

Abstract
In DNA, 2′-deoxyguanosine (dG) is susceptible to oxidative modification by reactive oxygen species (ROS) yielding many products, one of which is 8-oxo-7,8-dihydro-2′-deoxyguanosine (dOG). Interestingly, dOG is stable but much more labile toward oxidation than dG, furnishing 5-guanidinohydantoin-2′-deoxyribose (dGh) that is favored in the duplex context or spiroiminodihydantoin-2′-deoxyribose (dSp) that is favored in the oxidation of single-stranded contexts. Previously, exposure of DNA to ionizing radiation found ~50% of the dOG exists as a tandem lesion with an adjacent formamide site. The present work explored oxidation of dOG in a tandem lesion with a THF abasic site analog (F) that models the formamide on either the 5′ or 3′ side. When dOG was in a tandem lesion, both dGh and dSp were observed as oxidation products. The 5′ versus 3′ side in which F resided influenced the stereochemistry of the dSp formed. Further, tandem lesions with dOG were found to be up to two orders of magnitude more reactive to oxidation than dOG in an intact duplex. When dOG is in a tandem lesion it is up to fivefold more prone to formation of spermine cross-links during oxidation compared to dOG in an intact duplex. Lastly, dOG, dGh, and each dSp diastereomer were synthesized as part of a tandem lesion in a duplex DNA to establish that dOG tandem lesions decrease the thermal stability by 12–13 °C, while dGh or either dSp diastereomer in a tandem lesion decrease the stability by >20 °C. The biological consequences of these results are discussed.
Graphical Abstract
8-Oxo-2′-deoxyguanosine in a tandem lesion context is two orders of magnitude more susceptible to oxidation than in a context of native DNA yielding hydantoin products.
Introduction
Cellular DNA is susceptible to oxidative modification by reactive oxygen species (ROS).1 In DNA, 2′-deoxyguanosine (dG) nucleotides are the most electron rich resulting in these sites being labile toward oxidation by ROS.2 Cellular studies suggest oxidative modification of dG in gene coding regions leads to mutations while modification of telomeres results in attrition, both of which are observed in stress-induced diseases.3–5 Alternatively, oxidative modification of dG in regulatory regions, such as gene promoters, may provide a signaling mechanism for cells to respond to ROS.6 Our molecular understanding of these cellular phenomena has been achieved by in vitro studies with model systems that provide details of oxidative changes to DNA. Furthermore, synthesis of oxidatively modified sites into model DNA strands allows the study of the structural impact and of how enzymes process these sites.
Oxidation of dG in model systems has led to the discovery of many oxidation pathways yielding end products. Typical oxidants used for oxidation of dG to determine the products include HO· generated by ionizing radiation or Fenton chemistry,7–11 CO3·-,12,13 Type I or II photooxidants,14–16 Cr(VI),17 or HOCl.18 Two-electron oxidation of dG yields 8-oxo-7,8-dihydro-2′-deoxyguanosine (dOG) and 5-carboxamido-5-formamido-2-iminohydantoin-2′-deoxyribose (d2Ih; Scheme 1).10 Four-electron oxidation of dG yields 2,5-diaminoimidazolone-2′-deoxyribose (dIz) that readily hydrolyzes to 2,2,4-triamino-2H-oxazol-5-one-2′-deoxyribose (dZ),19 while spiroiminodihydantoin-2′-deoxyribose (dSp) or 5-guanidinohydantoin-2′-deoxyribose (dGh) result from both direct four-electron oxidation of dG or two-electron oxidation of dOG (Scheme 1).20–22 Additional products observed during oxidation of dG include damage to the 2-deoxyribose sugar resulting in release of the guanine heterocycle, creation of a carbon-carbon covalent bond between the 5′ and 8 positions yielding 5′,8-cyclo-dG,23,24 or hydrolysis of the imidazole ring of dG initiated by oxidation has been reported to furnish 2,6-diamino-4-hydroxy-5-formamidopyrimidine-2′-deoxyribose (Fapy-dG; Scheme 1).25 Comparative studies have noted that the yields of these products are dependent on the nature of the oxidant, the reaction context, and the presence of reductants that model cellular glutathione concentrations.10,11,15,26 The utilization of liquid chromatography and high-accuracy mass spectrometry (LC-MS) can identify dG oxidation products from cellular DNA samples after nuclease and phosphatase digestion.4,27 The products dOG,4,7,27–29 dZ,28 dSp,4,29 dGh,4 and 5′,8-cyclo-dG30 have been detected and quantified by LC-MS methods from biological samples. Lastly, Fapy-dG has been detected in genomic DNA by gas-chromatography and high-accuracy mass spectrometry (GC-MS) following a harsh chemical derivatization protocol,25 in which the protocol has been demonstrated to generate dG lesions as artifacts.31 In cellular samples, the yields of dOG are almost always greatest, and therefore, this product is generally regarded as a biomarker of oxidative stress.27
Scheme 1.

Pathways and products observed from dG oxidation by ROS.
In vitro oxidation of model oligodeoxynucleotides (ODNs) or extracted genomic DNA with ionizing radiation or iron-Fenton chemistry under aerobic conditions have found a significant amount of dOG formed as part of a tandem lesion (i.e., two damaged sites adjacent to one another). The Box,32 Cadet,33 Greenberg,34 and Ravanat35 laboratories found that dOG tandem lesions are formed under aerobic conditions by initial HO· oxidation of a pyrimidine 5′ or 3′ to a dG nucleotide to yield dOG and a formamide-pyrimidine remnant (Scheme 2A). The HO·-oxidized pyrimidine reacts with O2 to generate a pyrimidine-peroxyl radical that subsequently reacts with C8 of dG to yield dOG, and the oxidized pyrimidine decomposes through multiple steps to yield a formamide remnant. The Ravanat laboratory exposed calf thymus DNA under aerobic conditions to HO· to find ~50% of dOG is formed as a tandem lesion with an adjacent formamide remnant;35 further, these tandem lesions are refractory to DNA repair resulting in the possibility that they may persist in DNA.35
Scheme 2.

(A) Pathway for formation of dOG as a tandem lesion with a formamide lesion, (B) and structure of dOG in a tandem lesion with a THF (F) abasic site analog.
The reduction potential of dOG is 0.55 V lower than that of dG causing a nearly 13 kcal/mol favorability in oxidation at dOG compared to dG;36 moreover, the formamide adjacent to dOG likely renders this site more solvent exposed causing dOG to be even more prone to oxidation by diffusible oxidants. How dOG reacts in the context of a tandem lesion has not been studied. The hydantoins dGh and dSp are formed by oxidation of dOG via the oxidants HO·,37 CO3·-,12,38,39 ·NO2,40 Na2IrCl6,22,39 K2IrBr6,39 K3Fe(CN)6,39 CoCl2/KHSO5,38 chromate complexes,26 and photooxidants when H2O is the nucleophile.20,41,42 The dGh lesion dominates in reactions conducted at pH < 5.7,22,39 or in dsODNs with dOG base paired with dC;26,39 in contrast, dSp yields dominate in reactions with pH > 5.7,39,42 in ssODN,26,39 and in G-quadruplex contexts.15 The mechanistic details to understand the pH dependency in the hydantoin yields were studied and established by density functional theory (DFT) to identify that a key intermediate leading to dGh or dSp has a titratable site that determines the reaction course.43 The hydantoins dGh and dSp are each diastereotopic. The dGh diastereomers readily interconvert and can further isomerize to the constitutional isomer iminoallantoin-2′-deoxyribose (dIa) at pH > 8.2 in DNA (Scheme 1),20,44 while the dSp diastereomers are stable, readily resolved by HPLC, and the absolute configurations for them are known.45–47 These previous works provide background knowledge for the current studies.
In the present work, dOG was synthesized into a model dsODN as part of a tandem lesion to interrogate how oxidation in this context impacts the hydantoin product yields when H2O is the nucleophile. Cellular DNA is surrounded by other potential nucleophiles that can compete with H2O to form adducts to the DNA heterocycles during oxidation reactions.48–50 One such nucleophile is spermine (Spm), and therefore, the yields of Spm adducts to dOG in the tandem lesion context were also measured after an oxidation reaction. The relative reactivity of dOG as part of a tandem lesion was evaluated and compared to values previously measured for dOG oxidation in an intact duplex. Lastly, thermal stabilities (i.e., Tm values) for dOG, dGh, or each dSp diastereomer as part of a tandem lesion in a model dsODN were measured.
Results and Discussion
A custom solid-phase synthesis protocol for ODNs with dOG adjacent to a formamide adduct was reported; however, the formamide was found to anomerize during ODN synthesis.51 In the present work, the formamide was modeled by a THF abasic site analog (F) for which a commercially available phosphoramidite is available that allows efficient solid-phase synthesis of highly pure ODNs with dOG and F tandem lesions (Scheme 2B). The formamide group is a poor nucleophile, and therefore, will not be competitive with H2O or Spm when dOG is oxidized to an electrophilic intermediate in the reactions reported. The dsODNs studied had dOG base paired with dC and an F 5′ to dOG (5′-F dsODN) or 3′ to dOG (3′-F dsODN; dOG = O). The complementary strand to dOG was synthesized with tails of a single nucleotide on both ends that allowed denaturing HPLC separation of the two strands followed by direct analysis of the products from dOG oxidation (ESI Fig. 1). The oxidation products of dOG in a ssODN context were readily separated by ion-exchange HPLC (ESI Fig. 1).39 By directly monitoring the oxidation products in the intact strand, we can more confidently quantify the yields, in contrast to nuclease digestion of the strands to nucleosides for analysis that can lead to incomplete digestion impacting the products quantified.
| 5′-F dsODN | 5′-TC | ATG | GGT | FOT | CGG | TAT | A-3′ |
| 3′-TAG | TAC | CCA | GCA | GCC | ATA | TT-5′ | |
| 3′-F dsODN | 5′-TC | ATG | GGT | COF | CGG | TAT | A-3′ |
| 3′-TAG | TAC | CCA | GCA | GCC | ATA | TT-5′ | |
| d(OG:C) dsODN | 5′-TC | ATG | GGT | COT | CGG | TAT | A-3′ |
| 3′-TAG | TAC | CCA | GCA | GCC | ATA | TT-5′ |
In the first set of studies, the dsODNs were allowed to react with the one-electron oxidant Na2IrCl6 (Ir(IV)) that is selective for oxidation of dOG.52 Oxidations were conducted in 20 mM NaPi (pH 7) buffer with 100 mM NaCl at 37 °C, 10 μM dsODN (dOG), and 100 μM Ir(IV) to achieve ~75% conversion of dOG to product on the basis of integration of the HPLC peak areas (ESI Fig. 1). As discussed below, the dsODNs studied with tandem lesions of dOG had Tm values > 54 °C and should be sufficiently stable at the reaction temperature studied. Comparisons were made to an intact dsODN without a tandem lesion (d(OG:C) dsODN) that was previously studied in our laboratory via the same approach.39 When the intact duplex d(OG:C) dsODN was oxidized, dGh was exclusively observed (Fig. 1A). In contrast, oxidation of 5′-F dsODN or 3′-F dsODN led to both dGh and dSp as oxidation products in roughly a 3:1 yield (Fig. 1A). To determine how important flexibility around the dOG site was on the hydantoin yield, the oxidations were repeated at 4 °C with 500 mM NaCl to decrease the flexibility of the tandem lesion (Fig. 1B). The new oxidations with 5′-F dsODN or 3′-F dsODN found an insignificant increase in the dGh yield. To further test the influence of F on the yields of dOG oxidation, additional studies were conducted with F in the complementary strand. The F modifications were placed 5′, across, or 3′ to the dOG, and oxidation of these new duplexes all provided dGh:dSp yields of nearly 3:1 (ESI Fig. 2). These results suggest local flexibility around dOG caused by F, and not the hole exposing the π face of dOG, allow both hydantoins to form during oxidation reactions. This observation is consistent with the favorability of dSp formation at pH 7 in contexts that are less sterically demanding than intact duplexes, such as ssODNs.26,39
Figure 1.
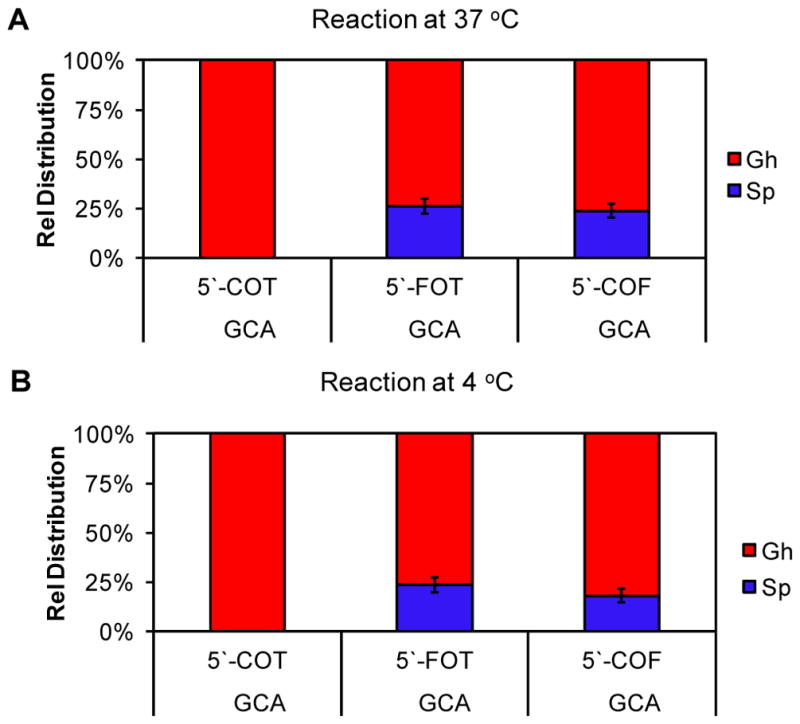
Product distributions observed when dOG was oxidized in the context of a tandem lesion. (A) Products observed with reactions in 20 mM NaPi and 100 mM NaCl at 37 °C, (B) and products observed with reactions in 20 mM NaPi and 500 mM NaCl at 4 °C. F = THF abasic site mimic; O = dOG
Previous mechanistic proposals (Scheme 3) allow comparisons to be made to address dSp and dGh formation when dOG was oxidized in the context of a tandem lesion.20,36,38,43 One-electron oxidation of dOG initially yields dOG·+ with a pKa of ~6.6.36 The mildly acidic dOG·+ can lose a proton to yield dOG·; a mixture of the radical cation and the neutral radical would exist at pH 7. Trapping of dOG·+ by nucleophilic H2O and loss of a second electron and proton yields the intermediate 5-HO-dOG. Alternatively, dOG· can be oxidized by a second electron to yield the electrophilic-quininoid compound dOGox. Trapping of dOGox by nucleophilic H2O also yields 5-HO-dOG. Low-temperature NMR analysis has confirmed the existence of the 5-HO-dOG intermediate.53 Next, protonation at N1 of 5-HO-dOG facilitates acid-catalyzed amide-bond hydrolysis, decarboxylation, and tautomerization to yield dGh; or, if N1 of 5-HO-dOG remains unprotonated, a 1,2-acyl shift yields the thermodynamically favorable dSp product. The mechanistic details proposed are supported by DFT calculations.43 In our previous work, we proposed that when dOG was base paired with dC, the reaction trajectory favored dGh because the dC base pair assisted in protonating the 5-HO-dOG intermediate at N1 to yield dGh when the duplex was intact.39 When dOG is more solvent exposed in the tandem lesion context, base pairing with dC must be compromised and minimizes trapping of the proton at N1 of 5-HO-dOG allowing dSp to form. The yield of dSp was not exclusive likely because the spirocyclic rings are sterically demanding and not favored during oxidations in duplex contexts, even if a tandem lesion was present.
Scheme 3.

Detailed mechanism proposed for oxidation of dOG to dGh and dSp.
The stereochemistry of dSp is chemically rationalized by the re versus si face reaction trajectory of H2O attack on dOG·+ or dOGox.45 When dOG is base paired with dC, the si face resides on the 5′ side and the re face resides on the 3′ side (Scheme 4), and therefore, H2O attack from the 5′ side would yield (S)-dSp and attack from the 3′ side would yield (R)-dSp (Scheme 4). Analysis of the products from the oxidation of the ODNs was achieved using a Dionex PA100 ion-exchange HPLC column that allows separation of the dSp-diastereomer containing ODNs; additionally, the absolute configuration for dSp in each peak was previously established on this HPLC column.45 This knowledge allows us to determine if having F either 5′ or 3′ to dOG during oxidation impacts the dSp diastereomer ratio by directing the side from which H2O attacks. Oxidation of the 5′-F dsODN or 3′-F dsODN at 37 °C led to the R versus S ratio of dSp to be nearly 1:1 (Fig. 2A). When the dSp diastereomer ratios were assessed for oxidations that occurred at 4 °C in 500 mM aqueous NaCl, ratios measured were consistent with the reaction trajectory analysis described. When the 5′-F dsODN was oxidized, the R versus S ratio was 1:2 favoring the S isomer of dSp, an expected result because the si face dOG was on the exposed 5′-side (Fig. 2B); in contrast, oxidation of the 3′-F dsODN furnished an R versus S isomer ratio of 2:1 that favors the R isomer, and again this was expected because the re face of dOG was solvent exposed on the 3′ side (Fig. 2B). When the duplex was stabilized by low temperature and high salt, the chiral reaction context influenced the stereochemical outcome for the formation of dSp. To support that it was the hole created by F flanking dOG that drove the stereoselectivity of dSp, the isomer ratio for the duplexes with F in the complementary strand were determined. The dSp diastereomer ratios for these duplexes were all nearly 1:1 (ESI Fig. 3), supporting the hole adjacent to dOG during oxidation directed H2O attack. In the cellular context, tandem lesions may be more stable than these ODN studies leading to a greater difference in the isomer ratio of dSp when dOG is oxidized in the context of a tandem lesion.
Scheme 4.

Attack trajectory of H2O on oxidized dOG determines the stereochemistry of dSp. (A) The re vs. si faces of dOG when base paired with dC in a duplex. (B) Attack trajectory of H2O on oxidized dOG determines the stereochemistry of the intermediate 5-HO-dOG that undergoes stereospecific 1,2-acyl shift to dSp.
Figure 2.
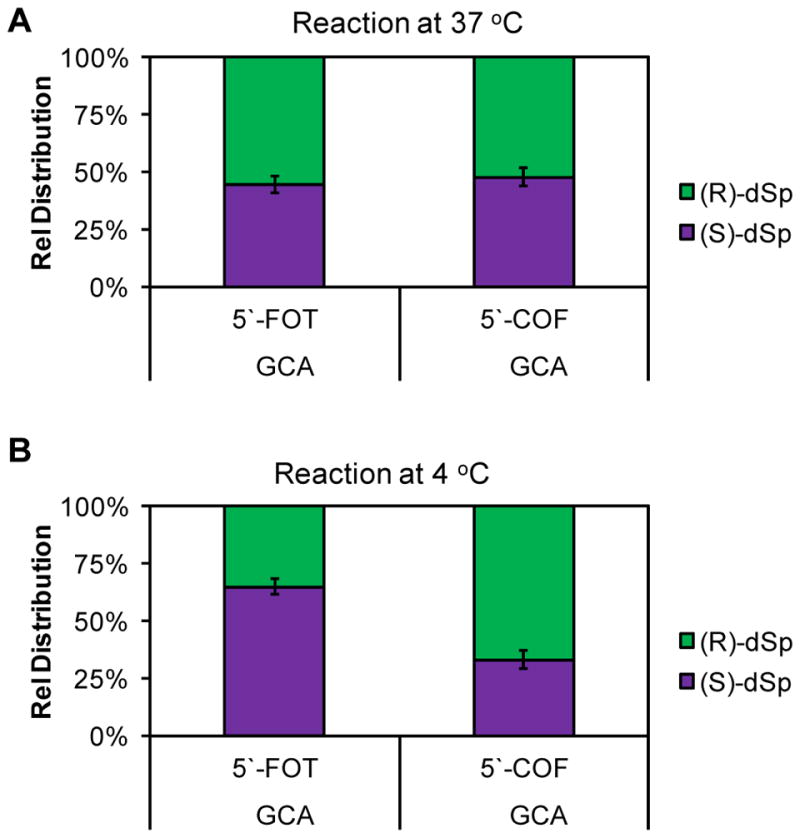
Ratios of dSp diastereomers observed when dOG was oxidized in the context of a tandem lesion. (A) The dSp ratios observed for oxidations in 20 mM NaPi at pH 7 and 100 mM NaCl at 37 °C, (B) and the dSp ratios observed for oxidations in 20 mM NaPi at pH 7 and 500 mM NaCl at 4 °C.
In the next set of studies, the relative reactivity of dOG in the context of a tandem lesion was measured in comparison to our previous results for dOG in the contexts of a ssODN, or an intact dsODN with dOG opposite dC.39 Ranking of the relative reactivities for dOG in the dsODN context was achieved using a dsODN with two dOGs (dsODN1 or dsODN2) that competed a d(OG:C) base pair in an intact region of the duplex versus a d(OG:C) base pair in a tandem lesion context. The dsODN with the two dOGs was 5′-32P labeled and then oxidized followed by piperidine treatment under conditions that yield nearly quantitative cleavage of dSp and dGh and negligible cleavage at dOG.54 The piperidine cleavage yields at the two sites were determined by denaturing polyacrylamide gel electrophoresis (PAGE) followed by quantification via storage-phosphor autoradiography. Further, the reactions were conducted under single-hit reaction conditions (<30% yield) to ensure only one reaction per strand occurred (ESI Fig. 4). Care was taken to place the more reactive dOG tandem lesion site closer to the 3′ side of the strand to avoid overestimating the reactivity. The Tm values for dsODN1 and dsODN2 were >45 °C; however, to ensure the duplexes were as stable as possible, the reactions were conducted at 4 °C with 500 mM NaCl present.
| dsODN1 | 5′-TTG | AGC | COT | CAG | ATG | TFO | TCG | CTG |
| 3′-AAC | TCG | GCA | GTC | TAC | TGC | AGC | GAC | |
| dsODN2 | 5′-TTG | AGC | COT | CAG | ATG | TCO | FCG | CTG |
| 3′-AAC | TCG | GCA | GTC | TAC | TGC | AGC | GAC |
Studies that competed dOG sites for Ir(IV) oxidation found that when dOG was base paired with dC in a tandem lesion it was tenfold more reactive toward oxidation than dOG in an intact context of the duplex (Fig. 3). Further, the relative reactivity of dOG in the context of a tandem lesion was similar in value to the relative increase in dOG reactivity previously observed in single-stranded contexts.39 Next, the dOG sites were competed for oxidation by the weaker one-electron oxidant K3Fe(CN)6 that is also selective for dOG oxidation.52 The weaker oxidant was found to favor oxidation of dOG in a tandem lesion by two orders of magnitude compared to dOG in an intact context (Fig. 3). The relative reactivity of dOG in the tandem lesion was not significantly influenced by the 5′ versus 3′ side in which the F resided (Fig. 3). If the observation of Ravanat and co-workers that ~50% of dOG is formed as part of a tandem lesion upon exposure to ionizing radiation holds in cells,35 then dOG in a tandem lesion would be up to two orders of magnitude more reactive toward oxidation by diffusible oxidants. When cellular DNA is exposed to ionizing radiation or iron-Fenton oxidative conditions, the present results suggest hydantoins may exist predominantly as tandem lesions. A predominance of hydantoins in tandem lesions could be problematic for quantitative studies of cellular DNA. The standard approach for quantification of modified DNA bases requires nuclease and phosphatase digestion of genomic DNA followed by LC-MS analysis.27 The reason for this concern is that hydantoins in oligodeoxynucleotides are poorly digested,55 and it is expected when dSp or dGh in tandem lesions would be even more poorly converted to nucleosides for LC-MS analysis. The low yields of dSp and dGh in vivo have been challenging enough to quantify,4,29 and the possibility that their digestion yields from DNA are not quantitative would lead to underestimation of their concentrations.
Figure 3.

Ranking of relative reactivities of dOG toward oxidation in the contexts studied. The reactions were conducted in 20 mM NaPi at pH 7 with 500 mM NaCl and 4 °C. Red = abasic site
Oxidation of dG or dOG yields electrophilic intermediates that are also trapped by available primary amine nucleophiles.48–50,56–59 The polyamine spermine (Spm) is associated with genomic DNA and has a cellular abundance in low millimolar concentrations.60 The next experiments quantified the yields of Spm adducts to oxidized dOG in the context of dsODNs that had intact flanking base pairs or dOG in a tandem lesion. Previously, our laboratory found dOG reacts with Spm to yield an initial adduct with a half-life of 12 h at 37 °C that decomposes to a 2′-deoxyribosylurea lesion (Fig. 4A).48 Therefore, to quantify the Spm adduct yield, the strand with dOG in the d(OG:C) dsODN, 5′-F dsODN, or 3′-F dsODN contexts was 5′-32P labeled to allow PAGE analysis directly after oxidation before the adduct decomposes. When Spm is adducted to DNA, the migration rate of the DNA during a gel electrophoresis experiment decreases to provide a direct readout of the yield using storage-phosphor autoradiography (Fig. 4B). The reactions were conducted with 10 μM dsODN (i.e., dOG) and 0, 10, 50, or 100 μM Spm present in 20 mM NaPi pH 7, 100 mM NaCl at 37 °C, and 120 μM Ir(IV). The adduct yields determined by PAGE analysis increased from 10 to 50 μM Spm and did not change significantly when going from 50 to 100 μM Spm. The Spm adduct yields reported in Figure 4C were determined with 100 μM Spm present during the oxidation. The Spm adduct yield was ~5% for the d(OG:C) dsODN context when dOG was flanked by canonical base pairs. When oxidation of dOG with Spm present was conducted with the 5′-F dsODN or 3′-F dsODN duplexes, the Spm adduct yield increased fivefold and fourfold, respectively, relative to the d(OG:C) dsODN context. On the basis of these results, when dOG is more solvent exposed in a tandem lesion, the ability to form cross-links to Spm during oxidative conditions increases significantly.
Figure 4.

Spermine adducts to dOG during an oxidation reaction form more favorably when dOG is more accessible in a tandem lesion. (A) Initial and final products of Spm addition to oxidized dOG. (B) Example PAGE for analyzing Spm adduct formation to dOG during an oxidation reaction. (C) Yields of Spm adducts to dOG. O = dOG
In the final study, 18-mer duplexes similar to those used in the product distribution experiments described above were synthesized with dOG, dGh, (R)-dSp, or (S)-dSp (Fig. 5 and ESI Fig. 5). The duplexes were intact or had an abasic site tandem lesion (i.e., F) on either the 5′ or 3′ side for measurement of the Tm values. The Tm values were determined on 5 μM duplex in 20 mM NaPi pH 7 with 100 mM NaCl. A dC was placed opposite dG or its oxidation products, as this would be the base pairing scheme most likely recognized by DNA repair proteins. The parent duplex had a Tm value of 67.1 ± 0.5 °C that decreased to 66.8 ± 0.6 °C when dOG replaced the centrally located dG of interest (Fig. 5). The negligible change in Tm value observed with dOG compared to dG in the position is consistent with literature reports.61,62 The Tm values were systematically evaluated to understand the progression of thermal stability decrease as the duplex was modified to have dGh or the dSp isomers synthetically incorporated in an intact duplex or as part of an abasic site tandem lesion. The first example described has dGh introduced into the duplex. The Tm value with only dGh decreased by 14 °C (Fig. 5 left side). A similar drop in Tm value was measured with an F introduced on either side of a d(OG:C) base pair (12–13 °C, Fig. 5 left side). Next, when dGh was in a tandem lesion with an F on the 5′ side, another 7 °C in stability was lost, and when the F was on the 3′ side of dGh the Tm value decreased by another 5 °C. Overall, the duplex with a 5′ F to dGh had a 21 °C lower Tm value than the parent duplex, and when dGh had an F on the 3′ side the Tm value decreased by 19 °C compared to the parent duplex. The interesting observation was that the introduction of either dGh or an F alone caused the greatest decrease in Tm value (12–14 °C; Fig. 5 left side), and addition of the second lesion resulted in a smaller overall decrease to the Tm value (5–7 °C; Fig. 5 left side). The changes in Tm values for a single dGh, F, or dOG, or dOG in tandem with an abasic site are similar in magnitude to literature reports.62–65 The present studies demonstrate that dGh in tandem lesions with an abasic site are highly duplex destabilizing (Tm decrease of >19 °C).
Figure 5.

Plot of Tm values measured for dOG, dGh, and the dSp diastereomers in the context of an intact dsODN or in the context of an abasic site tandem lesion (F) on either the 5′ or 3′ side of the oxidized dG nucleotide. Each reported value was measure in triplicate to obtain an estimated error of <1 °C for each of the values reported.
Synthesis of either dSp diastereomer into the duplex in tandem with an adjacent F found a few interesting features. First, the presence of either isomer of dSp caused a 15 °C reduction in Tm value for the duplex (Fig. 5 middle and right columns). The finding that the dSp diastereomers yield similar decreases in Tm values is consistent with previous reports.61,66 When F was synthesized 5′ to either dSp diastereomer, a nearly 9 °C reduction in Tm value was observed. Overall a nearly 23 °C reduction in Tm value for either dSp diastereomer with a 5′-F tandem lesion was measured (Fig. 5 middle and right columns). When an F was synthesized on the 3′ side of either dSp diastereomer, another ~6 °C drop in Tm value was observed. The Tm studies found that duplexes with dSp and a 3′ F had ~4 °C higher Tm values than duplexes with dSp and a 5′ F. This observation suggests the dSp isomers are slightly better accommodated in the hole generated by the F on the 3′ side relative to the 5′ side. Computational modeling of dSp in the context of a duplex has been conducted,67 but the results of those studies did not aid in understanding the present observations. These Tm studies found the presence of tandem lesions with either dSp or dGh and a baseless site results in >19 °C reduction in thermal stability. The present observations that hydantoin-abasic site tandem lesions are highly helix distorting could aid in future studies that try to understand the preferred DNA repair pathway activated in the cellular context (i.e., base excision repair versus nucleotide excision repair) by these types of lesions.
Conclusions
Oxidative modification of dG in cellular DNA occurs as a result of oxidative stress to yield many products with dOG generally existing as a major product detected.27 In vitro studies that exposed DNA to ionizing radiation under aerobic conditions found nearly 50% of dOG formed exists as a tandem lesion with a 5′ or 3′ formamide-pyrimidine remnant from the oxidation event.35 Oxidative conditions favor further oxidation of dOG to hydantoin products because dOG has a 0.55 V lower reduction potential than dG.36 The present work studied the further oxidation of dOG in the context of a tandem lesion in dsODNs. For synthetic considerations, the model systems had dOG in tandem with the THF abasic site analog (i.e., F) that is expected to generate a hole in the duplex similar to a formamide lesion. The results of these studies indicate oxidation of dOG base paired with dC in a tandem lesion with F yields dGh and dSp as oxidation products regardless of whether the F was on the 5′ or 3′ side of dOG (Fig. 1). This observation is in contrast to our previous finding that oxidation of dOG base paired with dC in an intact dsODN exclusively yields dGh.39 The presence of the hole generated by F directs the side at which nucleophilic H2O attacks the oxidized dOG to influence the stereochemical outcome of dSp (Fig. 2). Conducting competitive oxidation reactions allowed us to determine that dOG in a tandem lesion is up to two orders of magnitude more reactive toward oxidation than dOG flanked by canonical base pairs (Fig. 3). This finding suggests that a majority of hydantoin lesions in DNA may exist as tandem lesions when cells are exposed to ionizing radiation or iron-Fenton chemistry. Tandem lesions with hydantoins are anticipated to impact nuclease and phosphatase digestion of genomic DNA prior to LC-MS analysis that could lead to an underestimation of the dGh/dSp cellular levels. Finally, oxidation studies conducted with the polyamine Spm present showed that dOG in a tandem lesion provided 4–5 fold higher yields of Spm adducts in oxidation reactions (Fig. 4). The findings of these studies identify tandem lesions with dOG increase the solvent exposure at this reactive site to alter the oxidation products observed, increase the reactivity toward oxidation, and increase yields of Spm adducts.
Previous studies have demonstrated that dOG in tandem lesions are refractory to DNA repair and may persist in cells.35,68,69 Thus, dOG tandem lesion sites may be preferentially oxidized to yield dGh or dSp as tandem lesions. The hydantoins in ssODNs, dsODNs, as bulges, or in G-quadruplex contexts are excellent substrates for the NEIL family of DNA glycosylases that initiate base excision repair,70–73 in addition to being substrates for nucleotide excision repair.73,74 However, studies to determine if hydantoins as part of a tandem lesion impact the repair pathway have not been conducted. On the basis of the Tm studies in the present work (Fig. 5), when hydantoins are part of a tandem lesion they are more helix destabilizing than when they are not part of a tandem lesion. The increase in helix destabilization could dramatically influence the dominant DNA repair pathway activated for removal of these lesions, or the tandem lesion could be refractory to DNA repair as observed for dOG as part of a tandem lesion. Future studies are needed to better understand DNA repair of these lesions.
Experimental Section
Synthesis, purification, and preparation of the ODNs
The ODNs were synthesized by solid-phase synthesis using standard protocols and commercially available phosphoramidites by the DNA/Peptide core facility at the University of Utah. All ODNs were cleaved and deprotected following protocols established by the manufacturer of the phosphoramidites. The cleaved and deprotected ODNs were purified on a semi-preparative ion-exchange HPLC column running mobile phases A = 1:9 CH3CN:ddH2O and B = 1.5 M NaOAc pH 7 in 1:9 CH3CN:ddH2O with a flow rate of 3 mL/min while monitoring the ODN elution by the absorbance at 260 nm. The method was initiated at 1% B followed by a linear increase to 100% B over 30 min. The purification salts were removed by dialysis against ddH2O for 36 h at 4 °C while changing the ddH2O at least 4 times. The dialyzed ODNs were lyophilized to dryness and resuspended in ddH2O. The ODN concentrations were determined by UV-vis spectroscopy by measuring the absorbance at 260 nm to use Beer’s law to calculate the concentration. The extinction coefficients for each strand were calculated using the nearest neighbor approximation model, with dG replacing dOG and a nucleotide was omitted for F.
For the experiments requiring the dOG-containing strand to be 5′-32P labeled, the labeling reactions were conducted following literature protocols.39 Preparation of the dsODNs for the oxidation studies was achieved by mixing the purified dOG-containing strand and its complementary strand in 1:1.25 ratios, respectively, in the appropriate volume of buffer to achieve the desired dsODN concentration. The mixture was heated to 90 °C for 5 min and then slowly cooled to room temperature over 3 h. Preparation of the dsODNs for the Tm studies was achieved by mixing the purified dOG-containing strand and its complementary strand in a 1:1 ratio in the desired buffer with an appropriate volume to achieve the desired dsODN concentration followed by the same heating and cooling procedure described above.
Method for conducting the oxidation reactions to determine dOG oxidation product distributions
The dsODNs were prepared in 100 μL total volume with 10 μM dsODN in buffered solution with 20 mM NaPi pH 7 and 100 or 500 mM NaCl. The samples were thermally equilibrated at 4 or 37 °C for 30 min prior to the reaction. The reactions were commenced by a bolus addition of Na2IrCl6 to achieve a 100 μM solution of the oxidant. After allowing the reaction to progress for 30 min, the reaction was quenched by adding EDTA to a final concentration of 10 mM. The quenched reactions were then analyzed by HPLC to determine the product distributions.
The HPLC analysis for determination of the product distributions was performed by first denaturing the dsODNs via denaturing HPLC conditions to purify the oxidized strand away from the complementary strand. Next, the strand mixture with dOG, dGh, or the dSp diastereomers was resolved using a Dionex DNAPac PA100 ion-exchange HPLC column (250 × 4 mm, 5 μm). The elution profile for dOG, dGh, and dSp on this column was previously established.44,45 The denaturing HPLC was conducted on a C18 reversed-phase HPLC column with the mobile phases A = 100 mM TEAA pH 7 and 1 M urea in ddH2O, and B = 1:1 CH3CN:ddH2O with 100 mM TEAA and 1 M urea and a flow rate of 1 mL/min with the column heated to 65 °C while monitoring the elution profile by the UV absorbance at 260 nm. The method consisted of starting at 10% B followed by a linear increase to 65% B over 60 min. Because the oxidized strand is two nucleotides shorter than the complementary strand, it was baseline resolved and could be HPLC purified. The purified oxidized strands were dialyzed overnight against ddH2O followed by lyophilization to dryness and then resuspension in 100 μL of ddH2O. The resuspended sample was analyzed on the ion-exchange column with the mobile phases A = 1.5 M NaOAc pH 7 in 1:9 CH3CN:ddH2O and B = 1:9 CH3CN:ddH2O with a flow rate of 1 mL/min while monitoring the elution by the UV absorbance at 260 nm. The method was initiated at 15% B and increased linearly to 100% B over 30 min. The peak areas for the resolved dOG, dGh, and dSp isomers were integrated and normalized to determine the product distributions. Triplicate trials were conducted to determine the standard deviations from the mean that were reported as the errors in the figures.
Oxidation reactions analyzed by denaturing PAGE were conducted in 50 μL of dsODN in a buffered solution. Prior to annealing, the samples were doped with 20,000 cpm of 5′-32P labeled strand that was used for visualization by storage-phosphor autoradiography. In the assays to compete two dOGs for oxidant, the buffer consisted of 20 mM NaPi and 500 mM NaCl. Prior to the reaction, the dsODN sample (10 μM dsODN or 20 μM dOG) was thermally equilibrated at 4 °C followed by a bolus addition of 7 μM Na2IrCl6 or 5 mM K3Fe(CN)6. The reactions were allowed to progress for 30 min followed by quenching with EDTA that was added to a 10 mM final concentration. Next, the samples were dialyzed against ddH2O overnight at 4 °C before piperidine cleavage. The piperidine cleavage reactions were previously demonstrated to yield quantitative cleavage at dGh and the dSp isomers while not cleaving dOG.54 Briefly, the dialyzed samples were lyophilized to dryness and resuspended in fresh 1 M piperidine in ddH2O with 200 mM β-mercaptoethanol. The solutions were heated at 90 °C for 2 h, after which the samples were lyophilized to remove the piperidine prior to PAGE analysis. The piperidine cleaved samples were suspended in 20 μL of ddH2O with 0.025% each of bromophenol blue and xylene cyanol and then 10 μL of the sample was separated on a 20% denaturing PAGE. After electrophoresing the samples at 75 W for 2 h, the gel was placed under a storage-phosphor screen and left for 18 h prior to visualization by storage-phosphor autoradiography. The visualized bands were quantified using Imagequant software. The oxidation reactions gave single-hit conductions (<30% yield) on the basis of the gel analysis. Each reaction was conducted in triplicate to establish the standard deviation of the measurements that were reported as the errors.
For the reactions conducted to determine the amount of Spm adducts formed at dOG during oxidation, dsODNs were annealed with 20,000 cpm of 5′-32P labeled dOG-containing strand, in buffer with 20 mM NaPi pH 7 and 100 mM NaCl with a dsODN concentration of 10 μM and a volume of 50 μL. Prior to commencement of the reaction, Spm was added to a final concentration of 0, 10, 50, or 100 μM, and the samples were thermal equilibrated at 37 °C before a bolus of Na2IrCl6 was added to a final concentration of 120 μM. After allowing the reactions to progress for 30 min, they were quenched with EDTA that was added to a final concentration of 10 mM. The samples were lyophilized to dryness, and then suspended in 20 μL of ddH2O with 0.025% each of bromophenol blue and xylene cyanol. Next, 10 μL of the sample was loaded on to a 20% denaturing PAGE and electrophoresed at 75 W for 2 h. The electrophoresed gel was placed under a storage-phosphor screen for 18 h, and then the bands were visualized using storage-phosphor autoradiography and quantified using Imagequant software.
Procedure to synthesize dGh and dSp in a ssODN
The hydantoins were synthesized from the purified dOG-containing ssODNs. Synthesis of dGh was achieved by placing the dOG-containing ssODN in ddH2O to achieve a 10 μM solution with a volume of 100 μL. The sample was thermally equilibrated at 4 °C for 30 min prior to a bolus addition of Na2IrCl6 to achieve a final concentration of 120 μM. After 30 min incubation, the reaction was quenched with EDTA to obtain a final concentration of 10 mM. The dGh-containing strands were purified by ion-exchange HPLC as described above for the product analysis. Synthesis of dSp-containing ssODNs was conducted by placing the dOG-containing ssODN in a buffered solution with 20 mM NaPi at pH 8 to obtain a 100 μL solution with 10 μM ssODN. The sample was thermally equilibrated at 65 °C for 10 min followed by a bolus addition of Na2IrCl6 to obtain a final concentration of 120 μM. After 30 min incubation, the reaction was quenched by addition of EDTA to a final concentration of 10 mM. The dSp-containing ssODNs were purified by ion-exchange HPLC via the method described above for the product analysis.
Method to measure the Tm values
The dsODNs to be analyzed were annealed at 5 μM in a buffered solution of 20 mM NaPi pH 7 and 100 mM NaCl with a volume of 125 μL. Next, the annealed samples were loaded into a Tm analysis cuvette with a volume of 125 μL and thermally equilibrated at 20 °C for 5 min in a temperature controlled UV-vis spectrometer. After the thermal equilibration, the samples were heated at 1 °C per min up to 90 °C. At each 1 °C increase in temperature, the samples were thermally equilibrated for 30 s before taking an absorbance reading at 260 nm. A control reaction of only buffer was also analyzed by the same method to be used to subtract from the experimental data. The background subtracted data were plotted, and the Tm values were determined using a two-point average analysis. Each Tm curve was obtained in triplicate to determine the reported errors.
Supplementary Material
Acknowledgments
This work was supported by a National Institutes of Health grant (R01 CA090689). The oligodeoxynucleotides were provided by the DNA/Peptide core facility at the University of Utah that is supported in part by an NCI Cancer Center Support grant (P30 CA042014).
Footnotes
Electronic supplementary information (ESI) available: Outline of the analytical methods used for product analysis and ranking of relative reactivities, example HPLC, PAGE, and Tm data, chromatograms demonstrating purities for the synthesized single-stranded oligodeoxynucleotide. See DOI:XXX
References
- 1.Lonkar P, Dedon PC. Int J Cancer. 2011;128:1999–2009. doi: 10.1002/ijc.25815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Steenken S, Jovanovic SV. J Am Chem Soc. 1997;119:617–618. [Google Scholar]
- 3.O’Donovan A, Tomiyama AJ, Lin J, Puterman E, Adler NE, Kemeny M, Wolkowitz OM, Blackburn EH, Epel ES. Brain Behav Immun. 2012;26:573–579. doi: 10.1016/j.bbi.2012.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mangerich A, Knutson CG, Parry NM, Muthupalani S, Ye W, Prestwich E, Cui L, McFaline JL, Mobley M, Ge Z, Taghizadeh K, Wishnok JS, Wogan GN, Fox JG, Tannenbaum SR, Dedon PC. Proc Natl Acad Sci USA. 2012;109:E1820–E1829. doi: 10.1073/pnas.1207829109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Neeley WL, Essigmann JM. Chem Res Toxicol. 2006;19:491–505. doi: 10.1021/tx0600043. [DOI] [PubMed] [Google Scholar]
- 6.Fleming AM, Burrows CJ. DNA Repair (Amst) 2017;56:75–83. doi: 10.1016/j.dnarep.2017.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pouget JP, Douki T, Richard MJ, Cadet J. Chem Res Toxicol. 2000;13:541–549. doi: 10.1021/tx000020e. [DOI] [PubMed] [Google Scholar]
- 8.Reisz JA, Bansal N, Qian J, Zhao W, Furdui CM. Antioxid Redox Signal. 2014;21:260–292. doi: 10.1089/ars.2013.5489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lloyd RV, Hanna PM, Mason RP. Free Radic Biol Med. 1997;22:885–888. doi: 10.1016/s0891-5849(96)00432-7. [DOI] [PubMed] [Google Scholar]
- 10.Alshykhly OR, Fleming AM, Burrows CJ. J Org Chem. 2015;80:6996–7007. doi: 10.1021/acs.joc.5b00689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fleming AM, Muller JG, Ji I, Burrows CJ. Org Biomol Chem. 2011;9:3338–3348. doi: 10.1039/c1ob05112a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Crean C, Geacintov NE, Shafirovich V. Angew Chem, Int Ed. 2005;44:5057–5060. doi: 10.1002/anie.200500991. [DOI] [PubMed] [Google Scholar]
- 13.Douki T, Cadet J. Free Radic Res. 1996;24:369–380. doi: 10.3109/10715769609088035. [DOI] [PubMed] [Google Scholar]
- 14.Cadet J, Decarroz C, Wang SY, Midden WR. Israel J Chem. 1983;23:420–429. [Google Scholar]
- 15.Fleming AM, Burrows CJ. Chem Res Toxicol. 2013;26:593–607. doi: 10.1021/tx400028y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kino K, Saito I, Sugiyama H. J Am Chem Soc. 1998;120:7373–7374. [Google Scholar]
- 17.Sugden KD, Campo CK, Martin BD. Chem Res Toxicol. 2001;14:1315–1322. doi: 10.1021/tx010088+. [DOI] [PubMed] [Google Scholar]
- 18.Suzuki T, Friesen MD, Ohshima H. Chem Res Toxicol. 2003;16:382–389. doi: 10.1021/tx025638y. [DOI] [PubMed] [Google Scholar]
- 19.Cadet J, Berger M, Buchko GW, Joshi PC, Raoul S, Ravanat JL. J Am Chem Soc. 1994;116:7403–7404. [Google Scholar]
- 20.Ye Y, Muller JG, Luo W, Mayne CL, Shallop AJ, Jones RA, Burrows CJ. J Am Chem Soc. 2003;125:13926–13927. doi: 10.1021/ja0378660. [DOI] [PubMed] [Google Scholar]
- 21.Luo W, Muller JG, Rachlin EM, Burrows CJ. Org Lett. 2000;2:613–616. doi: 10.1021/ol9913643. [DOI] [PubMed] [Google Scholar]
- 22.Luo W, Muller JG, Rachlin EM, Burrows CJ. Chem Res Toxicol. 2001;14:927–938. doi: 10.1021/tx010072j. [DOI] [PubMed] [Google Scholar]
- 23.Chatgilialoglu C, Ferreri C, Terzidis MA. Chem Soc Rev. 2011;40:1368–1382. doi: 10.1039/c0cs00061b. [DOI] [PubMed] [Google Scholar]
- 24.Pogozelski WK, Tullius TD. Chem Rev. 1998;98:1089–1108. doi: 10.1021/cr960437i. [DOI] [PubMed] [Google Scholar]
- 25.Dizdaroglu M. Mutat Res Rev Mutat Res. 2015;763:212–245. doi: 10.1016/j.mrrev.2014.11.002. [DOI] [PubMed] [Google Scholar]
- 26.Gremaud JN, Martin BD, Sugden KD. Chem Res Toxicol. 2010;23:379–385. doi: 10.1021/tx900362r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Collins A. Free Radical Biol Med. 2003;34:1089–1099. doi: 10.1016/s0891-5849(03)00041-8. [DOI] [PubMed] [Google Scholar]
- 28.Matter B, Malejka-Giganti D, Csallany AS, Tretyakova N. Nucleic Acids Res. 2006;34:5449–5460. doi: 10.1093/nar/gkl596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hailer MK, Slade PG, Martin BD, Sugden KD. Chem Res Toxicol. 2005;18:1378–1383. doi: 10.1021/tx0501379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Krokidis MG, Terzidis MA, Efthimiadou E, Zervou SK, Kordas G, Papadopoulos K, Hiskia A, Kletsas D, Chatgilialoglu C. Free Rad Res. 2017;51:470–482. doi: 10.1080/10715762.2017.1325485. [DOI] [PubMed] [Google Scholar]
- 31.Cadet J, Douki T, Ravanat JL, Wagner JR. Bioanal Rev. 2012;4:55–74. [Google Scholar]
- 32.Box HC, Freund HG, Budzinski EE, Wallace JC, MacCubbin AE. Radiat Res. 1995;141:91–94. [PubMed] [Google Scholar]
- 33.Bourdat AG, Douki T, Frelon S, Gasparutto D, Cadet J. J Am Chem Soc. 2000;122:4549–4556. [Google Scholar]
- 34.San Pedro JMN, Greenberg MM. J Am Chem Soc. 2014;136:3928–3936. doi: 10.1021/ja412562p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bergeron F, Auvre R, Radicella JP, Ravanat JL. Proc Natl Acad Sci USA. 2010;107:5528–5533. doi: 10.1073/pnas.1000193107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Steenken S, Jovanovic SV, Bietti M, Bernhard K. J Am Chem Soc. 2000;122:2373–2374. [Google Scholar]
- 37.White B, Tarun MC, Gathergood N, Rusling JF, Smyth MR. Mol Biosyst. 2005;1:373–381. doi: 10.1039/b511756a. [DOI] [PubMed] [Google Scholar]
- 38.Niles JC, Wishnok JS, Tannenbaum SR. Chem Res Toxicol. 2004;17:1510–1519. doi: 10.1021/tx0400048. [DOI] [PubMed] [Google Scholar]
- 39.Fleming AM, Muller JG, Dlouhy AC, Burrows CJ. J Am Chem Soc. 2012;134:15091–15102. doi: 10.1021/ja306077b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shafirovich V, Cadet J, Gasparutto D, Dourandin A, Geacintov NE. Chem Res Toxicol. 2001;14:233–241. doi: 10.1021/tx000204t. [DOI] [PubMed] [Google Scholar]
- 41.Niles JC, Wishnok JS, Tannenbaum SR. Org Lett. 2001;3:963–966. [PubMed] [Google Scholar]
- 42.Luo W, Muller JG, Burrows CJ. Org Lett. 2001;3:2801–2804. doi: 10.1021/ol0161763. [DOI] [PubMed] [Google Scholar]
- 43.Munk BH, Burrows CJ, Schlegel HB. J Am Chem Soc. 2008;130:5245–5256. doi: 10.1021/ja7104448. [DOI] [PubMed] [Google Scholar]
- 44.Zhu J, Fleming AM, Orendt AM, Burrows CJ. J Org Chem. 2016;81:351–359. doi: 10.1021/acs.joc.5b02180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Fleming AM, Orendt AM, He Y, Zhu J, Dukor RK, Burrows CJ. J Am Chem Soc. 2013;135:18191–18204. doi: 10.1021/ja409254z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Karwowski B, Dupeyrat F, Bardet M, Ravanat JL, Krajewski P, Cadet J. Chem Res Toxicol. 2006;19:1357–1365. doi: 10.1021/tx060088f. [DOI] [PubMed] [Google Scholar]
- 47.Eckenroth BE, Fleming AM, Sweasy JB, Burrows CJ, Doublie S. Biochemistry. 2014;53:2075–2077. doi: 10.1021/bi500270e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hosford ME, Muller JG, Burrows CJ. J Am Chem Soc. 2004;126:9540–9541. doi: 10.1021/ja047981q. [DOI] [PubMed] [Google Scholar]
- 49.Perrier S, Hau J, Gasparutto D, Cadet J, Favier A, Ravanat JL. J Am Chem Soc. 2006;128:5703–5710. doi: 10.1021/ja057656i. [DOI] [PubMed] [Google Scholar]
- 50.Uvaydov Y, Geacintov NE, Shafirovich V. Phys Chem Chem Phys. 2014;16:11729–11736. doi: 10.1039/c4cp00675e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bourdat AG, Gasparutto D, Cadet J. Nucleic Acids Res. 1999;27:1015–1024. doi: 10.1093/nar/27.4.1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hickerson RP, Prat F, Muller JG, Foote CS, Burrows CJ. J Am Chem Soc. 1999;121:9423–9428. [Google Scholar]
- 53.McCallum JEB, Kuniyoshi CY, Foote CS. J Am Chem Soc. 2004;126:16777–16782. doi: 10.1021/ja030678p. [DOI] [PubMed] [Google Scholar]
- 54.Fleming AM, Alshykhly O, Zhu J, Muller JG, Burrows CJ. Chem Res Toxicol. 2015;28:1292–1300. doi: 10.1021/acs.chemrestox.5b00096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Chen X, Fleming AM, Muller JG, Burrows CJ. New J Chem. 2013;37:3440–3449. doi: 10.1039/C3NJ00418J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Xu X, Muller JG, Ye Y, Burrows CJ. J Am Chem Soc. 2008;130:703–709. doi: 10.1021/ja077102a. [DOI] [PubMed] [Google Scholar]
- 57.Xu X, Fleming AM, Muller JG, Burrows CJ. J Am Chem Soc. 2008;130:10080–10081. doi: 10.1021/ja803896d. [DOI] [PubMed] [Google Scholar]
- 58.Solivio MJ, Joy TJ, Sallans L, Merino EJ. J Inorg Biochem. 2010;104:1000–1005. [PubMed] [Google Scholar]
- 59.Silerme S, Bobyk L, Taverna-Porro M, Cuier C, Saint-Pierre C, Ravanat JL. Chem Res Toxicol. 2014;27:1011–1018. doi: 10.1021/tx500063d. [DOI] [PubMed] [Google Scholar]
- 60.Igarashi K, Kashiwagi K. Biochem Biophys Res Commun. 2000;271:559–564. doi: 10.1006/bbrc.2000.2601. [DOI] [PubMed] [Google Scholar]
- 61.Gruessner B, Dwarakanath M, Stewart E, Bae Y, Jamieson ER. Chem Res Toxicol. 2016;29:279–284. doi: 10.1021/acs.chemrestox.5b00453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Plum GE, Grollman AP, Johnson F, Breslauer KJ. Biochemistry. 2002;34:16148–16160. doi: 10.1021/bi00049a030. [DOI] [PubMed] [Google Scholar]
- 63.Kalam MA, Basu AK. Chem Res Toxicol. 2005;18:1187–1192. doi: 10.1021/tx050119r. [DOI] [PubMed] [Google Scholar]
- 64.Yennie CJ, Delaney S. Chem Res Toxicol. 2012;25:1732–1739. doi: 10.1021/tx300190a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Gelfand CA, Plum GE, Grollman AP, Johnson F, Breslauer KJ. Biochemistry. 1998;37:7321–7327. doi: 10.1021/bi9803372. [DOI] [PubMed] [Google Scholar]
- 66.Khutsishvili I, Zhang N, Marky LA, Crean C, Patel DJ, Geacintov NE, Shafirovich V. Biochemistry. 2013;52:1354–1363. doi: 10.1021/bi301566v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Jia L, Shafirovich V, Shapiro R, Geacintov NE, Broyde S. Biochemistry. 2005;44:13342–13353. doi: 10.1021/bi050790v. [DOI] [PubMed] [Google Scholar]
- 68.Cunniffe S, O’Neill P, Greenberg MM, Lomax ME. Mutat Res Fundam Mol Mech Mutagen. 2014;762:32–39. doi: 10.1016/j.mrfmmm.2014.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Pearson CG, Shikazono N, Thacker J, O’Neill P. Nucleic Acids Res. 2004;32:263–270. doi: 10.1093/nar/gkh150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Krishnamurthy N, Zhao X, Burrows CJ, David SS. Biochemistry. 2008;47:7137–7146. doi: 10.1021/bi800160s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Zhao X, Krishnamurthy N, Burrows CJ, David SS. Biochemistry. 2010;49:1658–1666. doi: 10.1021/bi901852q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Zhou J, Fleming AM, Averill AM, Burrows CJ, Wallace SS. Nucleic Acids Res. 2015;43:4039–4054. doi: 10.1093/nar/gkv252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Shafirovich V, Kropachev K, Anderson T, Liu Z, Kolbanovskiy M, Martin BD, Sugden K, Shim Y, Chen X, Min JH, Geacintov NE. J Biol Chem. 2016;291:5309–5319. doi: 10.1074/jbc.M115.693218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.McKibbin PL, Fleming AM, Towheed MA, Van Houten B, Burrows CJ, David SS. J Am Chem Soc. 2013;135:13851–13861. doi: 10.1021/ja4059469. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.