

Abstract
Hepatocellular carcinoma (HCC) is the third most deadly cancer in the US, with a meager 5-year survival rate of less than 20%. Such unfavorable numbers are closely related to the heterogeneity of the disease and the unsatisfactory therapies currently used to manage patients with invasive HCC. Outside of the clinic, gene therapy research is evolving to overcome the poor responses and toxicity associated with standard treatments. The inadequacy of gene delivery vectors, including poor intracellular delivery and cell specificity, are major barriers in the gene therapy field. Herein, we described a non-viral strategy for effective and cancer-specific DNA delivery to human HCC using biodegradable Poly(Beta-Amino Ester) (PBAE) nanoparticles (NPs). Varied PBAE NP formulations were evaluated for transfection efficacy and cytotoxicity to a range of human HCC cells as well as healthy human hepatocytes. To address HCC heterogeneity, nine different sources of human HCC cells were utilized. The polymeric NPs composed of 2-((3-aminopropyl)amino)ethanol end-modified poly(1,5-pentanediol diacrylate-co-3-amino-1-propanol) (‘536’) at a 25 polymer-to-DNA weight-to-weight ratio led to high transfection efficacy to all of the liver cancer lines, but not to hepatocytes. Each individual HCC line had a significantly higher percentage of exogenous gene expression than the healthy liver cells (P < 0.01). Notably, this biodegradable end-modified PBAE gene delivery vector was not cytotoxic and maintained the viability of hepatocytes above 80%. In a HCC/hepatocyte co-culture model, in which cancerous and healthy cells share the same micro-environment, 536 25 w/w NPs specifically transfected cancer cells. PBAE NP administration to a subcutaneous HCC mouse model, established with one of the human lines tested in vitro, confirmed effective DNA transfection in vivo. PBAE-based NPs enabled high and preferential DNA delivery to HCC cells, sparing healthy hepatocytes. These biodegradable and liver cancer-selective NPs are a promising technology to deliver therapeutic genes to liver cancer.
Keywords: Nanoparticle, Gene Therapy, Cancer, Hepatocellular
Graphical abstract
Introduction
Hepatocellular carcinoma (HCC), which accounts for approximately 90%[1] of all primary liver malignancies, is the sixth most commonly diagnosed cancer and the second main cause of cancer death worldwide[2]. In the U.S., the incidence rates are on the rise and HCC is currently considered the fastest-growing cause of cancer mortality in the country[3–5], rating an overall 5-year survival of less than 20%[3, 4]. Such poor outcomes can be related to a number of factors, including late diagnosis, inefficacy of standard therapies[6], and heterogeneity of the disease.
As for other cancer types, chemotherapeutic drugs are main components of the treatment approaches for HCC. The off-target toxicity of anticancer drugs, however, is particularly critical in HCC cases. HCC patients, especially those with more invasive disease, often present some stage of liver failure. If the treatment for HCC promotes destruction of fully active and healthy hepatocytes, patients die due to progression of liver failure. In fact, failure caused by the injury to normal tissue is the main cause of death among patients treated with transarterial chemoembolization (TACE)[7], a palliative intervention considered the standard treatment for patients with invasive intrahepatic disease and no extrahepatic spread[6, 8]. Drug resistance represents another drawback of chemotherapeutic-based strategies to treat HCC and essentially chemo-refractory and/or recurrent tumors are common outcomes of systemic or local chemotherapy. [9].
Gene therapy research is evolving to overcome the poor responses and toxicity associated with standard treatments. Emerging gene-based strategies can be tuned to fit each disease or patients’ needs, avoiding treatment resistance, improving cancer-targeting and limiting hepatocyte damage. The efficacy of gene therapy studies, however, has been greatly affected by the inadequacy of delivery vectors, which, in addition to safety considerations, have shown poor selectivity in vector targeting and ineffective delivery.
Despite the fact that viruses are powerful vectors with high transfection efficacy[10], their application in the clinical setting raises important safety concerns[10–13]. Non-viral vectors, on the other hand, can be near non-immunogenic and safer, but have shown unsatisfactory transfection efficacy[14–16]. The optimization of non-viral vectors for gene therapy is of great interest, since they are relatively inexpensive to produce, have a large DNA carrying capacity, and allow easy modifications[17]. Poly(beta-amino ester) (PBAE) is a class of cationic polymers designed for non-viral gene delivery[18, 19]. PBAEs with varying chemical structures and properties are synthesized from a library of backbone, side chain, and end-cap units, which enables high throughput screening and optimization of PBAEs for particular applications[18, 20]. Unlike many other cationic polymers, PBAEs can be degraded by hydrolysis, releasing the nucleic acid cargo after endosomal scape and reducing cytotoxicity[19]. Certain PBAE NPs have also demonstrated intrinsic biomaterial-mediated cell specificity, delivering DNA to cancer cells while avoiding healthy tissues. The engineering of polymer and NP properties is the key to improve selective and effective internalization of therapeutic genes into target cells. Herein, we describe a non-viral strategy for effective, non-hepatotoxic and cancer-specific DNA delivery to human HCC using biodegradable PBAE NPs.
Materials and Methods
PBAE Synthesis and Characterization
PBAE polymers were synthesized by combining backbone, side chain and end-cap monomer analogs through a two-step reaction (Figure 1A), as previously reported by us[21, 22]. In summary, a diacrylate backbone (“B”) and an amino-alcohol side chain (“S”) were mixed at varied B:S molar ratios and the reaction allowed to proceed for 24 hours under magnetic stirring (1000 rpm) at 90°C. Next, the resultant base polymer (BS) and an amine-containing end-cap monomer (“E”) were separately dissolved in anhydrous tetrahydrofuran (THF), and the solutions combined at a 10-fold molar excess of end group to diacrylate-termini. The end-capping reaction mix was then stirred at 400 rpm for 1 hr at room temperature and polymers precipitated with anhydrous diethyl ether for purification. The resultant suspension was vortexed for approximately 20 seconds and spun down at 1000 rpm for 5 minutes at 4°C. After centrifugation, the supernatant was removed and the precipitated polymer was washed once more with ether. Remaining traces of ether were removed by placing the polymer under vacuum with desiccant for 5 days. Ether-purified polymers were dissolved in anhydrous DMSO to a final BS concentration of 100 mg/mL and stored with desiccant at −20°C.
Fig. 1.

A. Synthesis of end-capped PBAE polymers. A diacrylate backbone monomer (B), an amino-alcohol sidechain monomer (S) and an amine containing end-capping molecule (E) were conjugated through a two-step process. B. Chemical structures of B, S and E monomers used in the synthesis of PBAE polymers for this study.
The following monomers were utilized in the synthesis of PBAE polymers (Figure 1B): 1,4-butanediol diacrylate (Alfa Aesar, Ward Hill, MA); 1,5-pentanediol diacrylate (Monomer-Polymer and Dajac Labs, Trevose, PA); 3-amino-1-propanol (Alfa Aesar); 4-amino-1-butanol (Alfa Aesar); 5-amino-1-pentanol (Alfa Aesar); 1,3-diaminopentane (TCI America, Portland, OR); 2-Methyl-1,5-diaminopentane (TCI America); 2-(3-aminopropylamino)ethanol (Sigma-Aldrich, St. Louis, MO); and 1-(3-aminopropyl)-4-methylpiperazine (Alfa Aesar). For the purpose of PBAE polymers’ nomenclature, monomers used in the synthesis of the BS base polymer were denominated by a letter representing their function in the PBAE structure, i.e., B for backbone and S for side chain, followed by the number of carbons in their hydrocarbon chain. End-capping monomers were sequentially numbered according to similarities in their amine structures. The PBAE polymer was referred as the BSE monomers found on its structure; for example, the polymer synthesized from monomers B5 and S3 and end-capped with E6 was denominated “B5S3E6” or “536”.
Gel permeation chromatography (GPC, Waters 2414 Refractive Index Detector, Milford, MA) was used to characterize all PBAE polymers used in this study (results summarized in Supplemental Fig. 1). The structure of 2-((3-aminopropyl)amino)ethanol end-modified poly(1,5-pentanediol diacrylate-co-3-amino-1-propanol) (536), chosen as the optimal PBAE polymer for subsequent studies, was analyzed by Proton Nuclear Magnetic Resonance (1H-NMR) using a Bruker Avance III 500 MHz NMR spectrometer in CDCI 3.
Cell Culture
A human hepatocyte line (THLE-3 [ATCC® CRL-11233™]) and eight HCC cell lines (Hep3b [ATCC® HB-8064™], HepG2 [ATCC® HB-8065™], C3A, SK-HEP-1, PLC/PRF/5, SNU-387, SNU-423, SNU-475 [ATCC® TCP-1011™]) were purchased from ATCC (Manassas, VA). The cell culture method followed the protocol provided by ATCC for each line. Briefly, Hep3b, HepG2, PLC/PRF/5, SK-HEP-1 and C3A were cultured in Minimum Essential Media (MEM) supplemented with 10% heat inactivated FBS, 100 U/mL Penicillin/100μg/mL Streptomycin, 100 uM of MEM non-essential amino acids solution, and 1 mM of sodium pyruvate. Complete growth media of RPMI Medium 1640 with 10% heat inactivated FBS and 100 U/mL Penicillin/100μg/mL Streptomycin was used to culture SNU-475, SNU-387 and SNU-423. The human hepatocyte line was cultured in Bronchial Epithelial Cell Growth Medium (BEBM) enriched with the additives accompanying the kit (BEGM Bullet Kit [CC3170]; Lonza/Clonetics Corporation, Walkersville, MD), except Gentamycin-Amphotericin and Epinephrine, and further supplemented with 10% FBS, certified, US origin from Gibco™ (16000044, Thermo Fisher Scientific, Waltham, MA), 100 U/mL Penicillin/100μg/mL Streptomycin, 5 ng/mL of human epidermal growth factor (EGF), and 70 ng/mL of O-phosphorylethanolamine. The flasks used for sub-culturing of THLE-3 cells were pre-coated with a solution of 0.01 mg/mL of human fibronectin, 0.03 mg/mL of bovine collagen type I, and 0.01 mg/mL of bovine serum albumin dissolved in BEBM basal medium. Coated flasks were allowed to incubate overnight at 37°C and excess solution was removed from the vessels immediately before use. The HCC line Huh-7 was kindly provided by Dr. Phuoc Tran’s laboratory from the Johns Hopkins University School of Medicine. Huh-7 cells were cultured in high glucose Dulbecco’s Modified Eagle Medium with 10% heat inactivated FBS and 100 U/mL Penicillin/100μg/mL Streptomycin. All cell cultures were maintained in a humidified incubator, at 37°C, with 5% CO2.
Transfections
All the cell lines were separately seeded in tissue culture-treated 96-well plates at a density of 10,000 cells/well and allowed to adhere to the plates for 24 hrs. Hepatocytes were seeded in plates treated with pre-coating mix (described above). Immediately before transfection, the growth medium of each cell line was renewed (100 μL/well).
To form NPs, individual PBAE polymers and pEGFP-N1 (eGFP) plasmid DNA [purchased from Clontech Laboratories Inc. (Mountain View, CA) and amplified by Aldevron (Fargo, ND)] were separately diluted in 25 mM sodium acetate buffer (NaAc; pH 5) and subsequently combined at 25, 50 and 75 polymer-to-DNA weight-to-weight (w/w) ratios, to a final DNA dosage of 600 ng/well (30 ng/μL of DNA in NP mix). Polymer and DNA were allowed to complex for 10 minutes at room temperature and the NP mix (20 μL/well) was then added to the culture media covering the cell monolayer. Cells were incubated with NPs at 37°C for 2 hours, following which period the NP-containing medium was replaced with fresh culture medium. Lipofectamine™ 2000 (Invitrogen, Carlsbad, CA), jetPRIME™ (Polyplus-transfection SA, Illkirch-Graffenstaden, France), and 25kDa branched polyethylenimine (PEI, Sigma-Aldrich) were used as positive control transfection reagents. For these transfections, positive control and eGFP were separately diluted in buffer/media and then combined for NP formation. Lipofectamine™ 2000 was diluted in Opti MEM™ (Thermo Fisher Scientific, Waltham, MA), jetPRIME™ in the supplied buffer, and PEI in 150 mM NaCl. After complexation, 20 μL/well of NP mix (600 ng of DNA per well) were added to 100 μL of media covering the cells, similar to the procedure described for PBAE NPs. Transfections with PBAE and positive controls were performed side-by-side. Six replicates (3 for each assay, i.e., MTS and flow cytometry) were tested for each polymer and condition.
The average hydrodynamic size and zeta potential of 536 25 w/w NP were measured using Malvern Zetasizer Nano ZS (Malvern Instruments, Malvern, UK). Shape and size of 536 25 w/w NPs were also recorded by transmission electronic microscopy (TEM, Philips/FEI BioTwin CM120 TEM, Eindhoven, Netherlands).
For co-culture experiments, Huh-7 cells were pre-transfected with a red fluorescent protein (RFP) using the Piggybac transposon/transposase system to enable permanent RFP expression. The Piggybac transposon carrying mRFP-Ruby (ex. max.: 585 nm, em. max.: 604 nm) maker was purchased from System Biosciences (PB-CMV-MCS-EF1-RFP cDNA, Palo Alto, CA) and the Piggybac transposase plasmid was kindly provided by Dr. Karl Wahlin of Dr. Donald Zack’s laboratory in the Department of Ophthalmology at the Johns Hopkins University School of Medicine. The transfection was performed in T175 tissue culture-treated vessels using the polymer 536 at 25 w/w ratio, with 5/6 of the DNA dose corresponding to transposon and 1/6 to transposase. The total DNA dose and volumes of media and NP mix were simply scaled up from 96-well plates according to the surface area. After four weeks in culture to allow transient expression to fade, RFP positive cells were sorted via fluorescent-activated cell sorting (FACS) using a yellow green laser (561 nm). The sorting procedure was repeated twice until 99% purity was reached. To maintain this high purity level, RFP+ cells were frozen two days after the second FACS and only thawed two days before seeding for co-culture experiments.
Finally, RFP+ Huh-7 cells and THLE-3 hepatocytes (RFP negative) were seeded together in 96-well plates at 10,000 cells/well (5,000 cells of each line). THLE-3 complete growth medium was used for cells in co-culture. Twenty-four hours after seeding, cells were transfected with eGFP using the same procedure described for separate cultures.
Cell Viability Assay
In order to assess cell viability, the metabolic activity of transfected and non-transfected cells were measured using MTS assay from Promega (CellTiter 96 AQueous Nonradioactive Cell Proliferation Assay; Madison, WI) 24 hours post-transfection. After preparation according to the manufacturer’s protocol, the absorbance at 490 nm was recorded for each well using the Synergy 2 Multi-mode Reader/Gen5™ software (Biotek, Winooski, VT). The background absorbance was subtracted and the average metabolic activity of cells from each condition was then calculated relative to the untreated controls.
Assessment of Transfection Efficacy and Specificity
Forty-eight hours after transfection, representative bright-field and fluorescence microscopy images were taken for each of the conditions tested. For quantitative purposes, the level of eGFP expression for each replicate and condition was evaluated using the BD Accuri C6 Flow Cytometer (BD Biosciences, San Jose, CA) connected to a HyperCyt™ autosampler (IntelliCyt Corporation, Albuquerque, NM). Immediately before flow, cells were detached from the transfection plates using a trypsin-ETDA solution, mixed with 1:200 v/v ratio of PI (propidium iodide):PBS with 2% FBS (flow buffer with PI), transferred to round-bottom plates, centrifuged and re-suspended in flow buffer with PI. Using the FlowJo™ software v.10.1r7 (Ashland, OR), dead cells (PI+, FL3-H 610±20 nm) were excluded and live single cells analyzed for the percentage (eGFP positive %) and intensity of eGFP expression (geometric mean) using the FL1-H channel (533±30 nm). For co-culture experiments, flow cytometry was performed without PI staining using a green (488 nm) and yellow-green (561 nm) lasers.
Assessment of Cell Doubling Time
To establish the cellular growth rate, cells from each line (HCCs and THLE-3) were seeded in 12 well plates at 71,250 cells/well. Fifteen wells were prepared per line, allowing the analysis to be performed in triplicate for each time point (15, 45, 70, 94 and 116 hours). At the established time points, three wells per line were trypsinized and individually counted using a hemocytometer.
Electroporation of DNA
For electroporation studies, cells were harvested from cell culture flasks, washed with culture media not containing antibiotics, and aliquoted in 1.5 mL tubes at 100.000 cells/tube. After centrifugation, the cell pellet was resuspended with Resuspension Buffer R (Invitrogen) containing 10% of eGFP-N1 DNA plasmid solution to a final DNA concentration of 0.1 μg/μL. Using the Microporator MP-100 and 10 μL Neon™ tips (Invitrogen), 10,000 cells were electroporated at a time with a single pulse of 1300 or 1950 V per 20 s. Cells were then seeded in triplicate at 10,000 cells/well in 96 well plates pre-filled with 100 μl/well of antibiotic-free media. Flow cytometry was performed using the same protocol described above 48 hours after the electroporation procedure.
Cellular Uptake
Uptake of 536-DNA NPs was evaluated using flow cytometry analysis of labeled-DNA. eGFP-N1 plasmid DNA was labeled with Cy5 dye (ex.: 650 nm, em.: 670 nm) using the Label IT™ Nucleic Acid Labeling Kit, Cy5 (Mirus Bio, Madison, WI). NPs were formed and delivered to cells (seeded at 10,000 cells/well in 96 well plates) as described above. Two hours later, two washes of 50 μg/mL heparin were performed to remove traces of non-internalized DNA and cells were harvested for flow cytometry (640 nm laser, FL4-H channel 675±25 nm).
In vivo Studies
In vivo experiments included in this study were approved by the Institutional Animal Use and Care Committee (IACUC) of the Johns Hopkins University. For establishment of the xenograft model, fifteen athymic nude mice (female, 4 weeks old) were injected subcutaneously in the right upper flank with 3×106 Huh-7 cells suspended in 100 μL of Matrigel™ HC (Corning Life Sciences, Tewksbury MA) mixed with complete growth medium (1:1 v/v ratio). Animals were kept anesthetized during the inoculation using 2.5% isoflurane in oxygen (2 L/min). Four weeks after cell injection, the seven animals that developed tumors (average of 1.2 cm in diameter) were randomized into two groups: 4 mice for PBAE NP and 3 mice for PBS injection.
To enable in vivo imaging, a luciferase expression plasmid was used to form NPs. Luciferase-pcDNA3 plasmid DNA [purchased from Addgene (Cambridge, MA) and amplified by Aldevron (Fargo, ND)] and the polymer 536 at a 25 w/w ratio were combined (described above) 10 minutes prior to injection. Each animal received an intratumoral injection of 100 μL of NP solution in NaAc, with a total of 40 μg of DNA. To avoid leakage, particles were injected slowly and in multiple sites of the tumor. After 6, 24 and 48 hours of NP administration, bioluminescence images were captured using Xenogen IVIS™ Spectrum In Vivo Imaging (Caliper Life Sciences, Waltham, MA) upon intraperitoneal administration of D-Luciferin Potassium Salt at 150 mg/kg body weight (Gold Biotechnology, St. Louis, MO). Animals were imaged after 10 minutes of luciferin injection and were kept anesthetized with 2% isoflurane in oxygen (2 L/min) for the entire period. The average radiance from regions of interest (ROI) was measured using the Living Image software (Caliper Life Sciences).
Statistical Analysis
All experimental conditions were tested in triplicates and the results described as mean ± standard error of the mean (SEM). Two-tailed Student’s t-test was used for paired comparisons and one-way ANOVA followed by Dunnett’s post-hoc test for many-to-one comparisons. ANOVA and Bonferroni’s post-hoc test was applied for determination of the best formulation conditions among the positive controls.
Results
PBAE and Positive Control NP Screening
In order to find the most suitable PBAE formulation for DNA transfection to human HCC, ten end-capped PBAE polymers were evaluated for eGFP delivery. These polymers were complexed with eGFP-N1 plasmid to form NPs at three polymer-to-DNA w/w ratios (25, 50 and 75). While the amount of plasmid remained the same (600 ng/well) throughout all NP formulations, a range of polymer concentrations were evaluated to optimize w/w and balance high efficacy with cellular viability. PEI 25 kDa, jetPRIME™ and Lipofectamine™ 2000, also evaluated over a broad concentration range, were used as positive controls. All NP formulations were tested in nine different sources of HCC cells to address the genetic heterogeinity of human HCCs. Additionally, to evaluate cancer-selectivity and cytotoxicity to non-cancerous liver cells, NP screening was also performed on a healthy human hepatocyte line.
Due to the importance of minimizing damage to the liver parenchyma and mitigating progression of liver failure, a viability assay was used to evaluate off-target cytotoxicity to hepatocytes and narrow NPs down to optimal formulations. A minimal post-transfection metabolic activity of 80% was set as a threshold for the healthy human hepatocyte THLE-3 cells (Figure 2). Except for polymer 446, which was neither toxic nor effective in any of the concentrations tested, all other PBAE polymers at 50 and/or 75 w/w ratios resulted in unacceptable toxicity for THLE-3 hepatocyte cells. At 25 w/w ratios, four PBAE structures, 447, 456, 536 and 547, were observed to cause 20% or less cytotoxicity to the hepatocyte line. Among the positive controls, jetPRIME™ and PEI 25 kDa, at their two lowest concentrations (1:0.5 and 1:1 DNA-to-polymer w/v ratio for jetPRIME™ and 1 and 2 polymer-to-DNA w/w ratio for PEI), maintained the viability of THLE-3 above 80% after treatment. Lipofectamine™ 2000 was highly toxic to THLE-3 cultures at all formulations evaluated, even at a relatively low concentration of 1:3 Lipofectamine™ 2000-to-DNA w/w ratios, in which case THLE-3 viability was below 62% (61.6 ± 0.4).
Fig. 2.

Viability of hepatocytes (THLE-3) 24 hours after transfection with a broad range of PBAE structures and concentrations. PBAE polymers were evaluated in comparison to three commercially available non-viral transfection vectors (positive controls) at a range of dosages. Viability of cells treated with polymer 536 at a 25 polymer-to-DNA w/w ratio remained above 80%. Among the positive controls, only jetPRIME™ and PEI 25kDa at relatively low concentrations enabled comparably high viability. Results from each condition were obtained as the average metabolic activity relative to untreated controls. The proportions of Lipofectamine™ 2000-to-DNA and jetPRIME™-to-DNA in each condition are expressed as weight-to-weight and weight-to-volume ratios, respectively.
As a general trend, cytotoxicity to hepatocytes and transfection efficacy to HCC cells increased with increasing polymer concentrations. Most PBAEs led to 20% or higher exogenous eGFP expression only at 50 and/or 75 w/w ratios, toxic concentrations to THLE-3 cells. This same trend was also observed among the positive controls. At 25 w/w ratio, polymers 446 and 547, non-toxic for THLE-3 cells, led to low transfection efficacy to all cancer lines. The polymer 456 25 w/w was only able to promote higher transfection in four out of the nine lines. At such low concentration, the PBAE structure referred to as 536 was the only polymer capable of effectively transfecting all the HCC cell lines while maintaining THLE-3 viability. jetPRIME™ at 1:0.5 and 1:1, even though nearly non-toxic to hepatocytes, were unable to transfect cancer cells. PEI 1 and 2 w/w had similar delivery capacities, with the latter leading to slightly higher eGFP expression than the former. Therefore, 536 at 25 w/w was chosen as the optimal NP due to its high transfection efficacy to a heterogenic HCC population and its low cytotoxicity to hepatocytes. Among the positive controls that showed high hepatocyte cytotoxicity (PEI 4 and 6 w/w, jetPRIME™ 1:2 and 1:3 w/v, and Lipofectamine™ 2000 at all concentrations), only transfection with jetPRIME at 1:2 w/v resulted in a comparably high eGFP expression to all HCC cell lines. Figure 3 shows results from the full transfection screen performed for hepatocytes and two representatives of HCC cancer populations. Results from the remaining HCC lines can be found in the Figure 3 of the supplementary material.
Fig. 3.

Transfection efficacy screen of all PBAE and positive control NPs to A. Two representatives of HCC cell lines and the THLE-3 hepatocytes. EGFP expression was measured using flow cytometry 48 hours after transfection and analyzed for the percentage (positive %) and intensity (geometric mean) of GFP expression. Geometric mean results from each condition are relative to untreated controls. Norm.: Normalized. * P < 0.05 for statistically significant differences between PBAE polymers with superior transfection efficacy and the most effective among positive controls for each cell line.
536-based NP’s Specificity and Transfection Efficacy to HCC cells
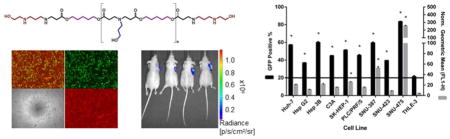
The optimal 536 PBAE NP formulation was compared side by side with the positive control that showed the least unsatisfactory results from the screen (PEI 25 kDa 2 w/w). 536 25 w/w NPs led to consistent and effective transfection of eGFP DNA to all cancer cells, which was also specific over healthy hepatocytes (P < 0.01) (Figure 4). The eGFP expression % ranged from 36.9% ± 0.4 to 83.5% ± 0.5 in HCC cells and was approximately 20% for the THLE-3 line. PEI 25kDa 2 w/w NPs, on the other hand, while also specific for the cancer lines, did not promote effective transfection in a consistent manner throughout the HCC cells. Out of the nine cancer lines, only 2 reached eGFP expression (positive %) levels comparable to the ones achieved with 536 25 w/w NPs. Interestingly, some slow dividing cells, such as SNU-387 and SNU-475 (doubling times for all cell lines can be viewed under Supplemental Figure 6) were poorly transfected with the PEI polymer (7.8 ± 0.1 and 12.9 ± 0.1 for SNU-387 and SNU-475, respectively) while they had high (60 ± 1 and 83.5 ± 0.5, respectively) eGFP expression levels with the 536 polymer. Notably, the THLE-3 doubling time was an intermediate value (53 ± 5 hours) between the HCC cells with the fastest and slowest growth rates (ranging from 24 ± 0.3 hours to 76 ± 4 hours). These results demonstrate that the cell specificity of cancer cells compared to healthy cells is unlikely to be due to differences in cell growth rate and the breakdown of the nuclear membrane as a gene delivery barrier.
Fig. 4.

Transfection efficacy of the optimal PBAE (536 25 w/w) and positive control (PEI 25kDa 2 w/w) NP formulations for all cell lines. Transfection of eGFP DNA with 536 25 w/w NPs was effective and consistent to all cancer cells. In addition, the eGFP expression in all HCC lines was also specific over healthy hepatocytes. While also cancer-selective, PEI 25kDa 2 w/w NPs did not promote consistently high eGFP expression to all HCC cells. A. EGFP expression of all cell lines 48 hours after transfection with 536 25 w/w (left) or PEI 25kDa 2 w/w (right). B. Flow cytometry gating and microscopy images of two representatives of HCC lines and THLE-3 hepatocytes treated with 536 25 w/w (left column) or PEI 25kDa 2 w/w (right column). * P < 0.01
To analyze the effects of different rates of plasmid transcription and translation in cancer vs. non-cancer cells, we electroporated the plasmid without the use a polymeric gene delivery particle. Following electroporation, the eGFP expression (eGFP positive %) in THLE-3 cells was 38% ± 3 (Figure 5A), higher than the expression observed in Huh-7 HCC cells (P < 0.05) and not significantly different from four other lines (Hep3B, HepG2, PLC/PRF/5, and C3A). These results demonstrate that the cancer cell specificity is not based on transcriptional targeting of the plasmid to only cancer cells or to dramatically different rates of transcription and translation between the cancerous and healthy cells.
Fig. 5.

A. Levels of eGFP expression following electroporation for HCC cells and hepatocytes. B. Cy5 signal from all nine cancer cell populations and THLE-3 hepatocytes showing cellular uptake of 536 NPs. * P < 0.05 for statistically significant differences between hepatocytes and each individual HCC line.
To analyze cellular uptake, Cy5 fluorescently labeled-DNA was utilized for tracking. The cellular uptake of the NPs, as measured by the percentage of cells that positively show Cy5 fluorescence following delivery of the labeled plasmid, show significantly less uptake in the THLE-3 cells (81% ± 5) when compared to all of the nine HCC lines (P < 0.05; Figure 5B). Overall, the intensity of the Cy5 signals (geometric mean) was also lower in the THLE-3 cells than in the HCC lines (reaching statistical significance in six of the nine cell lines and with no cancer cell line having reduced cellular uptake of the 536 NPs as compared to the healthy hepatocytes). Taken together, these results indicate that the delivery vector material type and its consequent cellular uptake may play an essential role in the observed biomaterial-mediated cancer-targeting phenomenon.
Under fluorescence microscopy visualization, co-cultures of Huh-7 (RFP positive) and THLE-3 (RFP negative) cells indicated an impressive co-expression of GFP and RFP following transfection with 536 25 w/w NPs carrying eGFP (Figure 6A). This finding was also observed with flow cytometry (Figure 6B and C), which evidenced over 90% of eGFP signal in Huh-7 cells (95.37% ± 0.5), expression significantly higher than the observed in THLE-3 hepatocytes (35.6% ± 0.26, P < 0.01). When compared to separate culture experiments, the percentage of eGFP expression was 1.6-fold higher for both Huh-7 and THLE-3 cells. Despite also preserving cancer-specificity, treatment with PEI 25 kDa 2 w/w NPs to Huh-7/THLE-3 co-cultures did not cause improvement in the eGFP expression to Huh-7 cells while increasing it over 3-fold to hepatocytes (Supplemental Figure 5).
Fig. 6.

Both efficacy and cancer-specificity of 536 25 w/w NPs were preserved in co-culture of HCC (Huh-7) and hepatocytes (THLE-3). A. Microscopy images and B. Flow cytometry gating of a 536 25 w/w NP-treated co-culture representative 48 hours after eGFP transfection. Microscopy images show: merged eGFP and RFP channels (top left), eGFP channel only (top right), bright-field only (bottom left), and RFP channel only (bottom right). C. Percentage of GFP positive cells among Huh-7 (RFP positive) and THLE-3 (RFP negative) by flow cytometry analysis. Co-cultures were plated and transfected in triplicate. * P < 0.05
Characterization of the Polymer 2-((3-aminopropyl)amino)ethanol end-modified poly(1,5-pentanediol diacrylate -co-3-amino-1-propanol) (536) and 536 25 w/w NPs
On GPC, the weight average of polymer 536 was 8229 Da (relative to monodisperse polystyrene standards) with a polydispersity of 1.77. The structure of the 536 polymer is shown in Figure 7A and its H1- NMR spectrum in Supplemental Figure 4. Shape and size of 536 25 w/w NPs were visualized under TEM and are shown in Figure 7B. 536 25 w/w NPs had a hydrodynamic size of 157 ± 3 nm (Figure 7C) and 85 ± 2 nm when diluted 1:1 (v/v) in phosphate buffered saline (PBS) 1x and 1:1 (v/v) in NaAc, respectively. The zeta potential of these particles was 18 ± 0.3 mV in 1:1 (v/v) of PBS 1x.
Fig. 7.

A. Chemical structure of the polymer 2-((3-aminopropyl)amino)ethanol end-modified poly(1,5-pentanediol diacrylate-co-3-amino-1-propanol) (536). B. TEM image and C. Physical characterization (size and zeta-potential) of 536-based NPs carrying eGFP plasmid DNA at the 25 w/w ratio. Imaging, sizing and zeta-potential measurement were carried out with NPs prepared following the same methods described for transfection experiments and immediately after NP complexation. A 1:1 v/v dilution of NP mix to PBS 1x was performed for sizing and zeta-potential measurements.
In vivo Study
Athymic nude mice bearing subcutaneous human HCC xenografts were employed to test the ability of the leading PBAE NP formulation to deliver DNA to HCC tumors in vivo. Bioluminescence performed at 6, 24 and 48 hours following intratumoral NP injection showed that 536 25 w/w NPs can successfully transfect HCC tumors in vivo (Figure 8). The luminescence signal started to appear as early as 6 hours after NP administration (1.90E+04 ± 2.98E+03) and became significantly higher than the background at 24 hours post-injection (536 25 w/w NPs = 5.06E+04 ± 7.13E+03, PBS = 1.24E+03 ± 1.96E+02, P < 0.05).
Fig. 8.

Effective DNA delivery in vivo using an optimal PBAE formulation. A. Bioluminescence images of subcutaneous Huh-7 xenograft mice at 6, 24 and 48 hours following intratumoral injection of 536 25 w/w NPs or PBS. B. Summary analysis of the average radiance for treated and control animals at the three recorded time points. * P < 0.05
Discussion
Viral methods have been widely explored for gene delivery due to the efficacy of their naturally evolved transformation mechanisms. While many ongoing clinical trials have used viral gene delivery to treat HCC[23–28], viral therapeutics raise safety concerns, including immunogenicity and tumorigenicity following insertional mutagenesis [29]. Non-viral polymer- and lipid-based gene delivery methods do not pose these risks, but they have traditionally been limited by poor transfection efficacy. Recently, biodegradable PBAE NPs have shown promising transfection rates in several cancer cell lines, suggesting their potential for gene delivery to HCC[30–32]. This study characterized PBAE-DNA NP delivery to HCC by evaluating transfection efficacy, cytotoxicity, and biomaterial-mediated specificity in vitro, in single and co-culture systems. The capacity of PBAE-DNA NPs as a delivery vector to transfect HCC tumors was also tested in vivo.
Following screening of a PBAE NP library, a leading formulation (polymer 536 at 25 polymer-to-DNA w/w ratio) demonstrated to effectively transfect nine human HCC cell lines with low cytotoxicity to human hepatocytes, and superiority over PEI and other commercially available transfection reagents (Lipofectamine™ 2000 and jetPRIME™). jetPRIME™ succeeded in transfecting HCC cells only at relatively high concentrations, which caused critical viability loss in heathy human liver cells. Lipofectamine™ 2000 failed to promote effective transfection to the majority of cancer cells while also causing significant toxicity to human hepatocytes at all concentrations, results consistent with previous study demonstrating significant toxicity of Lipofectamine™ 2000 to murine hepatocytes[21]. PEI 25kDa preserved viability of hepatocytes at lower concentrations, but was unable to address the HCC heterogeneity issue, failing to consistently transfect all HCC lines. Using 536 25 w/w NPs, high transfection rates were possible and consistent across all the tested human HCC cell lines. The robust nature of the results strengthens the clinical significance of this study because, like many cancers, HCC is a heterogeneous disease. Genetic profiles of HCC tumors vary between patients, and these differences can dramatically affect the treatment response[33]. Mutations and changes in expression profiles have also been shown to induce chemoresistance in HCC. For instance, 29% of HCC have mutations in TP53, which affects G1 checkpoint regulation and induces drug resistance[34]. Huh-7, an HCC cell line with mutated TP53, was transfected effectively by leading PBAE formulations, suggesting that these particles can target cells that are resistant to chemotherapies.
NPs are attractive options for cancer drug delivery because they have potential for specificity through active and passive tumor targeting. PBAEs in particular have shown biomaterial-mediated targeting to cancer cells, which can be optimized by tuning polymer properties such as molecular weight, charge, and hydrophobicity[35]. Small changes to the base-polymer (BS) hydrocarbon chain can drastically modify the polymer’s structure-activity relationship. While hydrophobicity can be directly associated with transfection capacity, increments in its levels can cause increased cytotoxicity. A balance between hydrophobicity and hydrophilicity of the polymer structure can promote effective transfection and also preserve cell viability. Thus, the B5S3 (or 53) base polymer structure, with five carbons between acrylate groups in the B backbone monomer and three carbons and a hydroxyl group in the S side chain, can lead to optimized transfection and viability outcomes by maintaining ideal hydrophobicity levels[36, 37]. Secondary amine groups present in the E6 end-cap monomer of the 536 polymer can have high endosomal buffering capacity, unlike primary amines (e.g.: E4), a characteristic that has shown to also favor transfection efficacy[38]. Overall, the combination of a favorable end-cap monomer, structural balance and hydrolytically degradable ester groups show that the 536 polymer is well-designed for gene delivery. The leading PBAE NP formulation in this study, 536 25 w/w, transfected nine human HCC cell lines at a significantly higher rate than human hepatocytes when the cells were cultured separately. Further, in a co-culture of Huh-7 HCC cells and THLE3 hepatocytes, 536 25 w/w NPs preferentially transfected the cancer line over healthy cells. These results, particularly in a micro-environment shared by human cancerous and non-cancerous cells, show potential for biomaterial-mediated targeting of HCC in vivo. Other PBAE polymers have been previously reported to preferentially transfect cancer cells[21, 30], but the mechanism behind this phenomenon remains unknown. In this study, we demonstrated that the cell growth rate seems not to be a determining factor, since the measurement of the doubling times showed that THLE-3 hepatocytes grow both faster than some of the HCC cell lines and slower than some of the HCC cell lines. Similarly, the lack of clear gene delivery specificity with the plasmid following electroporation among the whole panel of nine human HCC lines compared to the hepatocytes indicates that transcriptional targeting and transcription and translation rates are not the main driver for the observed robust cancer specific gene delivery.
On the other hand, PBAE NP uptake did demonstrate selectivity, with inferior cellular uptake in hepatocytes when compared to cancer populations. Corroborative findings of Kim et al. [39] reveal that the specific cellular uptake pathways employed by PBAE NPs can also be critical. In this work, although the preferential uptake mechanism for PBAE NPs in human triple-negative breast cancer cells was observed to be caveolae-mediated, uptake through the clathrin pathway was suggested to be more efficient, leading to relatively higher transfection levels [39]. Sunshine et al. [38] and Bhise et al. [40] suggest that the end-capping of PBAEs is a critical parameter for increasing the cellular uptake of PBAE NPs as well and this is an intriguing area of research for future studies.
To investigate biocompatibility and gene delivery capacity of PBAE NPs in vivo, 536 25 w/w NPs were injected locally in a subcutaneous xenograft model in athymic nude mice. After delivering luciferin systemically, luciferase activity was observed at the tumor site, suggesting that the particles effectively transfected tumor cells. 536 25 w/w NPs may be well suited for other in vivo applications, including systemic and/or transarterial delivery as, due to their small size (< 200 nm), they could potentially localize to tumor sites by the enhanced permeability and retention (EPR) effect[41–44] as has been described for similar polymeric NPs. The possibility for transarterial delivery is of particular interest in the case of HCC, since the differential vascularization between normal parenchyma (portal vein) and HCC (hepatic artery) enables another level of cancer-selectivity (regio-selectivity) through the approach of the hepatic artery, a benefit that is already been exploited in the clinical setting[45]. Because PBAE 536 NPs show promising transfection efficacy, low cytotoxicity, and HCC specificity, they may be an enabling gene delivery technology for HCC therapy.
Conclusions
Top PBAE-based NP formulations were observed to enable high and preferential DNA delivery to varied human HCC cells, sparing healthy hepatocytes and preserving cell viability. The polymer referred to as 536 at a 25 polymer-to-DNA w/w ratio was highly consistent in its cancer-selectivity, leading to significantly higher transfection to a heterogeneous range of human HCC lines when individually compared to healthy parenchymal cells and when evaluated in an in vivo-mimicking HCC/hepatocyte co-culture model. This same NP formulation could also achieve significant gene delivery to human HCC in vivo. These findings suggest that biomaterial-mediated DNA delivery using synthetic PBAEs is a potentially viable strategy for liver cancer targeting and treatment.
Supplementary Material
Acknowledgments
We thank Dr. Karl Wahlin, Dr. Donald Zack, and Dr. Phuoc Tran and their and laboratories for providing essential materials for this study. We also thank Dr. Hao Zang, who performed flow cytometry-based sorting of mRFP-Ruby positive cells.
Funding: This work was supported by the National Institutes of Health [grant numbers R01EB022148, R01EB016721, P30EY001765, and CA058236].
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.El–Serag HB, Rudolph KL. Hepatocellular carcinoma: epidemiology and molecular carcinogenesis. Gastroenterology. 2007;132:2557–2576. doi: 10.1053/j.gastro.2007.04.061. [DOI] [PubMed] [Google Scholar]
- 2.Ferlay J, Soerjomataram I, Dikshit R, Eser S, Mathers C, Rebelo M, Parkin DM, Forman D, Bray F. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer. 2015;136:E359–E386. doi: 10.1002/ijc.29210. [DOI] [PubMed] [Google Scholar]
- 3.Hashem BES. Hepatocellular carcinoma. N Engl J Med. 2011;365:1118–1127. doi: 10.1056/NEJMra1001683. [DOI] [PubMed] [Google Scholar]
- 4.Ries LAG, Eisner MP, Kosary CL, Hankey BF, Miller BA, Clegg L. Surveillance, Epidemiology, and End Results (SEER) Program SEER* Stat Database: Incidence—SEER 9 Regs Public-Use, Nov 2004 Sub (1973–2002) National Cancer Institute, Division of Cancer Control and Population Sciences, Surveillance Research Program, Cancer Statistics Branch; 2005. [Google Scholar]
- 5.Venook AP, Papandreou C, Furuse J, de Guevara LL. The incidence and epidemiology of hepatocellular carcinoma: a global and regional perspective. Oncologist. 2010;15:5–13. doi: 10.1634/theoncologist.2010-S4-05. [DOI] [PubMed] [Google Scholar]
- 6.Forner LJA, Bruix J. Hepatocellular carcinoma. Lancet. 2012;379:1245–1255. doi: 10.1016/S0140-6736(11)61347-0. [DOI] [PubMed] [Google Scholar]
- 7.Huang YS, Chiang JH, Wu JC, Chang FY, Lee SD. Risk of hepatic failure after transcatheter arterial chemoembolization for hepatocellular carcinoma: predictive value of the monoethylglycinexylidide test. Am J Gastroenterol. 2002;97:1223–1227. doi: 10.1111/j.1572-0241.2002.05709.x. [DOI] [PubMed] [Google Scholar]
- 8.Llovet BAJM, Bruix J. Hepatocellular carcinoma. Lancet. 2003;362:1907–1917. doi: 10.1016/S0140-6736(03)14964-1. [DOI] [PubMed] [Google Scholar]
- 9.Asghar U, Meyer T. Are there opportunities for chemotherapy in the treatment of hepatocellular cancer? J Hepatol. 2012;56:686–695. doi: 10.1016/j.jhep.2011.07.031. [DOI] [PubMed] [Google Scholar]
- 10.Hatefi A, Canine BF. Perspectives in vector development for systemic cancer gene therapy. Gene Ther Mol Biol. 2009;13:15–19. [PMC free article] [PubMed] [Google Scholar]
- 11.Clément N, Knop DR, Byrne BJ. Large-scale adeno-associated viral vector production using a herpesvirus-based system enables manufacturing for clinical studies. Hum Gene Ther. 2009;20:796–806. doi: 10.1089/hum.2009.094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Avila MA, Berasain C, Sangro B, Prieto J. New therapies for hepatocellular carcinoma. Oncogene. 2006;25:3866–3884. doi: 10.1038/sj.onc.1209550. [DOI] [PubMed] [Google Scholar]
- 13.Thomas CE, Ehrhardt A, Kay MA. Progress and problems with the use of viral vectors for gene therapy. Nat Rev Genet. 2003;4:346–358. doi: 10.1038/nrg1066. [DOI] [PubMed] [Google Scholar]
- 14.Zhu L, Mahato RI. Lipid and polymeric carrier-mediated nucleic acid delivery. Expert Opin Drug Deliv. 2010;7:1209–1226. doi: 10.1517/17425247.2010.513969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Glover DJ, Glouchkova L, Lipps HJ, Jans DA. Overcoming barriers to achieve safe, sustained and efficient non-viral gene therapy. Adv Gene Mol Cell Ther. 2007;1:126–140. [Google Scholar]
- 16.Pack DW, Hoffman AS, Pun S, Stayton PS. Design and development of polymers for gene delivery. Nat Rev Drug Discov. 2005;4:581–593. doi: 10.1038/nrd1775. [DOI] [PubMed] [Google Scholar]
- 17.Bhise NS, Shmueli RB, Gonzalez J, Green JJ. A novel assay for quantifying the number of plasmids encapsulated by polymer nanoparticles. Small. 2012;8:367–373. doi: 10.1002/smll.201101718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shmueli RB, Sunshine JC, Xu Z, Duh EJ, Green JJ. Gene delivery nanoparticles specific for human microvasculature and macrovasculature. Nanomedicine. 2012;8:1200–1207. doi: 10.1016/j.nano.2012.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sunshine JC, Peng DY, Green JJ. Uptake and transfection with polymeric nanoparticles are dependent on polymer end-group structure, but largely independent of nanoparticle physical and chemical properties. Mol Pharm. 2012;9:3375–3383. doi: 10.1021/mp3004176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tzeng SY, Green JJ. Subtle changes to polymer structure and degradation mechanism enable highly effective nanoparticles for siRNA and DNA delivery to human brain cancer. Adv Healthc Mat. 2013;2:468–480. doi: 10.1002/adhm.201200257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tzeng SY, Higgins LJ, Pomper MG, Green JJ. Biomaterial-mediated cancer-specific DNA delivery to liver cell cultures using synthetic poly (beta-amino esters) J Biomed Maters Res A. 2013;101:1837. doi: 10.1002/jbm.a.34616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tzeng SY, Green JJ. Subtle changes to polymer structure and degradation mechanism enable highly effective nanoparticles for siRNA and DNA delivery to human brain cancer. Adv Healthc Mater. 2013;2:468–480. doi: 10.1002/adhm.201200257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lu ZR, Ye F, Vaidya A. Polymer platforms for drug delivery and biomedical imaging. J Control Release. 2007;122:269–277. doi: 10.1016/j.jconrel.2007.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Iyer AK, He J, Amiji MM. Image-guided nanosystems for targeted delivery in cancer therapy. Curr Med Chem. 2012;19:3230–3240. doi: 10.2174/092986712800784685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bao G, Mitragotri S, Tong S. Multifunctional nanoparticles for drug delivery and molecular imaging. Annu Rev Biomed Eng. 2013;15:253–282. doi: 10.1146/annurev-bioeng-071812-152409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li S, Goins B, Zhang L, Bao A. Novel multifunctional theranostic liposome drug delivery system: construction, characterization, and multimodality MR, near-infrared fluorescent, and nuclear imaging. Bioconjug Chem. 2012;23:1322–1332. doi: 10.1021/bc300175d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Xie J, Chen K, Huang J, Lee S, Wang J, Gao J, Li X, Chen X. PET/NIRF/MRI triple functional iron oxide nanoparticles. Biomaterials. 2010;31:3016–3022. doi: 10.1016/j.biomaterials.2010.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Koning GA, Krijger GC. Targeted multifunctional lipid-based nanocarriers for image-guided drug delivery. Anticancer Agents Med Chem. 2007;7:425–440. doi: 10.2174/187152007781058613. [DOI] [PubMed] [Google Scholar]
- 29.Nayerossadat N, Maedeh T, Ali PA. Viral and nonviral delivery systems for gene delivery. Adv Biomed Res. 2012;1:27. doi: 10.4103/2277-9175.98152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Guerrero-Cázares H, Tzeng SY, Young NP, Abutaleb AO, Quiñones-Hinojosa A, Green JJ. Biodegradable polymeric nanoparticles show high efficacy and specificity at DNA delivery to human glioblastoma in vitro and in vivo. ACS Nano. 2014;8:5141–5153. doi: 10.1021/nn501197v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kamat CD, Shmueli RB, Connis N, Rudin CM, Green JJ, Hann CL. Poly(beta-amino ester) nanoparticle-delivery of p53 has activity against small cell lung cancer in vitro and in vivo. Mol Cancer Ther. 2013;12:405–415. doi: 10.1158/1535-7163.MCT-12-0956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tzeng SY, Hung BP, Grayson WL, Green JJ. Cystamine-terminated poly(beta-amino ester)s for siRNA delivery to human mesenchymal stem cells and enhancement of osteogenic differentiation. Biomaterials. 2012;33:8142–8151. doi: 10.1016/j.biomaterials.2012.07.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jeng K-S, Chang C-F, Jeng W-J, Sheen IS, Jeng C-J. Heterogeneity of hepatocellular carcinoma contributes to cancer progression. Crit Rev Oncol Hematol. 94:337–347. doi: 10.1016/j.critrevonc.2015.01.009. [DOI] [PubMed] [Google Scholar]
- 34.Asghar U, Meyer T. Are there opportunities for chemotherapy in the treatment of hepatocellular cancer? J Hepatol. 2012;56:686–695. doi: 10.1016/j.jhep.2011.07.031. [DOI] [PubMed] [Google Scholar]
- 35.Bishop CJ, Abubaker-Sharif B, Guiriba T, Tzeng SY, Green JJ. Gene delivery polymer structure-function relationships elucidated via principal component analysis. Chem Commun (Camb) 2015;51:12134–12137. doi: 10.1039/c5cc04417k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sunshine JC, Sunshine SB, Bhutto I, Handa JT, Green JJ. Poly (β-amino ester)-nanoparticle mediated transfection of retinal pigment epithelial cells in vitro and in vivo. PloS one. 2012;7:e37543. doi: 10.1371/journal.pone.0037543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sunshine JC, Akanda MI, Li D, Kozielski KL, Green JJ. Effects of base polymer hydrophobicity and end-group modification on polymeric gene delivery. Biomacromolecules. 2011;12:3592–3600. doi: 10.1021/bm200807s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sunshine JC, Peng DY, Green JJ. Uptake and transfection with polymeric nanoparticles are dependent on polymer end-group structure, but largely independent of nanoparticle physical and chemical properties. Mol Pharm. 2012;9:3375–3383. doi: 10.1021/mp3004176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kim J, Sunshine JC, Green JJ. Differential polymer structure tunes mechanism of cellular uptake and transfection routes of poly (β-amino ester) polyplexes in human breast cancer cells. Bioconjug Chem. 2013;25:43–51. doi: 10.1021/bc4002322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bhise NS, Gray RS, Sunshine JC, Htet S, Ewald AJ, Green JJ. The relationship between terminal functionalization and molecular weight of a gene delivery polymer and transfection efficacy in mammary epithelial 2-D cultures and 3-D organotypic cultures. Biomaterials. 2010;31:8088–8096. doi: 10.1016/j.biomaterials.2010.07.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Maeda H, Wu J, Sawa T, Matsumura Y, Hori K. Tumor vascular permeability and the EPR effect in macromolecular therapeutics: a review. J Control Release. 2000;65:271–284. doi: 10.1016/s0168-3659(99)00248-5. [DOI] [PubMed] [Google Scholar]
- 42.Petros RA, DeSimone JM. Strategies in the design of nanoparticles for therapeutic applications. Nat Rev Drug Discov. 2010;9:615–627. doi: 10.1038/nrd2591. [DOI] [PubMed] [Google Scholar]
- 43.Blanco E, Shen H, Ferrari M. Principles of nanoparticle design for overcoming biological barriers to drug delivery. Nat Biotechnol. 2015;33:941–951. doi: 10.1038/nbt.3330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.He C, Hu Y, Yin L, Tang C, Yin C. Effects of particle size and surface charge on cellular uptake and biodistribution of polymeric nanoparticles. Biomaterials. 2010;31:3657–3666. doi: 10.1016/j.biomaterials.2010.01.065. [DOI] [PubMed] [Google Scholar]
- 45.Vogl TJ, Naguib NNN, Nour-Eldin NEA, Rao P, Emami AH, Zangos S, Nabil M, Abdelkader A. Review on transarterial chemoembolization in hepatocellular carcinoma: palliative, combined, neoadjuvant, bridging, and symptomatic indications. Eur J Radiol. 2009;72:505–516. doi: 10.1016/j.ejrad.2008.08.007. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.