

Abstract
Eicosanoids are a group of bioactive lipids, shown to be important mediators of neutrophilic inflammation, and selective targeting of their function confers therapeutic benefit in a number of diseases. Neutrophilic airway diseases, including cystic fibrosis, are characterized by excessive neutrophil infiltration into the airspace. Understanding the role of eicosanoids in this process may reveal novel therapeutic targets. The eicosanoid hepoxilin A3 (HxA3) is a pathogen-elicited, epithelial-produced neutrophil chemoattractant that directs trans-epithelial migration in response to infection. Following HxA3-driven trans-epithelial migration, neutrophil chemotaxis is amplified through neutrophil production of a second eicosanoid, leukotriene B4 (LTB4). The rate-limiting step of eicosanoid generation is the liberation of arachidonic acid by phospholipase A2 (PLA2), and the cytosolic PLA2α (cPLA2α) isoform has been specifically shown to direct LTB4 synthesis in certain contexts. Whether cPLA2α is directly responsible for neutrophil synthesis of LTB4 in the context of Pseudomonas aeruginosa-induced neutrophil trans-epithelial migration has not been explored. Human and mouse neutrophil-epithelial co-cultures were employed to evaluate the role of neutrophil-derived cPLA2α in infection-induced trans-epithelial signaling using pharmacological and genetic approaches. Primary human airway basal stem cell-derived epithelial cultures and micro-Optical Coherence Tomography (μOCT), a new imaging modality that captures two- and three-dimensional real-time dynamics of neutrophil trans-epithelial migration, were applied. Evidence from these studies suggests that cPLA2α expressed by neutrophils, but not epithelial cells, plays a significant role in infection-induced neutrophil trans-epithelial migration by mediating leukotriene B4 synthesis during migration, which serves to amplify the magnitude of neutrophil recruitment in response to epithelial infection.
Keywords: Neutrophils, Lung, Phospholipase A2, Inflammation
Introduction
Prostacyclins, prostaglandins, leukotrienes, lipoxins, and hepoxilins are inflammatory modulating lipid mediators, which are collectively termed eicosanoids. Eicosanoids are the product of oxygenation of arachidonic acid and play a significant role in multiple pathological processes including atherosclerosis, asthma, and rheumatoid arthritis(1–3). Pharmacological targeting of eicosanoid production is a well-established therapeutic tool. Aspirin, which inhibits the enzyme responsible for generating prostaglandins called cyclo-oxygenase-2 (COX-2), is a treatment in atherosclerosis(4). Zileuton and montelukast are therapeutic agents commonly used to treat allergies and asthma by targeting leukotriene synthesis(5, 6). Eicosanoids also play a key role in neutrophilic diseases, such as sepsis(7) and pneumonia(8, 9), but precise pharmacologic targeting of these pathways has not been adopted as clinical therapies. Gaining a better understanding of the mechanisms that underlie these processes may provide more focused therapeutic targets.
The generation of eicosanoids is regulated by phospholipases A2 (PLA2), a family of enzymes responsible for releasing arachidonic acid from cellular membranes to serve as substrate for eicosanoid synthesis. There are several isoform classes of the PLA2 family of enzymes: secretory PLA2, cytosolic PLA2 (cPLA2), calcium-independent PLA2 (iPLA2), lysosomal PLA2, platelet activating factor acetylhydrolase (PAF-AH), and bacterial PLA2, each with a unique physiologic role and eicosanoid generating capacity(10). There is emerging evidence that certain PLA2 isoforms associate with specific eicosanoid generating enzymes establishing selectivity towards eicosanoid species generation under certain physiological contexts(10). For example, cPLA2α governs proximity to 5-lipoxygenase (5-LOX) and 5-LOX-activating protein (FLAP), resulting in LTB4 production(11), while in other contexts, cPLA2α will co-localize with COX-1 and COX-2 leading to efficient generation of prostaglandins(12–14). In order to stimulate arachidonic acid release from membranes, cPLA2α translocates from the cytosol to the intracellular membrane in response to calcium where cPLA2α is phosphorylated by MAPKs, enhancing its catalytic activity(15). Once activated, cPLA2α liberates arachidonic acid from membrane phospholipids, which triggers downstream synthetic enzymes to oxygenate arachidonic acid, generating a variety of eicosanoids.
Multiple eicosanoids have been shown to play key roles in neutrophil recruitment to the airway(16–19). Neutrophils migrate from circulation, across the endothelium, interstitium and epithelial layer to reach infectious insults within the airway. In certain diseases, like acute respiratory distress syndrome (ARDS) or cystic fibrosis (CF), this neutrophilic inflammatory response is excessive, contributing directly to airway damage(20, 21). Both epithelial cells and neutrophils rely on eicosanoid signaling to mediate and coordinate the trans-epithelial passage of neutrophils in response to pathogenic infection of the epithelium. Pulmonary epithelial cells infected with Pseudomonas aeruginosa release the 12-lipoxygenase generated eicosanoid, hepoxilin (HxA3), in an apically-directed manner, generating a chemotactic gradient that drives neutrophils from the interstitium across the epithelium and into the airway(9). We previously assessed the role of cPLA2α in epithelial production of eicosanoids and demonstrated that infection of lung epithelial cells with Pseudomonas aeruginosa elicits the generation of multiple eicosanoids, including prostaglandin E2 (PGE2) and HxA3(10). Although cPLA2α drives epithelial production of PGE2 in response to infection, epithelial cell-derived cPLA2α did not appear to exert any influence on HxA3 release or neutrophil trans-epithelial migration(16, 22).
After migrated neutrophils reach the airspace in response to HxA3, the migrated neutrophils produce a second eicosanoid, LTB4, which augments neutrophil trans-epithelial migration(19). LTB4 is generated by oxidation of arachidonic acid by 5-LOX in association with FLAP(23). cPLA2α has been demonstrated to play a direct role in LTB4 production(11), as cPLA2α-derived arachidonic acid affects the conformation of FLAP to enhance its ability to bind 5-LOX, favoring the production of LTB4. cPLA2α is prominently expressed in lung epithelium, endothelium, fibroblasts, macrophages and neutrophils(24, 25).
As cPLA2α exhibits selective eicosanoid generating capacity favoring PGE2 and LTB4, but not HxA3 synthesis, we sought to investigate whether migrated neutrophil LTB4 synthesis is dependent on the cPLA2α isoform. Understanding inflammatory mechanisms of different PLA2 isoforms in the context of neutrophilic responses is critical to identify appropriate therapeutic targets and may provide important clinical insight. Utilizing a range of models of neutrophil trans-epithelial migration, we sought to determine whether cPLA2α expressed in neutrophils contributes to bacterial induced neutrophil trans-epithelial migration.
Materials and Methods
Growth and maintenance of epithelial cells
The H292 cell line is a human pulmonary epithelial cell line derived from a patient with mucoid pulmonary carcinoma. MLE12 cells are a murine epithelial cell line. H292 and MLE12 cells were maintained in DMEM/F12 (1:1) culture medium with 10% heat inactivated fetal bovine serum and antibiotics. Both cell lines were seeded onto inverted 24- or 96-well 3μm Transwell inserts (Corning Life Sciences) until adherent (4 hours), and then re-inverted into a Transwell receiver plate. H292 monolayers were maintained in culture media for >7 days to ensure mature monolayers for migration assays. MLE12 monolayers were maintained in culture media for 3–5 days prior to migration assays.
Human airway basal stem cell isolation, culture and mucociliary differentiation on air-liquid interface
Human airway basal stem cells were isolated from discarded human airways harvested from the New England Organ Bank or Department of Pathology (Massachusetts General Hospital) under an IRB-approved protocol (#2010P001354), as previously described(26). The trachea and mainstem bronchi were dissected and cleared of connective tissues and blood cells, and epCAM+ epithelial stem cells were isolated. These cells were cultured in complete SAGM (Lonza, Cat. CC-3118) on plates pre-coated with laminin-enriched 804G-conditioned medium. Growth on 3.0 μm Transwells, required for neutrophil trans-epithelial assays, involved a two-step coating process: the side of the membrane for cell seeding was pre-coated with 804G medium only and the opposite side was pre-coated with 804G medium containing 5% Matrigel (BDBiosciences, 354230). To seed the airway basal stem cells, the Transwell inserts were removed, flipped, and airway basal stem cells, with a seeding density of >6000 cells/mm2, were applied on the surface of flipped membranes for 6 hours. Inserts were then returned to their original orientation. The growth medium (complete SAGM) was added to both upper and lower chamber for 1–2 days to ensure that the cells were confluent before replacing with complete Pneumacult-ALI medium (StemCell Techonology, Cat. 05001)(27) in both the upper and lower chambers and cultured for another day. The following day, ALI medium was added only in the top chamber to initiate airway-liquid interface on the down-facing surface of the Transwell (count as day 0). The medium was changed every 1–2 days until differentiation was well established. Ciliogenesis was monitored by inverted-phase microscopy and could be observed after 11–15 days. ALIs used in experiments were cultured for 14–26 days to allow full maturation. Trans-epithelial electrical resistance (TEER) was assessed using a voltmeter prior to migration assays to ensure the establishment of polarized epithelial barrier (EVOM2, Epithelial Voltohmmeter, World Precision Instruments, Inc.).
Neutrophil isolation
Neutrophils were isolated from healthy volunteers under an Institutional Review Board-approved protocol (#1999P007782) at the Massachusetts General Hospital using an established isolation technique(28). Briefly, blood was drawn by venipuncture into a syringe containing anticoagulant, acid citrate/dextrose, then centrifuged at room temperature, 2000 rpm for 20 min without brake to allow layering of the buffy coat. Plasma and mononuclear cells were removed by aspiration. A 2% gelatin sedimentation technique, followed by wash with RBC lysis buffer allowed for removal of RBCs. Cells were then washed and resuspended in HBSS without calcium or magnesium (HBSS-) and suspended at a concentration of 5×10ˆ7 neutrophils/ml. This neutrophil isolation technique allows for isolation of functionally active neutrophils (>98%) at 90% purity(29).
Bacterial strains
Escherichia coli (MC1000) and Pseudomonas aeruginosa (PAO1) were grown aerobically in Luria-Bertani broth overnight at 37°C in a shaking incubator. Prior to the experiments, each bacterial suspension was washed and resuspended in HBSS to a concentration of 6×107 CFU/ml.
Neutrophil trans-epithelial migration
Assays were adapted from the neutrophil trans-epithelial migration model(28). In brief, epithelial monolayers were grown on the underside of Transwell filters with 3.0μm pores to accommodate neutrophil passage. This model allows for neutrophil migration from the basolateral to the apical compartment, toward chemoattractant gradients that are exogenously provided or endogenously generated by epithelial infection with the P. aeruginosa strain, PAO1, triggering epithelial secretion of HxA3. To interfere with epithelial signaling pathways, epithelial cells were treated for 2 hours with specific chemical inhibitors, washed, and infected with 25μl of 6×107 CFU/ml bacteria (MC1000 or PAO1) for 1 hour prior to undergoing neutrophil trans-epithelial migration. Additional uninfected controls were placed in wells containing chemoattractants to create an exogenous apical chemotactic gradient with fMLP (100nM, Sigma) IL-8 (100ng/ml; eBioscience, San Diego CA), or LTB4 (5ng/ml; Enzo Life Sciences, Farmingdale, NY), capable of driving neutrophil trans-epithelial migration. To interfere with neutrophil signaling pathways, neutrophils were incubated in chemical inhibitors for 30–60min at 37°C prior to placement in the basolateral compartment at a concentration of 2×105 neutrophils/96-well Transwell or 1×106 neutrophils/24-well Transwell. Neutrophils were not washed following treatment with chemical inhibitors due to concern that additional handling of cells would impact their activation state or alter relative counts prior to migration. Migration was allowed to progress for two hours while at 37°C, 5% CO2. After two hours, Transwells were discarded and migrated neutrophils were quantified using a neutrophil myeloperoxidase (MPO) activity assay(28), whereby the magnitude of MPO activity (OD@405 nm) measured using a colorimetric enzyme assay directly correlates with number of neutrophils migrated in a linear fashion (R2>0.99)(9, 29, 30). Standard curves were used for each condition of neutrophil pre-treatment to control for possible variability of MPO production between neutrophil groups. Figure 1 provides a cartoon schematic of the neutrophil trans-epithelial migration assays designed to target either the epithelial response or the neutrophil response. Data were displayed to reflect the magnitude of migration as “percent of PAO1 infected control” for comparative purposes.
Figure 1. Schematic drawing of trans-epithelial migration assay with chemical inhibition targeting epithelium or neutrophils.
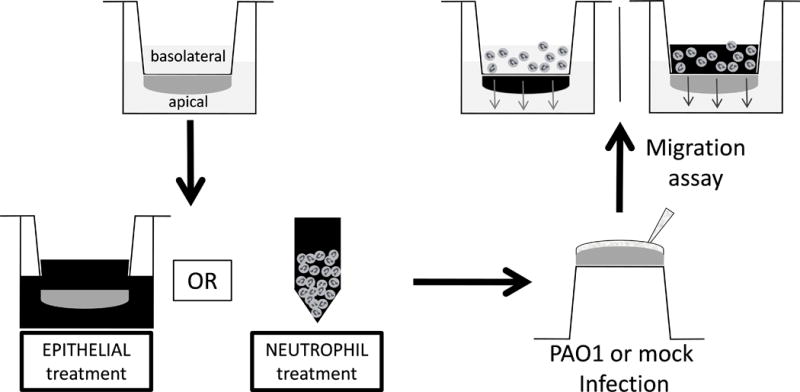
Epithelial cells are matured on the underside of a Transwell filter with 3μm pore size. Chemical inhibitors (represented by darkened background) are then applied to the epithelium for two hours prior to infection, or the neutrophils for 30–60 minutes prior to migration. Transwells are washed and infected with PAO1 or mock infection. Neutrophils, unwashed, are then applied to the basolateral surface and allowed to undergo trans-epithelial migration. The transwell with the darkened epithelium represents migration across epithelium pre-treated with chemical inhibitor and washed prior to infection. The transwell with the neutrophils in a darkened suspension indicate neutrophils pre-treated with chemical inhibitor then applied, unwashed, to the epithelium to measure trans-epithelial migration.
Drug treatment
Chemical inhibitors ONO-RS-082 (ONO, Enzo Life Sciences), cPLA2α inhibitor (cPLAi, Calbiochem), U0126 (Cell Signaling Technology), bromoenol lactone (BEL, Cayman Chemical), or diphenyleneiodonium (DPI, Sigma) were suspended as per manufacturers’ instructions and diluted to desired concentrations. Vehicle controls consisted of DMSO diluted with HBSS at a matched dilution factor.
Cell viability assay
Following treatment with HBSS, vehicle control, or chemical inhibitor, neutrophils were counted for viability. Neutrophils were stained with trypan blue, placed on a hemocytometer, then visualized under a microscope. Live and dead cells were manually counted to determine total viability.
Reactive oxidase species (ROS) assay
Neutrophils were incubated in lucigenin (Invitrogen) at a concentration of 5×107 cells/ml for 15 minutes on ice, then transferred to a black, clear bottom 96 well plate with 4×106 neutrophils/well. Inhibitor (BEL, DPI) or vehicle control was added for an additional 10 min treatment. Cells were stimulated with 10μM fMLP or HBSS, and ROS activity was quantified via luminescence and scintillation report by Top Count machine (Perkin Elmer).
Leukotriene measurement
Following trans-epithelial migration assays, Transwells were discarded and plates were spun 1800 rpm × 5 min in centrifuge. Supernatant was collected and stored at −80 degrees. LTB4 was measured by ELISA per manufacturer’s instructions (Cayman Chemical).
μOCT imaging
High-resolution micro-optical coherence tomography (μOCT) is a custom-built imaging technology providing optical resolutions of 2μm in the lateral/horizontal direction, and 1μm in the axial/vertical direction through analysis of reflectivity of a sample as a function of depth. μOCT imaging methods have been previously reported(31). The μOCT instrument was inverted with the imaging laser beam directed to the sample from below and required a custom holder with a transparent OCT-compatible bottom to hold the Transwell containing the epithelium and neutrophils at a distance of approximately 100μm from the glass bottom. μOCT captured time-lapse 3D imaging every 10 minutes over two hours immediately after the placement of neutrophils into the basolateral compartment. The sample was maintained near 37°C during migration by placing an incandescent heat source. 3D Viewer plugging on ImageJ was used to render 3D μOCT volume sequences. Neutrophil migration volume was determined by measuring the number of voxels in a region of interest exceeding a brightness threshold divided by the bright voxels measured from an isolated neutrophil.
Bone marrow collection
Experiments were performed according to the guidelines of the Institutional Animal Care and Use Committee of Massachusetts General Hospital. Femurs and tibias of cPLAα−/− mice on BALB/c background or 129Sv/C57BL/6 background, as well as inbred strain matched littermate control mice(32), were removed for the harvest of bone marrow. Bone marrow was flushed and collected in HBSS-. RBC were lysed with RBC lysis buffer then washed and resuspended in HBSS- to a concentration of 20×106 cells/ml. Trans-epithelial migration was assessed using single and mixed bone marrow migration across MLE epithelial cell monolayers(19). Mixed bone marrow from cPLAα−/− mice and wild type controls were also stained differentially with CFSE (BioLegend; San Diego, CA) then combined at a 1:1 ratio to assess potential for compensatory signaling. Migrated cells were collected and analyzed by flow cytometry. Data was collected on an Accuri C6 flow cytometer (BD Biosciences; San Jose, CA), and analyzed with FlowJo analysis software (FlowJo; Ashland, OR).
Statistics
Data was reported as means ± SD and individual conditions were compared by unpaired, two tail Student’s t-test to appropriate control. To analyze multiple comparisons, we used one-way ANOVA. In cases of significant difference, Bonferroni post-test analysis was used to assess differences within the group, or Dunnett’s test was used to compare individual conditions to control. A p value ≤ 0.05 was considered statistically significant, a statistically significant difference is indicated by (*) in each figure.
Results
Inhibition of epithelial and neutrophil sources of PLA2 disrupt Pseudomonas-mediated migration
Eicosanoids are critical mediators of neutrophil trans-epithelial migration and have been shown to be produced by both infected epithelial cells and migrating neutrophils. As phospholipase A2 (PLA2) activity plays a key role in eicosanoid generation(16, 22), the contribution of PLA2 activity was assessed in both cell types following epithelial infection with PAO1. Epithelial cells were pre-treated with escalating doses of ONO, a general PLA2 inhibitor that non-specifically targets all isoforms, for 2 hours prior to infection. Following a 1 hour infection, untreated neutrophils were placed in the basolateral compartment and allowed to migrate from the basolateral to the apical aspect of the epithelium for 2 hours. Epithelial PLA2 inhibition reduced PAO1-induced neutrophil trans-epithelial migration in a dose dependent manner (Figure 2A), consistent with previous studies(22). Inhibition of epithelial cells by PLA2 inhibitor (ONO) did not impact responses to HBSS buffer alone or to infection with non-pathogenic E. coli MC1000 (Figure 2B). The contribution of PLA2 activity in neutrophils was then assessed by treatment of neutrophils with PLA2 inhibitor (ONO) in escalating doses for 30 minutes prior to migration. ONO or vehicle pre-treated neutrophils were placed in the basolateral compartment of the Transwell and allowed to migrate across PAO1-infected epithelium. Inhibition of neutrophil PLA2 activity was also associated with decreased migration in response to PAO1-infected epithelium (Figure 2C), again, in a dose-dependent manner. As was observed with inhibition of epithelial PLA2 activity, interference of neutrophil PLA2 activity resulted in reduced migration to PAO1 infection without impacting responses to buffer or to infection with non-pathogenic E. coli strain, MC1000 (Figure 2D). Neutrophil treatment with ONO did not affect cell viability or disrupt neutrophil ability to produce MPO (Supplemental Figure 1). Additionally, the effects of PLA2 activity inhibition were additive when the epithelium and neutrophils were simultaneously treated with ONO (Figure 2E), suggesting PLA2 activity is functionally important in neutrophil trans-epithelial migration in both the epithelial and neutrophil signaling processes.
Figure 2. PLA2 activity is required in both the epithelium and the neutrophil for efficient neutrophil trans-epithelial migration in response to epithelial infection with PAO1.
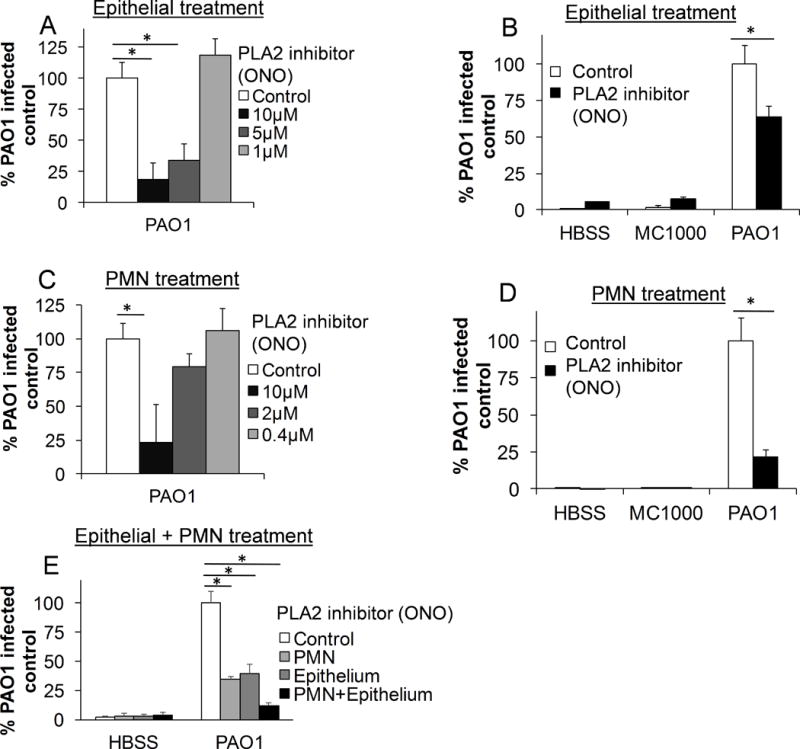
A) H292 epithelial monolayers, grown on inverted Transwells, were pretreated with the pan-PLA2 inhibitor, ONO, at multiple doses, or vehicle control (DMSO 1:1000). Epithelial monolayers were then infected apically with the pathogenic P. aeruginosa strain, PAO1, washed, and untreated neutrophils were provided to the basolateral surface. B) H292 monolayers were pretreated with the PLA2 inhibitor ONO (10μM) or vehicle control (DMSO 1:1000). Monolayers were infected with either PAO1, a non-pathogenic strain of E. coli, MC1000, or mock-infection (HBSS). Untreated neutrophils were provided to the basolateral compartment. C) Healthy primary human neutrophils were pretreated with multiple doses of the PLA2 inhibitor ONO or vehicle control (DMSO 1:1000) for 30 min. Untreated H292 epithelial monolayers were infected with PAO1, washed, and treated neutrophils were provided to the basolateral compartment. D) Neutrophils were pretreated for 30 min. with ONO (10μM) or vehicle control (DMSO 1:1000) and migrated in response to epithelial infection with PAO1, non-pathogenic MC1000, or mock infection (HBSS). E) Neutrophils and epithelial monolayers were treated for 30 min. and 2 hours, respectively, with ONO (10μM) separately or in combination, as above, prior to migration. For all experiments, relative neutrophil migration was assessed by myeloperoxidase activity in the apical compartment following 2h migration. Total myeloperoxidase activity was corrected for using standard curves generated for each drug treatment of neutrophil. Data are shown as mean corrected value +/− standard deviation. Experiments were performed on at least 3 occasions with n≥3 technical replicates. P values were calculated by ANOVA with Dunnett’s test (A, C, E) or paired Student’s t-test (B, D). P values ≤0.05 was consider statistically significant.
Inhibition of neutrophil-derived cPLA2α blunts Pseudomonas-mediated migration
Cytosolic PLA2, specifically the isoform cPLA2α, is expressed in both lung epithelium and neutrophils(24, 25) and has been associated with the synthesis of eicosanoids in a number of models(33–35). We previously investigated the PLA2 isoforms responsible for HxA3 generation, and found that cPLA2α did not appear to impact HxA3 generation despite the fact that it was expressed and operative in the epithelium, leading to the generation of PGE2(16). This selective eicosanoid generating capacity emphasizes the importance of critically evaluating isoform-specific contributions of PLA2 activity within each cellular context where eicosanoids are generated. Although cPLA2α was dispensable for epithelial generation of HxA3, we considered the possibility that the cPLA2α isoform might be required in the neutrophil to promote neutrophil trans-epithelial migration, given the requirement for general PLA2 activity (Figure 2C & D). Neutrophils were treated with escalating doses of cPLA2α-specific isoform inhibitor prior to assessment of trans-epithelial migration. cPLA2α inhibition in the neutrophil resulted in decreased trans-epithelial migration in response to apical epithelial infection with PAO1 in a dose dependent manner (Figure 3A). Migration to exogenous chemoattractant, IL-8, which does not trigger neutrophil LTB4 mediated augmentation, was not affected by cPLA2α inhibition (Figure 3B). Neutrophil treatment with cPLA2α inhibitor did not affect cell viability or prevent MPO production (Supplemental Figure 1). Prior studies have shown that cPLA2α inhibition in epithelial cells reduces the liberation of arachidonic acid and subsequent generation of certain eicosanoids, including PGE2. Inhibition of cPLA2α within the epithelium did not impact 12-lipoxygenase metabolites or neutrophil trans-epithelial migration(16). Consistent with these previous studies, we found that treatment of epithelial cells with cPLA2α inhibitor did not reduce PAO1 induced neutrophil trans-epithelial migration (Fig 3C). Inhibitors of cPLA2α did not impact responses to buffer alone or infection with MC1000, and it did not serve to inhibit neutrophil trans-epithelial migration towards an imposed IL-8 gradient. Thus, neutrophil cPLA2α activity is critical in mediating PAO1-induced neutrophil trans-epithelial migration whereas epithelial cPLA2α activity is dispensable for this process.
Figure 3. Neutrophil cPLA2α is required for efficient neutrophil trans-epithelial migration in response to epithelial infection with PAO1.
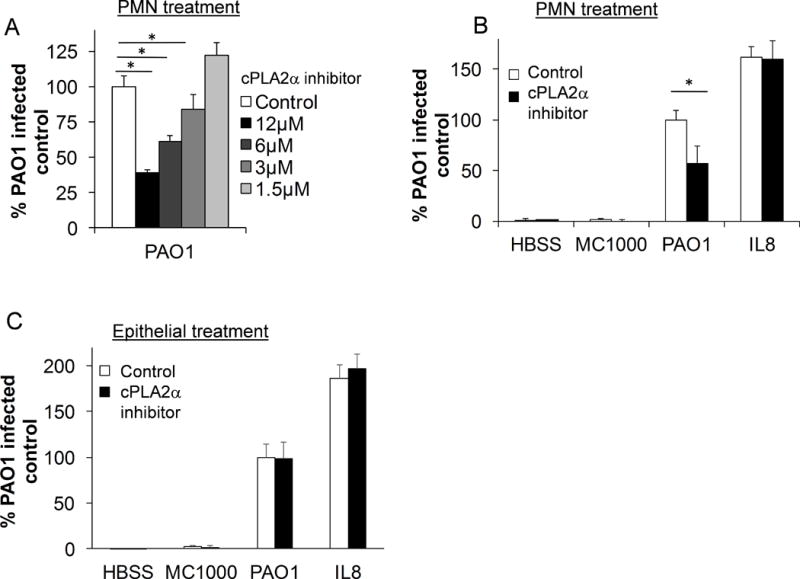
A) Healthy primary human neutrophils were pretreated for 60 min. with multiple doses of cPLA2α inhibitor or vehicle control (DMSO 1:500) and applied to the basolateral aspect of PAO1-infected H292 epithelial monolayers. B) Neutrophils were pretreated for 30 min. with cPLA2α inhibitor (6μM) or vehicle control (DMSO 1:1000) and migrated in response to PAO1 infection, non-pathogenic MC1000 or mock infection (HBSS), or towards an exogenous IL-8 gradient placed in the apical compartment of the Transwell. C) H292 monolayers were pretreated with cPLA2α inhibitor (6μM) or vehicle control (DMSO 1:1000) prior to infection with PAO1, MC1000 or HBSS. Untreated neutrophils were applied to the basolateral surface and allowed to migrate towards infection, or towards exogenous IL-8. For all experiments, following a 2h migration, relative neutrophil migration was assessed by total myeloperoxidase activity in the apical compartment and corrected for using standard curves generated for each drug treatment of neutrophil. Data are shown as mean corrected value +/− standard deviation. Experiments were performed on at least 3 occasions with n≥3 technical replicates. P values were calculated by ANOVA with Dunnett’s test (A) or paired Student’s t-test (B, C). P values ≤0.05 was consider significant.
Neutrophil-derived ERK1/2 is required for Pseudomonas-induced trans-epithelial migration
cPLA2α activity is regulated post-translationally through phosphorylation by the kinase ERK1/2 within a variety of cells, including neutrophils(24, 36–38). We tested whether inhibition of ERK in neutrophils would alter their ability to migrate across the PAO1-infected epithelium. Neutrophils were pre-treated with the ERK inhibitor U0126 in escalating doses prior to migration. Inhibition of ERK activity decreased migration across the PAO1-infected epithelium in a dose dependent manner (Figure 4A). Inhibition of PMN ERK activity revealed reduced migration to epithelial PAO1 infection but not an imposed chemotactic gradient of IL-8 (Figure 4B). Neutrophil treatment with the ERK inhibitor, U0126, did not reduce cell viability or MPO production (Supplemental Figure 1). In agreement with previous studies(39), no effect was observed on PAO1-induced neutrophil trans-epithelial migration following epithelial treatment with ERK inhibitor, U0126 (Figure 4C). These findings support the hypothesis that neutrophil-derived ERK1/2 is important for mediating PAO1-induced neutrophil trans-epithelial migration, most likely through kinase associated activation of cPLA2α since treatment of neutrophils with ERK1/2 inhibitor did not impair trans-epithelial migration towards a gradient of IL-8.
Figure 4. Activation of ERK pathways within neutrophils are required for PAO1-induced neutrophil trans-epithelial migration.
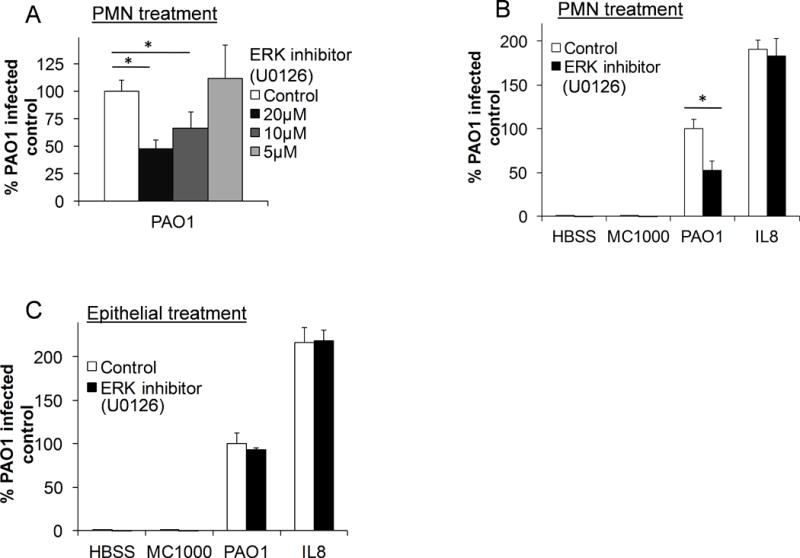
A) Healthy primary human neutrophils were pre-treated for 30 min. with escalating doses of the ERK inhibitor, U0126, or vehicle control (DMSO 1:1000) prior to migration across untreated H292 monolayers infected with PAO1. B) Neutrophils were pretreated for 30 min. with U0126 (20μM) or vehicle control (DMSO 1:1000) and allowed to migrate across an H292 monolayer infected with PAO1, MC1000, mock-infection (HBSS), or towards exogenous neutrophils chemoattractant, IL-8. C) H292 monolayers were pretreated with U0126 (20μM) or vehicle control (DMSO 1:1000) prior to infection with PAO1, MC1000 or mock-infection (HBSS). Untreated neutrophils migrated across the pretreated epithelium in response to infection, or towards exogenous IL-8. For all experiments, relative neutrophil migration into the apical compartment was assessed after 2h by total myeloperoxidase activity. Migration was corrected for using standard curves generated for each drug treatment of neutrophil. Data are shown as mean corrected value +/− standard deviation. Experiments were performed on at least 3 occasions with n≥3 technical replicates. P values were calculated by ANOVA with Dunnett’s test (A) or paired Student’s t-test (B, C). P values ≤0.05 was consider significant.
Neutrophil PLA2 activity required for trans-epithelial migration is isoform specific
To assess the specificity of neutrophil cPLA2α activity on the process of neutrophil trans-epithelial migration, we examined the effect of neutrophil treatment with an inhibitor of a distinct PLA2 isoforms. Calcium-independent PLA2 (iPLA2β and iPLA2γ) is present in both neutrophils(40, 41) and epithelial cells(42, 43). To assess the impact of iPLA2 isoforms on neutrophil trans-epithelial chemotaxis, neutrophils were pre-treated with iPLA2-specific inhibitor, bromoenol lactone (BEL), or vehicle control. There was no impact on migration in response to PAO1 infection over a range of treatment doses of BEL (Figure 5A). Neutrophil treatment with BEL did not reduce migration to IL-8 (Figure 5B), nor affect cell viability or disrupt MPO production (Supplemental Figure 1). Since BEL exerted no impact on any conditions in the migration assay, we sought to confirm that BEL was indeed functional at the dose examined. In neutrophils, iPLA2 isoforms have been previously demonstrated to mediate ROS production as pre-treatment with the iPLA2-specific inhibitor, BEL, abrogated this response(40, 41). Using a similar BEL pre-treatment dose, we confirmed a significant inhibition of fMLP-induced ROS activity, as measured by a ROS-sensitive probe emitting chemiluminescence (Figure 5C). The fMLP-induced ROS activity was completely abrogated by pre-treatment with the NADPH oxidase inhibitor, DPI, as expected(44) (Figure 5C). Taken together, these results indicate that neutrophil-derived signaling responsible for orchestrating chemotaxis is PLA2 activity selective, involving cPLA2α isoform activity, but not iPLA2 isoform activity.
Figure 5. Neutrophil-derived iPLA2 isoforms do not play a role in PAO1-induced neutrophil trans-epithelial migration.
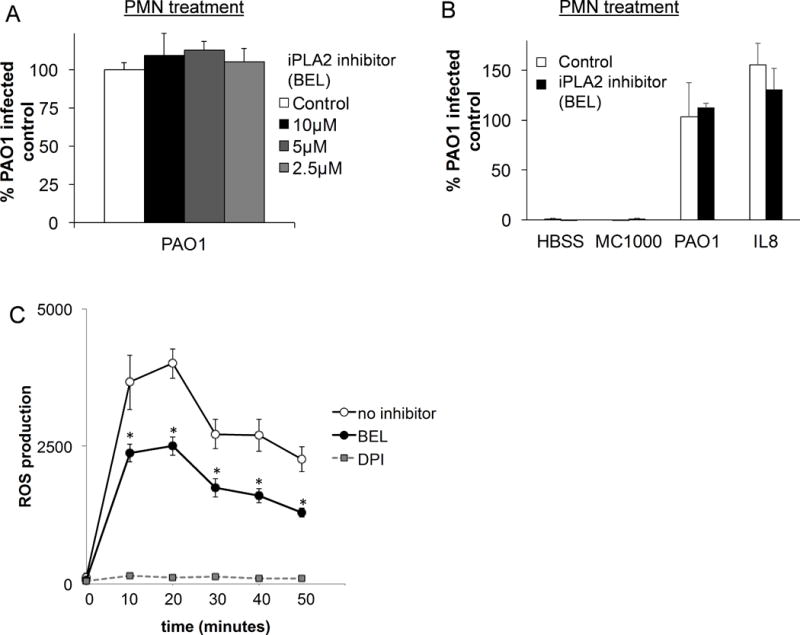
A) Healthy neutrophils were pretreated for 30 min. with multiple doses of BEL, an iPLA2 inhibitor, or vehicle control (DMSO 1:2500) prior to migration across a PAO1 infected H292 monolayer. B) Neutrophils were pretreated for 30 min. with BEL (10μM) or vehicle control (DMSO 1:500) prior to migration across an untreated H292 epithelium infected with PAO1, MC1000 or mock-infection (HBSS) or towards an IL-8 gradient. Migration was again assessed after 2h by comparison of total myeloperoxidase activity. Migration was corrected for using standard curves generated for each drug treatment of neutrophil. Data are shown as mean corrected value +/− standard deviation. C) Neutrophils were incubated in lucigenin then pretreated with BEL (10μM), DPI (2μM), an inhibitor of NADPH oxidase, or no inhibitor (HBSS) before stimulation with fMLP to trigger ROS release. ROS activity was quantified by chemoiluminescence activity. Experiments were performed on at least three occasions with n≥3 technical replicates. P values were calculated by ANOVA with Dunnett’s test (A), Bonferroni analysis (C), or paired Student’s t-test (B). P values ≤0.05 was consider significant.
Neutrophil-derived cPLA2α drives neutrophil release of LTB4
Having demonstrated that neutrophil-derived cPLA2α function is necessary for a robust migration response, we sought to assess whether this migration phenotype was mediated through cPLA2α-dependent generation of LTB4. To test this hypothesis, neutrophils were treated with cPLA2α inhibitor, ERK inhibitor (U0126), or vehicle control, then applied to the basolateral surface of epithelial monolayers, which were untreated and exposed to an imposed apical oriented gradient of IL-8, or infected with PAO1. Following a 2-hour incubation, supernatant from the apical compartment was collected and LTB4 was quantified by ELISA, while neutrophil migration was assessed by MPO activity. Substantial LTB4 release was observed in the context of PAO1-induced migration. Both LTB4 release and total neutrophil migration was significantly reduced by pre-treating neutrophils with either cPLA2α or ERK inhibitors (Figure 6A & B). Previous studies suggest that neutrophils represent the primary source of LTB4 in the epithelial-neutrophil co-culture(19). IL-8 induced neutrophil migration did not evoke significant LTB4 generation (Figure 6A), despite robust migration occurring (Figure 6B), consistent with previous studies(19). Because of the differences in total numbers of neutrophils migrating with and without inhibitor pre-treatment, we normalized LTB4 release relative to the number of transmigrating neutrophils. Inhibition of neutrophil-derived cPLA2α or ERK by pharmacological compounds decreased the relative LTB4 production per migrated neutrophil in the setting of PAO1-induced migration (Figure 6C). These findings support the role for cPLA2α activity in neutrophil-mediated LTB4 synthesis during migration. LTB4 release during PAO1-induced neutrophil trans-epithelial migration is necessary for augmenting the magnitude of migrating neutrophils, which are initiated to breach the epithelial barrier by epithelial-derived HxA3.
Figure 6. Neutrophil cPLA2α generates LTB4 production by the neutrophil following infection induced trans-epithelial migration.
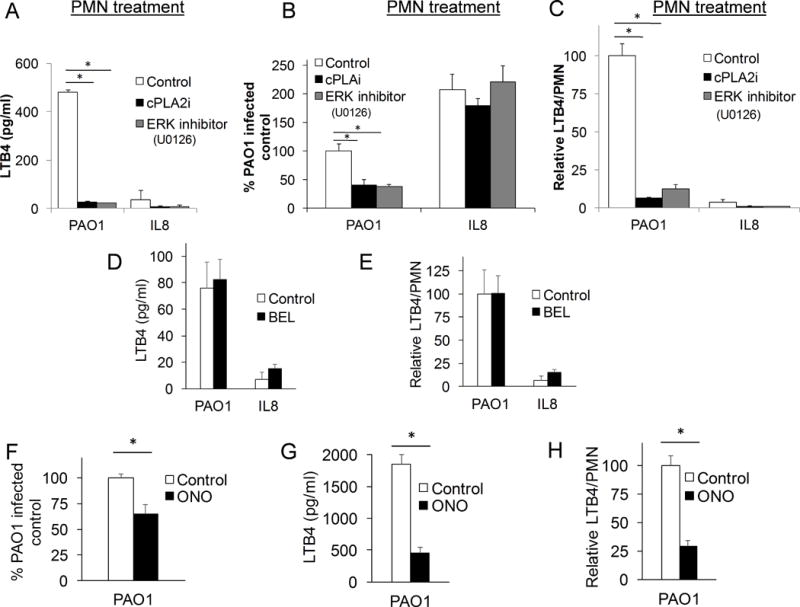
Healthy neutrophils were pretreated for 60 min. with cPLA2α inhibitor (12μM), ERK inhibitor, U0126 (20μM), or vehicle control (DMSO 1:500), then allowed to migrate across an untreated H292 monolayer infected with PAO1 or towards an apical gradient of IL-8. A) After a 2h migration, supernatant was collected from the apical compartment and LTB4 was quantified by ELISA. B) Relative migration was assessed after 2h migration by total myeloperoxidase activity and corrected for by standard curves. C) LTB4 production was adjusted by relative neutrophil migration to calculate relative LTB4 production per neutrophil. Healthy neutrophils were pretreated for 30 min. with iPLA2 inhibitor, BEL (10μM), then allowed to migrate across an untreated H292 monolayer infected with PAO1 or towards an apical gradient of IL-8. D) Supernatant following trans-epithelial migration of BEL-treated PMN was collected from the apical compartment after a 2h migration and LTB4 was quantified by ELISA. E) Relative LTB4 production, calculated as the LTB4/PMN generated as compared to that of PAO1-infected control (see Figure 5B for corresponding migration assay), was assessed. Healthy neutrophils were pretreated for 30 min. with general PLA2 inhibitor, ONO (10μM), then allowed to migrate across an untreated H292 monolayer infected with PAO1. F) Relative migration was assessed after 2h migration by total myeloperoxidase activity and corrected for by standard curves. G) Supernatant following trans-epithelial migration of ONO-treated PMN was collected from the apical compartment after a 2h migration and LTB4 was quantified by ELISA. H) Relative LTB4 production following ONO-treated PMN trans- epithelial migration was assessed. Data are shown as mean corrected value +/− standard deviation and experiments were performed on at least two occasions with n>3 technical replicates. P values involving multiple comparison were calculated by ANOVA with Dunnett’s test (A-C), or when comparing two conditions, p values were calculated by paired Student’s t-test (D-H). P values <0.05 was consider significant.
If PAO1-induced neutrophil trans-epithelial migration is augmented by neutrophil-derived LTB4 release through ERK1/2 activation of cPLA2α, then inhibition of iPLA2 isoforms (which do not impact PAO1-induced neutrophil trans-epithelial migration) would therefore not be expected to impact LTB4 release. Neutrophils pre-treated with BEL release equivalent amounts of LTB4 compared with vehicle control during PAO1-induced neutrophil trans-epithelial migration (Figure 6D). As described above, BEL does not impact the magnitude of PAO1-induced neutrophil trans-epithelial migration (Figure 5B). The LTB4 released per neutrophil between vehicle and BEL-treated neutrophils was also quite similar (Figure 6E). The general PLA2 inhibitor (ONO), on the other hand, significantly impaired neutrophil trans-epithelial migration (Figure 6F), along with LTB4 release (Figure 6G), when neutrophils were incubated with this compound prior to PAO1-induced neutrophil trans-epithelial migration. When evaluating LTB4 released per neutrophil (Figure 6H), a clear and significant decrease is associated with neutrophils pre-treated with the general PLA2 inhibitor (ONO). Thus, LTB4 release represents a critical functional attribute of PLA2 activity required for augmenting PAO1-induced neutrophil trans-epithelial migration and the specific isoform cPLA2α, rather than iPLA2 isoforms, exhibits this behavior in this context.
Neutrophils from cpla2α deficient mice display decreased migration in response to Pseudomonas epithelial infection
To further evaluate the role of neutrophil-derived cPLA2α in bacterial-induced neutrophil trans-epithelial migration, while avoiding concerns associated with pharmacological inhibition, a murine trans-epithelial migration assay system was employed to leverage the availability of mice with targeted gene deletions(19). Bone marrow was isolated from cpla2α−/− mice and littermate controls from a BALB/c background and used as a source of neutrophils in our assay. Mouse lung epithelial monolayers (MLE12) were grown on inverted transwells and infected with PAO1, as described previously. We have previously demonstrated that bone marrow neutrophils are selectively recruited across MLE12 epithelial monolayers in response to PAO1 infection(19). While bone marrow neutrophils from both cpla2α−/− and wild type controls migrated across MLE12 monolayers, cpla2α−/− neutrophil migration magnitude was significantly decreased in response to PAO1-induced migration (Figure 7A). Furthermore, migrated neutrophils of cpla2α−/− mice had minimal LTB4 production when compared to wild type neutrophils (Figure 7B). Defective migration of cpla2α−/− bone marrow cells was not observed when migration was driven by an applied gradient of LTB4 (5 ng/ml placed in the apical well) across uninfected MLE12 monolayers (Figure 7C). This result suggests that cpla2α−/− bone marrow neutrophils are capable of effectively responding to LTB4 signals, if provided, but since cpla2α−/− bone marrow cells are not capable of generating LTB4 during migration, the magnitude of migration is suppressed, as cpla2α−/− bone marrow cells must rely exclusively on epithelial-elicited HxA3 to move across the MLE12 barrier. If this hypothesis is correct, then mixing wild type and cpla2α−/− bone marrow cells prior to initiating migration would serve to rescue the defect in cpla2α−/− bone marrow cells because wild type cells are capable of providing the LTB4 gradient. To address this, wild type and cpla2α−/− bone marrow was differentially labeled and mixed in equal proportion. This mixed population was used as a source of neutrophils. In response to epithelial infection with PAO1, within the mixed population, cpla2α−/− migrated as effectively as wild type neutrophils as migrated ratios were similar to input ratios (Figure 7D). This suggests that the presence of wild type neutrophils rescues any defect in migration exhibited by cpla2α−/− neutrophils alone and is consistent with previous studies whereby neutrophils lacking the ability to synthesize LTB4 fail to augment trans-epithelial migration, but when mixed with neutrophils capable of synthesizing LTB4 are fully capable of responding to the LTB4 signal and migrating across the epithelium(19). Neutrophils isolated from cpla2α−/− mice on a 129Sv/C57BL6 background were also investigated to ensure these findings were not strain specific. Similar to the experiment using bone marrow from BALB/c mice, cpla2α−/− neutrophils exhibited reduced trans-epithelial migration in response to PAO1-infected MLE12 monolayers and failed to synthesize LTB4 (Supplemental Figure 2). Taken together, these data further support the importance of neutrophil expressed cPLA2α in neutrophil-mediated production of LTB4 and its role in coordinating maximal trans-epithelial migration.
Figure 7. PAO1-induced neutrophil trans-epithelial migration using a murine in-vitro Transwell co-culture model requires cPLA2α signaling from the neutrophil.
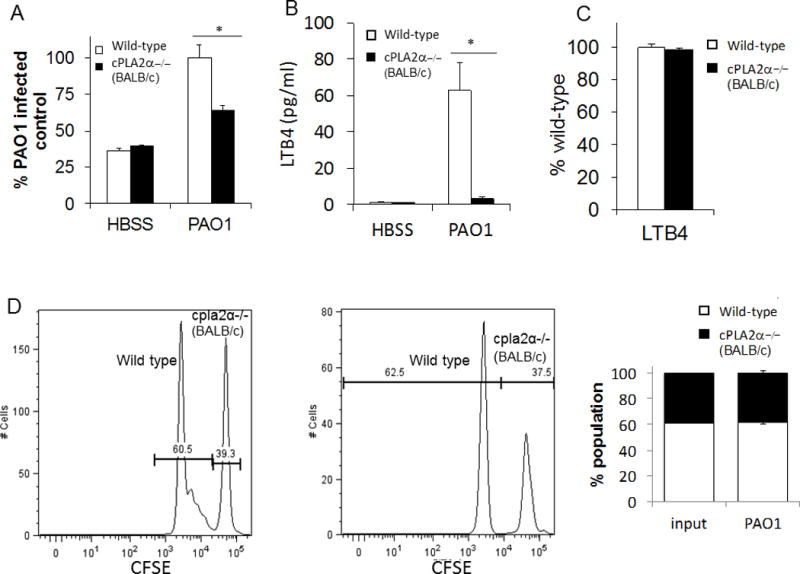
A) Total bone marrow was isolated from cPLA2α deficient mice on a BALB/c background and littermate controls then supplied to the basolateral compartment of MLE12 monolayers. Neutrophils were allowed to migrate across an MLE monolayer infected with PAO1 or mock-infection (HBSS). Relative neutrophil migration was assessed by myeloperoxidase activity in the apical compartment following 2h migration. Total myeloperoxidase activity was corrected for using standard curves generated for each bone marrow source. Data are shown as mean corrected value +/− standard deviation. B) Following migration, the apical compartment was sampled for the presence of LTB4 by ELISA. C) Neutrophils from cPLA2α deficient mice and littermate controls migrated across MLE12 monolayers towards an imposed gradient of LTB4 (5ng/ml). Total myeloperoxidase activity was corrected for using standard curves generated for each bone marrow source and data are shown as mean corrected value +/− standard deviation. D) Bone marrow from WT and cPLA2α −/− mice were differentially labeled with CFSE and allowed to undergo a combined migration in a near 1:1 ratio. The ratio of neutrophils collected from the apical compartment was assessed after two hours of migration across a PAO1-infected epithelium by flow cytometry. Representative histograms, gated on Ly6G+ neutrophils, and average proportion detected in multiple tests are shown. Experiments were performed on at least three occasions with n≥3 technical replicates. P values were calculated by paired Student’s t-test. P values ≤0.05 was consider significant.
Pseudomonas-induced neutrophil migration across human airway basal stem cell-derived airway epithelium cultured at air-liquid interface involves neutrophil-derived cPLA2α
In order to evaluate these mechanisms in a model that is more closely associated to the physiology present within the human airways, a primary human airway co-culture model was utilized. This model was recently developed ex vivo through the use of human airway basal stem cells cultured at an air-liquid interface (ALI)(26). ALI culturing of primary human airway cells allows for the generation of a functional mucociliary epithelial barrier(45, 46), which exhibit multi-cellular architecture, appropriate cell-specific morphology, and physiologic functioning that resembles the airway surface in vivo(26). In order to take advantage of this complex airway morphology, we also incorporated imaging with micro-optical coherence tomography (μOCT). This new imaging technique provides 1μm resolution, two- and three-dimensional perspectives that can be analyzed over time, and requires no fixation or staining. These attributes enable real-time imaging of micro-anatotomical processes at the mucosal surface(31). Human airway basal stem cells derived from a healthy donor and cultured under ALI conditions were infected on the apical surface with PAO1, and primary human neutrophils were applied to the basolateral compartment, as described with the previous co-culture models. During the two-hour migration time period, neutrophils can be seen migrating across the monolayer in clumps before dropping to the slide, which defines the bottom of the apical compartment (Figure 8A). To assess the effect of neutrophil-derived cPLA2α in PAO1-induced neutrophil migration across human airway basal stem cell-derived epithelium, neutrophils were treated with cPLA2α inhibitor or vehicle control before application to the basolateral compartment. Migration in response to infection was significantly reduced in the setting of cPLA2α inhibitor treated neutrophils, as observed by μOCT imaging (Figure 8B) (Supplemental video 1). This observable μOCT imaging-based decrease in neutrophil penetration of the epithelium with cPLA2α inhibited neutrophils can be quantified by utilizing a computer algorithm that assigns values representing the degree of neutrophil emergence into apical space over time (Figure 8C). Standard MPO assays also confirm total decreased migration across PAO1-infected human airway basal stem cell-derived epithelium when neutrophil cPLA2α is inhibited (Figure 8D). Lastly, neutrophils treated with cPLA2a inhibitor have decreased LTB4 production following migration across PAO1-infected human airway basal stem cell-derived epithelium (Figure 8E), even when corrected for numbers of neutrophils migrated (Figure 8F). Together, these findings support the mechanism that neutrophil cPLA2α augments migration across infected mature, primary basal stem cell-derived human airway mucosa through its involvement in synthesizing LTB4.
Figure 8. Neutrophil-derived cPLA2α plays a key role in migration across a human primary airway mucosal model.
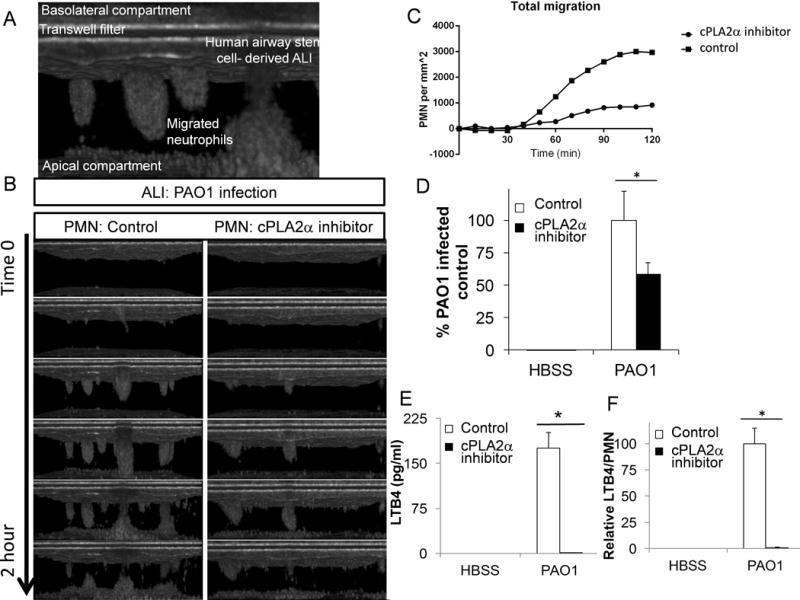
A) μOCT imaging displays trans-epithelial migration of neutrophils across a human airway basal stem cell-derived epithelium, grown in air-liquid interface. B) Neutrophils were pretreated for 60 min. with cPLA2α inhibitor (12μM) or vehicle control (DMSO 1:500) then allowed to migrate across a PAO1 infected mature epithelium derived from human airway basal stem cells grown in air-liquid interface. Migration was visualized by micro-OCT imaging. Representative images across the 2h migration period were selected. C) Neutrophil migration was quantified by neutrophil density per area (mm2) and plotted over time. D) Migration of neutrophils pretreated for 60 min. with cPLA2α inhibitor (12μM) or control (DMSO 1:500) across a mature human airway basal stem cell-derived epithelium in response to PAO1 infection or buffer (HBSS) was quantified by total myeloperoxidase activity. E) After the 2h migration, supernatant was collected from the apical compartment and LTB4 was quantified by ELISA. F) In the human airway basal stem cell-derived ALI model, LTB4 production was corrected for by neutrophil migrated and compared to display relative LTB4 production per neutrophil. Data are shown as mean corrected value +/− standard deviation. Experiments were performed on at least three occasions with n≥3 technical replicates. P values were calculated by paired Student’s t-test. P values ≤0.05 was consider significant.
Discussion
Trans-epithelial migration, the final step in a neutrophil’s journey from circulation to the airway, is a key micro-anatomical event that occurs during the inflammatory response at mucosal surfaces. Eicosanoids have been shown to be central to the process of neutrophilic breach of the epithelial mucosal barrier, delivering neutrophils to the site of infection(17, 19). In certain diseases, however, this neutrophilic response may be maladaptive, worsening rather than resolving disease. Therefore, understanding the mechanisms driving eicosanoid-mediated neutrophil trans-epithelial migration may offer novel therapeutic targets to modulate the inflammatory process.
Cystic fibrosis is a classic example of a disease plagued by chronic airway inflammation, with bouts of neutrophil infiltration that perpetuate pulmonary disease. Corticosteroids, which target PLA2 activity on the lipid membrane by inhibiting ERK-mediated phosphorylation(47), have been shown to reduce neutrophil inflammation in cystic fibrosis, but the numerous side effects outweigh the potential benefit(48). Non-steroidal anti-inflammatories, such as ibuprofen, have been shown to reduce eicosanoid production and reduce airway inflammation in cystic fibrosis(49), however much like corticosteroids, the side effects often make long-term treatment intolerable(50). LTB4 activity has been targeted by blocking the LTB4 receptor (BTL1) with BLT1 antagonists given orally to CF patients, however, negative outcomes were documented, halting further examination of this approach(51, 52). Precise targeting of specific signaling pathways in the relevant tissue location may be the key to providing effective therapeutic benefit without the accompanying adverse effects.
cPLA2α has been implicated in pulmonary disease, including asthma, acute lung injury and fibrosis(15). cPLA2α has been suggested to mediate allergic responses, including airway hyperreactivity and anaphylaxis in mice following allergic stimulation(53). cPLA2α also appears to play a role in mediating acute lung injury in mouse models. Following insult by endotoxin or acid aspiration, cPLA2α deficient mice are protected from protein leakage and neutrophil infiltration into the lung(33). Additionally, cPLA2α has been suggested to play a role in pulmonary fibrosis, as cPLA2α deficient mice are protected from the inflammatory response and fibrosis brought on by bleomycin exposure(33). Pre-clinical studies have assessed the effect of a cPLA2α inhibitor, PF-5212372, on animal models of allergic asthma, but have not considered neutrophilic responses(54, 55). Therefore, cPLA2α has been shown to play a role in the pulmonary disease, but a precise mechanism of action has not been described. Understanding its role in neutrophil trans-epithelial migration through LTB4 amplification may help us better delineate underlying mechanisms of disease.
Our data suggest that neutrophil cPLA2α plays a key role in neutrophil-epithelial cross talk when tested using both human and mouse epithelial co-culture models. Human neutrophils treated with chemical inhibition targeting PLA2 reveal that this enzyme activity is required for neutrophil signaling in PAO1-induced neutrophil trans-epithelial migration. Further, chemical inhibition of either the PLA2 subtype, cPLA2α, or the kinase required for activation of cPLA2α, ERK, reduces neutrophil trans-epithelial migration, suggesting that neutrophil-derived cPLA2α plays a key role in bacterial triggered neutrophil trans-epithelial migration. Chemical inhibition of related PLA2 isoforms of the iPLA2 class within the neutrophil did not impact neutrophil migration function. To examine our findings without the requirement of a pharmacological agent, we used mice genetically deficient in cPLA2α to assess trans-epithelial migration in the setting of infection. Mouse epithelium relies on release of HxA3 to initiate bacterial induced trans-epithelial migration(19, 56). We found that neutrophils from cPLA2α −/− mice had reduced migration across the infected mouse epithelium, with corresponding reduced levels of LTB4 production. When responding to an exogenously applied gradient of LTB4, no difference was observed between cPLA2α −/− and wild type-derived neutrophils, indicating neutrophils deficient in cPLA2α were not inherently impaired in their ability to migrate. Further, migration of a mixed population of cPLA2α −/− and wild type-derived neutrophils across infected mouse epithelial monolayers revealed that the presence of wild type neutrophils rescued the defective migration of cPLA2α −/−neutrophils as both populations were equally represented following migration. These results support the hypothesis that cPLA2α drives neutrophil production of LTB4, which augments the magnitude of neutrophil trans-epithelial migration. As long as the LTB4 signal is provided, cPLA2α −/− neutrophils can migrate across the epithelium with equal efficiency as wild type cells. As cPLA2α is highly conserved between mice and human, sharing 95% amino acid identity(57), function is highly likely to be preserved across species, further supporting the role of neutrophil cPLA2α in trans-epithelial signaling.
Investigating precise mechanisms of trans-epithelial migration in vivo is difficult as it is technically challenging to tease apart the signaling patterns specific to each step of the migratory process, including trans-endothelial migration and migration through the extracellular matrix. To simulate the inflamed human airway, we used primary human airway basal stem cells grown in air-liquid interface culturing conditions, which recapitulates the human trans-epithelial migration step in a more physiologic manner when paired with human neutrophils. Epithelium derived from human airway basal stem cells does not rely on cell immortalization and incorporates functional pulmonary features of mucous production and beating cilia, thus providing appropriate physiological context for these basic cellular mechanisms. Using this primary cell system, we found that blocking cPLA2α in neutrophils resulted in decreased trans-epithelial migration across P. aeruginosa-infected epithelium, as compared to controls. Our data further supports our hypothesis that neutrophil cPLA2α plays a key role in P. aeruginosa-induced neutrophil trans-epithelial migration in humans.
Using a novel cellular imaging technique, μOCT, we were able to captured detailed cellular mechanisms governing neutrophil transmigratory behavior. μOCT uses optical interference with a reference beam to determine the depths of back-scattered light generated by a sample, creating a cross-sectional map of optical reflectance. μOCT has been successfully used in real time to characterize airway microanatomy, including cilia beat function and mucous layer depth in a detailed, three-dimensional manner (31, 58). Imaging with this modality is unique because it does not require sample labeling or manipulation, and provides cellular dynamics in real-time imaging. With μOCT, we readily observed the impact of cPLA2α inhibition on neutrophil migration. Neutrophils migrated across the epithelial barrier in organized clusters at various sites throughout the mucosal surface. When pre-treated with cPLA2α inhibitor, the number of clusters per mm2 appeared to decrease, however it is unclear whether the neutrophil cluster size was impacted to any significant degree. The dynamic migratory process unveiled by μOCT imaging reveals a wealth of novel visual information that warrants further investigation. In addition, μOCT image-based data can be converted to quantitative metrics using computer algorithms that assign values to defined observations. Basic quantitative metrics such as total migration was demonstrated herein, however, the opportunity exists to expand and develop novel measurements based on neutrophil speed, cluster numbers or size, cluster formation and detachment dynamics, among any number of other possibilities(30).
In summary, neutrophil-derived cPLA2α plays a key role in amplifying the total neutrophilic response to bacterial-induced neutrophil migration by participating in the synthesis of LTB4 during the migratory process. The specific PLA2 isoform required for the generation of HxA3 within the epithelium following bacterial infection remains an open question that will be important to discern in future studies. cPLA2α has been implicated in mediating disease processes but has not been assessed specifically in the context of neutrophil recruitment. Furthermore, understanding the role of cPLA2α may be of benefit to specific diseases associated with neutrophil inflammation such as cystic fibrosis or ARDS. Targeting pathways involved in neutrophil-epithelial crosstalk may offer therapeutic benefit, but a more detailed understanding of the mechanisms involved is required. Identification of the contribution of cPLA2α in neutrophil signaling offers an additional key piece of information in the study of neutrophilic airway epithelial breach.
Supplementary Material
Acknowledgments
We thank Eileen O’Leary, BWH Renal Division Lab Manager, for technical assistance. We thank Rhianna M. Hibbler and Kevin S. Gipson, members of the Mucosal Immunology & Biology Research Center, for critical and editorial review of the manuscript.
Footnotes
This work has been supported by the NIH/NIAID (R01 AI095338), NIH/NHLBI (R01 HL116756, R01 HL118185), NIH/DK/NIDDK (R37 DK39773), as well as the Cystic Fibrosis Foundation (PAZOS13F0, MOU16G0, TEARNE07XX0, & HURLEY16G0)
References
- 1.Brouwers H, von Hegedus J, Toes R, Kloppenburg M, Ioan-Facsinay A. Lipid mediators of inflammation in rheumatoid arthritis and osteoarthritis. Best practice & research Clinical rheumatology. 2015;29:741–755. doi: 10.1016/j.berh.2016.02.003. [DOI] [PubMed] [Google Scholar]
- 2.Fanning LB, Boyce JA. Lipid mediators and allergic diseases. Annals of allergy, asthma & immunology: official publication of the American College of Allergy, Asthma, & Immunology. 2013;111:155–162. doi: 10.1016/j.anai.2013.06.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Oates JA, FitzGerald GA, Branch RA, Jackson EK, Knapp HR, Roberts LJ., 2nd Clinical implications of prostaglandin and thromboxane A2 formation (1) The New England journal of medicine. 1988;319:689–698. doi: 10.1056/NEJM198809153191106. [DOI] [PubMed] [Google Scholar]
- 4.Raju N, Sobieraj-Teague M, Hirsh J, O’Donnell M, Eikelboom J. Effect of aspirin on mortality in the primary prevention of cardiovascular disease. The American journal of medicine. 2011;124:621–629. doi: 10.1016/j.amjmed.2011.01.018. [DOI] [PubMed] [Google Scholar]
- 5.Hasday JD, Meltzer SS, Moore WC, Wisniewski P, Hebel JR, Lanni C, Dube LM, Bleecker ER. Anti-inflammatory effects of zileuton in a subpopulation of allergic asthmatics. American journal of respiratory and critical care medicine. 2000;161:1229–1236. doi: 10.1164/ajrccm.161.4.9904026. [DOI] [PubMed] [Google Scholar]
- 6.Leff JA, Busse WW, Pearlman D, Bronsky EA, Kemp J, Hendeles L, Dockhorn R, Kundu S, Zhang J, Seidenberg BC, Reiss TF. Montelukast, a leukotriene-receptor antagonist, for the treatment of mild asthma and exercise-induced bronchoconstriction. The New England journal of medicine. 1998;339:147–152. doi: 10.1056/NEJM199807163390302. [DOI] [PubMed] [Google Scholar]
- 7.Baenkler M, Leykauf M, John S. Functional analysis of eicosanoids from white blood cells in sepsis and SIRS. Journal of physiology and pharmacology: an official journal of the Polish Physiological Society. 2006;57(Suppl 12):25–33. [PubMed] [Google Scholar]
- 8.Benfield TL, van Steenwijk R, Nielsen TL, Dichter JR, Lipschik GY, Jensen BN, Junge J, Shelhamer JH, Lundgren JD. Interleukin-8 and eicosanoid production in the lung during moderate to severe Pneumocystis carinii pneumonia in AIDS: a role of interleukin-8 in the pathogenesis of P. carinii pneumonia. Respiratory medicine. 1995;89:285–290. doi: 10.1016/0954-6111(95)90089-6. [DOI] [PubMed] [Google Scholar]
- 9.Tamang DL, Pirzai W, Priebe GP, Traficante DC, Pier GB, Falck JR, Morisseau C, Hammock BD, McCormick BA, Gronert K, Hurley BP. Hepoxilin A(3) facilitates neutrophilic breach of lipoxygenase-expressing airway epithelial barriers. J Immunol. 2012;189:4960–4969. doi: 10.4049/jimmunol.1201922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hurley BP, McCormick BA. Multiple roles of phospholipase A2 during lung infection and inflammation. Infection and immunity. 2008;76:2259–2272. doi: 10.1128/IAI.00059-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bair AM, Turman MV, Vaine CA, Panettieri RA, Jr, Soberman RJ. The nuclear membrane leukotriene synthetic complex is a signal integrator and transducer. Mol Biol Cell. 2012;23:4456–4464. doi: 10.1091/mbc.E12-06-0489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Grewal S, Herbert SP, Ponnambalam S, Walker JH. Cytosolic phospholipase A2-alpha and cyclooxygenase-2 localize to intracellular membranes of EA.hy.926 endothelial cells that are distinct from the endoplasmic reticulum and the Golgi apparatus. FEBS J. 2005;272:1278–1290. doi: 10.1111/j.1742-4658.2005.04565.x. [DOI] [PubMed] [Google Scholar]
- 13.Cohen W, Fencl MM, Tulchinsky D. Amniotic fluid cortisol after premature rupture of membranes. J Pediatr. 1976;88:1007–1009. doi: 10.1016/s0022-3476(76)81064-5. [DOI] [PubMed] [Google Scholar]
- 14.Pardue S, Rapoport SI, Bosetti F. Co-localization of cytosolic phospholipase A2 and cyclooxygenase-2 in Rhesus monkey cerebellum. Brain Res Mol Brain Res. 2003;116:106–114. doi: 10.1016/s0169-328x(03)00262-6. [DOI] [PubMed] [Google Scholar]
- 15.Leslie CC. Cytosolic phospholipase A(2): physiological function and role in disease. Journal of lipid research. 2015;56:1386–1402. doi: 10.1194/jlr.R057588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hurley BP, Pirzai W, Mumy KL, Gronert K, McCormick BA. Selective eicosanoid-generating capacity of cytoplasmic phospholipase A2 in Pseudomonas aeruginosa-infected epithelial cells. American journal of physiology Lung cellular and molecular physiology. 2011;300:L286–294. doi: 10.1152/ajplung.00147.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mrsny RJ, Gewirtz AT, Siccardi D, Savidge T, Hurley BP, Madara JL, McCormick BA. Identification of hepoxilin A3 in inflammatory events: a required role in neutrophil migration across intestinal epithelia. Proceedings of the National Academy of Sciences of the United States of America. 2004;101:7421–7426. doi: 10.1073/pnas.0400832101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mumy KL, Bien JD, Pazos MA, Gronert K, Hurley BP, McCormick BA. Distinct isoforms of phospholipase A2 mediate the ability of Salmonella enterica serotype typhimurium and Shigella flexneri to induce the transepithelial migration of neutrophils. Infection and immunity. 2008;76:3614–3627. doi: 10.1128/IAI.00407-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pazos MA, Pirzai W, Yonker LM, Morisseau C, Gronert K, Hurley BP. Distinct cellular sources of hepoxilin A3 and leukotriene B4 are used to coordinate bacterial-induced neutrophil transepithelial migration. J Immunol. 2015;194:1304–1315. doi: 10.4049/jimmunol.1402489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Grommes J, Soehnlein O. Contribution of neutrophils to acute lung injury. Mol Med. 2011;17:293–307. doi: 10.2119/molmed.2010.00138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yonker LM, Cigana C, Hurley BP, Bragonzi A. Host-pathogen interplay in the respiratory environment of cystic fibrosis. Journal of cystic fibrosis: official journal of the European Cystic Fibrosis Society. 2015;14:431–439. doi: 10.1016/j.jcf.2015.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hurley BP, Williams NL, McCormick BA. Involvement of phospholipase A2 in Pseudomonas aeruginosa-mediated PMN transepithelial migration. American journal of physiology Lung cellular and molecular physiology. 2006;290:L703–L709. doi: 10.1152/ajplung.00390.2005. [DOI] [PubMed] [Google Scholar]
- 23.Peters-Golden M, Henderson WR., Jr Leukotrienes. The New England journal of medicine. 2007;357:1841–1854. doi: 10.1056/NEJMra071371. [DOI] [PubMed] [Google Scholar]
- 24.Ghosh M, Tucker DE, Burchett SA, Leslie CC. Properties of the Group IV phospholipase A2 family. Progress in lipid research. 2006;45:487–510. doi: 10.1016/j.plipres.2006.05.003. [DOI] [PubMed] [Google Scholar]
- 25.Schaloske RH, Dennis EA. The phospholipase A2 superfamily and its group numbering system. Biochimica et biophysica acta. 2006;1761:1246–1259. doi: 10.1016/j.bbalip.2006.07.011. [DOI] [PubMed] [Google Scholar]
- 26.Mou H, Vinarsky V, Tata PR, Brazauskas K, Choi SH, Crooke AK, Zhang B, Solomon GM, Turner B, Bihler H, Harrington J, Lapey A, Channick C, Keyes C, Freund A, Artandi S, Mense M, Rowe S, Engelhardt JF, Hsu YC, Rajagopal J. Dual SMAD Signaling Inhibition Enables Long-Term Expansion of Diverse Epithelial Basal Cells. Cell stem cell. 2016 doi: 10.1016/j.stem.2016.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Neuberger T, Burton B, Clark H, Van Goor F. Use of primary cultures of human bronchial epithelial cells isolated from cystic fibrosis patients for the pre-clinical testing of CFTR modulators. Methods Mol Biol. 2011;741:39–54. doi: 10.1007/978-1-61779-117-8_4. [DOI] [PubMed] [Google Scholar]
- 28.Kusek ME, Pazos MA, Pirzai W, Hurley BP. In vitro coculture assay to assess pathogen induced neutrophil trans-epithelial migration. Journal of visualized experiments: JoVE. 2014:e50823. doi: 10.3791/50823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.McCormick BA, Hofman PM, Kim J, Carnes DK, Miller SI, Madara JL. Surface attachment of Salmonella typhimurium to intestinal epithelia imprints the subepithelial matrix with gradients chemotactic for neutrophils. The Journal of cell biology. 1995;131:1599–1608. doi: 10.1083/jcb.131.6.1599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chu KK, Kusek ME, Liu L, Som A, Yonker LM, Leung H, Cui D, Ryu J, Eaton AD, Tearney GJ, Hurley BP. Illuminating dynamic neutrophil trans-epithelial migration with micro-optical coherence tomography. Sci Rep. 2017;8:45789. doi: 10.1038/srep45789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liu L, Chu KK, Houser GH, Diephuis BJ, Li Y, Wilsterman EJ, Shastry S, Dierksen G, Birket SE, Mazur M, Byan-Parker S, Grizzle WE, Sorscher EJ, Rowe SM, Tearney GJ. Method for quantitative study of airway functional microanatomy using micro-optical coherence tomography. PloS one. 2013;8:e54473. doi: 10.1371/journal.pone.0054473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bonventre JV, Huang Z, Taheri MR, O’Leary E, Li E, Moskowitz MA, Sapirstein A. Reduced fertility and postischaemic brain injury in mice deficient in cytosolic phospholipase A2. Nature. 1997;390:622–625. doi: 10.1038/37635. [DOI] [PubMed] [Google Scholar]
- 33.Nagase T, Uozumi N, Ishii S, Kume K, Izumi T, Ouchi Y, Shimizu T. Acute lung injury by sepsis and acid aspiration: a key role for cytosolic phospholipase A2. Nat Immunol. 2000;1:42–46. doi: 10.1038/76897. [DOI] [PubMed] [Google Scholar]
- 34.Rubin BB, Downey GP, Koh A, Degousee N, Ghomashchi F, Nallan L, Stefanski E, Harkin DW, Sun C, Smart BP, Lindsay TF, Cherepanov V, Vachon E, Kelvin D, Sadilek M, Brown GE, Yaffe MB, Plumb J, Grinstein S, Glogauer M, Gelb MH. Cytosolic phospholipase A2-alpha is necessary for platelet-activating factor biosynthesis, efficient neutrophil-mediated bacterial killing, and the innate immune response to pulmonary infection: cPLA2-alpha does not regulate neutrophil NADPH oxidase activity. J Biol Chem. 2005;280:7519–7529. doi: 10.1074/jbc.M407438200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Uozumi N, Kume K, Nagase T, Nakatani N, Ishii S, Tashiro F, Komagata Y, Maki K, Ikuta K, Ouchi Y, Miyazaki J, Shimizu T. Role of cytosolic phospholipase A2 in allergic response and parturition. Nature. 1997;390:618–622. doi: 10.1038/37622. [DOI] [PubMed] [Google Scholar]
- 36.Geijsen N, Dijkers PF, Lammers JJ, Koenderman L, Coffer PJ. Cytokine-mediated cPLA(2) phosphorylation is regulated by multiple MAPK family members. FEBS Lett. 2000;471:83–88. doi: 10.1016/s0014-5793(00)01373-9. [DOI] [PubMed] [Google Scholar]
- 37.Hazan-Halevy I, Seger R, Levy R. The requirement of both extracellular regulated kinase and p38 mitogen-activated protein kinase for stimulation of cytosolic phospholipase A(2) activity by either FcgammaRIIA or FcgammaRIIIB in human neutrophils. A possible role for Pyk2 but not for the Grb2-Sos-Shc complex. J Biol Chem. 2000;275:12416–12423. doi: 10.1074/jbc.275.17.12416. [DOI] [PubMed] [Google Scholar]
- 38.Hazan-Halevy I, Levy T, Wolak T, Lubarsky I, Levy R, Paran E. Stimulation of NADPH oxidase by angiotensin II in human neutrophils is mediated by ERK, p38 MAP-kinase and cytosolic phospholipase A2. J Hypertens. 2005;23:1183–1190. doi: 10.1097/01.hjh.0000170381.53955.68. [DOI] [PubMed] [Google Scholar]
- 39.Hurley BP, Siccardi D, Mrsny RJ, McCormick BA. Polymorphonuclear cell transmigration induced by Pseudomonas aeruginosa requires the eicosanoid hepoxilin A3. J Immunol. 2004;173:5712–5720. doi: 10.4049/jimmunol.173.9.5712. [DOI] [PubMed] [Google Scholar]
- 40.Mikami S, Aiboshi J, Kobayashi T, Kojima M, Morishita K, Otomo Y. Discrete roles of intracellular phospholipases A2 in human neutrophil cytotoxicity. J Trauma Acute Care Surg. 2015;79:238–246. doi: 10.1097/TA.0000000000000730. [DOI] [PubMed] [Google Scholar]
- 41.Tithof PK, Olivero J, Ruehle K, Ganey PE. Activation of neutrophil calcium-dependent and -independent phospholipases A2 by organochlorine compounds. Toxicol Sci. 2000;53:40–47. doi: 10.1093/toxsci/53.1.40. [DOI] [PubMed] [Google Scholar]
- 42.Lindbom J, Ljungman AG, Lindahl M, Tagesson C. Expression of members of the phospholipase A2 family of enzymes in human nasal mucosa. The European respiratory journal. 2001;18:130–138. doi: 10.1183/09031936.01.00054701. [DOI] [PubMed] [Google Scholar]
- 43.Vasta V, Meacci E, Catarzi S, Donati C, Farnararo M, Bruni P. Sphingosine 1-phosphate induces arachidonic acid mobilization in A549 human lung adenocarcinoma cells. Biochimica et biophysica acta. 2000;1483:154–160. doi: 10.1016/s1388-1981(99)00183-3. [DOI] [PubMed] [Google Scholar]
- 44.Cross AR, Jones OT. The effect of the inhibitor diphenylene iodonium on the superoxide-generating system of neutrophils. Specific labelling of a component polypeptide of the oxidase. Biochem J. 1986;237:111–116. doi: 10.1042/bj2370111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Whitcutt MJ, Adler KB, Wu R. A biphasic chamber system for maintaining polarity of differentiation of cultured respiratory tract epithelial cells. In vitro cellular & developmental biology: journal of the Tissue Culture Association. 1988;24:420–428. doi: 10.1007/BF02628493. [DOI] [PubMed] [Google Scholar]
- 46.Yamaya M, Finkbeiner WE, Chun SY, Widdicombe JH. Differentiated structure and function of cultures from human tracheal epithelium. The American journal of physiology. 1992;262:L713–724. doi: 10.1152/ajplung.1992.262.6.L713. [DOI] [PubMed] [Google Scholar]
- 47.Bailey JM. New mechanisms for effects of anti-inflammatory glucocorticoids. Biofactors. 1991;3:97–102. [PubMed] [Google Scholar]
- 48.Eigen H, Rosenstein BJ, FitzSimmons S, Schidlow DV. A multicenter study of alternate-day prednisone therapy in patients with cystic fibrosis. Cystic Fibrosis Foundation Prednisone Trial Group. The Journal of pediatrics. 1995;126:515–523. doi: 10.1016/s0022-3476(95)70343-8. [DOI] [PubMed] [Google Scholar]
- 49.Mogayzel PJ, Jr, Naureckas ET, Robinson KA, Mueller G, Hadjiliadis D, Hoag JB, Lubsch L, Hazle L, Sabadosa K, Marshall B, C. Pulmonary Clinical Practice Guidelines Cystic fibrosis pulmonary guidelines. Chronic medications for maintenance of lung health. American journal of respiratory and critical care medicine. 2013;187:680–689. doi: 10.1164/rccm.201207-1160oe. [DOI] [PubMed] [Google Scholar]
- 50.Konstan MW. Ibuprofen therapy for cystic fibrosis lung disease: revisited. Curr Opin Pulm Med. 2008;14:567–573. doi: 10.1097/MCP.0b013e32831311e8. [DOI] [PubMed] [Google Scholar]
- 51.Doring G, Bragonzi A, Paroni M, Akturk FF, Cigana C, Schmidt A, Gilpin D, Heyder S, Born T, Smaczny C, Kohlhaufl M, Wagner TO, Loebinger MR, Bilton D, Tunney MM, Elborn JS, Pier GB, Konstan MW, Ulrich M. BIIL 284 reduces neutrophil numbers but increases P. aeruginosa bacteremia and inflammation in mouse lungs. Journal of cystic fibrosis: official journal of the European Cystic Fibrosis Society. 2014;13:156–163. doi: 10.1016/j.jcf.2013.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Konstan MW, Doring G, Heltshe SL, Lands LC, Hilliard KA, Koker P, Bhattacharya S, Staab A, Hamilton A, Investigators, and B. I. T. Coordinators of A randomized double blind, placebo controlled phase 2 trial of BIIL 284 BS (an LTB4 receptor antagonist) for the treatment of lung disease in children and adults with cystic fibrosis. Journal of cystic fibrosis: official journal of the European Cystic Fibrosis Society. 2014;13:148–155. doi: 10.1016/j.jcf.2013.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Irvin CG, Tu YP, Sheller JR, Funk CD. 5-Lipoxygenase products are necessary for ovalbumin-induced airway responsiveness in mice. The American journal of physiology. 1997;272:L1053–1058. doi: 10.1152/ajplung.1997.272.6.L1053. [DOI] [PubMed] [Google Scholar]
- 54.Hewson CA, Patel S, Calzetta L, Campwala H, Havard S, Luscombe E, Clarke PA, Peachell PT, Matera MG, Cazzola M, Page C, Abraham WM, Williams CM, Clark JD, Liu WL, Clarke NP, Yeadon M. Preclinical evaluation of an inhibitor of cytosolic phospholipase A2alpha for the treatment of asthma. J Pharmacol Exp Ther. 2012;340:656–665. doi: 10.1124/jpet.111.186379. [DOI] [PubMed] [Google Scholar]
- 55.McKew JC, Lee KL, Shen MW, Thakker P, Foley MA, Behnke ML, Hu B, Sum FW, Tam S, Hu Y, Chen L, Kirincich SJ, Michalak R, Thomason J, Ipek M, Wu K, Wooder L, Ramarao MK, Murphy EA, Goodwin DG, Albert L, Xu X, Donahue F, Ku MS, Keith J, Nickerson-Nutter CL, Abraham WM, Williams C, Hegen M, Clark JD. Indole cytosolic phospholipase A2 alpha inhibitors: discovery and in vitro and in vivo characterization of 4-{3-[5-chloro-2-(2-{[(3,4-dichlorobenzyl)sulfonyl]amino}ethyl)-1-(diphenylmethyl)-1H-indol-3-yl]propyl}benzoic acid, efipladib. J Med Chem. 2008;51:3388–3413. doi: 10.1021/jm701467e. [DOI] [PubMed] [Google Scholar]
- 56.Bhowmick R, Tin Maung NH, Hurley BP, Ghanem EB, Gronert K, McCormick BA, Leong JM. Systemic disease during Streptococcus pneumoniae acute lung infection requires 12-lipoxygenase-dependent inflammation. J Immunol. 2013;191:5115–5123. doi: 10.4049/jimmunol.1300522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Clark JD, Lin LL, Kriz RW, Ramesha CS, Sultzman LA, Lin AY, Milona N, Knopf JL. A novel arachidonic acid-selective cytosolic PLA2 contains a Ca(2+)-dependent translocation domain with homology to PKC and GAP. Cell. 1991;65:1043–1051. doi: 10.1016/0092-8674(91)90556-e. [DOI] [PubMed] [Google Scholar]
- 58.Birket SE, Chu KK, Liu L, Houser GH, Diephuis BJ, Wilsterman EJ, Dierksen G, Mazur M, Shastry S, Li Y, Watson JD, Smith AT, Schuster BS, Hanes J, Grizzle WE, Sorscher EJ, Tearney GJ, Rowe SM. A functional anatomic defect of the cystic fibrosis airway. American journal of respiratory and critical care medicine. 2014;190:421–432. doi: 10.1164/rccm.201404-0670OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.