

Abstract
3D printed biomaterials with spatial and temporal functionality could enable interfacial manipulation of fluid flows and motile cells. However, such dynamic biomaterials are challenging to implement since they must be responsive to multiple, biocompatible stimuli. Here, we show stereolithographic printing of hydrogels using noncovalent (ionic) crosslinking, which enables reversible patterning with controlled degradation. We demonstrate this approach using sodium alginate, photoacid generators and various combinations of divalent cation salts, which can be used to tune the hydrogel degradation kinetics, pattern fidelity, and mechanical properties. This approach is first utilized to template perfusable microfluidic channels within a second encapsulating hydrogel for T-junction and gradient devices. The presence and degradation of printed alginate microstructures were further verified to have minimal toxicity on epithelial cells. Degradable alginate barriers were used to direct collective cell migration from different initial geometries, revealing differences in front speed and leader cell formation. Overall, this demonstration of 3D printing using non-covalent crosslinking may enable adaptive and stimuli-responsive biomaterials, which could be utilized for bio-inspired sensing, actuation, drug delivery, and tissue engineering.
Introduction
Additive manufacturing (“3D-printing”) may enable designer biomaterial architectures that direct microfluidic flows1, 2 or functionally interface with living cells.3, 4 Previously, bulk hydrogels based on crosslinked, hydrophilic polymers have been engineered with spatial or temporal complexity,5, 6 but this functionality remains nascent for 3D-printed structures. An important design consideration is that dynamic biomaterials must be responsive to at least two physicochemical stimuli – the first to pattern the desired structures and additional orthogonal stimuli that trigger a temporal response. An intriguing possibility is to utilize noncovalent intermolecular interactions, which permit stimuli-responsive and reversible behaviors.7
Photopatterning techniques such as stereolithography8, 9 and two photon polymerization10 utilize selective illumination of light-sensitive photoinitiators to generate free radicals that initiate polymerization or crosslinking.11 In these approaches, a computer-aided design (CAD) is first deconstructed into a vertical stack of evenly spaced layers.12 Next, these layers are written one by one through the manipulation of a focused laser or sample with a motorized stage. Such photopatterning approaches are advantageous since they permit automated and rapid fabrication with spatial resolution on the order of the laser beam width (~250 μm for stereolithography, ~1 μm for two photon polymerization). Moreover, photopatterning has been broadly applied to a range of biomaterials, including synthetic polymers such as poly(ethylene glycol)13–22 and naturally-derived polymers such as gelatin,23 hyaluronic acid24 and agarose.25
Alginate hydrogels display physiochemical properties that are advantageous for 3D printing and triggered degradation.26 Alginates are naturally-derived anionic polysaccharides consisting of 1,4-linked β-d mannuronic acid (M) and α-l-guluronic acid (G) units in alternating or similar blocks.27 Under physiological temperature and pH, alginate can be ionically crosslinked by divalent cations,28–32 then dissociated when these cations are chelated.33–35 These alginate hydrogels display varying mechanical and physical properties when crosslinked with various divalent cations.28, 36–39 Alternatively, methacrylated alginate can be covalently crosslinked through the addition of photoinitiators.40, 41 Recently, Raghavan et al. demonstrated a variation on these approaches by using ultraviolet (UV) illumination of a photoacid generator to trigger proton release, which subsequently reacted with an insoluble cation salt to generate free cations.42 Instead, alginate can be crosslinked by light-triggered uncaging of calcium,43, 44 or by controlled release of calcium-filled liposomes.45 Finally, alginate can be 3D printed using nozzle-based extrusion of filaments46–49 or droplets.50
Degradable biomaterials can be utilized as sacrificial templates to pattern microfluidic channels within a second, encapsulating material.1 Such microfluidic devices can be utilized to manipulate interfacial transport, including controlled mixing or gradient formation.51 In particular, gradients within hydrogels can be generated by diffusion between a source and a sink channel.52–55 For instance, sacrificial templates have been prepared by nozzle extrusion of temperature-sensitive triblock copolymers (e.g. Pluronic F127),56 gelatin,57 as well as water-soluble carbohydrate glass,58 agarose,59 and alginate.60, 61 These extrusion-based approaches are compatible with a wide range of biomaterials, given appropriate optimization of nozzle diameter and rheology.62 Alternatively, stereolithography has been utilized for direct writing of microfluidic channels in photopolymerizable poly(ethylene glycol) (PEG),20–22 but not for selectively degradable templates. In general, previous PEG hydrogels have been formulated to respond to only a single physiochemical stimulus, which permits either photopolymerization or photodegradation,63 but not both. As a consequence, PEG-based hydrogels are not usable as sacrificial templates. Lastly, microfluidic channels in hydrogels have been patterned by needle molding,64 or soft lithography-like approaches using micromolded hydrogels,65–67 which can be challenging to scale up.
A second application for stimuli-responsive biomaterials is to direct cell migration by manipulating the spatial microenvironment. For example, the removal of localized barriers68, 69 permits cells to migrate individually63, 70–72 or collectively as multicellular groups73–88 into unoccupied regions. The initial geometry of these multicellular groups can drive emergent behaviors, including the formation of finger-like strands guided by leader cells.74, 84 An analogous phenomenon can occur after biochemical stimulation of an epithelial-mesenchymal transition (EMT),89 associated with a weakening of cell-cell junctions and a transition from collective to individual migration.90 In order to better understand this interplay of multicellular geometry and collective migration, dynamic photopatterned hydrogels represent a powerful tool to define the initial conditions.
Here, we show for the first time 3D stereolithographic printing of hydrogels based on noncovalent (ionic) crosslinking. Specifically, we show that 3D alginate hydrogels can be vertically patterned through the selective illumination of photoacid generators in the presence of insoluble divalent cation salts, inspired by Raghavan et al.42 We adapt this approach for 3D printing by systematically exploring combinations of divalent cations that result in optimized mechanical properties with minimal feature distortion. We show that the degradation kinetics of these alginate hydrogels can be tuned through the use of divalent cations at varying concentrations and combinations. We then utilize 3D printed alginate structures with triggered degradation for two case studies. First, we pattern sacrificial alginate templates for microfluidic mixers and gradient generators within agarose hydrogels. Second, we use 3D printed alginate structures with triggered degradation to characterize collective cell migration after the abrupt removal of geometric constraints. We envision this approach could be utilized for 3D biofabrication of natural and synthetic polyelectrolyte hydrogels, enabling labon-a-chip devices, soft sensors and actuators, as well as other biologically-inspired devices.
Materials & Methods
Preparation of Alginate Precursor Solution
Sodium alginate (W201502), barium carbonate (9237108), calcium carbonate (C5929), magnesium carbonate (13117), diphenyliodonium nitrate (127396), and ethylenediaminetetraacetic acid (ED) were all purchased from Sigma Aldrich. Sodium hydroxide pellets (S320-1), and Hyclone 1X 0.0067M Phosphate Buffered Saline (PBS) (SH30256FS) were purchased from Fisher Scientific. 3% (w/v) sodium alginate (Sigma W201502) was prepared in a dilute solution of PBS in deionized water (2% v/v of 1X PBS, final) and balanced to pH 12 using sodium hydroxide. 3% alginate stock solutions were stored at 4°C until use. Cation sources (barium carbonate, magnesium carbonate, and calcium carbonate) of various concentrations (Figure S1) and the photoacid generator diphenyliodonium nitrate were mixed into the alginate. Due to the insoluble nature of the cation carbonates, formulations were placed in a sonication bath and then vortexed thoroughly before use. For low miscibility samples, sonication and vortexing was repeated until aggregates were broken up and thoroughly mixed.
Stereolithographic Patterning of Alginate
Stereolithography was performed using a 3D Systems SLA 250/50 (Rock Hill, SC) with a 355nm 60W diode-pumped solid state UV laser (DPSS Lasers Inc., Santa Clara, CA). Parts were designed in SolidWorks 2016 (Dassault Systèmes), and converted to SLA files using 3D Lightyear File Preparation Software v1.5.2 (3D Systems). The system was customized with a holder for smaller volume samples. For instance, degradation studies and microfluidic hydrogels were prepared with ~1 mL of alginate precursor solution pipetted into a 35 mm Petri dish (Genesee Scientific 32-103). For pattern fidelity and cell migration studies, alginate precursor solution was pipetted into a 6-well plate containing 700 μL precursor solution/well, and a 12-well plate for cell viability studies containing 450 μL precursor solution/well (Genesee Scientific 25–105).
Alginate Degradation Measurements
Alginate disks (10 mm diameter, 1 mm height) were printed in 35 mm Petri dishes using precursor solutions of varying cation concentrations and compositions (Figure S1). After printing, alginate disks were briefly washed with deionized water to remove uncrosslinked alginate and were then imaged in brightfield using a benchtop digital microscope (Celestron Handheld Digital Microscope Pro 44380). This rinsing with DI water was relatively rapid to prevent premature dissolution of the alginate due to leaching of the crosslinking cations. Alginate disks were then immersed in 2 mL 0.1 M ethylenediaminetetraacetic acid (EDTA) in 1X PBS at 25°C, 0.1 M EDTA at 37°C, or 0.5 M EDTA at 37°C. Samples were visually inspected every 5 min to check for complete degradation. Degradation times were normalized for sample overcure by dividing the degradation time by 1 + overcure (Figure S2). Overcure was determined using the images taken after printing and ImageJ by tracing a circle over the diameter of the printed part and using the measure function to determine the area of the traced circle. This actual printed area APrint was compared to the part area defined by the AutoCAD design ACAD by an overcure ratio: (APrint/ACAD) − 1 (Figure S2).
Pattern Fidelity Measurements
Spiral-shaped alginate structures were patterned to determine the minimum achievable lateral feature size for each formulation of alginate precursor solution. A spiral geometry was defined in Solidworks 2016 by two offset spirals described by: x(s) = (s/2) cos(s), y(s) = (s/2) sin (s). The start of each spiral was offset so that the initial separation between both spirals was 80 μm. The spiral pattern traced through two complete revolutions (4π) for a total length of ~44 mm. The height of the spiral structure was 1 mm. After printing, samples were rinsed with deionized water to wash away uncured alginate. The spiral structures were then imaged in brightfield using a benchtop digital microscope (Celestron Handheld Digital Microscope Pro 44380). Digital images were then loaded into MATLAB 2016b (Mathworks Inc., Natick, MA) and cropped to remove artifacts from stray reflections. Next, images were binarized by thresholding the pixel intensities and converting brighter regions (printed alginate) to 1 and darker regions (background) to 0. A median filter was used to remove noise, extraneous regions were removed, and holes were filled. Pattern fidelity was determined by quantitatively comparing the binarized object to the original CAD file. An image matching algorithm was used to determine the amount of overcure and undercure for each sample as compared to the original CAD file (Supplemental Information). For samples that displayed good pattern fidelity, further analysis was performed to determine the minimum feature size of each spiral (Figure S3). ImageJ was used to determine the largest diameter circle that could be enclosed in the tip of each spiral.
Next, flat alginate layers were patterned onto an inverted glass coverslip in order to determine the vertical resolution, based on a modified “windowpane” assay.15 From the Beer-Lambert equation, the thickness of a photopolymerized layer (cure depth) can be described as CD = DP ln (E/EC), for some material-dependent penetration depth DP, actual light exposure E, and threshold light exposure EC.8 The cure depth CD was empirically determined by varying the actual light exposure E for a given material formulation. A logarithmic fit of these two parameters can be used to extract the threshold light exposure EC (x-intercept) and the penetration depth DP (slope). Experimentally, a 35mm dish was covered by a glass coverslip (Fisher 12-545-88) and filled with 3% alginate containing 1:3 Ba2+:Mg2+ until the alginate came into contact with the coverslip. 0.4″ × 0.15″ (10.16 mm × 3.81 mm) rectangles with a height of 0.004″ (0.1 mm) were printed. The samples were cleaned with deionized water, and imaged using a Ramé-Hart contact angle goniometer (Ramé-Hart, Model 190). Height was measured in Image-J using the line segment tool and converted to appropriate units.
Shear Modulus Measurements based on Mechanical Indentation
Bulk samples (5 mL) were prepared in a 35 mm Petri dish for mechanical indentation. These samples were relatively thick (~5 mm) in order to reduce boundary effects from the bottom of the dish. These samples were then cured overnight under a 365 nm 15W 115V UV flood lamp (UVP XX Series UV Lamp, UVP95 0042 07). For indentation analysis, a custom-built mechanical indenter was used (Figure S4A). A 4 mm diameter steel ball bearing was used as the indenter. A 25 g compression/tension load cell was manufactured by Sensing Systems Corp., New Bedford, MA. Signal processing and linear actuator control were performed using LabVIEW (National Instruments), data acquisition was performed using a National Instruments M-Series USB Multifunction DAQ, and output voltages were read using an Omega DP41-B-A Ultra High Performance Meter. Samples were indented to a depth of 1 mm at a velocity of 1 mm/s. The indenter was held in place for 120 s, followed by retraction of the indenter at 1 mm/s. Custom MATLAB code was used to determine the peak force immediately after indentation (Figure S4B). This instantaneous load response (Pi) compression was solved numerically for by Chan et al.:95
Where R is the radius of the indenter, δ is depth of indentation, G is the shear modulus, and h is the thickness of the sample. fP accounts for deviations in the load response due to substrate stiffness with thin hydrogel samples and is given by:
For short times before appreciable solvent migration occurs, the hydrogel can be approximated as incompressible, with Poisson’s ratio ν = 0.5. Based on this assumption, the shear modulus G can be converted to an elastic modulus (E) (Figure S4C).
Stereolithographic Patterning of Stepped Alginate Microstructures
Alginate hydrogels with varying heights were patterned using a “bottom up approach” where the alginate precursor solution was added sequentially in layers, which were photopatterned separately. As a proof-of-concept, a stepped alginate ramp was designed to be 2 mm wide, which increased in height in steps of 175 μm for every 200 μm of length for a final height of 1.8 mm and a final length of 18 mm. For this microstructure, 700 μL of the 3:1 Ba2+:Mg2+ alginate formulation was pipetted into a 35 mm diameter dish for the first layer. For 3D parts, an additional 100 μL was added for each subsequent layer for a total of 5 layers (Supplemental Information). After printing, the ramp was washed with water and “post-cured” using 1X CaCl2 to mechanically strengthen the hydrogel.
Microfluidic Hydrogel Fabrication and Simulation
To pattern microfluidic biomaterials, sacrificial alginate template structures were first prepared in a 35 mm petri dish using the 3:1 Ba2+: Mg2+ formulation, then rinsed and post-cured with 1X CaCl2 (Figure S5). For the microfluidic T-junction device, the serpentine main channel was printed with a 350 μm height and length of 37 mm, while the source reservoirs were 4 mm in diameter and 1.4 mm high, and the sink reservoir was 6.3 mm in diameter and also 1.4 mm high. 1.5% (w/v) agarose was then prepared in 1X Hank’s Balanced Salt Solution (Fisher, 14-175-079), solubilized by mixing and heating to ~70°C in a microwave, then added to the dish so that the microfluidic alginate channel was encapsulated but the reservoirs remained exposed. Once the agarose had solidified, the alginate structures were degraded by submerging the microfluidic in 100 mM EDTA overnight on a rocker. To visualize the flow conditions, ~100 μL of deionized water and a 1:100 dilution of green food dye (McCormick & Co. Inc., 52100070889) were added separately to each source reservoir and allowed to flow and mix along the length of the microfluidic channel (Figure S6).
For the microfluidic gradient generator, parallel alginate microstructures were patterned to template “source” and “sink” channels with a length of 21 mm, width of 1.5 mm, and a separation of 2.0 mm (Figure S5, S7). A first set of channels was patterned with consistent heights of 100 μm. A second set of channels was patterned where one channel had a stepped profile of 500 μm height at the start and 100 μm height at the end, while the other channel remained at a constant 100 μm height (Figure S8). These channels were then encapsulated in 1.5% agarose, consistent with the previous microfluidic T-junction device. Reservoirs were then punched using a 3 mm diameter biopsy punch (World Precision Instruments, Inc., 504649). Alginate parts were again degraded overnight on a rocker using 100 mM EDTA. The source channel was filled with a 0.2% (w/v) solution of Evans Blue (T-1824) dye (Sigma, E2129), and the sink channel was filled with deionized water. The source and sink channels were replenished with ~ 50μL every 15 min. Diffusion was recorded using a benchtop digital microscope (Dino-Lite AM4115ZT) until the Evans Blue dye had reached the sink channel. Images were then analyzed in ImageJ. Intensity profiles were background substracted for ease of comparison.
COMSOL Multiphysics (COMSOL Multiphysics v5.1) simulations were used to analyze the transport of dye through the agarose hydrogel using Fick’s law (Supplemental Information). A twodimensional cross-section of the hydrogel was used for the simulation geometry, with a width of 9.5 mm and a height of 2 mm. For the even channel simulation, both source and sink channels had dimensions of 1.5 mm in width and 100 μm in height, and a separation of 2.0 mm. For the stepped channel simulation, both source and sink channels had widths of 1.5 mm and a separation of 2.0 mm, but the source channel had a height of 500 μm while the sink channel had a height of 100 μm. Dye concentrations in the source channel (2 mg/mL) and sink channel (0 mg/mL) were held constant. The top, sides, and bottom of the hydrogel were treated using no-flux boundary conditions.
Cell Viability Measurements
Cell viability was measured over the course of 24 h to account for potential toxicity from (1) cell confluence, (2) Ba2+ and Mg2+ crosslinked alginate, and (3) EDTA treatment. Four experimental conditions were evaluated: Cells only, Cells + EDTA, Cells + Alginate, and Cells + Alginate + EDTA. Cells were seeded either in an empty well or around a 3 mm × 3 mm alginate square, then grown to confluence. For conditions that involved EDTA treatment, the growth media was aspirated and cells were rinsed with 1X PBS. The PBS was then aspirated and 10 mM EDTA in 1X PBS was added for 20 min. The 10 mM EDTA solution was then aspirated and the wells rinsed again with 1X PBS. Finally, 1 mL of cell media supplemented with DRAQ7 (Abcam, 1:150 dilution) was added to stain for double stranded DNA in dead cells with far-red fluorescence. Cells were imaged on a Nikon TiE epifluorescence microscope with environmental control, immediately after EDTA treatment (0 h), 1 h after treatment, and 24 h after treatment. Cells were imaged either in the center of the well or immediately adjacent to the alginate square. Total cell counts were determined from the fluorescent cell nuclei (mCherry H2B) in the red channel (610 nm), while dead cell counts were determined from the DRAQ7-labeled nuclei in the far-red channel (670 nm), detected using Bitplane Imaris (Supplemental Information, Figure S9). Cell viability was calculated as the ratio of live cells to total cells.
Collective Cell Migration Measurements
Degradable alginate structures were used to investigate how collective cell migration occurred from different initial geometries. Degradable barriers were prepared using 700 μL of the 1:3 Ba2+: Mg2++ alginate formulation, pipetted into a tissue culture-treated six well plate. Flat and convex structures were printed and washed extensively with 1X PBS to remove any uncured alginate. To minimize potential contamination, parts were sterilized immediately after printing using a combination of UV flood light exposure for 30min, and immersion in 70% EtOH for 2 min, followed by extensive washing with 1X PBS. Printed samples were stored in 1X DMEM/F12 containing 1X Pen/Strep at 4°C.
Stably-transfected mammary epithelial cells (MCF-10A) with an ER-inducible Snail response element, as well as fluorescent protein expression in the nucleus (mCherry H2B) and cytoplasm (GFP) were a gift from G. Smolen and D. Haber (Massachusetts General Hospital).91 Cells were cultured in MCF-10A growth media: DMEM/F12 HEPES buffer (Fisher 11330057) containing 5% horse serum (Fisher 16050122), 20 ng/ml Animal-Free Recombinant Human Epidermal Growth Factor (PeproTech AF-100-15), 0.5 mg/mL hydrocortisone (Sigma H0888), 100 ng/mL cholera toxin (Sigma C8052), 10 μg/mL Insulin from bovine pancreas (Sigma I1882), and 1% Penicillin-Streptomycin (Fisher MT-30-002-CI).92 Next, cells were seeded uniformly throughout the well and allowed to proliferate to confluency over 24 h. Once the cells reached confluency, the alginate was degraded using 10 mM EDTA in 1X DMEM/F12 at pH 8 for ~20 min. After degradation, the EDTA was aspirated, the well washed gently with 1X PBS, fresh growth media was added, and the cells were allowed to recover for several hours before imaging. EMT was induced using growth media containing 500 nM hydroxytamoxifen to induce Snail expression, as described.91 For the uninduced control condition, growth media was supplemented with 0.05% DMSO.
After a few hours of cell recovery from EDTA exposure, cell migration was imaged for over 48 h using a Nikon Eclipse Ti fluorescence microscope, with light-guide coupled solid state illumination system (Lumencor Spectra-X3), sCMOS camera (Andor Neo), 10x Plan Fluor objective (NA 0.3), GFP/FITC Filter Set (Chroma 49002), TRITC/DSRed Filter Set (Chroma 49004), and equipped with NIS Elements software. Environmental controls were maintained at 37°C, 5% CO2, humidified. NIS Elements was used for the cell migration experiments, taking images every 12 h under consistent exposure times, camera gain/gamma control, and aperture. Cells were detected from their fluorescent nuclei using Bitplane Imaris (Supplemental Information). Cell coordinates were then imported into MATLAB, and the tightest single-region boundary around the monolayer was determined. The monolayer migration front was then determined by excluding the bottom and sides of the boundary based on cutoffs in x and y coordinates (Figure S10). For initially straight geometries, the mean position of the migration front was calculated as the average y value of all the cells comprising the front. For initially convex geometries, the motion of the front was determined from the lowest point of the migration front. Next, outliers (“leader cells”) were manually identified based on whether they satisfied at least four of the following criteria:74 1) Leader cells were located at the apex, dragging followers in a finger-like projection, 2) Leader cells displayed ruffled lamellipodia at the leading edge, 3) Leader cells were elongated in the direction of migration, 4) Leaders were at least twice as large as their followers and 5) Follower cells displayed a compact morphology and appeared to be in physical contact with their neighbors (Figure S11). Note that single cells breaking away from the migration front without attachments to followers were not counted as leader cells. Three different individuals independently counted leader cells and their counts were averaged together for each image. Statistical significance was analyzed using the two-sample Kolmogorov-Smirnov test, which makes no assumptions about the underlying statistical distribution of data.
Results
Photopatterning of Alginate Hydrogels using Photoacid Generators and Divalent Cation Salts
Our approach for 3D stereolithographic printing of degradable alginate hydrogels utilized local illumination of photoacid generators to dissociate cation salts, which resulted in ionic crosslinking of alginate (Figure 1A) and was inspired by Raghavan et al.42 First, an aqueous precursor solution consisting of 3% (w/v) sodium alginate was prepared with varying concentrations of divalent cation salts (YCO3, where Y could be calcium, barium or magnesium), and a photoacid generator (diphenyliodonium nitrate) (Figure 1Ai). Next, ~1 mL of the precursor solution was dispensed into a Petri dish and loaded into a stereolithography printer (3D Systems SLA 250 with a 60W solid state UV laser). A focused UV laser (λ = 355 nm) was then translated in a defined sequence relative to the stage, selectively illuminating the precursor solution based on a converted CAD file (Solidworks 2016, Dassault Systems) (Figure 1B). The spatially localized illumination of photoacid generators resulted in the formation of protons (H+), which dissolved the cation salts to generate free divalent cations (Figure 1C). These free divalent cations would then bind to the G units of nearby alginate polymers to form ionic crosslinks (Figure 1Aii). Subsequently, controlled degradation could be triggered using EDTA, which chelates divalent cations and breaks the crosslinks between alginate strands (Figure 1Aiii, 1D).
Figure 1.

(A) Approach for 3D printing and degradation of alginate microstructures. (i) Precursor solutions consisting of alginate, cation salts and photoacid generators were selectively illuminated with 355 nm UV light, resulting in (ii) direct writing due to ionic crosslinking, which could undergo (iii) triggered degradation by using EDTA to chelate cations. (B) Schematic of stereolithography apparatus. (C) Photopolymerization mechanism based on UV excitation of photoacid generators to dissociate cationic salts. (D) Degradation mechanism using EDTA to chelate cations.
Alginate Degradation Kinetics are Slowed by Increasing Ca2+ and Ba2+
We hypothesized that different cation types and concentrations in precursor solution would result in varying alginate degradation kinetics. To test this hypothesis, we prepared alginate disks (10 mm diameter, 1 mm in height) with 3% (w/v) sodium alginate in 1X PBS and varying concentrations and mixtures of MgCO3, CaCO3 and BaCO3 ranging from 8 mM to 61 mM (Figure 2A, Figure S1). Once completed, the alginate structures were rinsed in deionized water to remove uncrosslinked polymer, then immersed in three degradation solutions: 1) 0.1 M EDTA at 25°C, 2) 0.1 M EDTA at 37°C or 3) 0.5 M EDTA at 37°C, all in 1X PBS. It should be noted that EDTA has limited solubility at 25°C, so degradation solutions with concentration greater than 0.1 M EDTA were not utilized. Samples were visually inspected every 5 minutes until fully degraded (Figure 2A).
Figure 2.

(A) Approach to measure degradation kinetics of printed alginate disks (10 mm diameter, 1 mm height) in EDTA, and representative images of alginate degradation over 80 min. Scale bar = 1 cm. (B) Degradation kinetics as a function of ionic composition can be classified into three regimes based on “no printing,” “fast degradation,” (Fast) and “slow degradation” (Slow).
We found that the alginate degradation kinetics could be classified into three distinct regimes based on the divalent cation concentrations in the precursor solution (Figure 2B). For ease of comparison, a molar ratio of divalent cation concentration to alginate carboxyl groups of 0.18 was defined as “1X,” and multiples of 1X concentration were evaluated. First, there existed a regime where no printing occurred, likely due to insufficient crosslinking. This occurred for precursor solutions that contained only Mg2+, as well as only Ca2+ at lower concentrations (≤ 1X). Moreover, no printing was observed for the lowest concentration of Ba2+ considered (0.5X), and the mixture of 0.9X Mg2+/0.1X [Ba2+]. Second, there existed a regime where 3D printing was successful and subsequent degradation occurred relatively rapidly (~5 min) across the temperatures and EDTA concentrations considered. Within this fast degradation regime, precursor concentrations included only Ca2+ at high concentrations ≥ 2X, as well as mixtures of Ca2+/Ba2+ (0.9X/0.1X) and mixtures of Ba2+/Mg2+, where 0.25X ≤ Ba2+ ≤ 0.9X, in which total cation concentrations were 1X. Finally, a third regime existed where 3D printing occurred but degradation occurred relatively slowly (10 – 110 min). Within this slow degradation regime, precursor solutions contained only Ba2+ at concentrations ≥ 1X, as well as mixtures of Ba2+ and Ca2+, where 0.25X ≤ Ba2+ ≤ 0.9X, with total cation concentrations of 1X. Within this third regime, alginate hydrogels containing higher concentrations of Ba2+ ≥ 2X degraded much more slowly, over 25 min or more. Moreover, alginate hydrogels degraded more rapidly at increasing temperature and EDTA concentration (Figure 2B). It should be noted that the slow degrading formulations were associated with appreciable overcuring, which is examined quantitatively in the next section. To evaluate whether the slower degradation times resulted from larger than expected disks, we measured the disk area and used this to normalize the degradation time (Figure S2). We found that this normalization of degradation times made an appreciable difference in the slow degradation regime (from 10 –100 min to 5 – 80 min), but did not qualitatively affect the overall trends, indicating that degradation kinetics were primarily governed by cation concentration. Overall, the degradation kinetics across these three regimes could be summarized by the following: 1) Alginate could not be printed with only Mg2+ as the precursor, 2) Alginate could be printed with only Ca2+ as the precursor, which resulted in hydrogels that degraded relatively rapidly, 3) Alginate could also be printed at higher divalent cation concentrations or with a mixture containing Ba2+, resulting in progressively slower degradation kinetics.
Pattern Fidelity Worsens with Increasing Ba2+ Concentration
Next, we quantified the pattern fidelity of a printed alginate structure relative to the original CAD design. To characterize the lateral resolution of this technique, a flat spiral structure was designed with a starting width of 1.25 mm, which tapered to 80 μm over two complete revolutions and a distance of 44mm (Figure 3A). This structure was patterned by translation of the UV laser (Figure 3B), then imaged in brightfield using a Celestron digital microscope (Figure 3C). This grayscale image was thresholded into a binary image, which was used to determine the actual area of the printed alginate structure (Figure 3D). The pattern fidelity was quantified as the percentage difference between the bright areas of the thresholded image with the original CAD design.
Figure 3.

(A) Initial CAD design for a flat spiral structure. (B) Schematic showing 3D printing of spiral structure. (C) Representative brightfield image of a printed spiral. (D) Corresponding feature detection with overcure (red) and undercure (green). (E) Pattern fidelities for various precursor solutions with varying ion composition and concentration (n = 6 per condition). Error bars represent standard deviation. (F) Experimental setup for the modified “windowpane” test used to calculate the working curve. (G) The working curve for 3% alginate with 1:3 Ba2+: Mg2+ used to empirically determine the critical exposure (EC) and the penetration depth (DP). Trendline R2 = 0.9984. Error bars represent standard deviation.
Spiral patterns were prepared using precursor solutions containing only Ca2+, mixtures of Ba2+/Mg2+, Ba2+/Ca2+, or only Ba2+. We found that the best pattern fidelity occurred for precursor solutions with fast degradation kinetics, particularly 2X and 4X Ca2+ as well as 1:3 Ba2+:Mg2+, with percent overcure ranging from 30–50% for 2X Ca2+ and 1:3 Ba2+:Mg2+, and ~100% for 4X Ca2+. Overall, patterned features tended to be slightly wider than the intended CAD design, likely due to the Gaussian profile of the laser as well as some diffusion of activated PAG. Moreover, the patterned features were also slightly wider than the original design towards the center of the spiral, where the tapering was slightly smaller than the beam width. In contrast, increasing concentrations of Ba2+ only, or mixed with Mg2+ or Ca2+, resulted in progressively worse overcuring of up to ~100–500%. In these latter conditions, the spiral feature was bright and highly visible, but there was also a dimmer crosslinked region within the spiral, which remained solidified after patterning was complete (Figure 3E). To account for this overcured region during image analysis, the threshold was set to a lower value so that the overcured region would be segmented as part of the patterned structure, which likely overestimates the extent of overcuring for the Ca2+ and 1:3 Ba2+: Mg2+ precursor solutions. Qualitatively similar trends were observed when minimum feature size was measured, based on the largest diameter circle that would fit at the tip of the spiral (Figure S3). For instance, 1:3 Ba2+:Mg2+ as well as 2X Ca2+ formulations yielded spirals with average minimum feature sizes of 231 μm and 434 μm, respectively. In contrast, 4X Ca2+ yielded an average minimum feature size of 1.04 mm. Overall, alginate formulations with poor pattern fidelity correlated with longer degradation times as well as stiffer mechanical properties (Figure S4)93, suggesting higher levels of crosslinking. Importantly, the 1:3 Ba2+: Mg2+ alginate solution printed readily, displayed high pattern fidelity, and degraded rapidly in response to EDTA, and was thus chosen as the optimal formulation for 3D printing.
The vertical resolution achievable with the 1:3 Ba2+:Mg2+ formulation was then characterized by patterning flat alginate layers on an inverted coverslip, using a modified “windowpane” assay (Figure 3F).8, 16 Briefly, a working curve of layer thickness (cure depth CD) as a function of light exposure EMAX was measured in order to determine a material-dependent penetration depth DP and threshold exposure EC (Figure 3G). The best-fit logarithmic regression of CD vs EMAX yielded a penetration depth DP = 0.61 mm and a threshold exposure EC = 60 J/cm2 (R2 = 0.998). For comparison, 20% PEGDA has been reported to have a slightly smaller penetration depth of DP ~ 0.2–0.3 mm and a significantly lower threshold exposure EC ~ 10–40 mJ/cm2.16 Commercial photosensitive resins such as SOMOS BioClear (DSM) have comparable penetration depths of DP ~ 0.16 mm and threshold exposure EC = 11.5 mJ/cm2.94 The larger penetration depth and greater threshold exposures for alginate photopatterning are likely due to the additional reaction step of dissociating the cation salt prior to ionic crosslinking.
Improved Pattern Fidelity Correlates with Softer Alginate Stiffness
We further characterized the mechanical properties of bulk alginate hydrogels prepared from these precursor solutions, including Ba2+/Mg2+, Ba2+/Ca2+ and Ca2+. For bulk measurement conditions, 3D stereolithography printing was impractical. Instead, larger sample volumes (~5 mL) were prepared and crosslinked in bulk using a UV flood lamp. We utilized a custom-built mechanical indenter that displaced a 4 mm spherical probe to a fixed depth, then measured the corresponding stress relaxation of the hydrogel (Figure S4), based on a previously published approach.93 For short times, the hydrogel could be treated as incompressible and undergoing elastic deformation using modified Hertz contact mechanics. The instantaneous load response Pi can be approximated by , where R is the radius of the spherical indenter, δ is the indentation depth, G is the shear modulus, and fP accounts for deviations from Hertz mechanics due to the substrate stiffness in thin samples. Over longer times, stress relaxation processes become more prominent, which could result from viscoelastic relaxation due to conformational changes in the polymer chains, as well as poroelastic relaxation due to the transport of water molecules away from the deformed region.
A typical force indentation curve shows the initial indentation of the spherical probe, relaxation over 2 minutes, followed by retraction (Figure 4A). Based on this measurement, the instantaneous load response Pi was extracted from the peak of this curve at approximately 5 seconds. Since the spherical radius R, indentation depth δ and hydrogel thickness d were also known, the shear modulus G could be determined (Figure 4B). For 1:3 Ba2+:Mg2+, 2X Ca2+ and 4X Ca2+, the shear moduli were roughly consistent at G ~ 1.5–2.7 kPa. In contrast, 3:1 Ba2+:Ca2+ had an approximately two-fold higher shear modulus of G ~ 6.26 kPa, also with greater variability. Finally, when 1:3 Ba2+Mg2+ was post-cured in 1X CaCl2 to add mechanical strength for 3D parts, the shear modulus increased significantly to ~ 7.9 kPa. This shear modulus could be converted to instantaneous elastic modulus by assuming a Poisson’s ratio of ν = 0.5 (Figure S4B). These results are comparable with previous reports for 3% alginate crosslinked by Ca2+, including ~4 kPa reported by LeRoux et al,37 ~3 kPa reported by West et al.39 It should be noted that Kong et al. report a higher G ~ 15 kPa, but their experiments were crosslinked at much higher Ca2+ concentrations.38 Overall, the high mechanical stiffness observed for 3:1 Ba2+:Ca2+ coincided with increased overcure (Figure 3E) and slower degradation kinetics (Figure 1B) compared to other alginate formulations mechanically characterized (Figure 4B).
Figure 4.

(A) Mechanical measurements using spherical indentation with representative force relaxation trace. Shear modulus was calculated using the instantaneous load response (red circle). (B) Box and whisker plots of shear moduli for various precursor formulations, with mean shear modulus listed for each formulation. Data sets were compared using a Student’s t-test and statistically significant differences between conditions are indicated by * (p < 0.05) and ** (p < 0.01).
Stereolithographic Patterning of Alginate Microstructures with Variable Height
The optimized alginate formulation of 1:3 Ba2+:Mg2+ was then used to photopattern microstructures with variable height. As a proof of concept, a stepped ramp was designed with steps that were 2 mm wide and 175 μm tall, which increased over nine steps for a maximum height of 1.6 mm (Figure 5A). For this microstructure, a “bottom-up” approach was used where alginate precursor solution was added one layer at a time, then photopatterned.16 This procedure mimics the use of a recoater blade in conventional stereolithography, which applies new layers of photopolymerizable resin of constant thickness.8 In principle, this latter procedure is possible with alginate, but would be extremely wasteful of precursor solution since the entire part must be submerged. Following photopatterning, the stepped ramp was washed and post-cured with 0.18M CaCl2 for 1 h to add mechanical strength. The actual feature sizes were ~250 μm in step height, width of ~5 mm, and a final height of ~1.7 mm (Figure 5BC), which are likely due to some overcuring. For comparison, the layer thickness is typically 100 μm–150 μm reported for PEG-DA13, 15, 16 or GelMA,23 which can be attributed to the more efficient photocrosslinking reaction. This printed structure geometry demonstrates the approximate vertical spatial resolution (~250 μm) that can be achieved with this technique.
Figure 5.
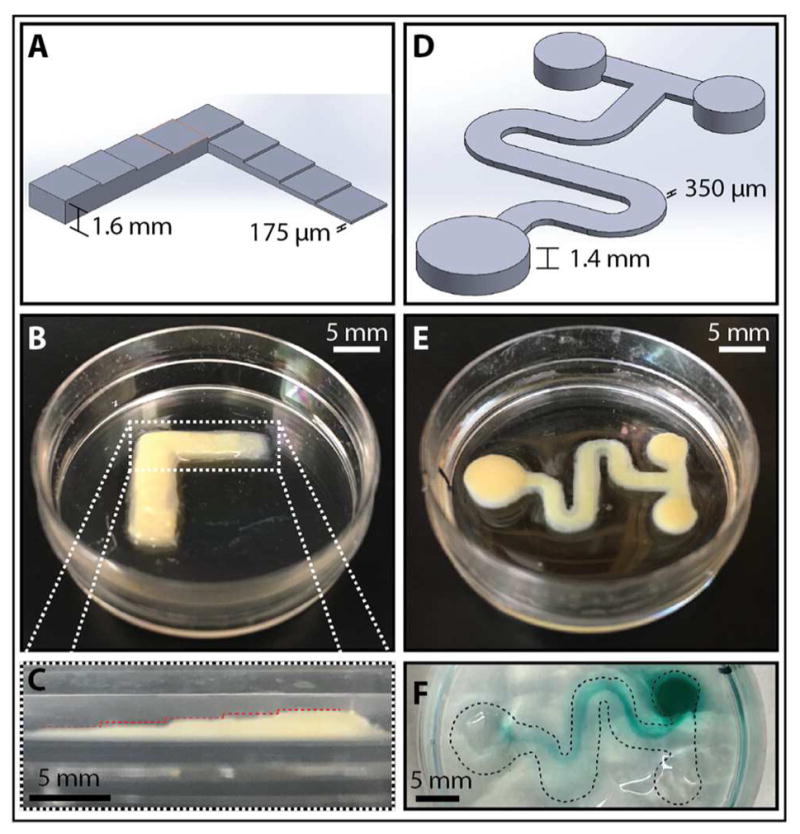
(A) CAD representation of the stepped ramp geometry starting with a height of 175 μm, increasing by 175 μm to a final height of 1.6 mm. (B) Printed alginate stepped ramp after post-curing with 1X CaCl2. (C) Cross-sectional view of the stepped ramp showing incremental height increases of ~250 μm (indicated by red lines). (D) CAD representation of a microfluidic mixer with 1.4 mm-high reservoirs and 350 μm-high mixing channel. (E) Printed microfluidic mixer showing raised reservoirs. (F) Microfluidic gravity-driven mixing of green food dye with water. Black dashed line marks the outline of the printed part.
Patterning and Degradation of Alginate Templates for Microfluidic Hydrogels
Next, alginate microstructures were printed as sacrificial templates for microfluidic channels in a second encapsulating hydrogel, incorporating features with tortuous geometries and variable heights (Figure S5). A microfluidic T-junction device was designed with a serpentine main channel that was 350 μm tall and 37 mm long. This channel was fed by inlet reservoirs that were 1.4 mm high and 4 mm in diameter, leading to an outlet reservoir that was 1.4 mm high and 6 mm in diameter (Figure 5D). This structure was again printed with the 1:3 Ba2+: Mg2++ alginate formulation, rinsed and postcured in 1X CaCl2 (Figure 5E). Subsequently, a warm solution of 1.5% agarose was added to encapsulate the main channel, while leaving the top of the inlet and outlet reservoirs exposed. After the agarose cooled to room temperature and solidified, 0.1M EDTA was added to degrade the alginate overnight. To visualize the flow within the serpentine channel, ~100 μL of diluted green food dye was added to one inlet reservoir, and ~100 μL of DI water was added to the other inlet reservoir, which resulted in gravity driven flow along the channel (Figure 5F, S6). No leakage of dye was observed, indicating that the agarose adhered tightly to the Petri dish around the alginate channels. It should be noted that the direct patterning of tall reservoirs required longer write times, but is useful for softer hydrogel materials that require gentle handling to prevent tearing.
A microfluidic gradient generator was also designed with two parallel “source” and “sink” channels, each with a width of 1.5 mm, a length of 21 mm, and a separation of 2.0 mm. A first set of even channels was patterned with both source and sink channels having consistent heights of 100 μm, using the procedure described previously for the microfluidic T-junction (Figure 6A). To demonstrate the robustness of this approach, comparable devices were also prepared by encapsulation in 10% gelatin or 2% silk fibroin (Figure S7). To visualize the diffusion-driven gradient, one inlet reservoir was loaded with 0.2% Evans Blue dye, while the other inlet reservoir was loaded with DI water. Over approximately 120 min, the dye diffused from source to sink and approached a steady state gradient between the two (Figure 6B). This concentration gradient across the cross-section of the microfluidic device was further modeled in COMSOL with a time-dependent diffusion equation assuming a constant concentration of dye at the source channel, zero dye at the sink channel, and no flux conditions at the boundaries. In this geometry, the dye diffused radially outwards from the source channel, saturating at the top by 60 min (Figure 6C). Between the source and sink channel, the dye diffused laterally in the x direction through 120 min. These simulated concentration profiles at z = 500 μm (Figure 6D) are in good agreement with the experimentally measured dye intensity (Figure 6B).
Figure 6.

Comparison of gradients generated between source and sink channels of even and uneven height, which are expected to establish spatially nonuniform concentration profiles both laterally and vertically. (A) Even 100 μm source and sink channels encapsulated in 1.5% agarose. (i) CAD design, (ii) Alginate printed template. (B) Evans Blue intensity (arbitrary units) as a function of time for a gradient between source and sink channels. Note that the intensity profiles at location 1 and 2 are comparable over time. (C) COMSOL simulations of Evans Blue diffusion in 1.5% agarose between even channels. (D) Simulated concentration profiles as a function of time at a height of z = 500 μm. (E) Uneven 100–500 μm source and 100 μm sink channels encapsulated in 1.5% agarose (i) CAD design, (ii) Alginate printed template. (F) Evans Blue intensity (arbitrary units) as a function of time for a gradient between source and sink channels. Note that the intensity profile is diffusing more rapidly at location 1 relative to location 2, due to the taller source channel at location 1. (G) COMSOL simulations of Evans Blue diffusion in 1.5% agarose between even channels. (H) Simulated concentration profiles as a function of time at a height of z = 500 μm.
More complex spatial gradients can be generated using microfluidic channels with varying height. As a proof-of-concept, an uneven set of channels was designed where the source channel had a stepped height of 500 μm near the inlet, which decreased down to 100 μm near the outlet (Figure 6E, S8). In this channel geometry, the dye front diffused more rapidly from the taller (500 μm) source channel relative to the shorter (100 μm source channel) (Figure 6F). Similarly, COMSOL modeling revealed that the dye also diffused radially from the tall source channel, reaching the top of the hydrogel by 60 min (Figure 6G). Due to the no-flux boundary conditions at the top, the dye diffusion became more pronounced laterally in the x direction (Figure 6G), relative to diffusion from short channels (Figure 6C). As a consequence, the simulated concentration profile advanced more rapidly from the tall source channel (Figure 6H), in good agreement with the experimental observations (Figure 6F). Overall, the improved control over microchannel height enabled by stereolithographic patterning can facilitate more complex spatial gradients that incorporate a vertical component.
Collective Cell Migration from Defined Geometries using Degradable Alginate Barriers
Alginate hydrogels with varying geometries were printed as degradable barriers that defined initial multicellular geometries, based on the previously optimized cation concentration of 1:3 Ba2+:Mg2+. These alginate barriers defined flat or convex geometries with a width of 1 mm and a central angle of 0° or 45°, respectively. Mammary epithelial cells (MCF-10A) were then plated around the alginate barriers and grown up to a monolayer of 80–90% confluency (Figure 7A). The alginate was then gently degraded with 10 mM EDTA in DMEM/F12 media for ~10 min, which was aspirated away and replaced with cell culture media. Cell exposure to these alginate microstructures and the subsequent degradation did not result in appreciable toxicity, as verified by DRAQ7 staining (Figure S7). This MCF-10A cell line was stably transfected with an inducible Snail construct with an estrogen receptor response element, which could be triggered by treatment with 4-hydroxytamoxifen (OHT). These cells also expressed fluorescent proteins in the nucleus (H2B mCherry) and cytoplasm (GFP).
Figure 7.

(A) Schematic of the experimental design for collective cell migration from initially straight or convex geometries. Alginate structures were printed, cells seeded around the printed structures, and allowed to grow to 90% confluency. Alginate structures were then degraded using 10mM EDTA, and cells allowed to migrate into the empty space over time. Red boxes indicate an example of the field of view imaged. Black arrows indicate direction of cell migration. (B–E) Representative fluorescence microscopy images of MCF-10A cell front migration at 12 hr timepoints. (B) DMSO control from initially straight geometries, (C) OHT treatment from initially straight geometries. (D) DMSO control from initially convex geometries, (E) OHT treatment from initially convex geometries.
Collective cell migration was characterized for initial geometries consisting of flat or convex triangles, as well as for OHT treatment relative to a DMSO control, at 12 h intervals over 24 h (Figure 7B–E). Qualitatively, monolayers in flat geometries treated with OHT tended to exhibit rougher migration fronts with multicellular, finger-like strands (Figure 7C), which advanced rapidly compared to the smoother fronts observed with the DMSO control (Figure 7B). Similarly, monolayers in convex geometries displayed rougher migration fronts for OHT treatment relative to DMSO controls, but advanced at comparable speeds (Figure 7D, E). For all conditions, the advancing monolayer fronts fully occupied the empty regions by 36 h – 48 h. Thus, cell migration and cell viability (Figure S9) appear to be minimally affected by the presence and degradation of printed alginate microstructures, indicative of good biocompatibility.
A preliminary analysis of the collective front motion reveals both curvature and EMT dependent behaviors. For instance, the initially straight migration front in the DMSO control conditions typically advanced ~500 μm in 24 h (Figure S10A), while the corresponding migration fronts after OHT treatment typically advanced farther by ~50 μm or more (Figure S10B), a statistically significant difference for initially straight geometries (Figure S10E). Instead, the initially convex fronts for the DMSO control and OHT treatment advanced by roughly the same distance of ~550 μm, based on the lowest point of the monolayer (Figure S10C, S10D). Moreover, collective fronts after OHT treatment exhibited more “leader cells” than DMSO controls (Figure S10F), as defined by multicellular, finger-like strands with a number of follower cells.74 These leader cells tended to be larger than their followers and elongated in the direction of migration, with ruffled lamellipodia at the leading edge (Figure S11). These complex and emergent behaviors suggest that mechanical cues from the initial geometry can affect molecular signaling and migration phenotype in epithelial monolayers, which will be explored more systematically in forthcoming work.
Discussion
In this article, we show for the first time that stereolithographic printing can be utilized to pattern hydrogels based on noncovalent (ionic) interactions. Alginate is advantageous since gelation and degradation can be controlled with divalent cations under relatively mild and biocompatible conditions (i.e. physiological temperature and pH).26 This ionic crosslinking occurs through the binding of divalent cations to guluronic acid (G-blocks), forming an “egg-box” conformation. Previous work has revealed subtle differences in the gelation, degradation and affinity of alginate for different cations. For instance, the concentration of divalent cations required for alginate gelation increased as Ba < Ca < Mg.30, 31 Indeed, monovalent cations generally do not induce gelation,26 while Mg2+ gels relatively slowly and weakly.32 Similar trends were observed for the degradation of alginate gels after EDTA treatment, with stability decreasing as Ba > Ca > Mg.35 It should be noted that divalent cations may also display some affinity for different blocks, with Ba2+ binding preferentially to mannuronic acid (M-blocks), and Ca2+ binding preferentially to alternating monomer regions (MG-blocks).36 Thus, alginate gelation and dissociation depends not only on divalent cation type, but also alginate monomer sequence. In this work, we investigated photopatterned alginate gels crosslinked with varying mixtures of Mg2+, Ca2+, and Ba2+. In agreement with previous reports, we show minimal alginate gelation with only Mg2+, as well as progressively slower degradation kinetics with increasing concentrations of Ca2+and Ba2+. Moreover, we find that mixtures of cations enable additional control over pattern fidelity, mechanical stiffness and degradation. Specifically, the addition of Ca2+ or Ba2+ results in stiffer gels, increased overcure and slower degradation. This result is consistent with the decreased concentration of Ca2+ or Ba2+ ions required for gelation,30, 31 as well as their affinity for EDTA.32 Unexpectedly, we found that alginates prepared with 1:3 Ba2+: Mg2+ were relatively soft, with excellent pattern fidelity and fast degradation kinetics. One possible explanation for this result is that the Mg2+ drives a “salting out” effect, which decreases the concentration of Ba2+ needed for gel formation. Qualitatively similar behaviors have been previously observed for the competition of Na+ and Ca2+ in promoting alginate gelation.30 Nevertheless, further molecular characterization will be needed to elucidate how mixtures of divalent cations govern alginate gelation and degradation.
Photopatterning approaches enable good spatial resolution under ambient conditions, which is governed by the optical properties of the precursor solution.9, 10 Using our experimental apparatus, we show that the spatial resolution is ~100 μm and appears to be limited by overcure. Several strategies may be employed to achieve finer spatial resolution and decrease overcure, based on a semi-empirical model of photopolymerization. In particular, the thickness of a photopolymerized layer is defined by CD = DP ln (E/EC), where DP is the penetration depth of the polymer, actual light exposure E, and threshold light exposure EC, based on the Beer-Lambert equation.7 It should be noted that the actual exposure E must exceed the threshold exposure EC in order to achieve sufficient mechanical strength. Nevertheless, one strategy to reduce overcure would be to decrease solidified layer thickness CD by decreasing the penetration depth DP. This could occur by adding compounds that adsorb UV light, including more photoacid generators or non-reactive dyes. Moreover, the cured linewidth for a scanned Gaussian laser is given by LW = B(CD/2DP)1/2, where B is the laser spot diameter for a Gaussian laser. An alternative strategy for finer features would be to alter the laser spot diameter using improved optics, digital micro-mirror systems, or two-photon polymerization.10 Finally, a selection of other divalent cation salts with decreased solubility may increase the reaction efficiency and permit lower exposures E. Future work will seek to optimize these crosslinking mechanisms, which we expect to be widely applicable for a variety of charged polyelectrolytes.
One advantage of 3D printing techniques is the ease of patterning microstructures with varying heights. We have demonstrated stereolithographic patterning of alginate hydrogels with multi-level architectures, including a ramp with nine steps, as well as templates for microfluidic channels connected with much taller reservoirs. Comparable microstructures with complicated vertical profiles are certainly possible with conventional photolithographic techniques, but are quite laborious in practice. We have further utilized multi-height structures to template microfluidic channels for complex spatial gradients within a second encapsulating hydrogel. An exciting future direction is to directly connect experimental and computational modeling by using the same digital design to prototype a physical structure, which could also serve as the basis for finite element modeling. One consideration is that the alginate hydrogels demonstrated here are relatively soft, with shear moduli of 1–5 kPa. As a consequence, these soft materials can deform or collapse under their weight, a common issue with 3D printing of hydrogels.4 We have shown that these hydrogels can be subsequently strengthened by a “post-cure” treatment with CaCl2, analogous to the post-curing required for the stereolithography of synthetic resins.8 Moreover, additional mechanical reinforcement may be possible by adding particles or fibers to form a composite hydrogel.6
Previous observations of collective cell migration into unoccupied regions (e.g. “wound-healing assays”) have also revealed emergent, geometry-dependent behaviors.77, 78, 80–82, 86–88 For instance, cells at the migration front may be “pulled” by supramolecular actomyosin cables (e.g. purse-string mechanism),88 which act to straighten out curved migration fronts. This mechanism is qualitatively consistent with our experimental observations, since monolayers with an initially convex geometry will be “pulled” forward faster than monolayers starting from an initially flat geometry. We further observe that EMT induction results in the emergence of leader cells and finger-like patterns of multicellular strands at a collective front. Classically, EMT has been associated with pathways that downregulate cell-cell adhesions (particularly E-cadherin), promoting cell scattering and dissemination.89 Nevertheless, it should be noted that leader cell formation is a relatively rare event, with only a few cells observed per condition. Further work is necessary to quantitatively elucidate this complex interplay of spatial geometry, biomechanical signaling and EMT.
Overall, stereolithographic 3D printing of ionically-crosslinked alginate hydrogels enables a wide range of biomaterials with spatial and temporal complexity. We have demonstrated this approach for patterning hydrogels with variable height profiles, which served to template microfluidic channels within a second encapsulating material. Moreover, these structures were used to investigate collective cell migration, since the removal of alginate barriers is relatively gentle relative to conventional “scratch” assays that can damage confluent cell monolayers and the underlying substrate.68, 69 Alginate is also advantageous since it can be manipulated under ambient conditions and is relatively bioinert,26 which can be controlled using chemical functionalization.27 The use of ionic crosslinking enables 3D biomaterials with reversible and “self-healing” properties,96 which could be used to dynamically tune hydrogel stiffness in order to mechanically interface with living cells and tissues.5 This stereolithographic printing approach could also be utilized for compartmentalized alginate architectures with tunable degradation kinetics for staged release of bioactive factors.26 Finally, these microfluidic hydrogels could be utilized for vascularized 3D microtissues that incorporate multiple cell types with controlled molecular gradients, flows, and forces.1 Further optimization of the spatial resolution, mechanical properties, and degradation kinetics of this 3D printing technique may permit a range of additional applications in biofabrication and biologically-inspired microdevices.
Conclusions
In this article, we show that 3D alginate hydrogels can be stereolithographically patterned through the use of photoacid generators and insoluble salts. We found that the pattern fidelity, mechanical stiffness and degradation kinetics of these alginate hydrogels can be tuned by the composition and concentration of divalent cations. In particular, alginate prepared with low concentrations of Ba2+ (~4 mM) or higher concentrations of Ca2+ (~30–60 mM) displayed the least overcure, relatively soft mechanical properties and fast degradation kinetics. An optimized precursor formulation was utilized to print a variety of alginate microstructures with variable height. As a proof-of-concept, we show that sacrificial alginate templates can be encapsulated within a second hydrogel to pattern microfluidic channels. Moreover, we show that degradable alginate barriers with varying geometries can direct collective cell migration. Remarkably, epithelial monolayers with initially convex geometries advanced faster than those with initially flat geometries. The induction of the EMT master regulator Snail resulted in roughening fronts with multicellular strands with further migration for initially flat geometries, but no significant effect for convex geometries. This gentle and controlled approach for templating cell monolayers may enable new insights into the interplay of initial geometry and cell migration. Moreover, we envision that this light-assisted direct write approach of noncovalent interactions will be broadly applicable for microphysiological systems, tissue engineering scaffolds, drug delivery systems, and other digital prototyping applications in biology and medicine.
Supplementary Material
Acknowledgments
We thank J. Jamieson (3D Systems) for his invaluable assistance in the setup and troubleshooting of the SLA 250 system, A. Landauer and C. Franck for assistance with hydrogel mechanical testing, G.A. Smolen and D.A. Haber for the gift of the MCF-10A cell line with inducible Snail construct, and F. Cui for assistance with COMSOL modeling. This work was supported by the Department of Education (GAANN Training Grant in Applications and Implications of Nanotechnology, P200A150037), the National Institutes of Health (T32 Training Grant in Environmental Pathology, 5T32ES007272-24), the Hibbitt Postdoctoral Fellowship, a Rhode Island Foundation (Medical Research Grant, 20144137), the COBRE Center for Cancer Research Development at Rhode Island Hospital (1P30GM110759-01A1), as well as Brown University (Karen T. Romer Undergraduate Teaching and Research Award, OVPR Research Seed Award, and Faculty Start-Up Funds).
References
- 1.Kinstlinger IS, Miller JS. Lab Chip. 2016;16:2025–2043. doi: 10.1039/c6lc00193a. [DOI] [PubMed] [Google Scholar]
- 2.Bhattacharjee N, Urrios A, Kang S, Folch A. Lab Chip. 2016;16:1720–1742. doi: 10.1039/c6lc00163g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bajaj P, Schweller RM, Khademhosseini A, West JL, Bashir R. Annu Rev Biomed Eng. 2014;16:247–276. doi: 10.1146/annurev-bioeng-071813-105155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Skardal A, Atala A. Ann Biomed Eng. 2015;43:730–746. doi: 10.1007/s10439-014-1207-1. [DOI] [PubMed] [Google Scholar]
- 5.Burdick JA, Murphy WL. Nat Commun. 2012;3:1269. doi: 10.1038/ncomms2271. [DOI] [PubMed] [Google Scholar]
- 6.Zhang YS, Khademhosseini A. Science. 2017;356:eaaf3627. doi: 10.1126/science.aaf3627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rybtchinski B. ACS nano. 2011;5:6791–6818. doi: 10.1021/nn2025397. [DOI] [PubMed] [Google Scholar]
- 8.Jacobs PF, Reid DT. Rapid prototyping & manufacturing: fundamentals of stereolithography. 1. Society of Manufacturing Engineers in cooperation with the Computer and Automated Systems Association of SME; Dearborn, MI: 1992. [Google Scholar]
- 9.Melchels FPW, Feijen J, Grijpma DW. Biomaterials. 2010;31:6121–6130. doi: 10.1016/j.biomaterials.2010.04.050. [DOI] [PubMed] [Google Scholar]
- 10.Hribar KC, Soman P, Warner J, Chung P, Chen S. Lab Chip. 2013 doi: 10.1039/c3lc50634g. [DOI] [PubMed] [Google Scholar]
- 11.Nguyen KT, West JL. Biomaterials. 2002;23:4307–4314. doi: 10.1016/s0142-9612(02)00175-8. [DOI] [PubMed] [Google Scholar]
- 12.Lantada AD, Morgado PL. Annu Rev Biomed Eng. 2012;14:73–96. doi: 10.1146/annurev-bioeng-071811-150112. [DOI] [PubMed] [Google Scholar]
- 13.Dhariwala B, Hunt E, Boland T. Tissue Eng. 2004;10:1316–1322. doi: 10.1089/ten.2004.10.1316. [DOI] [PubMed] [Google Scholar]
- 14.Hahn MS, Miller JS, West JL. Adv Mater. 2006;18:2679–2684. [Google Scholar]
- 15.Arcaute K, Mann BK, Wicker RB. Ann Biomed Eng. 2006;34:1429–1441. doi: 10.1007/s10439-006-9156-y. [DOI] [PubMed] [Google Scholar]
- 16.Chan V, Zorlutuna P, Jeong JH, Kong H, Bashir R. Lab Chip. 2010;10:2062. doi: 10.1039/c004285d. [DOI] [PubMed] [Google Scholar]
- 17.Seck TM, Melchels FPW, Feijen J, Grijpma DW. J Control Release. 2010;148:34–41. doi: 10.1016/j.jconrel.2010.07.111. [DOI] [PubMed] [Google Scholar]
- 18.Zhang AP, Qu X, Soman P, Hribar KC, Lee JW, Chen S, He S. Adv Mater. 2012;24:4266–4270. doi: 10.1002/adma.201202024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lin H, Zhang D, Alexander PG, Yang G, Tan J, Cheng AWM, Tuan RS. Biomaterials. 2013;34:331–339. doi: 10.1016/j.biomaterials.2012.09.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rogers CI, Qaderi K, Woolley AT, Nordin GP. Biomicrofluidics. 2015;9:016501. doi: 10.1063/1.4905840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Raman R, Bhaduri B, Mir M, Shkumatov A, Lee MK, Popescu G, Kong H, Bashir R. Adv Healthc Mater. 2015;5:610–619. doi: 10.1002/adhm.201500721. [DOI] [PubMed] [Google Scholar]
- 22.Urrios A, Parra-Cabrera C, Bhattacharjee N, Gonzalez-Suarez AM, Rigat-Brugarolas LG, Nallapatti U, Samitier J, DeForest CA, Posas F, Garcia-Cordero JL, Folch A. Lab Chip. 2016;16:2287–2294. doi: 10.1039/c6lc00153j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gauvin R, Chen YC, Lee JW, Soman P, Zorlutuna P, Nichol JW, Bae H, Chen S, Khademhosseini A. Biomaterials. 2012;33:3824–3834. doi: 10.1016/j.biomaterials.2012.01.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Seidlits SK, Schmidt CE, Shear JB. Adv Funct Mater. 2009;19:3543–3551. [Google Scholar]
- 25.Wylie RG, Ahsan S, Aizawa Y, Maxwell KL, Morshead CM, Shoichet MS. Nature Mat. 2011;10:799–806. doi: 10.1038/nmat3101. [DOI] [PubMed] [Google Scholar]
- 26.Wee S, Gombotz WR. Adv Drug Deliv Rev. 1998;31:267–285. doi: 10.1016/s0169-409x(97)00124-5. [DOI] [PubMed] [Google Scholar]
- 27.Augst AD, Kong HJ, Mooney DJ. Macromol Biosci. 2006;6:623–633. doi: 10.1002/mabi.200600069. [DOI] [PubMed] [Google Scholar]
- 28.Kuo CK, Ma PX. Biomaterials. 2001;22:511–521. doi: 10.1016/s0142-9612(00)00201-5. [DOI] [PubMed] [Google Scholar]
- 29.Zimmermann H, Wählisch F, Baier C, Westhoff M, Reuss R, Zimmermann D, Behringer M, Ehrhart F, Katsen-Globa A, Giese C. Biomaterials. 2007;28:1327–1345. doi: 10.1016/j.biomaterials.2006.11.032. [DOI] [PubMed] [Google Scholar]
- 30.Haug A, Smidsrød O. Acta Chem Scand. 1965;19:329–340. [Google Scholar]
- 31.Haug A, Smidsrød O. Acta Chem Scand. 1965;19:341–351. [Google Scholar]
- 32.Topuz F, Henke A, Richtering W, Groll J. Soft Matter. 2012;8:4877–4875. [Google Scholar]
- 33.Plouffe BD, Brown MA, Iyer RK, Radisic M, Murthy SK. Lab Chip. 2009;9:1507–1510. doi: 10.1039/b823523f. [DOI] [PubMed] [Google Scholar]
- 34.Delaney JT, Liberski AR, Perelaer J, Schubert US. Soft Matter. 2010;6:866–864. [Google Scholar]
- 35.Schweiger RG. Kolloid-Z. 1964;196:47–53. [Google Scholar]
- 36.Mørch ÝA, Donati I, Strand BL, Skjåk-Braek G. Biomacromolecules. 2006;7:1471–1480. doi: 10.1021/bm060010d. [DOI] [PubMed] [Google Scholar]
- 37.LeRoux MA, Guilak F, Setton LA. J Biomed Mater Res. 1999;47:46–53. doi: 10.1002/(sici)1097-4636(199910)47:1<46::aid-jbm6>3.0.co;2-n. [DOI] [PubMed] [Google Scholar]
- 38.Kong HJ, Lee KY, Mooney DJ. Polymer. 2002;43:6239–6246. [Google Scholar]
- 39.West ER, Xu M, Woodruff TK, Shea LD. Biomaterials. 2007;28:4439–4448. doi: 10.1016/j.biomaterials.2007.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rouillard AD, Berglund CM, Lee JY, Polacheck WJ, Tsui Y, Bonassar LJ, Kirby BJ. Tissue Eng Part C Methods. 2011;17:173–179. doi: 10.1089/ten.TEC.2009.0582. [DOI] [PubMed] [Google Scholar]
- 41.Jeon O, Bouhadir KH, Mansour JM, Alsberg E. Biomaterials. 2009;30:2724–2734. doi: 10.1016/j.biomaterials.2009.01.034. [DOI] [PubMed] [Google Scholar]
- 42.Javvaji V, Baradwaj AG, Payne GF, Raghavan SR. Langmuir. 2011;27:12591–12596. doi: 10.1021/la201860s. [DOI] [PubMed] [Google Scholar]
- 43.Cui J, Wang M, Zheng Y, Rodriguez Muniz GM, del Campo A. Biomacromolecules. 2013;14:1251–1256. doi: 10.1021/bm400022h. [DOI] [PubMed] [Google Scholar]
- 44.Chueh BH, Zheng Y, Torisawa YS, Hsiao AY, Ge C, Hsiong S, Huebsch N, Franceschi R, Mooney DJ, Takayama S. Biomed Microdevices. 2010;12:145–151. doi: 10.1007/s10544-009-9369-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Stowers RS, Allen SC, Suggs LJ. Proc Natl Acad Sci U S A. 2015;112:1953–1958. doi: 10.1073/pnas.1421897112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Khalil S, Sun W. J Biomech Eng. 2009;131:111002–111008. doi: 10.1115/1.3128729. [DOI] [PubMed] [Google Scholar]
- 47.Duan B, Hockaday LA, Kang KH, Butcher JT. J Biomed Mater Res. 2012;101A:1255–1264. doi: 10.1002/jbm.a.34420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mannoor MS, Jiang Z, James T, Kong YL, Malatesta KA, Soboyejo WO, Verma N, Gracias DH, McAlpine MC. Nano Lett. 2013;13:2634–2639. doi: 10.1021/nl4007744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Dubbin K, Hori Y, Lewis KK, Heilshorn SC. Adv Healthc Mater. 2016;5:2488–2492. doi: 10.1002/adhm.201600636. [DOI] [PubMed] [Google Scholar]
- 50.Boland T, Tao X, Damon BJ, Manley B, Kesari P, Jalota S, Bhaduri S. Mat Sci Eng C. 2007;27:372–376. [Google Scholar]
- 51.Beebe DJ, Mensing GA, Walker GM. Annu Rev Biomed Eng. 2002;4:261–286. doi: 10.1146/annurev.bioeng.4.112601.125916. [DOI] [PubMed] [Google Scholar]
- 52.Cheng SY, Heilman S, Wasserman M, Archer S, Shuler ML, Wu M. Lab Chip. 2007;7:763–769. doi: 10.1039/b618463d. [DOI] [PubMed] [Google Scholar]
- 53.Abhyankar VV, Toepke MW, Cortesio CL, Lokuta MA, Huttenlocher A, Beebe DJ. Lab Chip. 2008;8:1507–1515. doi: 10.1039/b803533d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mosadegh B, Huang C, Park JW, Shin HS, Chung BG, Hwang SK, Lee KH, Kim HJ, Brody J, Jeon NL. Langmuir. 2007;23:10910–10912. doi: 10.1021/la7026835. [DOI] [PubMed] [Google Scholar]
- 55.Chung S, Sudo R, Mack PJ, Wan CR, Vickerman V, Kamm RD. Lab Chip. 2009;9:269–275. doi: 10.1039/b807585a. [DOI] [PubMed] [Google Scholar]
- 56.Wu W, DeConinck A, Lewis JA. Adv Mater. 2011;23:H178–H183. doi: 10.1002/adma.201004625. [DOI] [PubMed] [Google Scholar]
- 57.Zhao L, Lee VK, Yoo SS, Dai G, Intes X. Biomaterials. 2012;33:5325–5332. doi: 10.1016/j.biomaterials.2012.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Miller JS, Stevens KR, Yang MT, Baker BM, Nguyen DHT, Cohen DM, Toro E, Chen AA, Galie PA, Yu X, Chaturvedi R, Bhatia SN, Chen CS. Nature Mat. 2012;11:768–774. doi: 10.1038/nmat3357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Bertassoni LE, Cecconi M, Manoharan V, Nikkhah M, Hjortnaes J, Cristino AL, Barabaschi G, Demarchi D, Dokmeci MR, Yang Y, Khademhosseini A. Lab Chip. 2014;14:2202–2211. doi: 10.1039/c4lc00030g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zhang Y, Yu Y, Chen H, Ozbolat IT. Biofabrication. 2013;5:025004–025011. doi: 10.1088/1758-5082/5/2/025004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Luo Y, Lode A, Gelinsky M. Adv Healthc Mater. 2013;2:777–783. doi: 10.1002/adhm.201200303. [DOI] [PubMed] [Google Scholar]
- 62.Kyle S, Jessop ZM, Al-Sabah A, Whitaker IS. Adv Healthc Mater. 2017 doi: 10.1002/adhm.201700264. [DOI] [PubMed] [Google Scholar]
- 63.Kloxin AM, Kasko AM, Salinas CN, Anseth KS. Science. 2009;324:59–63. doi: 10.1126/science.1169494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Chrobak KM, Potter DR, Tien J. Microvasc Res. 2006;71:185–196. doi: 10.1016/j.mvr.2006.02.005. [DOI] [PubMed] [Google Scholar]
- 65.Cabodi M, Choi NW, Gleghorn JP, Lee CSD, Bonassar LJ, Stroock AD. J Am Chem Soc. 2005;127:13788–13789. doi: 10.1021/ja054820t. [DOI] [PubMed] [Google Scholar]
- 66.Bettinger CJ, Cyr KM, Matsumoto A, Langer R, Borenstein JT, Kaplan DL. Adv Mater. 2007;19:2847–2850. doi: 10.1002/adma.200602487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Haessler U, Kalinin Y, Swartz MA, Wu M. Biomed Microdevices. 2009;11:827–835. doi: 10.1007/s10544-009-9299-3. [DOI] [PubMed] [Google Scholar]
- 68.Van Horssen R, ten Hagen TL. J Cell Physiol. 2011;226:288–290. doi: 10.1002/jcp.22330. [DOI] [PubMed] [Google Scholar]
- 69.Ashby WJ, Zijlstra A. Integr Biol (Camb) 2012;4:1338–1350. doi: 10.1039/c2ib20154b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Carter SB. Exp Cell Res. 1967;48:189–193. doi: 10.1016/0014-4827(67)90298-4. [DOI] [PubMed] [Google Scholar]
- 71.Guo Q, Wang X, Tibbitt MW, Anseth KS, Montell DJ, Elisseeff JH. Biomaterials. 2012;33:8040–8046. doi: 10.1016/j.biomaterials.2012.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Hoffmann JC, West JL. Integr Biol (Camb) 2013;5:817–827. doi: 10.1039/c3ib20280a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.van Horssen R, Galjart N, Rens JA, Eggermont AM, ten Hagen TL. J Cell Biochem. 2006;99:1536–1552. doi: 10.1002/jcb.20994. [DOI] [PubMed] [Google Scholar]
- 74.Poujade M, Grasland-Mongrain E, Hertzog A, Jouanneau J, Chavrier P, Ladoux B, Buguin A, Silberzan P. Proc Natl Acad Sci U S A. 2007;104:15988–15993. doi: 10.1073/pnas.0705062104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.DeForest CA, Anseth KS. Nat Chem. 2011;3:925–931. doi: 10.1038/nchem.1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Ng MR, Besser A, Danuser G, Brugge JS. J Cell Biol. 2012;199:545–563. doi: 10.1083/jcb.201207148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Anon E, Serra-Picamal X, Hersen P, Gauthier NC, Sheetz MP, Trepat X, Ladoux B. Proc Natl Acad Sci U S A. 2012;109:10891–10896. doi: 10.1073/pnas.1117814109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Rolli CG, Nakayama H, Yamaguchi K, Spatz JP, Kemkemer R, Nakanishi J. Biomaterials. 2012;33:2409–2418. doi: 10.1016/j.biomaterials.2011.12.012. [DOI] [PubMed] [Google Scholar]
- 79.Serra-Picamal X, Conte V, Vincent R, Anon E, Tambe DT, Bazellieres E, Butler JP, Fredberg JJ, Trepat X. Nat Phys. 2012;8:628–U666. [Google Scholar]
- 80.Vedula SR, Leong MC, Lai TL, Hersen P, Kabla AJ, Lim CT, Ladoux B. Proc Natl Acad Sci U S A. 2012;109:12974–12979. doi: 10.1073/pnas.1119313109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Salierno MJ, García AJ, del Campo A. Adv Funct Mater. 2013;23:5974–5980. [Google Scholar]
- 82.Kim JH, Serra-Picamal X, Tambe DT, Zhou EH, Park CY, Sadati M, Park JA, Krishnan R, Gweon B, Millet E, Butler JP, Trepat X, Fredberg JJ. Nat Mater. 2013;12:856–863. doi: 10.1038/nmat3689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Weiger MC, Vedham V, Stuelten CH, Shou K, Herrera M, Sato M, Losert W, Parent CA. PLoS One. 2013;8:e58859. doi: 10.1371/journal.pone.0058859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Reffay M, Parrini MC, Cochet-Escartin O, Ladoux B, Buguin A, Coscoy S, Amblard F, Camonis J, Silberzan P. Nat Cell Biol. 2014;16:217. doi: 10.1038/ncb2917. [DOI] [PubMed] [Google Scholar]
- 85.Streichan SJ, Hoerner CR, Schneidt T, Holzer D, Hufnagel L. Proc Natl Acad Sci U S A. 2014;111:5586–5591. doi: 10.1073/pnas.1323016111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Ravasio A, Cheddadi I, Chen T, Pereira T, Ong HT, Bertocchi C, Brugues A, Jacinto A, Kabla AJ, Toyama Y, Trepat X, Gov N, Neves de Almeida L, Ladoux B. Nat Commun. 2015;6:7683. doi: 10.1038/ncomms8683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Kollimada SA, Kulkarni AH, Ravan A, Gundiah N. PLoS One. 2016;11:e0153471. doi: 10.1371/journal.pone.0153471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Klarlund JK. Proc Natl Acad Sci USA. 2012;109:15799–15804. doi: 10.1073/pnas.1210992109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Christofori G. Nature. 2006;441:444–450. doi: 10.1038/nature04872. [DOI] [PubMed] [Google Scholar]
- 90.Wong IY, Javaid S, Wong EA, Perk S, Haber DA, Toner M, Irimia D. Nature Mat. 2014;13:1063–1071. doi: 10.1038/nmat4062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Javaid S, Zhang J, Anderssen E, Black JC, Wittner BS, Tajima K, Ting DT, Smolen GA, Zubrowski M, Desai R, Maheswaran S, Ramaswamy S, Whetstine JR, Haber DA. Cell Rep. 2013;5:1679–1689. doi: 10.1016/j.celrep.2013.11.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Debnath J, Muthuswamy SK, Brugge JS. Methods. 2003;30:256–268. doi: 10.1016/s1046-2023(03)00032-x. [DOI] [PubMed] [Google Scholar]
- 93.Chan EP, Hu Y, Johnson PM, Suo Z, Stafford CM. Soft Matter. 2012;8:1492–1498. [Google Scholar]
- 94. [accessed June 7, 2017];Somos BioClear DataSheet. https://www.dsm.com/content/dam/dsm/somos/en_US/documents/Brand-Status-Product-Datasheets/US English - A4/Somos BioClear Datasheet - US English A4.pdf.
- 95.Levick JR, Michel CC. Q J Exp Physiol Cogn Med Sci. 1973;58:67–85. doi: 10.1113/expphysiol.1973.sp002192. [DOI] [PubMed] [Google Scholar]
- 96.Huebsch N, Kearney CJ, Zhao X, Kim J, Cezar CA, Suo Z, Mooney DJ. Proc Natl Acad Sci USA. 2014;111:9762–9767. doi: 10.1073/pnas.1405469111. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.