

Abstract
Tumor cell metastasis to the brain involves cell migration through biochemically and physically complex microenvironments at the blood–brain barrier (BBB). The current understanding of tumor cell migration across the BBB is limited. We hypothesize that an interplay between biochemical cues and physical cues at the BBB affects the mechanisms of brain metastasis. We found that astrocyte conditioned medium (ACM) applied directly to tumor cells increased tumor cell velocity, induced elongation, and promoted actin stress fiber organization. Notably, treatment of the extracellular matrix with ACM led to even more significant increases in tumor cell velocity in comparison with ACM treatment of cells directly. Furthermore, inhibiting matrix metalloproteinases in ACM reversed ACM’s effect on tumor cells. The effects of ACM on tumor cell morphology and migration also depended on astrocytes’ activation state. Finally, using a microfluidic device, we found that the effects of ACM were abrogated in confinement. Overall, our work demonstrates that astrocyte-secreted factors alter migration and morphology of metastatic breast tumor cells, and this effect depends on the cells’ mechanical microenvironment.—Shumakovich, M. A., Mencio, C. P., Siglin, J. S., Moriarty, R. A., Geller, H. M., Stroka, K. M. Astrocytes from the brain microenvironment alter migration and morphology of metastatic breast cancer cells.
Keywords: extracellular matrix, tumor, actin cytoskeleton, blood–brain barrier, matrix metalloproteinases
Metastasis to the brain is one of the most deadly aspects of breast cancer, leading to a particularly poor prognosis for patients (1). During metastasis, breast tumor cells break away from the primary tumor, travel through the circulatory system, preferentially infiltrate the brain, and form secondary tumors that are notoriously difficult to treat (1). During metastasis, tumor cells encounter many heterogeneous microenvironments containing an array of biochemical and physical cues (2), both of which regulate the migration, mechanobiology, and signaling mechanisms used by tumor cells to navigate these environments (3). It has been shown that astrocytes in the brain microenvironment promote brain metastasis and facilitate tumor cell invasion (4–8). We hypothesize that astrocyte-secreted biochemical cues regulate the morphology and migration of metastatic breast tumor cells and that this effect depends on the astrocyte activation state, as well as the mechanical microenvironment of the tumor cells.
In healthy physiology, astrocytes provide support for neurons by maintaining an ionic, neurotransmitter, amino acid, and water balance in the brain (9). The end-feet of astrocytes support the blood–brain barrier (BBB) by assisting with the exchange of chemical signals between the circulatory system and the brain (9) and modulating the physiology of brain endothelial cells (9, 10). Although astrocytes are important regulators of brain homeostasis, they also play a role in brain metastasis. It has been reported that astrocyte-secreted serpins are involved in tumor cell survival during metastasis across the BBB in vivo (4). Another report demonstrated an increased invasiveness of tumor cells in response to astrocyte-conditioned medium (ACM), and they attributed this response to astrocyte-secreted matrix metalloproteinases (MMPs) (5). Others yet have shown that astrocytes and tumor cells can form gap junctions, which then transport molecular messages between the 2 cell types (8). Furthermore, it has been reported that extracellular vesicles secreted by astrocytes carry fibroblast growth factor (FGF)-2 and VEGF (11), which have been shown to enhance tumor cell proliferation (12). Together, these reports suggest that astrocytes interact with metastasizing tumor cells and thus influence brain metastasis.
Although there is evidence that astrocytes are involved in tumor cell metastasis across the BBB, it is unclear whether tumor cells react to signals from quiescent astrocytes or altered molecular signals from tumor cell–affected astrocytes. Secretion of MMPs by astrocytes does not necessarily require 2-way communication between tumor cells and astrocytes (5) and thus could be a part of the 1-way paracrine signaling of astrocytes to tumor cells that we explored in this study. MMPs are secreted by various cell types including both astrocytes and tumor cells, function to degrade the extracellular matrix, and are known to promote tumor cell splitting from a primary tumor, intravasation into the vasculature, and extravasation across capillary endothelial cells and the BBB (13). Thus, MMPs secreted by astrocytes could act on breast cancer cells, or their extracellular matrix (ECM), to potentiate the metastatic phenotype.
Tumor metastasis across the BBB likely causes BBB damage, which may be similar to the BBB damage that follows traumatic brain injury. A damaged BBB permits the infiltration of fibrinogen-bound TGF-β, which then activates astrocytes (14), leading to the formation of glial scars that primarily consist of chondroitin sulfate proteoglycans (CSPGs) (14) and altering the astrocyte secretome (15). The secretome of activated astrocytes is enriched in proinflammatory cytokines and other small molecules (16). In the case of cancer, similar activation of fibroblasts supports cancer cell growth, motility, and invasion (17), which suggests that astrocyte activation may also have a role in brain cancer metastases. Thus, we hypothesized that there would be differential effects of adding conditioned medium from untreated astrocytes vs. TGF-β-activated astrocytes to tumor cells.
In addition to biochemical cues, physical cues, such as physical confinement, also affect tumor cell migration. Metastasizing tumor cells encounter confined microenvironments when migrating through microtracks within tissues, through narrow brain capillaries that can be as small as 10 µm in diameter (18), through the brain endothelium, and along the blood vessel after extravasation across the endothelium (19). Thus, understanding the effect of confinement on tumor cell metastasis across the BBB is crucial. Prior studies by our laboratory demonstrated that an aquaporin-5-based mechanism can drive the migration of metastatic breast tumor cells in confined channels (20), suggesting that tumor cell migration in a 2-dimensional (2D) environment vs. confinement uses different mechanisms. Thus, the effects of astrocyte-secreted factors on tumor cells could be different depending on the mode of migration employed by the tumor cells. Hence, we hypothesize that physical confinement alters the effects of ACM on tumor cell migration in a microfluidic model where ACM serves as a potential chemoattractant.
To test our aforementioned hypotheses regarding the effects of astrocyte-secreted factors on metastatic breast tumor cell migration and morphology, we applied ACM (from untreated or TGF-β-treated astrocytes) to tumor cells or their ECM and quantified cell migration and morphology parameters on 2D substrates or in confined microchannels. In our work, ACM increased tumor cell migration and altered their morphology toward a more elongated and larger phenotype, with enhanced actin stress fiber organization. Furthermore, pretreating collagen substrates with ACM before seeding tumor cells resulted in significantly increased tumor cell velocity, compared with that of control cells, with increases in velocity that were more drastic than when cells were treated directly with ACM. In addition, ACM affected tumor cells differently, depending on the relative activation state of the astrocytes. We concluded that MMPs are likely to be at least partially responsible for the ACM-dependent increases in tumor cell migration. The effect of astrocyte-secreted factors was contingent on the physical microenvironment of the tumor cells.
MATERIALS AND METHODS
Cell culture
Human breast adenocarcinoma highly metastatic cells (MDA-MB-231), human breast adenocarcinoma metastatic cells (MDA-MB-468), human breast adenocarcinoma tumorigenic cells (MCF7), and human mammary gland noncancerous epithelial cells (MCF10A) were purchased from American Type Culture Collection (Manassas, VA, USA). MDA-MB-231 and MCF7 cells were cultured in medium consisting of DMEM with high glucose and pyruvate (Thermo Fisher Scientific, Waltham, MA, USA), 10% fetal bovine serum (HyClone Characterized; GE Healthcare, Pittsburgh, PA, USA, or Thermo Fisher Scientific), and 1% penicillin-streptomycin 10,000 U/ml. MDA-MB-468 cells were cultured similarly, but with DMEM/F12 and HEPES (Thermo Fisher Scientific) instead of DMEM. MCF10A cells were cultured in medium consisting of DMEM/F12, HEPES, 100 µg/ml endothelial growth factor (Millipore-Sigma, St. Louis, MO, USA) in 10 mM acetic acid, 1 mg/ml hydrocortisone (Millipore-Sigma) in 95% ethanol, 4 mg/ml insulin (Thermo Fisher Scientific), 5% horse serum, New Zealand origin (Thermo Fisher Scientific), and 1% penicillin-streptomycin 10,000 U/ml. Cells were washed with PBS (VWR, Radnor, PA, USA), and detached with 0.25% trypsin-EDTA (Thermo Fisher Scientific). MDA-MB-231 and MCF7 cells were used up to a passage of 30 after purchase, and MDA-MB-468 and MCF10A cells were used up to passage 10 after purchase. All cells were cultured at 37°C, 50% humidity, and 5% CO2:95% air.
ECM proteins and coatings
To study the role of different ECM proteins on the effect of ACM on cells, we examined several ECM conditions. Cells were grown on glass coated with 300 μl of 20 μg/ml of one of the following ECM proteins: type I collagen from rat tail, type IV collagen from human cell culture, fibronectin from human plasma, laminin from human fibroblasts, or 20 μg/ml poly-d-lysine hydrobromide (PDL; stored at 1 mg/ml and dissolved in sterile MilliQ water) (all from Millipore-Sigma) for 1 h at 37°C, depending on the experiment. After the incubation, wells with collagen type I or IV, or fibronectin were washed 3 times with PBS; wells with laminin were washed with HBSS (Thermo Fisher Scientific), and wells with PDL were washed with sterile MilliQ water.
ACM
All animal care and procedures were approved by the Institutional Animal Care and Use Committee [National Institutes of Health (NIH), National Heart, Lung, and Blood Institute], and performed in accordance with the NIH Guide for the Care and Use of Laboratory Animals. Pregnant C57Bl/6 mice were procured from Charles River Laboratories (Wilmington, MA, USA). Primary cortical astrocyte cultures were prepared from neonatal (1–3 d old) C57BL/6 mouse brains (15). In short, the cerebral cortices were dissected and dissociated into a single-cell suspension. Dissociated cells were seeded into T75 flasks and grown in DMEM with high glucose and pyruvate, 10% fetal bovine serum, and 1% penicillin-streptomycin 10,000 U/ml, at 37°C and 5% CO2:95% air atmosphere until the cells grew to confluence (10–14 d later). Flasks were shaken for 20 h (120 rpm, 37°C) to detach microglia, oligodendrocytes, and neurons from the more adherent astrocytes. After shaking, the medium was replaced with fresh medium, and again, 24 h after the end of shaking. After the second complete change of medium, ACM was harvested every 48 h and frozen until used. To harvest conditioned medium from reactive astrocytes, the purified astrocytes were plated onto 6-well plates or T-25 flasks in serum-containing medium. After once again reaching confluence, the astrocytes were incubated in one of 3 conditions: 1) in serum-free medium overnight and then treated with 10 ng/ml TGF-β (Peprotech, Rocky Hill, NJ, USA) in serum-free medium for 5 d before harvest, 2) in serum-free medium overnight and treated with 10 ng/ml TGF-β in serum-containing medium for 5 d before harvest, and 3) in serum-containing medium overnight and treated with 10 ng/ml TGF-β in serum-containing medium for 5 d before harvest. Activation of astrocytes was confirmed (data not shown) in astrocytes cultured in serum-free medium overnight and then treated with TGF-β (condition 1) by Western blots for GFAP in astrocytes and CSPGs in ACM (21). The different conditioned media collected from astrocytes were centrifuged at 300 g for 10 min, and the supernatant was filtered with a 40-μm nylon cell strainer (VWR) based on a published protocol (22). The debris pellet was discarded.
MMP inhibition
To inhibit MMPs in ACM, the broad-spectrum MMP inhibitor batimastat (BB94; Millipore-Sigma) was used (23, 24). Batimastat was diluted to 50 mM in DMSO (Millipore-Sigma) and frozen in aliquots. For experiments, the 50 mM stock of batimastat was diluted to 0.1, 1, or 5 μM in ACM. DMSO was used as a vehicle control.
Addition of exogenous MMP-2 and -9
For some experiments, MMP-2 from humans (Millipore-Sigma) was diluted to 5 mg/ml in water and then diluted to 100 and 40 ng/ml in control medium (25, 26). MMP-9 from humans (Millipore-Sigma) was diluted to 50 µg/ml in PBS and then diluted to 100 and 40 ng/ml in control medium (25, 26). The ECM was pretreated with MMP-2- or -9-containing medium. To increase the number of cells for the control condition (no MMPs), control data were pooled from both vehicle controls (water and PBS), given that they were run on the same well plate and displayed no significant differences between them.
2D migration assays
Glass bottomed 24-well cell culture plates (MatTek, Ashland, MA, USA) were coated with type I collagen, type IV collagen, fibronectin, laminin, or PDL, depending on the experiment. MDA-MB-231, MDA-MB-468, MCF7, or MCF10A cells (1 × 104 cells total) were plated in each well in experiments where the cells were later treated directly with ACM, depending on the experiment. All experiments comparing cell morphology and migration on different ECM molecules were performed with MDA-MB-231 cells only. All experiments comparing the response of different cell lines to ACM were performed with a type I collagen ECM. All other experiments were performed with MDA-MB-231 cells and a type I collagen ECM. Cells were incubated at 37°C, 50% humidity, and 5% CO2:95% air overnight. The next day, medium was changed to ACM, control medium, or ACM with batimastat, TGF-β, DMSO, water, PBS, MMP-2, or MMP-9, depending on the experiment. In some experiments, 10 ng/ml of TGF-β was directly added to ACM, control medium, and serum-free medium, to compare results with the ACM from TGF-β-treated astrocytes. For ACM pretreatment of ECM protein or PDL, ACM or control medium with DMSO, water, PBS, MMP-2, or MMP-9, depending on the experiment, was added directly to the ECM and incubated overnight at 37°C, 50% humidity and 5% CO2:95% air. On the following day, the ECM was washed 3 times with PBS, HBSS, or water, depending on the ECM, and 1 × 104 MDA-MB-231, MDA-MB-468, MCF7, or MCF10A cells were plated, depending on the experiment. Cells seeded on pretreated ECM were allowed to attach for 3 h before imaging. MDA-MB-231, MDA-MB-468, MCF7, or MCF10A cells (5 × 104 cells total) were plated in the same conditions as discussed above on ECM-coated glass-bottom dishes for staining with phalloidin.
Time-lapse and phase imaging
Images were acquired on an IX83 microscope (Olympus, Center Valley, PA, USA) with a ×10 objective. To maintain the cells alive during the imaging, a chamber calibrated to 37°C, 50% humidity, and 5% CO2:95% air was used on the microscope stage. Images were taken at 5–10 min intervals. On the following day, a collection of phase-contrast images was taken with a ×20 objective.
Microfabrication
We have reported the procedure for fabricating microfluidic devices containing channels of various widths (20, 27, 28). All fabrication was carried out in the University of Maryland microfabrication facility by using photolithographic methods. In brief, a mask was designed in AutoCAD (Autodesk, San Rafael, CA, USA). Four-inch-diameter silicon wafers (University Wafer, Boston, MA, USA) were spin coated with SU-8 (negative photoresist) 3010 and 3025 in 2 layers (MicroChem, Westborough, MA, USA) for the cell seeding and confinement channels, respectively. For each layer, an EVG620 Mask Aligner (EV Group, Albany, NY, USA) was used to crosslink the photoresist with UV through the mask. The noncrosslinked photoresist was removed with SU-8 developer solution. After fabrication, the wafer surface was silanized with tridecafluoro-1,1,2,2,tetrahydrooctyl-1-trichlorosilane (97%) (Pfaltz & Bauer, Waterbury, CT, USA) overnight. This process resulted in a silicone wafer with raised channels of 50, 20, 10, 6, and 3 μm widths (data shown for 50, 10, and 3 μm widths), 200 μm length, and ∼10 μm height. Polydimethylsiloxane (PDMS) was mixed to a 10:1 ratio of elastomer base and elastomer curing agent from a Silicon Elastomer Kit (Robert McKeown Co., Branchburg, NJ, USA), degassed in a vacuum chamber for ∼1 h, and baked at 80°C until cured (∼2 h). The PDMS devices, along with 35- by 75-mm 1.5 glass coverslips, were then cleaned with ethanol, reverse osmosis-treated water, and then ethanol again. They were then treated with plasma using a plasma cleaner (Harrick Plasma, Ithaca, NY, USA) and pressed together for bonding. The fabrication process resulted in a chemotactic device with 4 inlet wells. Cells were seeded in the bottom-most well, and a chemoattractant was added to the top-most well.
Confinement assays
Microfabricated devices containing microchannels were incubated with type I rat tail collagen (20 μg/ml) for 1 h at 37°C and then washed 3 times with PBS. MDA-MB-231 cells (1 × 105 cells suspended in 25 μl of either serum-free medium or full medium, depending on the experimental condition) were then seeded into the inlet of the microfluidic device and incubated for 5–10 min to allow the cells to loosely attach. Then, the remaining medium was removed and replaced by 60–80 μl/well of either serum-containing medium in all wells of the device, or serum-free medium in all wells except the topmost one, which contained serum. In both cases, the top channel contained either serum-containing control medium or ACM. The following chemoattractant conditions were considered: serum alone (positive control, given that serum is a chemoattractant for MDA-MB-231 cells), serum-containing ACM alone, serum-containing ACM with serum as additional chemoattractant, and no chemoattractant (all wells with serum-containing control medium acted as the negative control). Two general setups were used: 1) tumor cells were seeded in serum-free medium, and serum-containing ACM or serum-containing control medium was used as a chemoattractant in the topmost channel. In some cases, cells were preincubated in ACM for 1–4 d and seeded similarly with serum-containing ACM as the chemoattractant, and 2) tumor cells were seeded in serum-containing control medium, and either serum-containing ACM or serum-containing control medium was used as the chemoattractant or negative control, respectively, in the topmost channel. The cells were then imaged live with time-lapse microscopy at 37°C, 50% humidity and 5% CO2:95% at 10-min intervals.
Cell staining
Cells were washed once with PBS and fixed with 3.7% formaldehyde (Thermo Fisher Scientific) for 10 min. After fixation, the cells were washed 3 times with PBS, permeabilized with 1% Triton X-100 (Millipore-Sigma) for 5 min at room temperature, washed again 3 times with PBS, blocked for nonspecific binding with 2.5% bovine serum albumin (BSA) (Millipore-Sigma) for at least an hour, and incubated with 1:500 Alexa Fluor 488 conjugated to phalloidin (Thermo Fisher Scientific) and 1:2500 Hoechst 33342 (Thermo Fisher Scientific) in PBS for 1 h at room temperature. After staining, the cells were washed 3 times with PBS.
Confocal microscopy
Stained samples were imaged via an LSM 710 confocal microscope (Zeiss, Oberkochen, Germany) with a ×60 oil immersion objective, or an FV3000 confocal microscope (Olympus) in resonance scanning mode with deconvolution, with the appropriate filter and a ×100 silicone immersion objective. Confocal images were reconstructed in ImageJ software (NIH; https://imagej.nih.gov) into a 3-dimensional image with a maximum-intensity projection. Intensities of corresponding control medium–ACM image pairs were adjusted equally in ImageJ, as noted in the figure caption.
Data analysis
Data were analyzed with ImageJ software. The morphology of the cells was measured by first manually tracing the outlines of cells imaged via phase-contrast microscopy with a ×20 objective, as we have described in Stroka et al. (29). In brief, the cell’s inverse aspect ratio was calculated by dividing the length of the minor axis of the cell by the length of the major axis of the cell. Cell circularity was defined as 4πA/P2, where A is the area of the cell and P is the perimeter of the cell, with both of the cell projections in phase-contrast images. Cell solidity was defined as the ratio Acell/Aconvex, where Acell is the area of the cell and Aconvex is the convex area of the cell.
Positions of cells both in 2D and in confinement were evaluated by tracking them with the Manual Tracking ImageJ plugin. The approximate centroid of each cell was tracked over 300–670 min (for 2D assays, as stated in detail in figure captions) or 15 h (for confinement assays). Five- to 10-min time intervals were used between images, as stated in the figure captions. Cells were not tracked if they divided, went out of the frame, or were significantly obstructed by an artifact or another cell. MCF7 and MCF10A cells that formed clusters were not analyzed; only single cells were selected for analyses. Migration analysis was started on the frames where cells appeared to have attached to the surface. A custom MatLab (MathWorks, Natick, MA, USA) code was used to calculate velocities of the cells using output from the ImageJ tracking. The average velocity of cells in 2D was quantified by calculating the instantaneous velocities between every set of 2 points and then averaging the individual instantaneous velocities over all time points. The mean square displacement (MSD) was reported for 500 min, and the diffusion coefficient was obtained from a Langevin-type fit (30), carried out in MatLab to the MSD curve over 200–268 min. MSD was calculated as the average of the square of the distance traveled over a sliding time average. The sliding time average was obtained by averaging the MSDs for every time interval and thus obtaining the MSD at each time point. The Langevin-type equation used was Eq. 1:
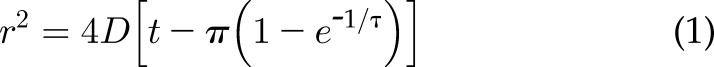 |
where D is the diffusion (or random motility) coefficient, r2 is the 2D MSD, t is time, and τ is the persistence time (30).
In confinement, speed was calculated in the same way as 2D velocity, with no consideration for whether the cell was moving toward or away from the chemoattractant. The chemotactic index (CI) was calculated by dividing the end-to-end distance of a cell from the beginning and end of tracking by the sum of the distances between all the points (Eq. 2):
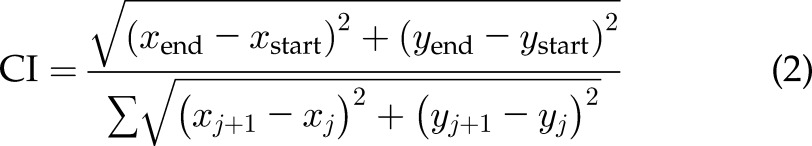 |
The chemotactic index ranges from 0 to 1, where 1 indicates that the cell is moving completely straight (i.e., persistently). To compare the increase in velocities of ACM-treated conditions when cells were treated directly and when collagen was pretreated with ACM, the percentage change was calculated by obtaining the difference between the velocities of cells in the ACM-treated conditions and the velocities of cells in the control medium–treated conditions and dividing by the reference value, which is the control medium–treated condition. This method was used in each of 3 trials for the ACM-treated cells and collagen sets of data (Eq. 3):
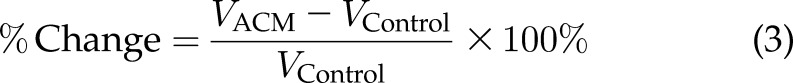 |
where V is velocity.
Statistics
Three independent trials were conducted for each experiment, unless otherwise stated in the figure captions. Cells from all 3 experiments were pooled. Normality was assumed because of the large sample size, which allows for parametric statistical analyses. For comparison of more than 2 groups of data, 1-way ANOVA was performed (GraphPad, La Jolla, CA, USA), with the Tukey post hoc test for multiple comparisons. A 2-sided Welch’s t test was used to compare means of 2-category results in GraphPad. A frequency analysis in GraphPad was used to generate histograms. A 95% confidence interval with P < 0.05 was used as the significance cutoff for all statistical tests. Cumulative distribution functions were tested for statistical significance with the 2-sample Kolmogorov-Smirnov test, with P < 0.05 indicating significantly different distributions.
RESULTS
ACM alters metastatic breast tumor cell morphology and migration
Astrocyte-secreted factors can reportedly enhance tumor cell invasion and metastatic potential (5), although the details of these effects are poorly understood. To corroborate previous reports regarding ACM-induced cell migration and identify whether these effects are accompanied by changes in tumor cell morphology, MDA-MB-231 cells were seeded on type I collagen–coated glass, allowed to attach overnight, and on the following day, culture medium was replaced with ACM and incubated overnight (∼16 h; Fig. 1A). Type I collagen was chosen as a substrate for preliminary experiments, because cell migration and morphology on this ECM have been well characterized in the literature (31–34). Phase-contrast images revealed that ACM-treated cells were larger and more elongated than control cells (Fig. 1B); this observation was confirmed quantitatively, with ACM-treated cells having significantly larger cell areas (Fig. 1C) and aspect ratios (Fig. 1D).
Figure 1.

A) The experimental setup, showing MDA-MB-231 cells plated on type I collagen on glass on d 1, and medium replaced with ACM on d 2. B) Representative phase-contrast images of MDA-MB-231 cells treated with control medium or ACM. Scale bar, 20 µm. C, D) Cell areas (C) and circularities (D), inverse aspect ratios, and solidities of the MDA-MB-231 cells treated with control medium or ACM. n(control) = 1800 cells and n(ACM) = 2045 cells. E) Trajectories of all the cells treated with control medium or ACM (5-min time intervals). F) Cumulative distribution function plot of cell velocities when treated with control medium or ACM. n(control) = 272 cells and n(ACM) = 300 cells. G) Average cell velocities tracked for 500 min with a 5-min time interval. H, I) MSDs over 500 min (H), and diffusion coefficients (I) obtained from a Langevin-type fit to the MSD plot over 200 min, of cells treated with control medium or ACM. n(control) = 272 cells and n(ACM) = 300 cells; 5-min time interval. In all plots, bars indicate mean of pooled data from N ≥ 3 independent experiments, and error bars represent sem. Statistical analysis performed with an unpaired Student’s t test followed by Welch’s correlation. ****P < 0.00005.
The trajectories of tumor cells treated with control medium or ACM overnight on type I collagen–coated glass indicated that the population of ACM-treated cells migrated over a larger area than control medium–treated cells (Fig. 1E). Cumulative distribution functions of velocities for control and ACM-treated cells were significantly different when tested with the 2-sample Kolmogorov-Smirnov test, with a shift toward increased velocity for cells treated with ACM (Fig. 1F). Further, the average velocity of ACM-treated cells was significantly greater (Fig. 1G) than those of control medium–treated cells, and the ACM-treated cells also had larger MSD vs. time (Fig. 1H) and a larger diffusion coefficient (Fig. 1I) in comparison with control cells.
Media conditioned by astrocytes were depleted of nutrients, including serum, in addition to being enriched in other soluble factors produced by the astrocytes. To evaluate the possible effects of the concentration of soluble factors in ACM on tumor cell morphology and migration, ACM was diluted to 5, 10, 20, 50, and 75% in serum-containing control medium. Serum-free medium was included as a control to test a nutrient-deficient condition. Tumor cells were seeded on type I collagen–coated glass and allowed to attach overnight. On the following day, culture medium was replaced with ACM at various dilutions, control medium, or serum-free medium, and incubated overnight. Tumor cell areas increased with increased ACM concentration (Fig. 2A), suggesting the presence of a soluble factor influencing tumor cell morphology. Areas, circularities, inverse aspect ratios, and solidities (Fig. 2B) were all significantly different from the control medium at ACM concentrations above 20% ACM. The velocities of the tumor cells were higher in concentrations of 50% ACM and above compared to the control (Fig. 2C). The diffusion coefficient of ACM-treated cells also tended to increase with ACM concentration, although this relationship was not statistically significant (Fig. 2D). Although tumor cells treated with serum-free medium had morphologies similar to those in 100% ACM (Fig. 2B), they did not display increased migration potential and further showed no differences in velocity or diffusion coefficient in comparison with cells treated with serum-containing control medium (Fig. 2C, D). Hence, we conclude that the effect of ACM on cell migration is related to soluble factors present in ACM and not to serum depletion.
Figure 2.

A, B) Areas (A) and circularities, inverse aspect ratios, and solidities (B) of the MDA-MB-231 cells (on type I collagen) treated with control medium or 5–100% ACM. n(serum free) = 682; n(0% ACM) = 1010; n(5% ACM) = 689; n(10% ACM) = 663; n(20% ACM) = 634; n(50% ACM) = 1025; n(75% ACM) = 476; and n(100% ACM) = 896 cells. C, D) Velocities (C) and diffusion coefficients (D), obtained from a Langevin-type fit to MSD curves over 200 min. n(serum free) = 96; n(0% ACM) = 157; n(5% ACM) = 102; n(10% ACM) = 109; n(20% ACM) = 83; n(50% ACM) = 165; n(75% ACM) = 54; n(100% ACM) = 187 cells. Time intervals, 5–6.7 min. In all plots, individual points indicate mean of pooled data from N ≥ 3 independent experiments, and error bars represent sem. Note that some error bars not visible because the size of the point marker is larger than the error bars. Statistical analysis with a 1-way ANOVA and Tukey’s post hoc analysis. *P < 0.05, **P < 0.005, ****P < 0.00005.
ACM-induced changes in ECM alter metastatic breast tumor cell migration
Our results herein and the work of others indicate that ACM applied directly to MDA-MB-231 tumor cells can increase tumor cell migration potential. However, no studies have yet evaluated the effects of astrocyte-secreted factors on the ECM. Thus, type I collagen was pretreated with ACM before seeding MDA-MB-231 cells (Fig. 3A). In these experiments, the area of the tumor cells did not increase on ACM-treated type I collagen, as it did when cells were treated directly with ACM. Of additional morphology parameters, inverse aspect ratio significantly increased when the collagen was pretreated with ACM (Fig. 3C), which was opposite the effect when cells were treated directly with ACM. Meanwhile, area (Fig. 3B) and other morphology parameters (Fig. 3C) did not change significantly. However, most notably, we observed a substantial increase in tumor cell migration on ACM-treated type I collagen substrates, with cell trajectories showing tumor cells on ACM-treated type I collagen migrating over a larger area than cells on control medium-treated type I collagen (Fig. 3D). Cumulative distribution functions of velocities of cells migrating on control and ACM-treated ECM were significantly different when tested with the 2-sample Kolmogorov-Smirnov test, with a shift toward increased velocity for cells on ACM-treated ECM (Fig. 3E). Quantitatively, there was a significant increase in tumor cell average velocity (Fig. 3F), MSD (Fig. 3G), and diffusion coefficient (Fig. 3H). This increase in migration was even greater than when cells were treated directly with ACM, as indicated by the comparison of a percentage increase in velocity of 36% for ACM-treated cells vs. 92% for ACM-treated type I collagen, as compared to control-medium treatments (Fig. 3I).
Figure 3.

A) Experimental setup, showing type I collagen plated and treated with ACM on d 1 and MDA-MB-231 cells plated in control medium on d 2. B, C) Quantification of cell areas (B) and circularities, inverse aspect ratios, and solidities (C) of MDA-MB-231 cells plated on control medium or ACM-treated type I collagen. n(control) = 716 cells; and n(ACM) = 742 cells. D) Trajectories of all the cells on collagen treated with control medium or ACM. n(control) = 223 cells; and n(ACM) = 201 cells. E) Cumulative distribution function plot of cell velocities on control- or ACM-treated type I collagen. F–H) Quantification of average cell velocities (F), MSDs with 5-min intervals over 500 min (G), and diffusion coefficients (H), obtained from a Langevin-type fit to MSD curves over 200 min, of MDA-MB-231 cells on control medium or ACM-treated type I collagen. I) Comparison of percentage increase in velocity between ACM-treated and control medium-treated conditions of the ACM-treated cells and ACM-treated type I collagen experiments. In all plots, bars indicate mean of pooled data from N ≥ 3 independent experiments, and error bars represent sem. Statistical analysis performed with an unpaired Student’s t test followed by Welch’s correlation. **P < 0.005, ***P < 0.0005, ****P < 0.00005.
Cells of varying tumorigenic and metastatic potential respond uniquely to ACM
To determine whether cell response to ACM is conserved over multiple cell lines, we repeated migration and morphology experiments on several additional cell lines, including: 1) the weakly metastatic human breast tumor cell line MDA-MB-468, 2) the tumorigenic but nonmetastatic breast tumor cell line MCF7, and 3) the normal breast epithelial cell line MCF10A. We selected these additional cell lines to evaluate the effects of ACM on an additional metastatic breast tumor cell line, and to evaluate whether ACM can alter the morphology and/or increase the migration of nonmetastatic breast tumor cells, or even normal breast epithelial cells. Migration assays on both type I collagen (and also on fibronectin; data not shown) revealed that MCF7 and MDA-MB-468 cells were nonmigratory (velocities <10 µm/h, which was mostly found to be small random displacements in place rather than true migration), and addition of ACM to either the cells or their ECM did not promote migration. Meanwhile, direct treatment of MCF10A cells with ACM led to a statistically significant increase in cell migration velocity (Fig. 4A), whereas MCF10A cells plated on ACM-treated type I collagen substrates demonstrated no change in cell migration velocity (Fig. 4B). Thus, for MCF10A cells, the percentage change in velocity from control to ACM was significantly more positive for the condition in which cells were directly treated with ACM, in comparison with ACM-treated ECM (Fig. 4C). These results differ from MDA-MB-231 cells (Fig. 3I), suggesting that the effects of ACM (either applied directly to cells or by treatment of ECM) on cell migration may be dependent on cell type and degree of tumorigenicity or metastatic potential.
Figure 4.

A, B) Velocities of MCF10A cells on type I collagen when the cells were treated with control medium or ACM (A) [n(control) = 100 cells; n(ACM) = 110 cells] and seeded on type I collagen pretreated with control medium or ACM (B) (n(control) = 118 cells; n(ACM) = 118 cells). Statistical analysis performed with an unpaired Student’s t test followed by Welch’s correlation. *P < 0.05. C) Percentage change in velocities of cells of MCF10A cells when treated with control medium or ACM or seeded on type I collagen that was pretreated with control medium or ACM. Cell migration was analyzed over 5 h with 10-min intervals between frames. D, E) Areas (D) and corresponding circularities, inverse aspect ratios, and solidities (E) of MCF10A, MCF7, and MDA-MB-468 cells on type I collagen when the cells were treated with control medium or ACM. n(control, MCF10A) = 120 cells; n(ACM, MCF10A) = 69 cells; n(control, MCF7) = 206 cells; n(ACM, MCF7) = 161; n(control, MDA-MB-468) = 492 cells; n(ACM, MDA-MB-468) = 380 cells. F, G) Areas (F) and corresponding circularities, inverse aspect ratios, and solidities (G) of MCF10A, MCF7, and MDA-MB-468 cells seeded on type I collagen that was pretreated with control medium or ACM. n(control, MCF10A) = 158 cells; n(ACM, MCF10A) = 139 cells; n(control, MCF7) = 562 cells; n(ACM, MCF7) = 389 cells; n(control, MDA-MB-468) = 646 cells; and n(ACM, MDA-MB-468) = 700 cells. Note that for all experiments, only single cells were analyzed, and cells that formed clusters were excluded. In all plots, bars indicate mean of pooled data from N = 2 independent experiments. Error bars represent sem. Statistical analysis was performed with a Tukey’s multiple comparison post hoc test. *P < 0.05, **P < 0.005, ****P < 0.00005.
Meanwhile, for all additional cell lines, we observed mostly similar trends in morphologic changes (i.e., same trends as MDA-MB-231 cells on type I collagen in Figs 1 and 3) in cells treated directly with ACM and also cells plated on ACM-treated type I collagen substrates (Fig. 4D–G). More specifically, direct treatment of cells with ACM resulted in statistically significant increases in cell area, along with decreases in inverse aspect ratio, circularity, and solidity for MCF10A and MDA-MB-468 cell lines (Fig. 4D, E; similar to MDA-MB-231 cells in Fig. 1B–D). Meanwhile, direct treatment of MCF7 cells with ACM resulted in no difference in cell area or solidity, but decreased inverse aspect ratio and circularity (Fig. 4D, E). We also observed some minor (or no) changes in cell morphologic parameters in cells plated on ACM-treated type I collagen substrates, but these were much less significant than in cells treated directly with ACM (Fig. 4F, G), which is also consistent with what we observed in MDA-MB-231 cells (Fig. 3B, C). Hence, the effects of ACM (either direct treatment of cells or treatment of the collagen substrate) on cell morphology seem to be conserved across multiple breast cell lines and cell lines with various tumorigenic and metastatic potential.
Cell response to ACM is conserved on multiple ECM proteins
To test whether the response of cells was unique to type I collagen or whether it was conserved across multiple ECM proteins, we repeated the morphology and migration experiments using MDA-MB-231 cells plated on several additional ECM proteins that are present in the BBB microenvironment, including fibronectin, laminin, and type IV collagen. The migration assays demonstrated that there was no significant increase in MDA-MB-231 cell velocity when cells (on fibronectin, laminin, or collagen IV) were treated directly with ACM (Fig. 5A), unlike on type I collagen, where an increase in cell migration velocity was observed (Fig. 1G). However, for fibronectin and laminin, there was a statistically significant increase in cell migration velocity on substrates pretreated with ACM (Fig. 5B). The more pronounced increase in migration on ACM-treated fibronectin and laminin substrates (in comparison with cells treated directly with ACM; Fig. 5C) is consistent with our migration results on type I collagen (Fig. 3I). We did not observe any statistically significant changes in migration velocity for any of the conditions on collagen IV (Fig. 5A, B), suggesting that there may be differential migration-induced effects of the ACM on various ECM or basement membrane proteins.
Figure 5.

A, B) Velocities of MDA-MB-231 cells on type IV collagen, fibronectin, laminin, and PDL when the cells were treated with control medium or ACM [A; n(control, type IV collagen) = 127 cells; n(ACM, type IV collagen) = 152 cells; n(control, fibronectin) = 133 cells; n(ACM, fibronectin) = 142 cells; n(control, laminin) = 118 cells; n(ACM, laminin) = 131 cells; n(control, PDL) = 73 cells; and n(ACM, PDL) = 58 cells] and seeded on ECM pretreated with control medium or ACM [B; n(control, type IV collagen) = 131 cells; n(ACM, type IV collagen) = 155 cells; n(control, fibronectin) = 141 cells; n(ACM, fibronectin) = 151; n(control, laminin) = 116 cells; n(ACM, laminin) = 129 cells; n(control, PDL) = 62 cells; and n(ACM, PDL) = 83 cells]. C) Percentage change in velocities of cells of MDA-MB-231 cells when seeded on type IV collagen, fibronectin, laminin, or PDL and treated with control medium or ACM or seeded on ECM that was pretreated with control medium or ACM. Cell migration was analyzed over 5 h with 10-min intervals between frames. D, E) Areas (D) and corresponding circularities, inverse aspect ratios, and solidities (E) of MDA-MB-231 cells on type IV collagen, fibronectin, laminin, and PDL when the cells were treated with control medium or ACM. n(control, type IV collagen) = 578 cells; n(ACM, type IV collagen) = 578 cells; n(control, fibronectin) = 704 cells; n(ACM, fibronectin) = 559 cells; n(control, laminin) = 436 cells; n(ACM, laminin) = 363 cells; n(control, PDL) = 220 cells; and n(ACM, PDL) = 208 cells. F, G) Areas (F) and corresponding circularities, inverse aspect ratios, and solidities (G) of MDA-MB-231 cells seeded on ECM pretreated with control medium or ACM. n(control, type IV collagen) = 560 cells; n(ACM, type IV collagen) = 642 cells; n(control, fibronectin) = 526 cells; n(ACM, fibronectin) = 733 cells; n(control, laminin) = 556 cells; n(ACM, laminin) = 585 cells; n(control, PDL) = 162 cells; and n(ACM, PDL) = 225 cells. In plots for type IV collagen, fibronectin, and laminin, bars indicate mean of pooled data from N ≥3 independent experiments whereas in plots for PDL, bars indicate mean of pooled data from N ≥ 2 independent experiments. Error bars represent sem. Statistical analysis was performed using a Tukey’s multiple comparison post hoc test. *P < 0.05, **P < 0.005, ***P < 0.0005, ****P < 0.00005.
Notably, MDA-MB-231 cells plated on each of fibronectin, laminin, and type IV collagen and treated directly with ACM demonstrated significant morphologic changes (similar to results on type I collagen), including increases in cell area and decreases in inverse aspect ratio, circularity, and solidity (Fig. 5D, E). Meanwhile, morphologic changes in MDA-MB-231 cells plated on ACM-treated fibronectin, laminin, and type IV collagen substrates were either not significantly different, or were less so than for the case of cells directly treated with ACM (e.g., P closer to 0.05) (Fig. 5F, G). These results are also consistent with our results for type I collagen (Fig. 3B, C).
We also performed morphology and migration experiments on substrates coated with PDL, which enhances electrostatic interactions between cells and the surface but does not promote integrin binding. We added this coating to the experimental setup to (indirectly) test the hypothesis that ACM treatment of the ECM or basement membrane alters integrin binding to the substrate. MDA-MB-231 cells plated on PDL and treated with ACM demonstrated statistically significant changes in morphologic parameters, including increased area and decreased inverse aspect ratio, circularity, and solidity (Fig. 5D, E). Meanwhile, cells plated on ACM-treated PDL demonstrated no changes in any morphologic parameter (Fig. 5F, G). Therefore, we speculate that ACM-induced changes in cell morphology are not integrin dependent. MDA-MB-231 cells plated on PDL and treated directly with ACM demonstrated no change in cell migration velocity (Fig. 5A) and trended toward decreased velocities (though not statistically significant) on ACM-treated PDL, in contrast with the increased velocities observed on other ACM-treated ECM substrates (Fig. 5B, C). Hence, we speculate that the migratory response of cells to ACM treatment of the ECM is dependent on integrin binding, though further work is needed to confirm this hypothesis.
Direct treatment of cells with ACM alters actin cytoskeleton organization
Given the significant alterations in cell morphology observed in response to direct treatment with ACM, we hypothesized that the actin cytoskeleton would also demonstrate concomitant changes in organization in response to ACM. Hence, cells were plated on ECM-coated glass substrates, fixed, permeabilized, stained for actin, and imaged via laser scanning confocal microscopy. Indeed, ACM-treated MDA-MB-231 cells displayed more prominent actin stress fibers in comparison with control medium–treated cells on type I collagen (Fig. 6A, B) and also on collagen IV, fibronectin, and laminin substrates (Fig. 6C). Meanwhile, ACM did not alter cell actin organization on PDL, where only a diffuse actin network was observed for both control and ACM-treated cells (Fig. 6C). There was also a (visual) increase in actin stress fiber alignment in ACM-treated MCF10A cells and a minimal (visual) increase in stress fiber content in MCF7 cells (Fig. 6D). ACM did not appear to alter the actin organization of MDA-MB-468 cells (Fig. 6D).
Figure 6.

A) Stitched images of a large area of MDA-MB-231 cells treated with control medium or ACM and stained for actin and the nucleus on type I collagen. B) Images of control medium-treated and ACM-treated MDA-MB-231 cells stained for actin and the nucleus on type I collagen. C) Images of control medium-treated and ACM-treated MDA-MB-231 cells on type IV collagen, fibronectin, laminin, or PDL and stained for actin and the nucleus. D) Images of MCF10A, MCF7, and MDA-MB-468 on type I collagen, stained for actin and the nucleus. Images obtained with a ×100 (A, B) or ×60 objective of a confocal microscope. The intensity of the images was adjusted equally between each set of conditions, to best view the actin arrangement. Scale bars, 20 µm.
Astrocyte activation via TGF-β does not enhance the effect of ACM
As previously discussed, the astrocyte secretome changes when astrocytes become reactive, which occurs in response to injury or insult, such as cancer or stroke (35). To date, no studies have systematically evaluated the effects of soluble factors from quiescent and activated astrocytes on tumor cell morphology and migration. We and others have previously shown that different serum conditions in conjunction with TGF-β treatment can be used to alter the activation state of astrocytes (15, 36, 37). Specifically, we have shown that TGF-β in combination with serum starvation induces a time-dependent increase in KCa3.1 protein expression, and at 5 d, induces expression of the glial fibrillary acidic protein (GFAP; marker protein of reactive astrogliosis) and the production of CSPGs. Thus, 3 ACM regimens were used, with or without TGF-β treatment: 1) astrocytes were cultured in serum-free medium overnight and then treated with TGF-β in serum-free medium for 5 d, 2) astrocytes were cultured in serum-free medium overnight and then treated with TGF-β in serum-containing medium for 5 d, and 3) astrocytes were cultured in serum-containing medium overnight and then treated with TGF-β in serum-containing medium for 5 d before harvest. The addition of serum was necessary because the ACM used for other experiments contained serum. We note that, in each of the experiments, astrocytes remained attached to the substrate, maintained confluence, and appeared viable. Astrocytes demonstrate strong contact inhibition and thus stop dividing once they reach confluence; hence, the total number of astrocytes was comparable in all experiments.
MDA-MB-231 tumor cells were cultured on type I collagen–coated glass overnight, and on the following day, culture medium was replaced with ACM from TGF-β-treated astrocytes, ACM from untreated astrocytes, or control medium and incubated overnight. Tumor cell morphology was not significantly different after treatment with ACM from a 5-d culture of TGF-β-activated astrocytes, in comparison with ACM from untreated astrocytes (Fig. 7A, B). However, ACM from TGF-β-treated serum-starved astrocytes resulted in significantly decreased tumor cell velocities in comparison with ACM from untreated serum-starved astrocytes (Fig. 7C). Without astrocyte serum starvation, TGF-β treatment in astrocytes had no effect on tumor cell velocity. Meanwhile, there was no significant difference in tumor cell diffusion coefficient after tumor cells were treated with ACM from TGF-β-treated vs. untreated astrocytes for any of the serum conditions (Fig. 7D).
Figure 7.

Serum free: astrocytes were cultured without serum with or without TGF-β; serum free O/N + serum: astrocytes were cultured without serum overnight, and medium was changed to medium with serum, with or without TGF-β; full: astrocytes cultured in full medium (with serum) with or without TGF-β. All ACM was conditioned for 5 d. A, B) Areas (A) and circularities, inverse aspect ratios, and solidities (B) of the MDA-MB-231 cells treated with control medium or ACM. n(serum free – TGF-β) = 626; n(serum free + TGF-β) = 634; n(serum free O/N + serum – TGF-β) = 531; n(serum free O/N + serum + TGF-β) = 569; n(full – TGF-β) = 728; n(full + TGF-β) = 638 cells. C, D) Velocities (C) (cells tracked for 670 min with 6.7 min intervals) and diffusion coefficients (D) obtained from a Langevin-type fit to MSD curves over 268 min. n(serum free – TGF-β) = 96; n(serum free + TGF-β) = 93; n(serum free O/N + serum – TGF-β) = 73; n(serum free O/N + serum + TGF-β) = 69; n(full – TGF-β) = 102; n(full + TGF-β) = 93 cells. In all plots, bars indicate mean of pooled data from N independent experiments, and error bars represent sem (N ≥ 3 for all conditions, except the serum free O/N + serum condition, where N = 2). Statistics obtained with a 1-way ANOVA and Tukey’s post hoc analysis. *P < 0.05.
However, TGF-β itself can promote epithelial–mesenchymal transition and induce directional migration in breast cancer cells (38). Thus, to confirm that the observed effects on tumor cell morphology and migration were not simply caused by free TGF-β in the ACM, 10 ng/ml TGF-β was added to serum-free medium, control medium, and 2 d ACM, and migration experiments were performed on MDA-MB-231 tumor cells cultured on type I collagen–coated glass. TGF-β addition resulted in increased circularity and inverse aspect ratio for tumor cells in serum free medium and 2 d ACM and increased area for tumor cells in control medium (Fig. 8A, B); however, the relative trends between TGF-β-treated and untreated tumor cells did not correspond to the changes seen in tumor cells treated with ACM from TGF-β-treated and untreated astrocytes, where there was no significant change in any morphology parameter for any condition (Fig. 7A, B). Furthermore, addition of TGF-β resulted in no changes in velocities (Fig. 8C) or diffusion coefficients (Fig. 8D) of tumor cells in serum-free medium, control medium, or ACM. Thus, we conclude that free TGF-β did not cause the decrease in tumor cell velocity measured when tumor cells were treated with ACM from TGF-β-activated astrocytes (Fig. 7C, D). Instead, changes in cell migration in response to ACM from TGF-β-activated astrocytes are likely a result of the altered astrocyte secretome in response to TGF-β.
Figure 8.

A, B) Areas (A) and circularities, inverse aspect ratios, and solidities (B) of MDA-MB-231 cells treated with control medium, serum-free medium, or ACM, with or without 10 ng/ml TGF-β. n(serum free – TGF-β) = 264; n(serum free + TGF-β) = 466; n(control – TGF-β) = 284; n(control + TGF-β) = 408; n(ACM – TGF-β) = 159; n(ACM + TGF-β) = 383 cells. C, D) Velocities (C) and diffusion coefficient (D) obtained from a Langevin-type fit to MSD curves over 200 min. n(serum free – TGF-β) = 54; n(serum free + TGF-β) = 66; n(control – TGF-β) = 49; n(control + TGF-β) = 71; n(ACM – TGF-β) = 33; n(ACM + TGF-β) = 74 cells. Time intervals, 5–6.7 min for 500–670 min. Statistics obtained with a 1-way ANOVA and Tukey’s post hoc analysis. In all plots, bars indicate the mean of pooled data from N independent experiments, and error bars represent sem (N ≥3 for all conditions, except the ACM condition, where n = 2). *P < 0.05, **P < 0.005.
Inhibiting MMPs in ACM reverses its effect on metastatic breast tumor cells
Some studies have implicated MMPs in tumor cell migration changes (39) and also in the astrocyte secretome (5, 40). Thus, MMPs could be one astrocyte-secreted factor that may potentiate the observed phenotypic changes in tumor cells in response to ACM. To test this hypothesis, a broad-spectrum MMP inhibitor, batimastat, was used to inhibit all types of MMPs present in ACM. MDA-MB-231 tumor cells were cultured on type I collagen–coated glass overnight, and on the following day, culture medium was replaced with control medium with vehicle control, ACM with vehicle control, or ACM with various concentrations of batimastat, and incubated overnight. In these experiments, as the concentration of batimastat in the ACM increased from 0.1 to 5 µM, the areas of tumor cells decreased to a size similar to the areas of cells treated with control medium (Fig. 9A). Similarly, as batimastat concentration increased, tumor cells also returned to control-like values of circularity, inverse aspect ratio, and solidity (Fig. 9B). Notably, batimastat treatment also dose dependently decreased the migration parameters of velocity (Fig. 9C) and diffusion coefficient (Fig. 9D) of cells treated with ACM. The results of MMP inhibition studies in ACM suggest that MMPs are implicated in the phenotypic changes of tumor cells treated with ACM.
Figure 9.

A, B) Areas (A) and circularities, inverse aspect ratios, and solidities (B) for MDA-MB-231 cells treated with control medium or ACM with DMSO, as well as with ACM with 0.1, 1, and 5 µM batimastat. n(control + DMSO) = 445; n(ACM + DMSO) = 343; n(ACM + 0.1 μM batimastat) = 328; n(ACM + 1 μM batimastat) = 325; n(ACM + 5 μM batimastat) = 360 cells. C, D) Velocities (cells tracked every 5 min for 500 min) (C) and diffusion coefficients (D) obtained from a Langevin-type fit to MSD curves over 200 min. n(control + DMSO) = 64; n(ACM + DMSO) = 71; n(ACM + 0.1 μM batimastat) = 73; n(ACM + 1 μM batimastat) = 71; n(ACM + 5 μM batimastat) = 73 cells. Statistics obtained with a 1-way ANOVA and Tukey’s post hoc analysis. In all plots, bars indicate mean of pooled data from N ≥3 independent experiments. Error bars represent sem. *P < 0.05, **P < 0.005, ****P < 0.00005.
There is evidence in the literature that MMP-2 and -9 both can both cleave type I collagen (41, 42) and both of these MMPs are reportedly contained within ACM (5). Thus, we added MDA-MB-231 cells to type I collagen–treated substrates that were pretreated with exogenous MMP-2 or -9 at 40 or 100 ng/ml. These concentrations were chosen based on an MMP-2 and -9 activity assay against gelatin (26), as well as a study with exogenous MMP-2 and -9 (25). Treatment of cells on MMP-2- or -9-treated ECM substrates resulted in increased (but not statistically significant) migration velocities similar to those with ACM (e.g., for 40 and 100 ng/ml MMP-2 and 40 ng/ml MMP-9; Fig. 10A), whereas morphology was mostly unaffected by MMP-2 or -9 (Fig. 10B, C). These results suggest that MMP-2 or -9 or both in ACM are at least partially responsible for the ACM-induced effects on migration of cells plated on ACM-treated type I collagen substrates, although the lack of statistical significance in velocity data (Fig. 10A) suggests that other mechanisms are likely also at play.
Figure 10.

A) Velocities of MDA-MB-231 cells on type I collagen that was pretreated with control medium, ACM, or control medium with exogenous MMP-2 or -9. n(control) = 80 cells; n(ACM) = 120 cells; n(control + 40 ng/ml MMP-2) = 120 cells; n(control + 100 ng/ml MMP-2) = 120 cells; n(control + 40 ng/ml MMP-9) = 80 cells; n(control + 100 ng/ml MMP-9) = 120 cells. Cell migration was analyzed over 5 h with 10-min intervals between frames. B, C) Areas (B) and corresponding circularities, inverse aspect ratios, and solidities (C) of MDA-MB-231 cells seeded on type I collagen that was pretreated with control medium, ACM, or control medium with exogenous MMP-2 or -9. n(control) = 321 cells; n(ACM) = 449 cells; n(control + 40 ng/ml MMP-2) = 379 cells; n(control + 100 ng/ml MMP-2) = 366 cells; n(control + 40 ng/ml MMP-9) = 256 cells; n(control + 100 ng/ml MMP-9) = 317 cells. Statistical analysis was performed with a Tukey’s multiple-comparison post hoc test. In all plots, bars indicate mean of pooled data from N ≥2 independent experiments, and error bars represent sem. *P < 0.05, **P < 0.005.
ACM does not serve as a chemoattractant for metastatic breast tumor cells in confined microchannels
During metastasis across the BBB, tumor cells are exposed, not only to biochemical cues, but also to physical cues such as confinement, because tumor cells have to travel through very narrow capillaries and then cross the BBB. Our previous work has suggested that tumor cells in confinement migrate via an alternate mechanism based on water permeation (20), as compared to cell migration in 2D, which is driven by actin polymerization. Other recent work has continued to explore potential mechanisms guiding tumor cell migration in 3-dimensional matrices (43, 44). Thus, an open question is whether ACM alters tumor cell migration differently in confinement vs. 2D. In addition, it was also important to address whether ACM acts as a chemoattractant for tumor cells. Hence, we used microfluidic devices containing microchannels of various widths to expose tumor cells to various degrees of physical confinement and to a chemoattractant (e.g., serum or ACM). Two different conditions were used: in condition A, cells were seeded in serum-containing control medium with no chemotactic gradient or with ACM as the sole chemoattractant (Fig. 11A); and in condition B, cells were seeded in serum-free medium with serum-containing control medium or serum-containing ACM on the other side of the channels, such that either serum or serum and ACM-containing soluble factors were the chemoattractants (Fig. 11B). Condition A was used to determine whether ACM by itself acts as a chemoattractant. Condition B was used to determine whether ACM enhances the effects of serum as a chemoattractant. In both conditions, cells were seeded in the collagen-coated devices, and medium was changed to one of the conditions described above. Cells migrated through the narrow channels toward the opposite side (Fig. 11C), and cell velocity and chemotactic index were calculated (Fig. 11D). In both cases, tumor cells’ speed showed a biphasic relationship with channel width, with cells having smallest speeds in 50- and 3-μm channels and largest speeds in the 10-µm channels. However, when ACM was the sole chemoattractant (condition A), there was no significant difference in cell speed (Fig. 11E) or chemotactic index (Fig. 11F). On the other hand, when the ACM was used as a chemoattractant in conjunction with serum (condition B), tumor cells were significantly slower in 10-μm channels when migrating toward the serum-containing ACM from serum-free medium (Fig. 11G) but had similar chemotactic indices in all channels (Fig. 11H). When tumor cells were first cultured in ACM, they were no longer affected by the ACM and serum gradient and migrated from serum-free medium at almost the same speed toward serum-containing ACM as toward serum-containing control medium gradient (Fig. 11G). Together, these results suggest that ACM does not serve as a chemoattractant for the tumor cells, either with or without serum in the seeding medium, because using ACM as a chemotactic source did not enhance the speed of the tumor cells in confinement, as it did in 2D. Furthermore, these results could suggest that the different migration modes used by tumor cells in 2D and in confinement are affected differently by astrocyte-secreted factors, which is important for understanding these interactions in physiologic microenvironments.
Figure 11.

A) Diagram of experimental setup where ACM serves as the sole chemoattractant (condition A). B) Diagram of experimental setup where ACM and serum are chemoattractants (condition B). Cells cultured in control medium or ACM. C) Image sequence taken of a single cell at 10-min intervals (indicated by arrows) traveling through a 6-µm channel with serum as a chemoattractant. The arrows indicate the same cell and are relocated from the leading to the trailing edge of cell because of space constraints at the end of the channel. D) Chemotactic index calculation. E, F) Speeds (E) and chemotactic indices (F) of cells traveling through microchannels of 3, 10, and 50 μm widths from control medium toward ACM or control medium (condition A). n(50-µm channels with control medium everywhere) = 152; n(50-µm channels with ACM as chemoattractant) = 212; n(10-µm channels with control medium everywhere) = 97; n(10-µm channels with ACM as chemoattractant) = 233; n(3-µm channels with control medium everywhere) = 18; and n(3-µm channels with ACM as chemoattractant) = 57 cells. G, H) Speeds (G) and chemotactic indices (H) of cells traveling through microchannels of 3, 10, and 50 μm widths from serum-free medium toward ACM or control medium (condition B). n(50-µm channels with control chemoattractant) = 200; n(50-µm channels with ACM as chemoattractant) = 158; n(50-µm channels with ACM as chemoattractant and cells pretreated in ACM) = 205; n(10-µm channels with control chemoattractant) = 183; n(10-µm channels with ACM as chemoattractant) = 126; n(10-µm channels with ACM as chemoattractant and cells pretreated in ACM) = 122; n(3-µm channels with control chemoattractant) = 18; n(3-µm channels with ACM as chemoattractant) = 23; and n(3-µm channels with ACM as chemoattractant and cells pretreated in ACM) = 63 cells. The tracking time interval was 10 min for up to 15 h. Statistical analysis was performed with a Tukey’s multiple-comparison post hoc test. In all plots, bars indicate the mean of pooled data from N ≥ 3 independent experiments, and error bars represent the sem. In some conditions, cells migrated into 3-µm channels in only 2 of 3 trials. ****P < 0.00005.
DISCUSSION
Mounting evidence suggests that astrocytes are implicated in increasing metastatic tumor cell invasiveness at the BBB (4–8). We investigated whether the 1-way biochemical communication from astrocytes to tumor cells (and/or their ECM) influences tumor cell migration and whether these effects are accompanied by alterations in tumor cell morphology and cytoskeletal organization. An additional goal was to probe the role of MMPs in the resulting phenotypic and behavioral changes of tumor cells. We found that: 1) application of ACM directly to MDA-MB-231 metastatic breast tumor cells or MCF10A normal breast epithelial cells increased cell velocity and induced an enlarged, elongated morphology; 2) pretreating type I collagen, fibronectin, or laminin ECM substrates with ACM before seeding MDA-MB-231 tumor cells resulted in significantly increased tumor cell velocity when compared to control cells, with increases in velocity that were more drastic than treating cells directly with ACM; 3) a broad-spectrum MMP inhibitor added to ACM reversed the effect of ACM on MDA-MB-231 tumor cell migration and morphology; 4) applying conditioned medium from TGF-β-activated astrocytes reduced MDA-MB-231 tumor cell migration velocity in comparison with medium from untreated astrocytes; and 5) applying ACM as a sole chemoattractant did not affect MDA-MB-231 tumor cell migration speed, but ACM in combination with serum as a chemoattractant reduced tumor cell migration speed in the 10 µm wide channels.
Our results suggest that astrocytes have a significant effect on MDA-MB-231 metastatic breast tumor cells before any potential signaling from tumor cells to astrocytes and reactive gliosis, indicating that astrocytes may play an important role in tumor cell recruitment and not just extravasation. A report (45) showed that changes in cell morphology on a 2D surface correlate with metastatic potential. The specific trends seem to depend on tumor cell type, but they suggest that quantitative cell shape parameters can provide useful information about other aspects of cell phenotype. In our study, astrocyte-secreted factors in ACM increased MDA-MB-231 and MCF10A cell migration velocity and altered their morphology to be larger and more elongated, with enhanced stress fiber formation on 2D ECM-coated substrates, all suggesting a more mesenchymal and metastatic phenotype (46). Thus, ACM can enhance the migratory potential of (already) migratory metastatic breast tumor cells and also normal breast epithelial cells. Our migration results are in agreement with past studies, where ACM increased tumor cell migration in wound healing and cell migration assays (5). Furthermore, we show that tumor cell morphology and actin cytoskeleton organization are also modulated by ACM, which could contribute to facilitation of migration in ACM-treated cells. We confirmed that serum depletion was not the cause of these results, because dilution of ACM with fresh serum-containing medium, down to 50%, also led to changes that were significantly different from those obtained with control medium. Thus, we conclude that there are soluble factors secreted by astrocytes that increase tumor cell velocity and alter their morphology. The altered morphology and migration potential of tumor cells during exposure to ACM indicate that secreted factors by astrocytes could affect tumor cell phenotype, even without any exposure of the astrocytes to the tumor cell or injury.
We found that ACM from astrocytes activated by TGF-β resulted in slower migration of tumor cells in comparison with untreated astrocytes, which is consistent with the literature showing that ACM from IFN-γ- and TNF-α-activated astrocytes decreases the proliferation of tumor cells, when compared to ACM from unreactive astrocytes (6). Because astrocytes generally become reactive in response to injury and their secretome changes as a result, it is significant to note that this astrocyte phenotype appears to be less conducive to tumor cell invasion, even though tumor cells are likely to cause injury and activate surrounding astrocytes. Because astrocytes need serum starvation for TGF-β to increase reactivity, as confirmed via Western blot, and Wang et al. (5) showed no significant difference in tumor cell invasion and metastasis in ACM, with and without serum, the fact that the most significant results occurred in the serum-free activation conditions is still indicative of TGF-β-induced reactivity playing a role in astrocytes’ effect on tumor cells. We speculate that once astrocytes become reactive, they also become increasingly protective of the brain microenvironment by producing biochemical cues to reduce tumor cell migration. However, in other studies, activation of astrocytes via recombinant IL-1β and subsequent coculture with MDA-MB-231 cells led to an enriched population of cancer stem cells in the culture. It is possible that the specific signaling leading to astrocyte activation, whether it is through injury-modulated TGF-β stimulation, or through tumor cell-modulated activation of Notch signaling in astrocytes (7), leads to different effects on tumor cells; future investigations could elucidate this relationship.
The astrocyte secretome contains an array of biochemical factors (47), many of which can influence cell migration. A study has implicated astrocyte-secreted MMPs in ACM-mediated effects on tumor cells (5). In the current study, to further explore the mechanisms that alter tumor cell phenotype when exposed to ACM, we used a broad-spectrum MMP inhibitor, batimastat, to inhibit MMP activity in ACM. In these experiments, we observed a dose-dependent decrease in cell velocity with increased batimastat treatment, with the highest dose nearly returning ACM-treated tumor cells to their velocity and morphology from the control condition. These results are consistent the earlier study, where inhibition of MMPs with another broad-spectrum inhibitor, ONO-4817, reduced cell invasion in a cell migration assay in a dose-dependent manner (5). Thus, we provide further evidence that MMPs produced by astrocytes could be at least partly responsible for the ACM-induced alterations in tumor cell migration and morphology. Furthermore, we provide preliminary evidence that addition of exogenous MMP-2 or -9 to ECM substrates can produce similar effects to ACM on cell migration. However, because tumor cells also secrete MMPs, application of an MMP inhibitor to the ACM may also inhibit MMPs produced by the tumor cells, thus confounding interpretation of the results. It has been found that TGF-β1 increases MMP production by astrocytes (48); however, the TGF-β treatment of astrocytes actually decreased the velocity of the subsequent ACM-treated tumor cells as compared to vehicle control-treated ACM. This suggests that MMPs are not the only, and potentially not the dominant, molecular mechanism at play in astrocyte-induced increased cell migration. Future analysis should be aimed at exploring the molecular interplay of the astrocyte-tumor cell signaling and the role of MMPs.
Perhaps most notably, we discovered that pretreating the ECM with ACM resulted in a significant increase in MDA-MB-231 tumor cell migration velocity in comparison with untreated ECM, which was even greater for type I collagen, laminin, and fibronectin substrates than the velocity increase when tumor cells were treated with ACM directly. Because the ACM was removed from the ECM before seeding cells, it seems that astrocyte-secreted factors in the ACM in some way alter the ECM, leading to enhanced migration. It is likely that the MMPs produced by astrocytes and contained in the ACM degrade, or reorganize the ECM, or both, because MMPs have collagenase and gelatinase activity. However, minimal change in morphology was observed when the ECM substrates were pretreated with ACM, suggesting that morphologic changes alone are not responsible for the increased migration. This finding also suggests that there are different mechanisms involved in the ACM-induced phenotypical changes in tumor cell morphology and migration, respectively. Alternatively, it is possible that other biochemical factors, such as IL-6, TNF-α, and IL-1β (49), within the ACM, bind to the ECM and subsequently enhance tumor cell migration through adhesion-based signaling mechanisms. However, if this were the case, we would have expected to observe increased cell velocity over time during the overnight migration assays, because, at later time points, the ECM would have been coated in the same ACM factors. Instead, cell velocity was mostly consistent during the entire time lapse sequence (data not shown), suggesting that this latter mechanism has no effect. Meanwhile, that cells plated on PDL (which promotes adhesion through electrostatic interactions between cells and the substrate rather than through integrin binding) did not exhibit increased migration in response to ACM, in comparison with cells plated on ECM proteins (i.e., type I collagen, fibronectin, and laminin) suggests that integrin-based adhesions are altered by ACM treatment. Although it is unclear by which mechanisms the ACM alters the ECM to enhance migration, these findings suggest that tumor cell-ECM interactions are critical for the effect that ACM has on tumor cells and provide a logical link to the MMP pathways because MMPs are known to degrade ECM proteins. Future work will focus on delineating this relationship and also will evaluate whether ACM can affect the ECM in a 3-dimensional matrix.
Furthermore, in earlier work, we have shown that tumor cells can use an alternate, water permeation–based migration mechanism in confinement in comparison with a 2D surface where migration is actin driven (20). Thus, we found it important to observe the interplay between effects of ACM and the physical cue of confinement. As discussed above, metastatic tumor cells experience confinement at the BBB when traveling through microtracks of the basement membrane in the brain, as well as when migrating through narrow brain capillaries, which can be as small as 10 µm in diameter (18), through the brain endothelium, and along the blood vessel after extravasation across the endothelium (19). The biphasic trend of tumor cell migration velocity with respect to channel width could be related to an optimal balance between effects of contact guidance and nuclear deformation in intermediate width channels. That is, in wide channels, no nuclear deformation is necessary, but cells are not completely guided by contact with both walls of the channels. In narrow channels, cells are able to receive contact guidance from channel wall on both sides, but the nucleus must significantly deform for cells to squeeze through the channels. Our work suggests that physical confinement and introduction of ACM as a chemoattractant contrasts the effects that ACM has on 2D surfaces, because cells exposed to ACM as a chemoattractant in confined microchannels were not preferentially attracted to the ACM over control medium. In addition, when the ACM gradient was combined with a serum gradient, tumor cells actually moved significantly slower toward the ACM in comparison with control medium. Furthermore, preculturing cells in ACM resulted in their migrating toward ACM at a similar speed as toward control medium. Future work will focus on which mechanisms (i.e., chemotaxis, confinement, and contact guidance by edges of the channels) are responsible for the altered behavior of the tumor cells in the microchannels, because the literature has shown that tumor cells can be attracted toward astrocytes in Boyden chamber coculture assays (6). Indeed, new assays are necessary to understand the physiologically relevant interaction of tumor cells and astrocytes and how cells respond to the complex interplay of biochemical and physical cues at the BBB.
CONCLUSIONS
This work demonstrates that astrocyte-secreted factors affect the migration, morphology, and actin cytoskeleton organization of metastatic breast tumor cells and also that of normal breast epithelial cells, which suggests that paracrine signaling between astrocytes that have not been exposed to metastatic breast tumor cells could enhance metastasis at the BBB, although this effect could be abrogated if tumor cells experienced physical confinement. When exposed to ACM, tumor cells become more elongated, larger, and faster, which suggests an increase in metastatic phenotype. Secretion of MMPs by astrocytes could be at least partially responsible for the changes observed in the tumor cell phenotype, and future work will continue to explore this hypothesis. Our work also found that ACM from TGF-β-activated astrocytes results in a lower tumor cell velocity than tumor cells treated with ACM from unreactive astrocytes, without significant changes in tumor cell morphology. Perhaps most notably, we found that treating the ECM substrate with ACM before seeding tumor cells has a greater effect on MDA-MB-231 tumor cell velocity than treating tumor cells directly with ACM, indicating that astrocyte-secreted factors can alter the cells’ ECM. Finally, we showed that ACM may not serve as a chemoattractant to tumor cells in unconfined or confined microenvironments that also subject tumor cells to chemotaxis and contact guidance, suggesting that astrocytes may not attract tumor cells to the brain or help them move through confined spaces, but simply alter general morphology and migration properties. Regardless, our work provides support that astrocytes have a clear effect on tumor cell migration, morphology, and actin cytoskeleton arrangement in 1-way paracrine communication, and we show that astrocyte-secreted factors can also act indirectly on cells through their ECM to induce changes in morphology and migration. Further work should continue to examine the specific astrocyte-secreted factors influencing tumor cells and their ECM and also the molecular mechanisms and pathways by which astrocyte secreted factors act on tumor cells.
ACKNOWLEDGMENTS
The authors thank The University of Maryland Core Imaging Facility for providing resources for confocal imaging; the University of Maryland Micro and Nano Fabrication Laboratory (FabLab) for providing photolithography resources; Mary Doolin for help with editing custom MatLab code; and Gregory Dawson for help with data analysis. This study was supported by a Burroughs Wellcome Career Award at the Scientific Interface (to K.M.S.), the Fischell Department of Bioengineering (University of Maryland), and the U.S. National Institutes of Health, National Heart Lung and Blood Institute (NHLBI) Intramural Research Program (to C.P.M. and H.M.G.). The authors declare no conflicts of interest.
Glossary
- 2D
2-dimensional
- ACM
astrocyte-conditioned medium
- BBB
blood–brain barrier
- BSA
bovine serum albumin
- CSPG
chondroitin sulfate proteoglycan
- ECM
extracellular matrix
- FBS
fetal bovine serum
- FGF
fibroblast growth factor
- GFAP
glial fibrillary acidic protein
- MMP
matrix metalloproteinase
- MSD
mean square displacement
- PDL
poly-d-lysine hydrobromide
- ROCK
rho-associated kinase
- TJ
tight junction
AUTHOR CONTRIBUTIONS
M. A. Shumakovich and K. M. Stroka designed the research with support from C. P. Mencio and H. M. Geller; M. A. Shumakovich, J. S. Siglin, and R. A. Moriarty performed the experiments with support of C. P. Mencio, who isolated and cultured the astrocytes and provided the ACM; M. A. Shumakovich, J. S. Siglin, and R. A. Moriarty analyzed the data, and all authors interpreted the data; M. A. Shumakovich wrote paper, with methods on ACM collection written by C. P. Mencio, and parts of the introduction were written by J. S. Siglin; and all authors edited the paper and reviewed and approved the final version.
REFERENCES
- 1.Witzel I., Oliveira-Ferrer L., Pantel K., Müller V., Wikman H. (2016) Breast cancer brain metastases: biology and new clinical perspectives. Breast Cancer Res. 18, 8–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Balkwill F. R., Capasso M., Hagemann T. (2012) The tumor microenvironment at a glance. J. Cell Sci. 125, 5591–5596 [DOI] [PubMed] [Google Scholar]
- 3.Fokas E., Engenhart-Cabillic R., Daniilidis K., Rose F., An H. X. (2007) Metastasis: the seed and soil theory gains identity. Cancer Metastasis Rev. 26, 705–715 [DOI] [PubMed] [Google Scholar]
- 4.Valiente M., Obenauf A. C., Jin X., Chen Q., Zhang X. H. F., Lee D. J., Chaft J. E., Kris M. G., Huse J. T., Brogi E., Massagué J. (2014) Serpins promote cancer cell survival and vascular co-option in brain metastasis. Cell 156, 1002–1016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wang L., Cossette S. M., Rarick K. R., Gershan J., Dwinell M. B., Harder D. R., Ramchandran R. (2013) Astrocytes directly influence tumor cell invasion and metastasis in vivo. PLoS One 8, e80933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hohensee I., Chuang H., Grottke A., Werner S., Horn S., Lamszus K., Bartkowiak K., Witzel I., Matschke J., Glatzel M., Jücker M., Pukrop T., Pantel K., Wikman H. (2017) PTEN mediates the cross talk between breast and glial cells in brain metastases leading to rapid disease progression. Oncotarget 8, 6155–6168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Xing F., Kobayashi A., Okuda H., Watabe M., Pai S. K., Pandey P. R., Hirota S., Wilber A., Mo Y. Y., Moore B. E., Liu W., Fukuda K., Iiizumi M., Sharma S., Liu Y., Wu K., Peralta E., Watabe K. (2013) Reactive astrocytes promote the metastatic growth of breast cancer stem-like cells by activating notch signalling in brain. EMBO Mol. Med. 5, 384–396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chen Q., Boire A., Jin X., Valiente M., Er E. E., Lopez-Soto A., Jacob L. S., Patwa R., Shah H., Xu K., Cross J. R., Massagué J. (2016) Carcinoma-astrocyte gap junctions promote brain metastasis by cGAMP transfer. Nature 533, 493–498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Abbott N. J., Rönnbäck L., Hansson E. (2006) Astrocyte-endothelial interactions at the blood-brain barrier. Nat. Rev. Neurosci. 7, 41–53 [DOI] [PubMed] [Google Scholar]
- 10.Abbott N. J. (2002) Astrocyte-endothelial interactions and blood-brain barrier permeability. J. Anat. 200, 629–638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Proia P., Schiera G., Mineo M., Ingrassia A. M. R., Santoro G., Savettieri G., Di Liegro I. (2008) Astrocytes shed extracellular vesicles that contain fibroblast growth factor-2 and vascular endothelial growth factor. Int. J. Mol. Med. 21, 63–67 [PubMed] [Google Scholar]
- 12.Liang Y., Brekken R. A., Hyder S. M. (2006) Vascular endothelial growth factor induces proliferation of breast cancer cells and inhibits the anti-proliferative activity of anti-hormones. Endocr. Relat. Cancer 13, 905–919 [DOI] [PubMed] [Google Scholar]
- 13.Löffek S., Schilling O., Franzke C. W. (2011) Series “matrix metalloproteinases in lung health and disease”: biological role of matrix metalloproteinases: a critical balance. Eur. Respir. J. 38, 191–208 [DOI] [PubMed] [Google Scholar]
- 14.Schachtrup C., Ryu J. K., Helmrick M. J., Vagena E., Galanakis D. K., Degen J. L., Margolis R. U., Akassoglou K. (2010) Fibrinogen triggers astrocyte scar formation by promoting the availability of active TGF-β after vascular damage. J. Neurosci. 30, 5843–5854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang H., Katagiri Y., McCann T. E., Unsworth E., Goldsmith P., Yu Z.-X., Tan F., Santiago L., Mills E. M., Wang Y., Symes A. J., Geller H. M. (2008) Chondroitin-4-sulfation negatively regulates axonal guidance and growth. J. Cell Sci. 121, 3083–3091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sochocka M., Diniz B. S., Leszek J. (2016) Inflammatory response in the CNS: friend or foe? [E-pub ahead of print] Mol. Neurobiol. 10.1007/212035-016-0297-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kuzet S.-E., Gaggioli C. (2016) Fibroblast activation in cancer: when seed fertilizes soil. Cell Tissue Res. 365, 607–619 [DOI] [PubMed] [Google Scholar]
- 18.Preston S. D., Steart P. V., Wilkinson A., Nicoll J. A., Weller R. O. (2003) Capillary and arterial cerebral amyloid angiopathy in Alzheimer’s disease: defining the perivascular route for the elimination of amyloid β from the human brain. Neuropathol. Appl. Neurobiol. 29, 106–117 [DOI] [PubMed] [Google Scholar]
- 19.Friedl P., Alexander S. (2011) Cancer invasion and the microenvironment: plasticity and reciprocity. Cell 147, 992–1009 [DOI] [PubMed] [Google Scholar]
- 20.Stroka K. M., Jiang H., Chen S. H., Tong Z., Wirtz D., Sun S. X., Konstantopoulos K. (2014) Water permeation drives tumor cell migration in confined microenvironments. Cell 157, 611–623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yu Z., Yu P., Chen H., Geller H. M. (2014) Targeted inhibition of KCa3.1 attenuates TGF-β-induced reactive astrogliosis through the Smad2/3 signaling pathway. J. Neurochem. 130, 41–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Taylor A. R., Robinson M. B., Milligan C. E. (2007) In vitro methods to prepare astrocyte and motoneuron cultures for the investigation of potential in vivo interactions. Nat. Protoc. 2, 1499–1507 [DOI] [PubMed] [Google Scholar]
- 23.Miller M. A., Meyer A. S., Beste M. T., Lasisi Z., Reddy S., Jeng K. W., Chen C.-H., Han J., Isaacson K., Griffith L. G., Lauffenburger D. A. (2013) ADAM-10 and -17 regulate endometriotic cell migration via concerted ligand and receptor shedding feedback on kinase signaling. Proc. Natl. Acad. Sci. USA 110, E2074–E2083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nosoudi N., Nahar-Gohad P., Sinha A., Chowdhury A., Gerard P., Carsten C. G., Gray B. H., Vyavahare N. R. (2015) Prevention of abdominal aortic aneurysm progression by targeted inhibition of matrix metalloproteinase activity with batimastat-loaded nanoparticles. Circ. Res. 117, e80–e89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rolli M., Fransvea E., Pilch J., Saven A., Felding-Habermann B. (2003) Activated integrin alphavbeta3 cooperates with metalloproteinase MMP-9 in regulating migration of metastatic breast cancer cells. Proc. Natl. Acad. Sci. USA 100, 9482–9487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Isaksen B., Fagerhol M. K. (2001) Calprotectin inhibits matrix metalloproteinases by sequestration of zinc. Mol. Pathol. 54, 289–292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Balzer E. M., Tong Z., Paul C. D., Hung W. C., Stroka K. M., Boggs A. E., Martin S. S., Konstantopoulos K. (2012) Physical confinement alters tumor cell adhesion and migration phenotypes. FASEB J. 26, 4045–4056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tong Z., Balzer E. M., Dallas M. R., Hung W.-C., Stebe K. J., Konstantopoulos K. (2012) Chemotaxis of cell populations through confined spaces at single-cell resolution. PLoS One 7, e29211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Stroka K. M., Wong B. S., Shriver M., Phillip J. M., Wirtz D., Kontrogianni-Konstantopoulos A., Konstantopoulos K. (2016) Loss of giant obscurins alters breast epithelial cell mechanosensing of matrix stiffness. Oncotarget 8, 54004–54020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Smith L. A., Aranda-Espinoza H., Haun J. B., Hammer D. A. (2007) Interplay between shear stress and adhesion on neutrophil locomotion. Biophys. J. 92, 632–640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kapoor A., Sen S. (2012) Synergistic modulation of cellular contractility by mixed extracellular matrices. Int. J. Cell Biol. 2012, 471591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kim J. A., Kim H. N., Im S.-K., Chung S., Kang J. Y., Choi N. (2015) Collagen-based brain microvasculature model in vitro using three-dimensional printed template. Biomicrofluidics 9, 024115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wolf K., Alexander S., Schacht V., Coussens L. M., von Andrian U. H., van Rheenen J., Deryugina E., Friedl P. (2009) Collagen-based cell migration models in vitro and in vivo. Semin. Cell Dev. Biol. 20, 931–941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Xu X., Wang Y., Chen Z., Sternlicht M. D., Hidalgo M., Steffensen B. (2005) Matrix metalloproteinase-2 contributes to cancer cell migration on collagen. Cancer Res. 65, 130–136 [PubMed] [Google Scholar]
- 35.Liddelow S. A., Guttenplan K. A., Clarke L. E., Bennett F. C., Bohlen C. J., Schirmer L., Bennett M. L., Münch A. E., Chung W.-S., Peterson T. C., Wilton D. K., Frouin A., Napier B. A., Panicker N., Kumar M., Buckwalter M. S., Rowitch D. H., Dawson V. L., Dawson T. M., Stevens B., Barres B. A. (2017) Neurotoxic reactive astrocytes are induced by activated microglia. Nature 541, 481–487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Flanders K. C., Ren R. F., Lippa C. F. (1998) Transforming growth factor-betas in neurodegenerative disease. Prog. Neurobiol. 54, 71–85 [DOI] [PubMed] [Google Scholar]
- 37.Smith G. M., Strunz C. (2005) Growth factor and cytokine regulation of chondroitin sulfate proteoglycans by astrocytes. Glia 52, 209–218 [DOI] [PubMed] [Google Scholar]
- 38.McAndrews K. M., McGrail D. J., Ravikumar N., Dawson M. R. (2015) Mesenchymal stem cells induce directional migration of invasive breast cancer cells through TGF-β. Sci. Rep. 5, 16941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Egeblad M., Werb Z. (2002) New functions for the matrix metalloproteinases in cancer progression. Nat. Rev. Cancer 2, 161–174 [DOI] [PubMed] [Google Scholar]
- 40.Yin K.-J., Cirrito J. R., Yan P., Hu X., Xiao Q., Pan X., Bateman R., Song H., Hsu F.-F., Turk J., Xu J., Hsu C. Y., Mills J. C., Holtzman D. M., Lee J.-M. (2006) Matrix metalloproteinases expressed by astrocytes mediate extracellular amyloid-beta peptide catabolism. J. Neurosci. 26, 10939–10948 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bigg H. F., Rowan A. D., Barker M. D., Cawston T. E. (2007) Activity of matrix metalloproteinase-9 against native collagen types I and III. FEBS J. 274, 1246–1255 [DOI] [PubMed] [Google Scholar]
- 42.Aimes R. T., Quigley J. P. (1995) Matrix metalloproteinase-2 is an interstitial collagenase: inhibitor-free enzyme catalyzes the cleavage of collagen fibrils and soluble native type I collagen generating the specific 3/4- and 1/4-length fragments. J. Biol. Chem. 270, 5872–5876 [DOI] [PubMed] [Google Scholar]
- 43.Petrie R. J., Koo H., Yamada K. M. (2014) Generation of compartmentalized pressure by a nuclear piston governs cell motility in a 3D matrix. Science 345, 1062–1065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Skau C. T., Fischer R. S., Gurel P., Thiam H. R., Tubbs A., Baird M. A., Davidson M. W., Piel M., Alushin G. M., Nussenzweig A., Steeg P. S., Waterman C. M. (2016) FMN2 makes perinuclear actin to protect nuclei during confined migration and promote metastasis. Cell 167, 1571–1585.e18 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 45.Lyons S. M., Alizadeh E., Mannheimer J., Schuamberg K., Castle J., Schroder B., Turk P., Thamm D., Prasad A. (2016) Changes in cell shape are correlated with metastatic potential in murine and human osteosarcomas. Biol. Open 5, 289–299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Deakin N. O., Turner C. E. (2011) Distinct roles for paxillin and Hic-5 in regulating breast cancer cell morphology, invasion, and metastasis. Mol. Biol. Cell 22, 327–341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dowell J. A., Johnson J. A., Li L. (2009) Identification of astrocyte secreted proteins with a combination of shotgun proteomics and bioinformatics. J. Proteome Res. 8, 4135–4143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hsieh H. L., Wang H. H., Wu W. B., Chu P. J., Yang C. M. (2010) Transforming growth factor-β1 induces matrix metalloproteinase-9 and cell migration in astrocytes: roles of ROS-dependent ERK- and JNK-NF-κB pathways. J. Neuroinflammation 7, 88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Seike T., Fujita K., Yamakawa Y., Kido M. A., Takiguchi S., Teramoto N., Iguchi H., Noda M. (2011) Interaction between lung cancer cells and astrocytes via specific inflammatory cytokines in the microenvironment of brain metastasis. Clin. Exp. Metastasis 28, 13–25 [DOI] [PMC free article] [PubMed] [Google Scholar]