune response did not play a prominent role in tumor suppression in Usp18-deficient cells (42). Instead, lung cancers that formed in KrasLA2/+/Usp18−/−mice had decreased expression of USP18-regulated oncoproteins like cyclin D1. We hypothesize that decreased expression of KRAS and other USP18-regulated oncoproteins downstream of KRAS, such as cyclin D1 (35), antagonize Kras mutant lung cancer formation in KrasLA2/+/Usp18−/− mice. We also propose that destabilization of KRAS, cyclin D1 and other USP18-regulated oncoproteins downstream of KRAS contribute to the observed consequences of USP18 reduced expression on lung cancer cells. Notably, engineered expression of oncogenic KRAS can induce senescence in cancer cells (43,44,45). Given this, it is not surprising that proteins downstream of KRAS like cyclin D1 (35) can rescue growth of USP18-repressed lung cancer cells (data not shown). It is interesting to note that many proteins regulated by ISG15 and USP18 are involved in the KRAS signaling pathway (8,46). Simultaneous destabilization of KRAS and other USP18-regulated proteins downstream of KRAS may play a functional role in reducing formation of Kras mutant lung cancers. Recent work indicates that many growth-regulatory proteins are altered by USP18 loss (46). Future work will establish whether these or other growth-regulatory proteins are affected by USP18, if they are downstream of KRAS signaling and whether they are repressed in KrasLA2/+/Usp18−/− lung cancers.
Taken together, this study advances prior work by reporting that the DUB USP18 is a candidate enzyme for pharmacologic inhibition that triggers destabilization of the KRAS oncoprotein and possibly downstream signaling proteins. An optimal DUB inhibitor would simultaneously destabilize growth-promoting oncoproteins and thereby reduce tumorigenesis (47). Findings reported here and elsewhere provide a strong rationale for developing such an inhibitor of the DUB USP18. That inhibitor could act as a single agent or combined with other chemotherapeutic agents to combat lung cancers driven by the KRAS oncogene.
Supplementary Material
Implications.
Taken together, this study highlights a new way to combat the oncogenic consequences of activated KRAS in lung cancer by inhibiting the DUB USP18.
Acknowledgments
We thank Neus Bota-Rabassedas (MD Anderson Cancer Center) and Matthew Ung (Geisel School of Medicine) for their helpful consultation. We also thank Gary Ward (Dartmouth-Hitchcock) in the DartLab (Immune Monitoring and Flow Cytometry Shared Resource) for technical assistance.
Financial Support
This study was supported by the National Institutes of Health (NIH), National Cancer Institute (NCI) grants R01-CA087546 (ED) and R01-CA190722 (ED), a Samuel Waxman Cancer Research Foundation Award (ED), a UT-STARs award (ED), an American Cancer Society Clinical Research Professorship (ED), a grant from the Cancer Research and Prevention Institute of Texas (CPRIT:RP130059) (JFH) and an NIH Pathway to Independence Award (K99-CA188593) (KJC). The Immune Monitoring and Flow Cytometry Shared Resource was supported in part by a Norris Cotton Cancer Center Support Grant (P30CA023108-36) and an Immunology COBRE Grant (P30GM103415-14) from the National Institute of General Medical Sciences.
Footnotes
Disclosure of Potential Conflicts of Interest:
No potential conflicts of interests exist.
Author Contributions
Conception and design: ED, LMM, SJF, YL
Development of methodology: ED, LMM, SJF
Acquisition of data: LMM, LJT, VM, JRC, BM, PAV, HK, JR, MK, KJC, SH, FC
Analysis and interpretation of data: ED, LMM, SJF, YL, XL
Writing, review and/or revision of the manuscript: ED, LMM, SJF
Administrative, technical or material support: IW, SMH, JFH
Study supervision: ED, SJF
References
- 1.Baines AT, Xu D, Der CJ. Inhibition of Ras for cancer treatment: the search continues. Future Med Chem. 2011;3:1787–1808. doi: 10.4155/fmc.11.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2015. CA Cancer J Clin. 2015;65:5–29. doi: 10.3322/caac.21254. [DOI] [PubMed] [Google Scholar]
- 3.Macerecilli M, Caramella C, Faivre L, Besse B, Pianchard D, Polo V, et al. Does KRAS mutational status predict chemoresistance in advanced non-small cell lung cancer (NSCLC)? J Lung Cancer. 2014;83:383–388. doi: 10.1016/j.lungcan.2013.12.013. [DOI] [PubMed] [Google Scholar]
- 4.Gysin S, Salt M, Young A, McCormick F. Therapeutic strategies for targeting Ras proteins. Genes Cancer. 2011;2:359–372. doi: 10.1177/1947601911412376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Frezza M, Schmitt S, Dou QP. Targeting the ubiquitin-proteasome pathway: an emerging concept in cancer therapy. Curr Top in Med Chem. 2011;11:2888–2905. doi: 10.2174/156802611798281311. [DOI] [PubMed] [Google Scholar]
- 6.Guo Y, Dolinko AV, Chinyengetere F, Stanton B, Bomberger JM, Demidenko E, et al. Blockade of the ubiquitin protease UBP43 destabilizes transcription factor PML/RARα and inhibits the growth of acute promyelocytic leukemia. Cancer Res. 2010;70:9875–9885. doi: 10.1158/0008-5472.CAN-10-1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shah SJ, Blumen S, Pitha-Rowe I, Kitareewan S, Freemantle SJ, Feng Q, et al. UBE1L represses PML/RARα by targeting the PML domain for ISG15ylation. Mol Cancer Ther. 2008;7:905–914. doi: 10.1158/1535-7163.MCT-07-0515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Guo Y, Chinyengetere F, Dolinko AV, Lopez-Aguiar A, Lu Y, Galimberti F, et al. Evidence for the ubiquitin protease UBP43 as an antineoplastic target. Mol Cancer Ther. 2012;11:1968–1977. doi: 10.1158/1535-7163.MCT-12-0248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yuan WM, Krug RM. Influenza B virus NS1 protein inhibits conjugation of the interferon (IFN)-induced ubiquitin-like ISG15 protein. EMBO J. 2011;20:362–371. doi: 10.1093/emboj/20.3.362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhao C, Beaudenon SL, Kelley ML, Waddell MB, Yuan W, Schulman BA, et al. The UbcH8 ubiquitin E2 enzyme is also the E2 enzyme for ISG15, an IFN-alpha/beta-induced ubiquitin-like protein. Proc Natl Acad Sci (USA) 2004;101:7578–7582. doi: 10.1073/pnas.0402528101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wong JJ, Pung YF, Sze NS, Chin KC. HERC5 is an IFN-induced HECT-type E3 protein ligase that mediates type I IFN-induced ISGylation of protein targets. Proc Natl Acad Sci (USA) 2006;103:10735–10740. doi: 10.1073/pnas.0600397103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Malakhov MP, Malakhova OA, Kim KI, Ritchie KJ, Zhang DE. UBP43 (USP18) specifically removes ISG15 from conjugated proteins. J Biol Chem. 2002;277:9976–9981. doi: 10.1074/jbc.M109078200. [DOI] [PubMed] [Google Scholar]
- 13.Malakhova OA, Kim KI, Luo JK, Zou W, Kumar KG, Fuchs SY, et al. UBP43 is a novel regulator of interferon signaling independent of its ISG15 isopeptidase activity. EMBO J. 2006;25:2358–2367. doi: 10.1038/sj.emboj.7601149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fraile JM, Quesada V, Rodriguez D, Freije JM, Lopez-Otin C. Deubiquitinases in cancer: new functions and therapeutic options. Oncogene. 2012;31:2373–2388. doi: 10.1038/onc.2011.443. [DOI] [PubMed] [Google Scholar]
- 15.Liu X, Sempere LF, Ouyang H, Memoli VA, Andrew AS, Luo Y, et al. MicroRNA-31 functions as an oncogenic microRNA in mouse and human lung cancer cells by repressing specific tumor suppressors. J Clin Invest. 2010;120:1298–1309. doi: 10.1172/JCI39566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wislez M, Fujimoto N, Izzo JG, Hanna AE, Cody DD, Langley RR, et al. High expression of ligands for chemokine receptor CXCR2 in alveolar epithelial neoplasia induced by oncogenic kras. Cancer Res. 2006;66:4198–4207. doi: 10.1158/0008-5472.CAN-05-3842. [DOI] [PubMed] [Google Scholar]
- 17.Neri A, Knowles DM, Greco A, McCormick F, Dalla-Favera R. Analysis of RAS oncogene mutations in human lymphoid malignancies. Proc Natl Acad Sci (USA) 1988;85:9268–9272. doi: 10.1073/pnas.85.23.9268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hu S, Danilov A, Godek K, Orr B, Tafe LJ, Rodriguez-Canales J, et al. CDK2 inhibition causes anaphase catastrophe in lung cancer through the centrosomal protein CP110. Cancer Res. 2015;75:2029–2038. doi: 10.1158/0008-5472.CAN-14-1494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cho KJ, Park JH, Piggott AM, Salim AA, Gorfe AA, Parton RG, et al. Staurosporines disrupt phosphatidylserine trafficking and mislocalizes ras proteins. J Biol Chem. 2012;287:43573–43584. doi: 10.1074/jbc.M112.424457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Appolloni A, Prior IA, Lindsay M, Parton RG, Hancock JF. H-ras but not K-ras traffics to the plasma membrane through the exocytic pathway. Mol Cell Biol. 2000;20:2475–2487. doi: 10.1128/mcb.20.7.2475-2487.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Prior IA, Muncke C, Parton RG, Hancock JF. Direct visualization of Ras proteins in spatially distinct cell surface microdomains. J Cell Biol. 2003;160:165–170. doi: 10.1083/jcb.200209091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Choy E, Chiu VK, Silletti J, Feoktistov M, Morimoto T, Michaelson D, et al. Endomembrane trafficking of ras: the CAAX motif targets proteins to the ER and Golgi. Cell. 1999;98:69–80. doi: 10.1016/S0092-8674(00)80607-8. [DOI] [PubMed] [Google Scholar]
- 23.Petty WJ, Dragnev KH, Memoli VA, Ma Y, Desai NB, Biddle A, et al. Epidermal growth factor receptor tyrosine kinase inhibition represses cyclin D1 in aerodigestive tract cancers. Clin Cancer Res. 2004;10:7547–7554. doi: 10.1158/1078-0432.CCR-04-1169. [DOI] [PubMed] [Google Scholar]
- 24.Ma Y, Fiering S, Black C, Liu X, Yuan Z, Memoli VA, et al. Transgenic cyclin E triggers dysplasia and multiple pulmonary adenocarcinomas. Proc Natl Acad Sci (USA) 2007;104:4089–4094. doi: 10.1073/pnas.0606537104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.To MD, Quigley DA, Mao JH, Del Rosario R, Hsu J, Hodgson G, et al. Progressive genomic instability in the FVB/Kras(LA2) mouse model of lung cancer. Mol Cancer Res. 2011;9:1339–1345. doi: 10.1158/1541-7786.MCR-11-0219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ritchie KJ, Malakhov MP, Hetherington CJ, Zhou L, Little MT, Malakhova OA, et al. Dysregulation of protein modification by ISG15 results in brain cell injury. Genes Dev. 2002;16:2207–2212. doi: 10.1101/gad.1010202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Richer E, Prendergast C, Zhang DE, Qureshi ST, Vidal SM, Malo D. N-ethyl-N-nitrosourea-induced mutation in ubiquitin-specific peptidase 18 causes hyperactivation of IFN-alphaα/β signaling and suppresses STAT4-induced IFN-gamma production, resulting in increased susceptibility to Salmonella typhimurium. J Immunol. 2010;185:3593–3601. doi: 10.4049/jimmunol.1000890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rajalingam K, Schreck R, Rapp UR, Albert S. Ras oncogenes and their downstream targets. Biochim Biophys Acta. 2007;1773:1177–1195. doi: 10.1016/j.bbamcr.2007.01.012. [DOI] [PubMed] [Google Scholar]
- 29.Basters A, Geurink PP, Oualid FE, Ketscher L, Casutt MS, Krause E, et al. Molecular characterization of ubiquitin-specific protease 18 reveals substrate specificity for interferon-stimulated gene 15. FEBS J. 2014;281:1918–1928. doi: 10.1111/febs.12754. [DOI] [PubMed] [Google Scholar]
- 30.Burks J, Reed RE, Desai SD. ISGylation governs the oncogenic function of Ki-Ras in breast cancer. Oncogene. 2014;33:794–803. doi: 10.1038/onc.2012.633. [DOI] [PubMed] [Google Scholar]
- 31.Elad G, Paz A, Haklai R, Marciano D, Cox A, Kloog Y. Targeting of K-Ras 4B by S-trans, trans-farnesyl thiosalicylic acid. Biochim Biophys Acta. 1999;1452:228–242. doi: 10.1016/s0167-4889(99)00144-5. [DOI] [PubMed] [Google Scholar]
- 32.Zimmerman G, Papke B, Ismail S, Vartak N, Chandra A, Hoffmann M, et al. Small molecular inhibition of the KRAS-PDEδ interaction impairs oncogenic KRAS signaling. Nature. 2003;497:638–642. doi: 10.1038/nature12205. [DOI] [PubMed] [Google Scholar]
- 33.Van der Hoeven D, Cho KJ, Chiqurupati S, Parton RG, Hancock JF. Fendiline inhibits K-Ras plasma membrane localization and blocks K-Ras signal transmission. Mol Cell Biol. 2013;33:237–251. doi: 10.1128/MCB.00884-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Johnson L, Mercer K, Greenbaum D, Bronson RT, Crowley D, Tuveson DA, et al. Somatic activation of the K-ras oncogene causes early onset lung cancer in mice. Nature. 2001;410:1111–1116. doi: 10.1038/35074129. [DOI] [PubMed] [Google Scholar]
- 35.Klein EA, Assoian RK. Transcriptional regulation of the cyclin D1 gene at a glance. J Cell Sci. 2008;121:3853–3857. doi: 10.1242/jcs.039131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jancik S, Drabek J, Radzioch D, Hajduch M. Clinical relevance of KRAS in human cancers. J Biomed Biotechol. 2010;2010:1–13. doi: 10.1155/2010/150960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Komander D, Clague MJ, Urbe S. Breaking the chains: structure and function of the deubiquitinases. Nat Rev Mol Cell Biol. 2009;10:550–563. doi: 10.1038/nrm2731. [DOI] [PubMed] [Google Scholar]
- 38.Pant V, Lozano G. Limiting the power of p53 through the ubiquitin proteasome pathway. Genes Dev. 2014;28:1739–1751. doi: 10.1101/gad.247452.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lopez-Alcala C, Alvarez-Moya B, Villalonga P, Calvo M, Bachs O, Agell N. Identification of essential interacting elements in K-Ras/calmodulin binding and its role in K-Ras localization. J Biol Chem. 2008;283:10621–10623. doi: 10.1074/jbc.M706238200. [DOI] [PubMed] [Google Scholar]
- 40.Shukla S, Allam US, Ahsan A, Chen G, Krishnamurthy PM, Marsh K, et al. KRAS protein stability is regulated through SMURF2: UBCH5 complex-mediated β-TrCP1 degradation. Neoplasia. 2014;16:115–128. doi: 10.1593/neo.14184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tsai Y, Pestka S, Wang L, Runnels LW, Wan S, Lyu YL, et al. Interferon-β signaling contributes to Ras transformation. PLoS ONE. 2011;6:e24291. doi: 10.1371/journal.pone.0024291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chinyengetere F, Sekula DJ, Lu Y, Giustini AJ, Sanglikar A, Kawakami M, et al. Mice null for the deubiquitinase USP18 spontaneously develop leiomyosarcomas. BMC Cancer. 2015;15:886. doi: 10.1186/s12885-015-1883-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Abbas T, Dutta A. p21 in cancer: intricate networks and multiple activities. Nat Rev Cancer. 2009;9:400–414. doi: 10.1038/nrc2657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sarkisian CJ, Keister BA, Stairs DB, Boxer RB, Moody SE, Chodosh LA. Dose-dependent oncogene-induced senescence in vivo and its evasion during mammary tumorigenesis. Nat Cell Biol. 2007;9:493–505. doi: 10.1038/ncb1567. [DOI] [PubMed] [Google Scholar]
- 45.Collado M, Serrano M. Senescence in tumors: evidence from mice and humans. Nat Rev Cancer. 2010;10:51–57. doi: 10.1038/nrc2772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mustachio LM, Kawakami M, Lu Y, Rodriguez-Canales J, Mino B, Behrens C, et al. The ISG15-specific protease USP18 regulates stability of PTEN. Oncotarget. 2017;8:3–14. doi: 10.18632/oncotarget.13914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Farshi P, Deshmukh RR, Nwankwo JO, Arkwright RT, Cvek B, Liu J, et al. Deubiquitinases (DUBs) and DUB inhibitors: a patent review. Expert Opin Ther Pat. 2015;25:1191–1208. doi: 10.1517/13543776.2015.1056737. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.