

Abstract
Pathological angiogenesis in the eye is an important feature in the pathophysiology of many vision-threatening diseases, including retinopathy of prematurity, diabetic retinopathy, and age-related macular degeneration, as well as corneal diseases with abnormal angiogenesis. Development of reproducible and reliable animal models of ocular angiogenesis has advanced our understanding of both the normal development and the pathobiology of ocular neovascularization. These models have also proven to be valuable experimental tools with which to easily evaluate potential antiangiogenic therapies beyond eye research. This review summarizes the current available animal models of ocular angiogenesis. Models of retinal and choroidal angiogenesis, including oxygen-induced retinopathy, laser-induced choroidal neovascularization, and transgenic mouse models with deficient or spontaneous retinal/choroidal neovascularization, as well as models with induced corneal angiogenesis, are widely used to investigate the molecular and cellular basis of angiogenic mechanisms. Theoretical concepts and experimental protocols of these models are outlined, as well as their advantages and potential limitations, which may help researchers choose the most suitable models for their investigative work.—Liu, C.-H., Wang, Z., Sun, Y., Chen, J. Animal models of ocular angiogenesis: from development to pathologies.
Keywords: choroidal neovascularization, corneal angiogenesis, macular degeneration, retinal vasculature, retinopathy
Development of the vascular system occurs mainly via two processes: vasculogenesis and angiogenesis (1). Vasculogenesis is initiated with the clustering of primitive vascular precursor cells or hemangioblasts into tube-like endothelial structures that form the primary vascular plexus. Vasculogenesis is followed by angiogenesis, where endothelial cells in existing vessels sprout and form new vessels under the guidance and balance of numerous angiogenic stimulators and inhibitors (2, 3). Angiogenesis plays important roles in both physiologic development and pathologic events. In diseases, a disrupted supply of oxygen and nutrients drives dysregulated angiogenic growth factors that influence angiogenesis, whereas dysregulation of angiogenesis also reciprocally disrupts the delivery of oxygen and nutrients, which results in a disturbed balance between metabolic demand and supply, thereby impairing tissue function. Abnormal angiogenesis is associated with many diseases, including cancers, cardiovascular diseases, neurodegeneration, and proliferative retinopathies (4).
The eye is one of the most important and intricate sensory organs. Its high energy demand is met by a well-organized ocular vascular system that also clears metabolic waste. Pathologic conditions that affect blood vessels in the eye pose direct threats to normal vision. Abnormal ocular angiogenesis can occur in a broad spectrum of eye disorders, including retinal vessel occlusion, retinopathy of prematurity (ROP), diabetic retinopathy (DR), neovascular age-related macular degeneration (AMD), neovascular glaucoma, and corneal neovascularization secondary to chemical injury or infectious or inflammatory processes (5–7).
In the last several decades, multiple animal models were developed that mimic these vascular eye diseases with retinal, choroidal, or corneal angiogenesis. These works significantly advanced our understanding of the basic angiogenic mechanisms, including the roles of oxygen and VEGF, one of the most crucial angiogenic factors that drives blood vessel growth. Work in these preclinical models has also paved the way for evaluation of the efficacy and development of antiangiogenic therapies, including current anti-VEGF therapies for the treatment of neovascular AMD. Current therapeutic strategies focus mostly on the elimination of pathologic neovessels, which are major culprits for vision loss, with a majority of these therapies targeting interference of VEGF function (8–12). Yet the clinical use of anti-VEGF drugs may have systemic impacts on healthy blood vessels as a result of the roles of VEGF in physiologic angiogenesis (13, 14). The identification of additional antiangiogenic therapies that can be supplemental or independent of anti-VEGF therapies is dependent on basic experimental studies in reliable preclinical models of ocular angiogenesis. In this review, we outline the fundamental concepts and recent progress in animal models of ocular angiogenesis and discuss the advantages and potential limitations of these ocular angiogenesis models as well as future perspectives and clinical implications.
OCULAR VASCULATURE AND ITS DEVELOPMENT
The eye is composed of three concentric tissue layers that surround a transparent lens, with an outermost layer of sclera and cornea, a middle layer of heavily vascularized uvea divided into the choroid, ciliary body, and stroma of iris, and an innermost retinal layer that includes the neural retina and the retinal pigment epithelium (RPE; Fig. 1A). The major arterial input to the eye is provided by the ophthalmic artery, which is derived from the internal carotid artery and passes beneath the optic nerve through the optic canal into the inner orbital wall. It then divides into the central retinal artery, the short and long posterior ciliary arteries, and the anterior ciliary arteries.
Figure 1.
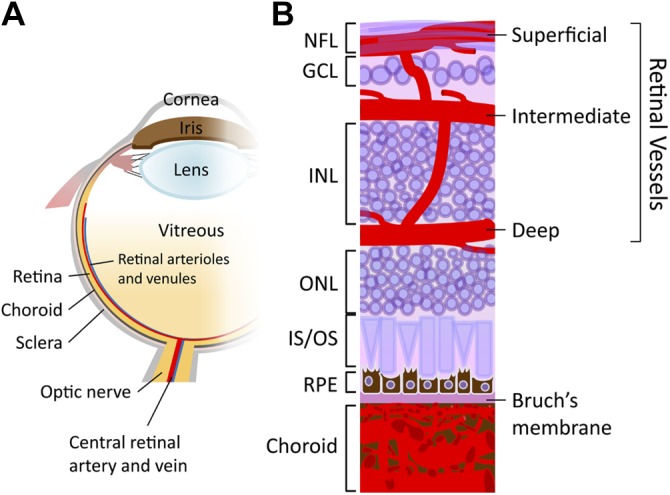
Schematic diagram of the ocular vasculature. A) Drawing showing a cross-section of an eyeball. B) An enlarged schematic illustration showing a cross-sectional view of the retinal and choroidal vasculature. Three interconnected layers of retinal vessels are embedded among retinal neurons: the superficial retinal vasculature lies in the nerve fiber layer (NFL) and the intermediate and deep retinal vascular networks along each side of the inner nuclear layer (INL). The choroidal vessels are located beneath RPE and Bruch’s membrane and supply oxygen and nutrients to the outer portion of the retina. GCL, ganglion cell layer; IS/OS, inner segment/outer segment of photoreceptor; ONL, outer nuclear layer.
Blood supply in the posterior segment of the eye
The human eye can be divided to two anatomic segments: the posterior segment in the back two thirds of the eye, including the vitreous humor, retina, choroid and optic nerve, and the anterior segment, the front third of the eye that consists of the cornea, iris, ciliary body, lens, and the anterior chamber that is filled with aqueous humor (Fig. 1A). During embryonic and fetal development, a transient vascular bed that surrounds the lens—the hyaloid vasculature that originates from the ophthalmic artery—provides blood supply to the developing eye, including the inner retina, and the lens during its maturation (15, 16). Hyaloid vessels first exist at the fourth week of gestation and reach maximum prominence at the ninth week in the human fetus. When the retinal vasculature develops during midgestation, hyaloid vessels then regress concurrently (16).
The retina is supplied by two blood sources: the central retinal artery, which is derived from the ophthalmic artery, and the choriocapillaris in the back of the retina, which is derived from the short posterior ciliary arteries. The central retinal artery travels along the inferior margin of the optic nerve sheath and enters the eye through the optic disk. The vessel then branches within the inner part of the retina to form nonfenestrated retinal vasculature, which is composed of three capillary layers: the superficial, intermediate, and deep layers that are located in the nerve fiber layer and along each side of the inner nuclear layer, respectively (17, 18) (Fig. 1B). Retinal vessels provide oxygenated blood to the inner retinal neurons up until the inner two thirds of the inner nuclear layer. In contrast, the retinal photoreceptor layer is avascular and relies on the choriocapillaris to supply oxygen by diffusion.
The choroid, a densely pigmented vascular membrane, sandwiches between the retina and the sclera. Choroidal capillaries (Fig. 1B) originate from both the short posterior ciliary arteries that supply the posterior portion of choroid and from the long posterior ciliary arteries that supply the anterior choroid, ciliary body, and iris. Choroidal vasculature has three interconnected distinct layers: the innermost choriocapillaris, which are characterized by an exceedingly fine capillary plexus adjacent to the Bruch’s membrane, the intermediate Sattler’s layer with gradually wider diameters of lumens, and the outermost Haller’s layer, which is composed mainly of small arteries and veins (16, 19, 20). The choriocapillaris is a highly fenestrated, sinusoidal vascular plexus and the site of the greatest blood flow in the body (21), comprising up to 85% of the blood volume in the eye to nourish the outer portion of retina and RPE (22).
Whereas the retinal vasculature completes its development before birth in humans, in rodents, the retinal vasculature develops postnatally (23, 24). In mice, retinal vascularization is initiated during the first postnatal week, matures by 3 wk, and is accompanied by simultaneous regression of hyaloid vessels (6). This postnatal development provides a major advantage of high accessibility and easy visualization to allow experimental manipulation, which has established the murine ocular vasculature as a widely used model system for studying angiogenesis both during development and in disease (6).
Vascular system in the anterior segment of the eye
Both the cornea and lens in the anterior segment of the eye are avascular to allow light to pass through. In the absence of blood vessels, the cornea receives oxygen that has been dissolved in the tears by diffusion. Nutrients that are supplied to the cornea are also transported via diffusion from the tears through the outside surface and from the aqueous humor through the inside surface, as well as from neurotrophins that are supplied by innervating nerve fibers. Similarly, the lens receives its nourishment for the aqueous humor via diffusion (25).
Conversely, the iris and ciliary body are supplied by the long posterior ciliary arteries and anterior ciliary arteries, respectively. Long posterior ciliary arteries—arising from the ophthalmic artery—pierce the posterior parts of the sclera, form an arterial circle around the circumference of the iris, and supply the ciliary body as well as the choroid. Anterior ciliary arteries are derived from the muscular branches of the ophthalmic artery and travel with the extraocular muscles, joining the arterial circle of the iris by piercing the sclera near the limbus (26).
ANIMAL MODELS OF OCULAR ANGIOGENESIS IN THE POSTERIOR SEGMENT
Pathologic angiogenesis in the posterior segment of the eye impacts retinal function and is a major feature in such diseases as proliferative retinopathies and neovascular AMD. Mouse models of oxygen-induced retinopathy (OIR) and laser-induced choroidal neovascularization (CNV) are two popular models with which to investigate pathologic neovascularization in the retina and choroid, respectively. In addition, transgenic knockout or knock-in mice with either deficient vascular development or spontaneous intraretinal or subretinal neovascularization also serve as great experimental tools with which to study the effects of specific genes in angiogenesis.
OIR
Aberrant retinal angiogenesis is one of the key features in many retinal diseases, including ROP and DR. Of note, ROP is the major cause of vision loss in premature neonates around the world, owing to incomplete development of retinal vascularization after premature birth and vascular suppressive effects by relative hyperoxia of the extrauterine environment compared with the in utero condition (27–29). Moreover, routine supplemental oxygen therapy for respiratory support also exacerbates the ocular vessel abnormalities in preterm infants (30, 31). Several common animal models of OIR were established in different species: feline (32), murine (33, 34), canine (35), and fish (36), all of which resemble ROP in humans. By exposure to consistent or cycling hyperoxia, OIR models successfully and reliably reproduce the two phases of the disease—an initial vaso-obliteration phase and a subsequent ischemia- and hypoxia-induced neovascularization phase. Exposure to high oxygen induces the cessation of growth or even loss of the existing retinal vasculature (6). After being returned to normoxic condition, inadequate vascular supply leads to retinal tissue ischemia and hypoxia, which results in increased levels of hypoxia-induced growth factors, including VEGF, that stimulate abnormal neovascularization at the junction between vascularized and avascular areas (37). Although the reproducibility of neovascularization varies across different model organisms, the ease of neovascularization induction, coupled with easy visualization and quantification, makes OIR models popular for the study mechanisms and potential therapeutics for ischemic proliferative retinopathies, including ROP and DR.
Feline and canine OIR models
In the early 1950s, the cat OIR model was the first model used to explore the effects of altering oxygen concentration on retinal vascular development (32). Kittens were exposed to 70–80% of oxygen for at least 4 d, which caused retinal vascular closure and obliteration, then returned to ambient air (21% oxygen), which resulted in hypoxia-induced vasoproliferation (32, 38). Similar studies were also performed in the less commonly used canine OIR model by the Lutty group (35, 39). Beagle puppies were exposed to 95–100% of oxygen for 4 d. The pattern and severity of the vascular reaction to hyperoxia in the puppy model is similar to those observed in the kitten model and in human infants (35, 39). Moreover, retinal detachment, which can be observed in the late severe stage of human ROP, occurs in the puppy model but not the kitten or murine OIR models (40).
Mouse OIR model: one of the most popular retinal angiogenesis models
The advantage of genetic manipulation in mice makes the mouse OIR model, described by Smith et al. (34), one of the most commonly used OIR models. Neonatal mice are exposed to 75% of oxygen for 5 d starting at postnatal day (P) 7, followed by 5 d in room air (Fig. 2A). Under hyperoxia from P7 to 12, retinal vessels regress in the central zone, which leads to vaso-obliteration, mimicking the initial phase of ROP, with maximal vaso-obliteration observed at P9 (41). From P9 to 12, slow revascularization begins in the vaso-obliterated areas (41, 42). At P12, after returning to normoxic condition, the vaso-obliterated area (Fig. 2B) becomes ischemic and hypoxic, which leads to up-regulation of hypoxia-inducible factor–dependent cascades (6, 43) that indicate an unmet oxygen demand. Hypoxia-inducible factor–responsive angiogenic factors, such as VEGF and erythropoietin, are triggered in the second phase of OIR (43–46), which leads to both physiologic revascularization of the vaso-obliteration area and uncontrolled compensatory pathologic neovascularization (Fig. 2B) that resembles human proliferative retinopathies (34). These pathologic neovessels—also called preretinal tufts—are disorganized, small-caliber vessels that grow at the border between vascular and avascular regions and that also protrude into the vitreous cavity (Fig. 2C, D). The extent of pathologic neovessels reaches the maximal severity at P17, the usual end point of this mouse model. Increased vascular leakage and breakdown of the blood-retinal barrier can also be observed. After P17, abnormal neovascularization starts a gradual regression spontaneously and completely disappears by approximately P25 (47). Both vaso-obliteration and neovascularization can be quantified as the percentage of total retinal areas in retinal flat mounts that are stained with isolectin B4 to visualize blood vessels (47, 48). Preretinal tufts that protrude into vitreous can also be visualized and quantified in retinal cross-sections (34) (Fig. 2D). Vascular leakage can be visualized by fluorescent angiography perfusion and quantified by using the Miles assay (29, 48–51). The mouse OIR model allowed some of the early work of investigating the role of VEGF in ROP (43), as well as of those factors that are critical in the third trimester and deficient after preterm birth, such as IGFs and ω-3 polyunsaturated fatty acids (52, 53). Inhibition of VEGF (45) or other angiogenic factors, such as erythropoietin (46); supplementing IGF-1 or ω-3 polyunsaturated fatty acids (52, 53); or modulation of microRNAs, such as miR-150 (54), are just a few examples of effective therapies against neovascularization in the mouse OIR model.
Figure 2.
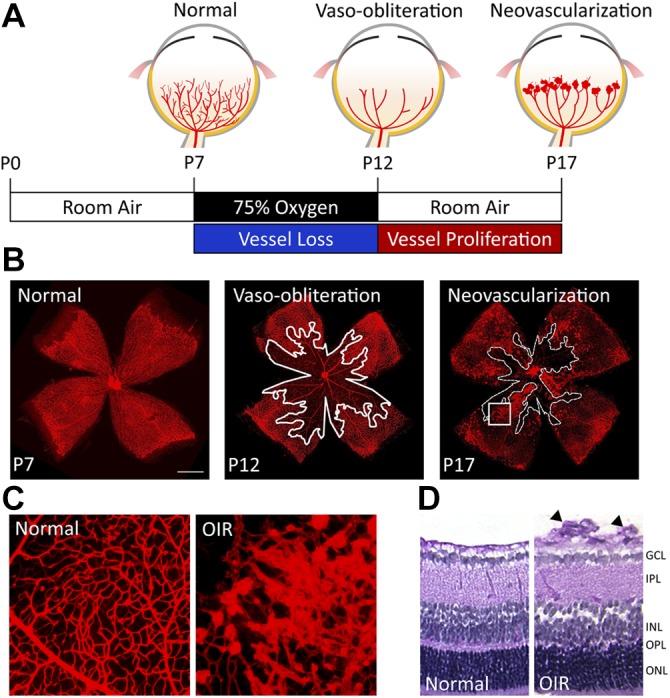
Mouse model of OIR. A) A schematic diagram depicting the mouse OIR procedure. Neonatal mice and their nursing mother are exposed to 75% oxygen from P7 to 12. Excessive oxygen suppresses the development of the retinal vasculature and leads to regression of the existing immature retinal vessels, which results in a central zone of vaso-obliteration. At P12, mice are returned to room air, and the relative hypoxia triggers both normal vessel regrowth toward the vaso-obliteration zone and pathologic retinal neovascularization at the border between the vascular and avascular zones, which also protrude toward the vitreous space. The levels of neovascularization reach maximum severity at P17. B) Representative images of retinal flat mounts with isolectin staining in the normoxic and OIR retinas show the normal retinal vasculature at P7 (left), vaso-obliteration at P12 (middle; the vessel loss area is highlighted with white outline), and pathologic neovascularization at P17 (right; the vessel loss area is highlighted with white dashed line). Scale bar, 1 mm. C) Magnified images of isolectin-stained retinal flat mounts show the normal retinal vasculature from normoxic retinas (left) and abnormal neovessels from OIR retinas (right; image enlarged from the rectangular white box region in panel B at P17). D) Cross-sections of mouse retinas with normoxia (left) and OIR (right) at P17. Preretinal neovascular tufts (arrowheads) arise from the superficial retinal vascular layer of the OIR retina and protrude into the vitreous space. GCL, ganglion cell layer; INL, inner nuclear layer; IPL, inner plexiform layer; ONL, outer nuclear layer; OPL, outer plexiform layer.
Rat OIR model: a model of peripheral severe ROP
Unlike the mouse model, which uses continuous oxygen exposure, the rat OIR model—developed by Penn et al. (33, 55)—utilizes alternating hyperoxia–hypoxia cycles, where the oxygen levels cycle between 50 and 10% every 24 h for the first 14 d after birth, followed by room air exposure through P20. Exposure to cycling levels of oxygen slow the retinal vascular development, which leads to a peripheral avascular zone. After returning to room air, pathologic neovessels grow at the boundary between vascularized and avascular areas in the midperipheral retina. The rat OIR model produces vaso-obliteration in the peripheral zone of the retina, which is geographically similar to the peripheral avascular zone that has been observed in human ROP, whereas in the mouse model, the central retinal vessels are obliterated (37, 56).
Previous clinical studies have shown that, in addition to continuous oxygen supplement, oxygen fluctuation also contributes to the incidence of severe ROP (57, 58). The partial pressure of dissolved arterial oxygen can fluctuate quickly in premature infants, which leads to the alternating occurrence of severe and extended hyperoxemia and hypoxemia (57) that may lead to a higher risk of developing ROP. The rat model recreates this oxygen tension fluctuation, which mirrors oxygen levels measured in preterm infants who develop severe ROP, whereas in the mouse model, the oxygen level remains constant (27). Overall, the rat model shows clinically relevant features of ROP, with delayed retinal vascular development and subsequent pathologic neovascularization, yet there is lack of genetic manipulation in rats, and the levels of observed neovascularization are relatively low compared with mouse OIR model, which limits its use in screening antiangiogenic compounds.
Zebrafish model of hypoxia-induced retinal angiogenesis
In addition to the mammal models, Cao et al. (36, 59) generated an adult zebrafish model of hypoxia-induced retinopathy. Adult fli1:EGFP transgenic zebrafish were placed in a hypoxic aquaria with 10% air saturation (820 ppb) for 3–12 d, and new vessel sprouts reached a plateau of maximal angiogenic responses at d 12. In a separate study, Wu and colleagues induced a model of ROP in zebrafish embryos by using CoCl2, a hypoxia-inducing agent, and a VEGF inducer, GS4012, to stimulate abnormal retinal angiogenesis. Fli1:EGFP zebrafish embryos were treated with CoCl2, followed by GS4012 at 1 d postfertilization, which significantly increased the number of vascular branch points and sprouts observed 2–4 d after treatment (60). These zebrafish models serve as additional models to supplement the mammal OIR models for angiogenic studies and drug screening.
OIR as a model for proliferative DR
The murine OIR model is not only an ideal experimental model for ROP, but is also considered a model for proliferative DR, as current rodent models of DR—either genetic or drug-induced—reproduce the early nonproliferative stage of DR, but do not develop the prominent proliferative stage (61), likely as a result of the short lifespan of rodents. DR is one of the most common complications of diabetes, yet the development of clinical DR takes decades. DR is characterized by a chronic and slow process of microvascular degeneration in the initial nonproliferative phase that, in some patients, advances to a subsequent proliferative phase, which results in abnormal vascular architecture and increased permeability (62). Chronic hyperglycemia induces biochemical changes in endothelial cells, which leads to their degeneration. Hypoxia and ischemia as a result of the initial microvascular degeneration is considered one of the factors that leads to increased VEGF, among other angiogenic growth factors, and results in the proliferative phase with neovascularization (63).
Pharmacologically induced models of DR, such as streptozotocin- and alloxan-induced type I diabetic mice (64, 65), do not fully simulate the DR disease process and lack the proliferative DR symptoms. With the introduction of ischemia and hypoxia in the retina, OIR models are considered to be compensatory models with which to study the angiogenesis aspect in proliferative DR (37); however, because of the lack of systemic diabetic characteristics, the progression of vascular abnormalities in OIR models is fundamentally different and does not mimic the hyperglycemia aspects in DR. Spontaneous hyperglycemia in animal models that carry an endogenous mutation, such as Ins2Akita, nonobese diabetic, db/db, and KKAy mice, develop relatively consistent systemic phenotypes of type 1 or 2 diabetes as well as retinopathies with varying extents of acellular capillaries and pericyte dropout (66–70), with modest levels of diabetic retinal angiogenesis reported in aging Ins2Akita mice (66).
Laser-induced CNV
CNV lesion refers to the abnormal new vessel growth from the choriocapillaris that extends through Bruch’s membrane into the sub-RPE and/or subretinal spaces. Left untreated, CNV may advance to a cicatricial stage, known as disciform scar, that results in fibrotic scar that underlies the retina and impairs vision (37). CNV is a feature of numerous eye diseases, such as neovascular AMD, pathologic myopia, pseudoxanthoma elasticum with angioid streaks, and ocular histoplasmosis syndrome (71, 72). Formation of CNV involves a primarily angiogenic process that is intertwined with both inflammation and proteolysis (73). Changes in pro- and antiangiogenic growth factors derived from RPE have been considered a primary underlying cause of CNV pathogenesis. Proangiogenic factors, such as VEGF, basic fibroblast growth factor (bFGF), and PDGF, can all stimulate endothelial proliferation in CNV, with VEGF being a main driver of both clinical and experimental CNV (74–76). Disruption or degradation of Bruch’s membrane is another early step of CNV initiation, which allows proliferating neovascular tissue derived from the choroid to invade the subretinal space, eliciting an inflammatory response. Many inflammatory pathways, including the pro- and ant-inflammatory cytokines, chemokines, and the widely studied complement cascade, are implicated in the formation of CNV. Macrophages release multiple inflammatory and angiogenic factors that attract and stimulate monocyte recruitment and endothelial cell migration and proliferation and, thus, are also critical for CNV pathogenesis (37). Moreover, the imbalance between proteolytic components, such as matrix metalloproteinases and tissue factors, may also impact the degradation of extracellular matrix and the formation of the fibrin matrix scaffold, which are essential for CNV initiation and the formation of the fibrovascular membrane (77, 78).
The ideal experimental models of CNV must be stable, efficient, and reproducible, and exhibit pathologic features that are similar to those observed in human CNV lesions. The laser-induced CNV model is one of the most widely used models that recapitulates the angiogenic aspect of neovascular AMD. The laser-induced CNV model is relatively rapid to develop and establish, and uses laser photocoagulation to injure the outer retina and disrupt the Bruch’s membrane, which leads to the formation of CNV that infiltrates the subretinal space (79).
Feline and primate models of laser-induced CNV
The cat model of subretinal neovascularization that was developed by Baum et al. (80) was one of the first animal models to demonstrate CNV lesions and retinal detachment induced by photocoagulation. In late 1970s, Ryan et al. (81) established a primate model of CNV by using an argon laser to induce the disruption of Bruch’s membrane in monkeys. This study showed that new choroidal vessels grew into the laser-injured subretinal areas with increased expression of proangiogenic factors, and provided evidence that inflammation played an important role in CNV formation (81, 82). Ultrastructural analysis of the neovascular CNV sprouts revealed primitive endothelial tubes surrounded by pericytes and entangled with loose basement membrane–like substances (83).
Rodent models of laser-induced CNV
The laser-induced CNV model has extended to rodents since the late 1980s (84–86) and is now used extensively in both the rat and mouse to study CNV mechanisms and evaluate antiangiogenic intervention (87–89). In the mouse model, laser photocoagulation is carried out with laser spots that are approximately 2 discs in diameter from the optic nerve (Fig. 3A). Formation of an acute vapor bubble indicates the successful rupture of Bruch’s membrane (79). CNV lesions reach maximal levels at d 7 postlaser, which can be visualized and quantified for leakage and lesion size by using fluorescein fundus angiography (Fig. 3B) and immunohistologic staining (Fig. 3C, D) (90). CNV then regresses and completely disappears approximately 1 mo postlaser. Subretinal fibrosis at the lesion sites can also be observed and continues to increase through d 35 postlaser (91). This fibrotic component of CNV can be used to study subretinal fibrosis, the progression of which is often considered a contributing factor in the lack of response to anti-VEGF therapies.
Figure 3.

Laser-induced CNV model. A) Scheme of laser-induced CNV in mice. Adult mice are anesthetized and pupils are dilated. Laser burns are produced in the 3, 6, 9, and 12 o’clock positions around the optic disc (OD), with the laser focused on the RPE. The presence of a subretinal bubble confirms that the laser impact caused the disruption of Bruch’s membrane and RPE, which is necessary for the induction of successful CNV lesions. At 1 and 2 wk after laser treatment, CNVs growing into the subretinal space are observed and analyzed by using fundus fluorescein angiography (FFA) and dissected choroidal flat mounts with isolectin staining. B) A representative image of FFA from a mouse on d 6 after laser burns shows the formation of CNV (green lesions, arrowheads) beneath the retinal vasculature (green). C) A fluorescence microscopy image of an isolectin-stained flat mount of the mouse choroid/RPE at 1 wk after laser treatment shows the extent of CNV lesions (arrowheads). Scale bars, 500 µm. D) Immunohistologic examination of a cross-sectioned mouse eye at 1 wk after laser photocoagulation shows extension of the CNV lesion (red, stained with isolectin; arrow) into the subretinal space between photoreceptors and RPE. Scale bars, 100 µm. GCL, ganglion cell layer; INL, inner nuclear layer; ONL, outer nuclear layer.
The mouse model of laser-induced CNV is reproducible, cost-effective, and relatively simple to establish with short experimental duration, and also allows genetic manipulation—its major advantages. Although the laser-induced CNV model is often referred to as a neovascular AMD model (92), as an acute injury model, in addition to the lack of macula in rodents, it also does not recapitulate the aging aspect of AMD. This model is also limited by the miniature size of the mouse eye and the artificial nature of the CNV induced via laser photocoagulation. In addition, lesion sizes often show considerable variation among animals or even within the same eye, likely as a result of technical variations in each laser application. Sufficient sample size and rigorous pre-established inclusion and exclusion criteria are necessary to obtain reliable results (90, 93, 94). Nevertheless, the laser-induced CNV model is one of the standard models with which to investigate choroidal angiogenesis and has been successful in predicting the therapeutic value of anti-VEGF therapies (95), which, together with the pioneering work in both OIR models and iris angiogenesis, has laid the experimental foundation for current anti-VEGF therapies (45, 95).
Surgically induced CNV
In addition to laser-induced CNV, another less commonly used CNV model uses a subretinal injection of Matrigel to induce CNV in rats and mice (96, 97). CNV can be observed 4 d after Matrigel injection and increases in size progressively after, up to 20 d in rats. Compared with laser-induced CNV, the Matrigel-induced model can be more convenient and feasible for researchers who are without access to laser photocoagulation. Moreover, transgenic animal models of CNV were also discovered, detailed in the following section.
Genetic models of abnormal intraretinal and subretinal angiogenesis
Pathological subretinal vessel formation may originate not only from the choroid, but sometimes also from the retinal vasculature. Abnormal intraretinal angiogenesis that invades the subretinal space with the formation of a retinal–choroidal anastomosis is characteristic of a distinct variant of neovascular AMD: retinal angiomatous proliferation (RAP). RAP is present in approximately 12–15% of patients with newly diagnosed neovascular AMD (98–101). Studying RAP requires animal models that exhibit intraretinal, subretinal, and choroidal angiogenic features that mimic the observations of these three stages of vasogenic sequence in human patients. Several genes have been discovered, including very-LDL receptor (VLDLR) and VEGF, in which mutations or dysregulation influence RAP pathogenesis (102–105). Transgenic mouse models with VLDLR knockout or VEGF overexpression were thus established, both of which demonstrate characteristic features of RAP-like lesions as well as CNV in the late stage in VLDLR-knockout mice (102, 104–106).
VLDLR germline-knockout mice
Mice that lack VLDLR were discovered in the last 2 decades as a unique spontaneous model of RAP and neovascular AMD (Fig. 4) (102, 106). As an 86-kDa transmembrane protein that was initially identified in endothelial cells (107, 108), VLDLR belongs to the LDL receptor family (109). In mammals, VLDLR was detected in the retina, especially in retinal endothelial cells and RPE (106). Genetic variations in VLDLR are significantly associated with AMD in humans (110), with the mixed dry and neovascular forms (106, 110).
Figure 4.
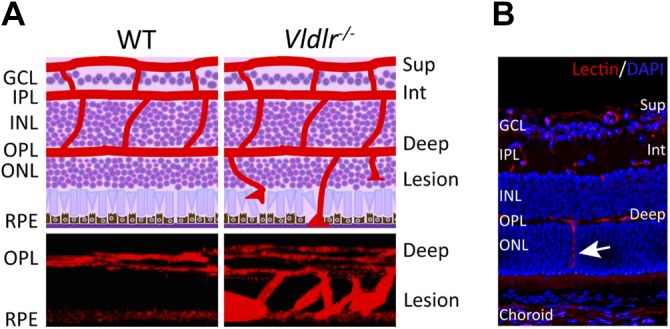
Vldlr−/− mouse model of retinal angiomatous proliferation. A) Schematic diagrams (top) show the retinal neuronal layers (purple) and 3 layers of retinal blood vasculature (superficial, intermediate, and deep; red) on cross-section of wild-type (WT) and Vldlr−/− mice, with blood vessels in Vldlr−/− retinas extending from the deep layer toward RPE. Three-dimensional reconstructed images of vascular lesions in the photoreceptor layer of Vldlr−/− retinas compared with the avascular photoreceptor layer in WT retinas (bottom). Blood vessels were stained with isolectin (red, vessel marker). B) Immunohistochemical staining with isolectin (red) and DAPI (blue, nuclear marker) on a Vldlr−/− mouse retinal cross-section shows abnormal vessel growth (arrows) that originates from the deep vessel layers through the normally avascular photoreceptor layer [outer nuclear layer (ONL)] and toward RPE [reprinted from Hua et al. (112), copyright owned by the Association for Research in Vision and Ophthalmology]. Deep, deep retinal vessel; GCL, ganglion cell layer; INL, inner nuclear layer; Int, intermediate retinal vessels; IPL, inner plexiform layer; OPL, outer plexiform layer; Sup, superficial retinal vessel.
In the 1990s, Herz and colleagues (111), by using targeted disruption, generated Vldlr germline-knockout (Vldlr−/−) mice, which were later characterized as a model of subretinal neovascularization and choroidal anastomosis by Heckenlively et al. (102). The abnormal neovessels in Vldlr−/− retinas sprout from the deep vascular layer in the outer plexiform layer and invade the subretinal space as early as P15 (Fig. 4) (102, 112). Subretinal hemorrhages and choroidal anastomosis are evident in Vldlr−/− eyes by age ∼2 mo and increased vascularity of the iris and ciliary body can be observed by age 8 mo (102), with secondary photoreceptor degeneration and subretinal fibrosis (106).
The spontaneous intraretinal neovascularization in Vldlr−/− mice has been attributed to the altered expression of angiogenic factors, most prominently VEGF and bFGF (103). VEGF levels in Vldlr−/− retinas were increased in the photoreceptor layer around the area of vascular lesion at age 4 wk. Expression of bFGF was also observed around Vldlr−/− lesions (103). Moreover, mice with Vldlr deficiency developed other pathologic features of wet AMD, such as degeneration of rod and cone photoreceptors and chronic inflammation in the retina and RPE (113, 114). The unique pathological features in this mouse model enabled numerous studies that investigated the potential mechanisms and treatments of AMD, RAP, and macular telangiectasia, including antioxidants, neurotrophic factors, resveratrol, anti-inflammation, cellular bioenergetics, and transcriptional control (112, 114–119), where dietary treatments with either antioxidants or resveratrol were demonstrated to be effective therapies to suppress Vldlr−/− neovascularization.
Rhodopsin/VEGF transgenic mice
VEGF was recognized as a critical inducer of pathological ocular neovascularization since the clinical discovery of elevated VEGF in the eyes of patients with proliferative retinopathy (29, 120). VEGF also regulates retinal vascular development by mediating the proliferation and migration of endothelial cells (121). Alternative splicing variants of VEGF have distinct roles in retinal angiogenesis (122). In humans, VEGF is produced as 3 isoforms, VEGF121, VEGF165, and VEGF189, whereas VEGF isoforms in mice are VEGR120, VEGF164, and VEGF188, with differences in their solubility, molecular mass, receptor binding, and ability for heparin sulfate binding (123). VEGF isoforms bind mainly to 2 tyrosine kinase receptors, VEGR receptor 1 (VEGFR1; Flt-1) and VEGFR2 (KDR/Flk-1), both of which are expressed in endothelial cells (123). VEGF165 also binds selectively to neuropilin-1 and neuropilin-2 coreceptors, which hare members of the semaphorin receptors (123). Early studies that used the mouse OIR model demonstrated that the inhibition of VEGF signaling via soluble Flt-IgG chimeric proteins reduced retinal neovascularization (45), which highlighted therapeutic value of VEGF inhibition in neovascular ocular diseases. Numerous studies have investigated anti-VEGF therapies that demonstrated efficacy in controlling ocular neovascularization in eye diseases (11, 12, 124–126). In addition, VEGF also maintains cellular homeostasis for vascular integrity in the eye (127, 128), and its role was further exemplified in transgenic overexpression model mice.
VEGF-overexpressing transgenic mice were developed by the Campochiaro group in the late 1990s by overexpressing VEGF in photoreceptor cells by incorporating a full-length cDNA of human VEGF under a bovine rhodopsin promoter (104, 105). This rhodopsin/VEGF transgenic mouse model (rho/VEGF mouse) shows focal areas of intraretinal and subretinal neovascularization with vascular leakage (104). In adult rho/VEGF retinas, blood vessels extend from the inner nuclear layer toward RPE to form a large plexus of blood vessels in the subretinal space. In addition, rho/VEGF retinas demonstrate substantial loss of photoreceptors and disorganized inner nuclear layer, which indicates severe photoreceptor degeneration (104). This model, which presents with features that are similar to RAP, demonstrates that overexpression of VEGF in photoreceptors is sufficient to induce intraretinal and subretinal neovascularization, rather than choroidal neovascularization, when RPE and Bruch’s membrane are intact. This model is useful for investigating early VEGF-induced changes that occur in the retina and ultimately lead to neovascularization, as well as for testing the effects of purported angiogenesis inhibitors.
JR5558 mice
The neoretinal vascularization 2 mouse, also called the JR5558 mouse, is another recently discovered spontaneous model that exhibits intraretinal and subretinal neovascularization and is VEGF-A dependent (129, 130). This novel mouse model harbors currently unknown genetic mutations (131) that seem to have a recessive mode of inheritance. Eye abnormalities include multiple areas of retinal depigmentation and neovascularization that are associated with vascular leakage. Retinal neovascularization originates from the retinal vascular plexus and grows toward the subretinal space, forming neovascular structures at the RPE and Bruch’s membrane interface and mimicking the early clinical presentation of RAP in humans. Of interest, vascular abnormalities in JR5558 mice are considerably similar to those observed in Vldlr−/− mice, which suggests that, perhaps, the underlying genetic variations may include, or be related to, VLDLR. Conversely, JR5558 mice may harbor a mutation of Rd8, a retinal degeneration gene (132); therefore, interpretation of retinal neuronal pathologies in aged JR5558 mice requires some caution. Nevertheless, this new model provides a useful tool for the study of RAP and AMD, as well as for evaluating potential therapeutics for both diseases, such as IL-18 immunotherapy and C-C chemokine receptor type 3 antagonists (130–135).
Together, these genetic models, combined with induced CNV models, may mimic several important pathologic features of AMD, such as CNV in wet AMD and RAP in the genetic mouse models (85–89, 102, 111, 136). Yet none of these models recreated all of the pathologic features of human AMD, which has risk factors with both genetic and environmental components. Combined with the fact that murine models are anatomically deficient of macula, these animal models, each with a disruption of a single gene or unknown genetic variations, have their limitations in thoroughly capturing the complexity of the multifactorial pathogenesis of neovascular AMD.
Genetic models of deficient intraretinal angiogenesis
Incomplete or deficient intraretinal angiogenesis has been observed in several rare human eye diseases, including familial exudative vitreoretinopathy (FEVR) and Norrie disease. FEVR is a hereditary disorder with retinal hypovascularization that is characterized by incomplete peripheral retinal vasculature, lack of deep retinal vascular layers, and, in less common cases, persistent hyaloid vessels (137). In some severe cases, compensatory preretinal neovascularization and fibrosis can occur, which may result in retinal detachment, retinal folds, and visual impairment. The related Norrie disease—an X-linked recessive disease with retinal hypovascularization—primarily affects the eyes in male infants at birth, or soon after birth, and may lead to blindness. In addition to ocular symptoms, some patients with Norrie disease also show mental retardation and hearing difficulty that may be associated with vascular defects in the inner ear (138, 139).
Genetic studies have linked both FEVR and Norrie disease to the Wnt signaling pathway, which plays a major role in both physiologic and pathologic angiogenesis (140). The most common mutations implicated in FEVR are in genes that encode the Wnt receptor, frizzled-4 (FZD4), and the coreceptors, LDL receptor–related protein 5 (LRP5) (141) and tetraspanin 12 (TSPAN12) (142). In contrast, Norrie disease is linked with mutations in the NDP (Norrin) gene that encodes the atypical high-affinity Wnt pathway ligand, norrin (143), with limited structural similarity to other Wnt ligands. In the canonical Wnt signaling pathway, ligands (Wnts or norrin) bind to a cell-surface receptor complex that consists of frizzled receptors and LRP5/6, which leads to the stabilization of cytoplasmic β-catenin by attenuating its phosphorylation. The nonphosphorylated β-catenin then translocates to the nucleus, where it associates with T-cell factor/lymphoid enhancer factor transcription factors to activate the transcription of Wnt target genes, including growth factors that participate in ocular angiogenesis (144, 145). Genetically engineered mice that lack Wnt signaling components, such as FZD4, LRP5, norrin, and TSPAN12, all exhibit similar disease-linked phenotypes in the ocular vasculatures (143, 146–151).
Mouse models of FEVR and Norrie disease: Lrp5-knockout and Norrin-knockout mice
Mice with genetic deficiency in Lrp5 (Lrp5−/−) or Norrin (Ndpy/−) show similar ocular vascular abnormalities that resemble human pathologies in FEVR and Norrie disease, respectively (143, 146–149). Lrp5−/− mice have delayed primary retinal angiogenesis, with more sparse vascular coverage at P7 (Fig. 5A) (148, 149, 152), and a complete absence of the secondary and tertiary vascular plexus in the adult (Fig. 5B, C) (148, 149, 152, 153), as well as persistent hyaloid vasculature (Fig. 5D) (146, 148, 149, 154). Remarkably similar, yet more severe, vascular defects are observed in Ndpy/− eyes, with intraocular hemorrhage in some cases (143, 147). The absence of inner retinal vessels in both Lrp5−/− and Ndpy/− eyes contributes to a secondary hypoxia, which leads to a compensative up-regulation of Vegfa and, consequently, has been associated with increased pathologic glomeruloid vascular structures in the superficial plexus (147, 149, 155). Deficient intraretinal vessels and hypoxia in the Lrp5−/− retina are also associated with impaired visual function with diminished b wave in electroretinography (149, 156).
Figure 5.
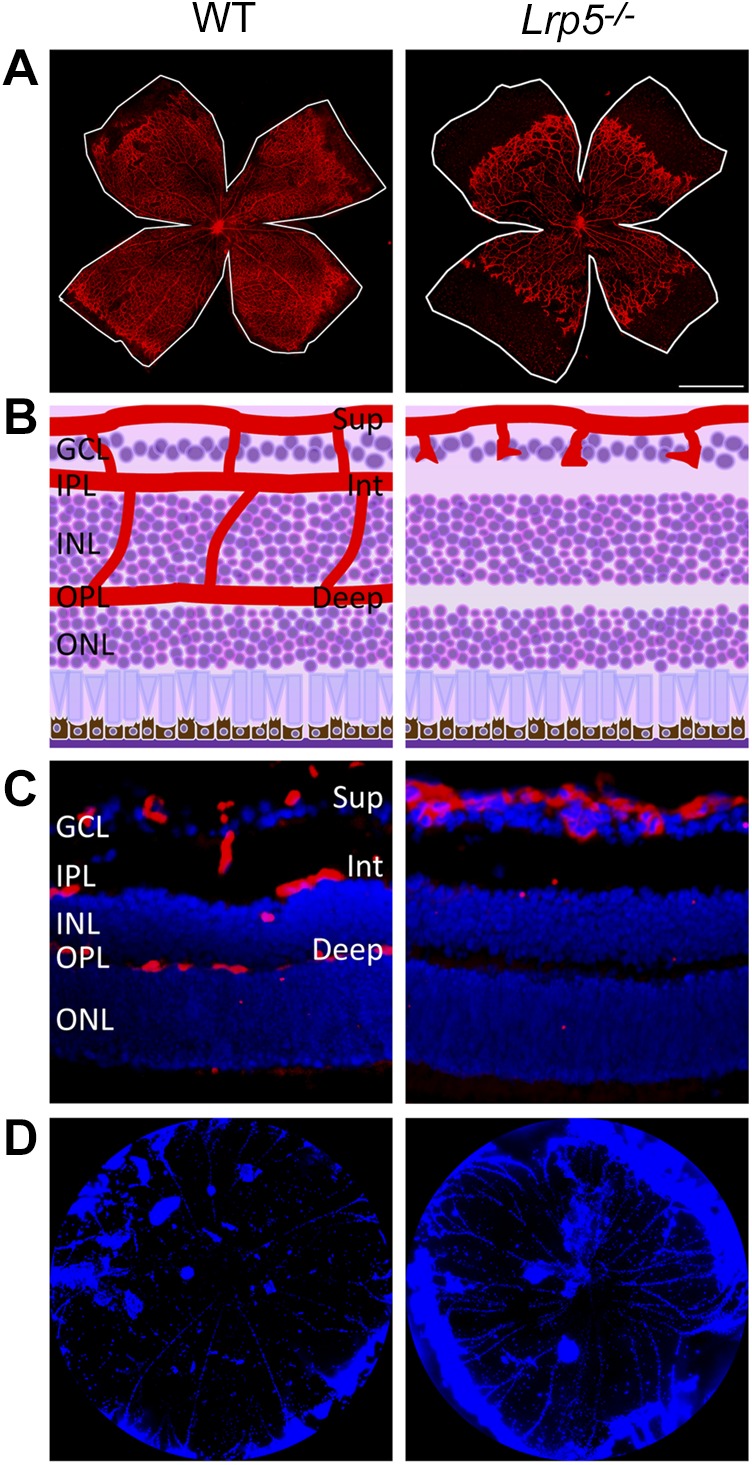
Lrp5−/− mouse model of deficient intraretinal angiogenesis. A) Isolectin-stained (red) mouse retinal flat mounts show delayed retinal vascular development in Lrp5−/− mice (right) compared with wild-type (WT; left) at P7. White lines indicate the edge of the retinal periphery. B) Schematic representation of the retinal layers and blood vasculature on cross-section of WT and Lrp5−/− mice. C) Immunofluorescence images showing the vasculature in WT and Lrp5−/− mouse retinas. Endothelial cells (red) and nucleus (blue) were visualized by staining with isolectin and DAPI, respectively. D) DAPI-labeled (blue) vitreous bodies that were isolated from WT (left) and Lrp5−/− (right) mice at P8. Lrp5−/− mice retain more persistent hyaloid vasculature compared with WT mice. Deep, deep retinal vessel; GCL, ganglion cell layer; INL, inner nuclear layer; Int, intermediate retinal vessels; IPL, inner plexiform layer; ONL, outer nuclear layer; OPL, outer plexiform layer; Sup, superficial retinal vessel.
Loss of Wnt signaling also causes a breakdown of the blood-retina barrier in both Lrp5−/− and Ndpy/− eyes (152, 157), with a substantial induction of an endothelial fenestration marker, plasma lemma vesicle–associated protein, and suppression of an endothelial tight junction protein, claudin5 (148, 157, 158). Furthermore, Lrp5−/− and Ndpy/− eyes both demonstrate persistent hyaloid vasculature, likely as a result of the incomplete vascularization of the intraretinal vasculature that demands more metabolic support from hyaloid, thereby impeding its regression (149). Both FZD4 and LRP5 are also expressed in hyaloid endothelial cells, which may directly induce apoptosis of hyaloid endothelial cells (146), potentially by responding to macrophage-derived Wnt7b-mediated programmed cell death (159).
In addition to LRP5 and Norrin, mutations in FZD4 and TSPAN12 have also been observed in human patients with FEVR (141, 142). Similarly, both Fzd4- and Tspan12-null mice demonstrate strikingly similar hypovascularized phenotypes (150, 151). Currently, there is no effective therapy to treat or restore the hypovascularization, although overexpression of Norrin restored the vascular defects in mouse retinas with Norrie disease (160), and pharmacologic activation of Wnt signaling with the Wnt activator, lithium, partially promoted the normalization of vasculature and visual function in Lrp5−/− eyes (149). Overall, mice with mutations in Norrin/FZD4/LRP5 signaling are valuable for the study of the basic mechanisms of intraretinal angiogenesis and for exploration of the pathophysiology and therapeutics for both FEVR and Norrie disease, as well as molecular control of blood-retinal barrier and hyaloid vessel regression.
Girdin-knockout mice
In addition to the Wnt signaling pathway, the PI3K/AKT pathway—essential in VEGF-mediated angiogenesis—also affects intraretinal vascular growth. Girdin, an AKT substrate that functions as an actin-binding protein, is important in VEGF-dependent endothelial cell migration and tubular formation (161, 162). Girdin-knockout (Girdin−/−) retinas showed more sparse vasculature coverage at P7 and incomplete intermediate and deep vascular layers at P18 (163). The Girdin-KISA/SA mouse model, with a knock-in point mutation (S1416A) that disrupts the AKT phosphorylation site of girdin (164), presents abnormal retinal angiogenesis that is similar to that in Girdin−/− mice. Together these Girdin mutant mice provide additional animal models with which to study the mechanisms of intraretinal angiogenesis, which may or may not be directly related to Wnt signaling.
ANIMAL MODELS OF OCULAR ANGIOGENESIS IN THE ANTERIOR SEGMENT: CORNEAL VASCULARIZATION MODELS
The cornea has been a proving ground for antiangiogenic strategies for decades because of its avascularity and easy accessibility (165). As one of few tissues that are completely devoid of blood vessels under normal conditions, the cornea is essential for optical clarity and optimal vision (166). The underlying mechanisms of its avascular characteristics have been extensively studied and its avascularity provides a unique system with which to evaluate the pro- and antiangiogenic potential of various factors. Both systemic and local antiangiogenic mediators have been suggested to contribute to the maintenance of corneal avascularity (166–169). Recent studies have suggested that soluble VEGFR1 (also known as sFlt-1), which is highly expressed in the cornea, serving as an endogenous VEGF-A trap (170, 171), and the ectopic expression of VEGFR3 (Flt-4) by the epithelium (172) are key modulators that inhibit blood vessel invasion of the corneal stroma. In addition, corneal avascularity is also associated with the integrity of the corneal epithelium. The corneal epithelium is endowed with extensive regenerative capacity in response to injury, which requires functional limbal stem cells (173, 174). The WNT7A-PAX6 (paired box protein 6) signaling pathway regulates the differentiation of limbal stem cells into corneal epithelial cells, the disturbance of which may cause metaplastic changes, including subepithelial corneal neovascularization (175).
Clinically associated with allograft failure and the loss of visual acuity, the etiologies of corneal neovascularization are typically secondary to two types of conditions: inflammation or hypoxia (176). Pathologic corneal neovascularization occurs when new blood vessels invade the cornea from the limbus, in some diseased conditions with hypoxia, infection, inflammation, and poor limbal barrier function, which leads to decreased corneal transparency and, ultimately, vision loss (177). Four general animal models of corneal neovascularization have been developed with which to investigate the pathogenesis and to evaluate novel antiangiogenesis drugs: the chemical injury model, the cornea suture model, the corneal micropocket assay, and the transgenic model.
Chemical burn model
Chemical burns are a common ocular emergency that requires immediate assessment and treatment and is easy to imitate in animal models. Alkali injuries occur more frequently and are more severe than acid injuries (178). The alkali burn corneal neovascularization model has been widely used in rats, mice, and rabbits (179–181), with some modification of an established procedure developed by Ormerod et al. (182). In this work, a paper disc soaked with NaOH (1 N) is applied for a short duration—typically 10–30 s—followed by intense washing with sterile saline solution (179, 180, 183). Corneal neovascularization is induced on the surface of cornea and can usually be evaluated at 7–14 d after the procedure (184), when sprouting vessels grow from limbal vasculature that points toward the injured area (Fig. 6A, E). The extent of vessel growth can be quantified by the area of the vessels on the cornea (179, 185). The chemical burn model is easy to perform and highly relevant to clinically scenarios, yet the limitations of the model include the extensively damaged corneal tissues after alkali burn, which may evoke a number of other processes, such as wound healing, epithelial proliferation, and inflammation, which can interfere with angiogenesis.
Figure 6.

Murine models of corneal angiogenesis. A) Chemical injury model by alkali burn. A filter paper disk that is saturated with 1 N NaOH or control saline solution is applied to the center of the cornea for 30 s, followed by rinsing with saline. At 7–14 d after the alkali burn, corneal angiogenesis can be observed as neovessels growing from the limbus toward the injured region. B) Suture injury model. The cornea is artificially perforated with 10.0 nylon sutures in the experimental animal. Corneal neovascularization can be triggered and observed within 2 wk after the procedure. C) Corneal micropocket assay. A micropocket with a pellet (yellow) that contains proangiogenic factors or vehicle control is created in the stroma of the cornea. At 5–7 d after pellet implantation, corneal neovascularization can be observed and evaluated. D) Transgenic spontaneous models of corneal neovascularization. Genetically engineered mice, such as knockout of Destrin, CD36, or soluble VEGFRs, as well as overexpression of PAX6 in corneas, develop spontaneous corneal neovascularization. E) Representative images of rat corneas that were treated with control solution (Ctrl) or alkali solution-soaked filter papers. The cornea with alkali burn shows neovessels (arrowheads) growing from limbal vessels toward the central corneal burn. F) Mouse corneas with pellet implantation into micropockets. No abnormal corneal angiogenesis is observed with the implant of control buffer–containing pellet, whereas the pellet with angiogenic factor (arrow) induces pathologic vessel growth (arrowheads) from the limbus. Red arrows in panels A–D represent the direction of blood vessel growth.
Suture injury model
Suture placement is another injury model with which to induce corneal neovascularization (165). One or more 10 nylon sutures are placed eccentrically in the corneal stroma 1.5 mm from the limbus. These nylon knots are left unburied and the threads remain in the corneal stroma, which induces inflammation and neovascularization (186). Corneal neovascularization is usually evaluated 7–14 d after the procedure, and new vessels grow from the limbal vasculature toward the suture placements (Fig. 6B) (183). Blood vessel growth is similar between suture and alkali burn-induced models, yet corneal lymphangiogenesis is more evident in the suture-induced model (183), which may represent an advantage for lymphangiogenesis studies.
Micropocket assay
The corneal micropocket assay that was originally developed in rabbit is a frequently used angiogenesis model (165). Later adapted for mice (187), this assay is performed by surgically creating a micropocket in the corneal stroma of the cornea 1.0–1.5 mm from the limbus and implanting a pellet in the micropocket (Fig. 6C). The pellet is made of slow-release hydron polymers [poly(2-hydroxyethyl methacrylate)] that contain ≥1 proangiogenic factors, which are gradually set free in the course of several days (179, 188, 189). The extent of corneal neovascularization is typically assessed 5–7 d after pellet implantation (Fig. 6F), and both VEGF-A and bFGF are the most commonly used angiogenic factors in pellets to stimulate vessel growth (189–191). The advantages of this model include its relative noninflammatory nature and the absence of corneal edema.
Genetic models with spontaneous corneal neovascularization
In the past two decades (192), several genetically engineered mice were discovered that develop spontaneous corneal neovascularization (193), such as deficiency of Destrin (194) and CD36 (195), overexpression of Pax6 (196), and conditional knockout of soluble VEGFR1 (171) and soluble VEGFR2 (197). Depletion of Destrin leads to both corneal hemangiogenesis and lymphangiogenesis starting at 4 wk (194), whereas loss of CD36 induces corneal angiogenesis that increases in severity with age at 1 yr (196). Corneal overexpression of Pax6 produces neovascularization that is associated with altered epithelial cell morphology and the invasion of immune cells (196). Of importance, soluble VEGFR1 is expressed in the cornea, which preserves its avascularity, and genetic disruption of soluble VEGFR1 in the cornea leads to corneal neovascularization (171). Similarly, both Destrin- and Pax6-deficient mice demonstrate corneal deficiency in soluble VEGFR1 (171). Loss of soluble VEGFR2, an alternative splice variant, also leads to spontaneous corneal lymphatic invasion, and administration of soluble VEGFR2 inhibited lymphangiogenesis, but not hemangiogenesis, in the corneal suture injury model (197). Together, these transgenic mouse models of corneal neovascularization are handy tools with which to identify and investigate multiple intertwined signaling pathways that maintain corneal avascularity. Disruption of these pathways may ultimately lead to corneal neovascularization.
SUMMARY
Ocular neovascularization is a major cause of visual impairment. Whereas each animal model has its unique features and strengths and its limitations (summarized in Table 1), these models have together served as experimental tools with which to explore the many aspects of ocular angiogenesis and have greatly advanced our current understanding of angiogenesis in general, including the roles of growth factors, fatty acids, inflammation, and signaling cascades, as well as transcriptional regulations, noncoding RNAs, and bioenergetic metabolism. Whereas these models will continue to be valuable for future studies, investigators must match their areas of interest and working hypotheses with the best suitable model. For instance, the OIR model is one of the most representative models for studying retinal and tissue ischemia and hypoxia-induced angiogenesis. For evaluating new antiangiogenic drugs, both the OIR and laser-induced CNV models are extensively used because they develop consistent neovascularization relatively fast, in days to weeks—in contrast to the transgenic models (summarized in Table 2), which may take months or even years to develop vascular lesions. Converesely, the corneal angiogenesis model is also widely used for screening potential antiangiogenic molecules, owing to its optical clarity and ease of visualization, and serves as a basis with which to investigate pathologic angiogenesis without preexisting vessels, whereas in the retina and choroid, changes in the preexisting vasculature are the major objects of the investigation of pathologic angiogenesis (193). Corneal angiogenesis models are fairly reproducible, cost-efficient, and fast (79), yet the 2-dimensional environment of the cornea may pose as a potential limitation as typical angiogenesis that occurs in other organs in humans is 3-dimensional (193).
TABLE 1.
Animal models of induced ocular angiogenesis
| Animal model | Species | Advantages | Limitations | Reference |
|---|---|---|---|---|
| OIR | Cat | Vessel loss and neovascularization are consistent, reproducible, and quantifiable | Maternal care and postnatal weight gain of the pups may affect development of vessel loss and neovascularization | 32 |
| Rat | 33 | |||
| Mouse | 34 | |||
| Dog | 35, 39 | |||
| Zebrafish | 36 | |||
| Laser-induced CNV | Cat | Relatively fast to develop and establish; reproducible and cost-effective | Variability of CNV response | 80 |
| Monkey | 81 | |||
| Rat | 85 | |||
| Rabbit | 84 | |||
| Mouse | 86 | |||
| Surgically induced CNV | Rat | Convenience and feasibility without access to laser | Variability of CNV response | 97 |
| Mouse | 96 | |||
| Alkali burn corneal neovascularization | Rabbit | Easy to perform; highly relevant to clinical scenarios | Extensive corneal damage after chemical burn; complex pathophysiologic processes may interfere with angiogenesis | 182 |
| Mouse | 183 | |||
| Rat | 180 | |||
| Corneal suture injury | Rabbit | Relevant to clinical scenarios in both angiogenesis and lymphangiogenesis | Variability of response; surgery may cause an inflammatory response | 165 |
| Mouse | 183 | |||
| Corneal micropocket | Rabbit | Relative noninflammatory nature; absence of corneal edema | Variability of response | 165 |
| Mouse | 187 |
TABLE 2.
Transgenic mouse models with spontaneous or deficient ocular angiogenesis
| Gene | Mouse model | Disease/pathologic feature | Reference |
|---|---|---|---|
| Spontaneous intraretinal/ subretinal angiogenesis | |||
| Vldlr | Vldlr−/− | RAP, AMD | 111, 102, 101, 114, 116, 119 |
| Vegf | rho/VEGF transgenic | RAP, AMD | 104 |
| Unknown | JR5558 (NRV2) | RAP, AMD | 129–131 |
| Deficient intraretinal angiogenesis | |||
| Lrp5 | Lrp5−/− | FEVR | 146, 148, 149, 153 |
| Norrin | Ndpy/− | Norrie disease | 143, 147, 153 |
| Fzd4 | Fzd4−/− | FEVR | 143, 153 |
| Tspan12 | Tspan12−/− | FEVR | 151 |
| Girdin (Ccdc88a) | Girdin−/− | Incomplete retinal vessel development | 163 |
| Girdin-KISA/SA | 164 | ||
| Spontaneous corneal angiogenesis | |||
| Destrin | Corn1 | Spontaneous corneal neovascularization | 194 |
| Cd36 | Cd36−/− | 195 | |
| Pax6 | Pax6 Tg | 196 | |
| Flt1 | NLS-Cre;flt-1loxP/loxP | 171 | |
| Kdr | LeCre;vegfr2loxP/loxP | 197 |
NRV2, neoretinal vascularization 2.
As the research interest in angiogenesis continues, there will be an increasing need for developing and choosing appropriate experimental models with the most reliability and reproducibility. Currently, none of these models successfully reproduces the diabetes-induced proliferative stage of retinal neovascularization in DR or the age-dependent choroidal neovascularization in AMD as seen in human patients. Overall, future development of such new animal models will be of tremendous experimental value for angiogenesis research in eye studies and beyond.
ACKNOWLEDGMENTS
This study was supported by U.S. National Institutes of Health (NIH), National Eye Institute Grant R01-EY024963, the Boston Children’s Hospital Ophthalmology Foundation, Massachusetts Lions Eye Research Fund, Inc. and BrightFocus Foundation (to J.C.), Knights Templar Eye Foundation Pediatric Ophthalmology Career-Starter Research Grants (to C.-H.L.), and Boston Children’s Hospital Faculty Career Development Grant OFD/BTREC/CTREC (to Y.S.). All animal work was approved by the Boston Children’s Hospital Animal Care and Use Committee and adhered to the Association for Research in Vision and Ophthalmology Statement for the Use of Animals in Ophthalmic and Vision Research. The authors declare no conflicts of interest.
Glossary
- AMD
age-related macular degeneration
- bFGF
basic fibroblast growth factor
- CNV
choroidal neovascularization
- DR
diabetic retinopathy
- FEVR
familial exudative vitreoretinopathy
- FZD4
frizzled-4
- LRP5
LDL receptor–related protein 5
- OIR
oxygen-induced retinopathy
- RAP
retinal angiomatous proliferation
- ROP
retinopathy of prematurity
- RPE
retinal pigment epithelium
- TSPAN12
tetraspanin 12
- VEGFR
VEGF receptor
- VLDLR
very-LDL receptor
- Vldlr−/−
VLDLR germline knockout
AUTHOR CONTRIBUTIONS
C.-H. Liu and J. Chen conceived of and designed the manuscript; C.-H. Liu, Z. Wang, Y. Sun, and J. Chen collected and organized data and figures; and all authors wrote, edited, and approved the manuscript.
REFERENCES
- 1.Wacker A., Gerhardt H. (2011) Endothelial development taking shape. Curr. Opin. Cell Biol. 23, 676–685 [DOI] [PubMed] [Google Scholar]
- 2.Gerhardt H., Golding M., Fruttiger M., Ruhrberg C., Lundkvist A., Abramsson A., Jeltsch M., Mitchell C., Alitalo K., Shima D., Betsholtz C. (2003) VEGF guides angiogenic sprouting utilizing endothelial tip cell filopodia. J. Cell Biol. 161, 1163–1177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Potente M., Gerhardt H., Carmeliet P. (2011) Basic and therapeutic aspects of angiogenesis. Cell 146, 873–887 [DOI] [PubMed] [Google Scholar]
- 4.Folkman J. (1995) Angiogenesis in cancer, vascular, rheumatoid and other disease. Nat. Med. 1, 27–31 [DOI] [PubMed] [Google Scholar]
- 5.Al-Latayfeh M., Silva P. S., Sun J. K., Aiello L. P. (2012) Antiangiogenic therapy for ischemic retinopathies. Cold Spring Harb. Perspect. Med. 2, a006411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Stahl A., Connor K. M., Sapieha P., Chen J., Dennison R. J., Krah N. M., Seaward M. R., Willett K. L., Aderman C. M., Guerin K. I., Hua J., Löfqvist C., Hellström A., Smith L. E. (2010) The mouse retina as an angiogenesis model. Invest. Ophthalmol. Vis. Sci. 51, 2813–2826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Afzal A., Shaw L. C., Ljubimov A. V., Boulton M. E., Segal M. S., Grant M. B. (2007) Retinal and choroidal microangiopathies: therapeutic opportunities. Microvasc. Res. 74, 131–144 [DOI] [PubMed] [Google Scholar]
- 8.Crawford Y., Ferrara N. (2009) VEGF inhibition: insights from preclinical and clinical studies. Cell Tissue Res. 335, 261–269 [DOI] [PubMed] [Google Scholar]
- 9.Smith L. E. (2008) Through the eyes of a child: understanding retinopathy through ROP the Friedenwald lecture. Invest. Ophthalmol. Vis. Sci. 49, 5177–5182 [DOI] [PubMed] [Google Scholar]
- 10.Elman M. J., Aiello L. P., Beck R. W., Bressler N. M., Bressler S. B., Edwards A. R., Ferris F. L. III, Friedman S. M., Glassman A. R., Miller K. M., Scott I. U., Stockdale C. R., Sun J. K. (2010) Randomized trial evaluating ranibizumab plus prompt or deferred laser or triamcinolone plus prompt laser for diabetic macular edema. Ophthalmology 117, 1064–1077.e1035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gragoudas E. S., Adamis A. P., Cunningham E. T. Jr, Feinsod M., Guyer D. R.; VEGF Inhibition Study in Ocular Neovascularization Clinical Trial Group (2004) Pegaptanib for neovascular age-related macular degeneration. N. Engl. J. Med. 351, 2805–2816 [DOI] [PubMed] [Google Scholar]
- 12.Rosenfeld P. J., Brown D. M., Heier J. S., Boyer D. S., Kaiser P. K., Chung C. Y., Kim R. Y.; MARINA Study Group (2006) Ranibizumab for neovascular age-related macular degeneration. N. Engl. J. Med. 355, 1419–1431 [DOI] [PubMed] [Google Scholar]
- 13.Penn J. S., Madan A., Caldwell R. B., Bartoli M., Caldwell R. W., Hartnett M. E. (2008) Vascular endothelial growth factor in eye disease. Prog. Retin. Eye Res. 27, 331–371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Weis S. M., Cheresh D. A. (2005) Pathophysiological consequences of VEGF-induced vascular permeability. Nature 437, 497–504 [DOI] [PubMed] [Google Scholar]
- 15.Fruttiger M. (2007) Development of the retinal vasculature. Angiogenesis 10, 77–88 [DOI] [PubMed] [Google Scholar]
- 16.Hartnett M. E. (2013) Pediatric Retina, Lippincott Williams & Wilkins, Philadelphia [Google Scholar]
- 17.Standring S. (2008) Gray’s Anatomy, 40th Ed., Elsevier, Atlanta, GA, USA [Google Scholar]
- 18.Netter F. H. (2006) Atlas of Human Anatomy, Elsevier Health Sciences, Amsterdam [Google Scholar]
- 19.Ross M. H., Pawlina W. (2005) Histology: A Text and Atlas With Correlated Cell and Molecular Biology, Lippincott Williams & Wilkins, Philadelphia [Google Scholar]
- 20.Paul Riordan-Eva E. T. C. (2011) Vaughan & Asbury’s General Ophthalmology, McGraw-Hill Professional, New York [Google Scholar]
- 21.Henkind P., Hansen R. I., Szalay J. (1979) Physiology of the human eye and visual system. In Ocular Circulation (Records R. E., ed.), pp. 98–155, Harper & Row, New York [Google Scholar]
- 22.Bela Anand-Apte J. G. H. (2011) Developmental anatomy of the retinal and choroidal vasculature. In The Retina and its Disorders (Besharse J., Bok D., eds.), Academic Press, Oxford [Google Scholar]
- 23.Dorrell M. I., Aguilar E., Friedlander M. (2002) Retinal vascular development is mediated by endothelial filopodia, a preexisting astrocytic template and specific R-cadherin adhesion. Invest. Ophthalmol. Vis. Sci. 43, 3500–3510 [PubMed] [Google Scholar]
- 24.Stone J., Itin A., Alon T., Pe’er J., Gnessin H., Chan-Ling T., Keshet E. (1995) Development of retinal vasculature is mediated by hypoxia-induced vascular endothelial growth factor (VEGF) expression by neuroglia. J. Neurosci. 15, 4738–4747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Moore K. L., Dalley A. F., Agur A. M. R. (2009) Clinically Oriented Anatomy, Lippincott Williams & Wilkins, Philadelphia [Google Scholar]
- 26.Kiel J. W. (2010) The Ocular Circulation, Morgan & Claypool, San Rafael, CA, USA: [PubMed] [Google Scholar]
- 27.Hartnett M. E., Penn J. S. (2012) Mechanisms and management of retinopathy of prematurity. N. Engl. J. Med. 367, 2515–2526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chen J., Smith L. E. (2007) Retinopathy of prematurity. Angiogenesis 10, 133–140 [DOI] [PubMed] [Google Scholar]
- 29.Chen J., Stahl A., Hellstrom A., Smith L. E. (2011) Current update on retinopathy of prematurity: screening and treatment. Curr. Opin. Pediatr. 23, 173–178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kinsey V. E. (1956) Retrolental fibroplasia; cooperative study of retrolental fibroplasia and the use of oxygen. AMA Arch. Opthalmol. 56, 481–543 [PubMed] [Google Scholar]
- 31.Sapieha P., Joyal J. S., Rivera J. C., Kermorvant-Duchemin E., Sennlaub F., Hardy P., Lachapelle P., Chemtob S. (2010) Retinopathy of prematurity: understanding ischemic retinal vasculopathies at an extreme of life. J. Clin. Invest. 120, 3022–3032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ashton N., Ward B., Serpell G. (1954) Effect of oxygen on developing retinal vessels with particular reference to the problem of retrolental fibroplasia. Br. J. Ophthalmol. 38, 397–432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Penn J. S., Tolman B. L., Lowery L. A. (1993) Variable oxygen exposure causes preretinal neovascularization in the newborn rat. Invest. Ophthalmol. Vis. Sci. 34, 576–585 [PubMed] [Google Scholar]
- 34.Smith L. E., Wesolowski E., McLellan A., Kostyk S. K., D’Amato R., Sullivan R., D’Amore P. A. (1994) Oxygen-induced retinopathy in the mouse. Invest. Ophthalmol. Vis. Sci. 35, 101–111 [PubMed] [Google Scholar]
- 35.McLeod D. S., Brownstein R., Lutty G. A. (1996) Vaso-obliteration in the canine model of oxygen-induced retinopathy. Invest. Ophthalmol. Vis. Sci. 37, 300–311 [PubMed] [Google Scholar]
- 36.Cao R., Jensen L. D., Söll I., Hauptmann G., Cao Y. (2008) Hypoxia-induced retinal angiogenesis in zebrafish as a model to study retinopathy. PLoS One 3, e2748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Grossniklaus H. E., Kang S. J., Berglin L. (2010) Animal models of choroidal and retinal neovascularization. Prog. Retin. Eye Res. 29, 500–519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Phelps D. L., Rosenbaum A. L. (1984) Effects of marginal hypoxemia on recovery from oxygen-induced retinopathy in the kitten model. Pediatrics 73, 1–6 [PubMed] [Google Scholar]
- 39.McLeod D. S., Crone S. N., Lutty G. A. (1996) Vasoproliferation in the neonatal dog model of oxygen-induced retinopathy. Invest. Ophthalmol. Vis. Sci. 37, 1322–1333 [PubMed] [Google Scholar]
- 40.McLeod D. S., D’Anna S. A., Lutty G. A. (1998) Clinical and histopathologic features of canine oxygen-induced proliferative retinopathy. Invest. Ophthalmol. Vis. Sci. 39, 1918–1932 [PubMed] [Google Scholar]
- 41.Lange C., Ehlken C., Stahl A., Martin G., Hansen L., Agostini H. T. (2009) Kinetics of retinal vaso-obliteration and neovascularisation in the oxygen-induced retinopathy (OIR) mouse model. Graefes Arch. Clin. Exp. Ophthalmol. 247, 1205–1211 [DOI] [PubMed] [Google Scholar]
- 42.Gu X., Samuel S., El-Shabrawey M., Caldwell R. B., Bartoli M., Marcus D. M., Brooks S. E. (2002) Effects of sustained hyperoxia on revascularization in experimental retinopathy of prematurity. Invest. Ophthalmol. Vis. Sci. 43, 496–502 [PubMed] [Google Scholar]
- 43.Pierce E. A., Avery R. L., Foley E. D., Aiello L. P., Smith L. E. (1995) Vascular endothelial growth factor/vascular permeability factor expression in a mouse model of retinal neovascularization. Proc. Natl. Acad. Sci. USA 92, 905–909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chen J., Connor K. M., Aderman C. M., Smith L. E. (2008) Erythropoietin deficiency decreases vascular stability in mice. J. Clin. Invest. 118, 526–533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Aiello L. P., Pierce E. A., Foley E. D., Takagi H., Chen H., Riddle L., Ferrara N., King G. L., Smith L. E. (1995) Suppression of retinal neovascularization in vivo by inhibition of vascular endothelial growth factor (VEGF) using soluble VEGF-receptor chimeric proteins. Proc. Natl. Acad. Sci. USA 92, 10457–10461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chen J., Connor K. M., Aderman C. M., Willett K. L., Aspegren O. P., Smith L. E. (2009) Suppression of retinal neovascularization by erythropoietin siRNA in a mouse model of proliferative retinopathy. Invest. Ophthalmol. Vis. Sci. 50, 1329–1335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Connor K. M., Krah N. M., Dennison R. J., Aderman C. M., Chen J., Guerin K. I., Sapieha P., Stahl A., Willett K. L., Smith L. E. (2009) Quantification of oxygen-induced retinopathy in the mouse: a model of vessel loss, vessel regrowth and pathological angiogenesis. Nat. Protoc. 4, 1565–1573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Stahl A., Connor K. M., Sapieha P., Willett K. L., Krah N. M., Dennison R. J., Chen J., Guerin K. I., Smith L. E. (2009) Computer-aided quantification of retinal neovascularization. Angiogenesis 12, 297–301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Vähätupa M., Prince S., Vataja S., Mertimo T., Kataja M., Kinnunen K., Marjomäki V., Uusitalo H., Komatsu M., Järvinen T. A., Uusitalo-Järvinen H. (2016) Lack of R-ras leads to increased vascular permeability in ischemic retinopathy. Invest. Ophthalmol. Vis. Sci. 57, 4898–4909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Fulton A. B., Akula J. D., Mocko J. A., Hansen R. M., Benador I. Y., Beck S. C., Fahl E., Seeliger M. W., Moskowitz A., Harris M. E. (2009) Retinal degenerative and hypoxic ischemic disease. Doc. Ophthalmol. 118, 55–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Xu Q., Qaum T., Adamis A. P. (2001) Sensitive blood-retinal barrier breakdown quantitation using Evans blue. Invest. Ophthalmol. Vis. Sci. 42, 789–794 [PubMed] [Google Scholar]
- 52.Connor K. M., SanGiovanni J. P., Lofqvist C., Aderman C. M., Chen J., Higuchi A., Hong S., Pravda E. A., Majchrzak S., Carper D., Hellstrom A., Kang J. X., Chew E. Y., Salem N. Jr, Serhan C. N., Smith L. E. (2007) Increased dietary intake of omega-3-polyunsaturated fatty acids reduces pathological retinal angiogenesis. Nat. Med. 13, 868–873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Smith L. E., Shen W., Perruzzi C., Soker S., Kinose F., Xu X., Robinson G., Driver S., Bischoff J., Zhang B., Schaeffer J. M., Senger D. R. (1999) Regulation of vascular endothelial growth factor-dependent retinal neovascularization by insulin-like growth factor-1 receptor. Nat. Med. 5, 1390–1395 [DOI] [PubMed] [Google Scholar]
- 54.Liu C. H., Sun Y., Li J., Gong Y., Tian K. T., Evans L. P., Morss P. C., Fredrick T. W., Saba N. J., Chen J. (2015) Endothelial microRNA-150 is an intrinsic suppressor of pathologic ocular neovascularization. Proc. Natl. Acad. Sci. USA 112, 12163–12168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Penn J. S., Rajaratnam V. S., Collier R. J., Clark A. F. (2001) The effect of an angiostatic steroid on neovascularization in a rat model of retinopathy of prematurity. Invest. Ophthalmol. Vis. Sci. 42, 283–290 [PubMed] [Google Scholar]
- 56.Madan A., Penn J. S. (2003) Animal models of oxygen-induced retinopathy. Front. Biosci. 8, 1030–1043 [DOI] [PubMed] [Google Scholar]
- 57.York J. R., Landers S., Kirby R. S., Arbogast P. G., Penn J. S. (2004) Arterial oxygen fluctuation and retinopathy of prematurity in very-low-birth-weight infants. J. Perinatol. 24, 82–87 [DOI] [PubMed] [Google Scholar]
- 58.Cunningham S., Fleck B. W., Elton R. A., McIntosh N. (1995) Transcutaneous oxygen levels in retinopathy of prematurity. Lancet 346, 1464–1465 [DOI] [PubMed] [Google Scholar]
- 59.Cao Z., Jensen L. D., Rouhi P., Hosaka K., Länne T., Steffensen J. F., Wahlberg E., Cao Y. (2010) Hypoxia-induced retinopathy model in adult zebrafish. Nat. Protoc. 5, 1903–1910 [DOI] [PubMed] [Google Scholar]
- 60.Wu Y. C., Chang C. Y., Kao A., Hsi B., Lee S. H., Chen Y. H., Wang I. J. (2015) Hypoxia-induced retinal neovascularization in zebrafish embryos: a potential model of retinopathy of prematurity. PLoS One 10, e0126750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Robinson R., Barathi V. A., Chaurasia S. S., Wong T. Y., Kern T. S. (2012) Update on animal models of diabetic retinopathy: from molecular approaches to mice and higher mammals. Dis. Model. Mech. 5, 444–456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lai A. K., Lo A. C. (2013) Animal models of diabetic retinopathy: summary and comparison. J. Diabetes Res. 2013, 106594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Crawford T. N., Alfaro D. V. III, Kerrison J. B., Jablon E. P. (2009) Diabetic retinopathy and angiogenesis. Curr. Diabetes Rev. 5, 8–13 [DOI] [PubMed] [Google Scholar]
- 64.Rerup C. C. (1970) Drugs producing diabetes through damage of the insulin secreting cells. Pharmacol. Rev. 22, 485–518 [PubMed] [Google Scholar]
- 65.Junod A., Lambert A. E., Stauffacher W., Renold A. E. (1969) Diabetogenic action of streptozotocin: relationship of dose to metabolic response. J. Clin. Invest. 48, 2129–2139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Han Z., Guo J., Conley S. M., Naash M. I. (2013) Retinal angiogenesis in the Ins2(Akita) mouse model of diabetic retinopathy. Invest. Ophthalmol. Vis. Sci. 54, 574–584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Barber A. J., Antonetti D. A., Kern T. S., Reiter C. E., Soans R. S., Krady J. K., Levison S. W., Gardner T. W., Bronson S. K. (2005) The Ins2Akita mouse as a model of early retinal complications in diabetes. Invest. Ophthalmol. Vis. Sci. 46, 2210–2218 [DOI] [PubMed] [Google Scholar]
- 68.Shaw S. G., Boden J. P., Biecker E., Reichen J., Rothen B. (2006) Endothelin antagonism prevents diabetic retinopathy in NOD mice: a potential role of the angiogenic factor adrenomedullin. Exp. Biol. Med. (Maywood) 231, 1101–1105 [PubMed] [Google Scholar]
- 69.Midena E., Segato T., Radin S., di Giorgio G., Meneghini F., Piermarocchi S., Belloni A. S. (1989) Studies on the retina of the diabetic db/db mouse. I. Endothelial cell-pericyte ratio. Ophthalmic Res. 21, 106–111 [DOI] [PubMed] [Google Scholar]
- 70.Ning X., Baoyu Q., Yuzhen L., Shuli S., Reed E., Li Q. Q. (2004) Neuro-optic cell apoptosis and microangiopathy in KKAY mouse retina. Int. J. Mol. Med. 13, 87–92 [PubMed] [Google Scholar]
- 71.Holz F. G., Pauleikhoff D., Klein R., Bird A. C. (2004) Pathogenesis of lesions in late age-related macular disease. Am. J. Ophthalmol. 137, 504–510 [DOI] [PubMed] [Google Scholar]
- 72.Campochiaro P. A. (2015) Molecular pathogenesis of retinal and choroidal vascular diseases. Prog. Retin. Eye Res. 49, 67–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Hageman G. S., Luthert P. J., Victor Chong N. H., Johnson L. V., Anderson D. H., Mullins R. F. (2001) An integrated hypothesis that considers drusen as biomarkers of immune-mediated processes at the RPE-Bruch’s membrane interface in aging and age-related macular degeneration. Prog. Retin. Eye Res. 20, 705–732 [DOI] [PubMed] [Google Scholar]
- 74.Amin R., Puklin J. E., Frank R. N. (1994) Growth factor localization in choroidal neovascular membranes of age-related macular degeneration. Invest. Ophthalmol. Vis. Sci. 35, 3178–3188 [PubMed] [Google Scholar]
- 75.Kvanta A., Algvere P. V., Berglin L., Seregard S. (1996) Subfoveal fibrovascular membranes in age-related macular degeneration express vascular endothelial growth factor. Invest. Ophthalmol. Vis. Sci. 37, 1929–1934 [PubMed] [Google Scholar]
- 76.Lopez P. F., Sippy B. D., Lambert H. M., Thach A. B., Hinton D. R. (1996) Transdifferentiated retinal pigment epithelial cells are immunoreactive for vascular endothelial growth factor in surgically excised age-related macular degeneration-related choroidal neovascular membranes. Invest. Ophthalmol. Vis. Sci. 37, 855–868 [PubMed] [Google Scholar]
- 77.Carmeliet P., Collen D. (1998) Tissue factor. Int. J. Biochem. Cell Biol. 30, 661–667 [DOI] [PubMed] [Google Scholar]
- 78.Steen B., Sejersen S., Berglin L., Seregard S., Kvanta A. (1998) Matrix metalloproteinases and metalloproteinase inhibitors in choroidal neovascular membranes. Invest. Ophthalmol. Vis. Sci. 39, 2194–2200 [PubMed] [Google Scholar]
- 79.Montezuma S. R., Vavvas D., Miller J. W. (2009) Review of the ocular angiogenesis animal models. Semin. Ophthalmol. 24, 52–61 [DOI] [PubMed] [Google Scholar]
- 80.Baum J. L., Wise G. N. (1965) Experimental subretinal neovascularization. Trans. Am. Ophthalmol. Soc. 63, 91–107 [PMC free article] [PubMed] [Google Scholar]
- 81.Ryan S. J. (1979) The development of an experimental model of subretinal neovascularization in disciform macular degeneration. Trans. Am. Ophthalmol. Soc. 77, 707–745 [PMC free article] [PubMed] [Google Scholar]
- 82.Miller H., Miller B., Ryan S. J. (1986) The role of retinal pigment epithelium in the involution of subretinal neovascularization. Invest. Ophthalmol. Vis. Sci. 27, 1644–1652 [PubMed] [Google Scholar]
- 83.Archer D. B., Gardiner T. A. (1981) Electron microscopic features of experimental choroidal neovascularization. Am. J. Ophthalmol. 91, 433–457 [DOI] [PubMed] [Google Scholar]
- 84.ElDirini A. A., Ogden T. E., Ryan S. J. (1991) Subretinal endophotocoagulation. A new model of subretinal neovascularization in the rabbit. Retina 11, 244–249 [PubMed] [Google Scholar]
- 85.Dobi E. T., Puliafito C. A., Destro M. (1989) A new model of experimental choroidal neovascularization in the rat. Arch. Ophthalmol. 107, 264–269 [DOI] [PubMed] [Google Scholar]
- 86.Tobe T., Ortega S., Luna J. D., Ozaki H., Okamoto N., Derevjanik N. L., Vinores S. A., Basilico C., Campochiaro P. A. (1998) Targeted disruption of the FGF2 gene does not prevent choroidal neovascularization in a murine model. Am. J. Pathol. 153, 1641–1646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Takahashi T., Nakamura T., Hayashi A., Kamei M., Nakabayashi M., Okada A. A., Tomita N., Kaneda Y., Tano Y. (2000) Inhibition of experimental choroidal neovascularization by overexpression of tissue inhibitor of metalloproteinases-3 in retinal pigment epithelium cells. Am. J. Ophthalmol. 130, 774–781 [DOI] [PubMed] [Google Scholar]
- 88.Yanagi Y., Tamaki Y., Obata R., Muranaka K., Homma N., Matsuoka H., Mano H. (2002) Subconjunctival administration of bucillamine suppresses choroidal neovascularization in rat. Invest. Ophthalmol. Vis. Sci. 43, 3495–3499 [PubMed] [Google Scholar]
- 89.Kwak N., Okamoto N., Wood J. M., Campochiaro P. A. (2000) VEGF is major stimulator in model of choroidal neovascularization. Invest. Ophthalmol. Vis. Sci. 41, 3158–3164 [PubMed] [Google Scholar]
- 90.Gong Y., Li J., Sun Y., Fu Z., Liu C. H., Evans L., Tian K., Saba N., Fredrick T., Morss P., Chen J., Smith L. E. (2015) Optimization of an image-guided laser-induced choroidal neovascularization model in mice. PLoS One 10, e0132643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Ishikawa K., Kannan R., Hinton D. R. (2016) Molecular mechanisms of subretinal fibrosis in age-related macular degeneration. Exp. Eye Res. 142, 19–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Lambert V., Lecomte J., Hansen S., Blacher S., Gonzalez M. L., Struman I., Sounni N. E., Rozet E., de Tullio P., Foidart J. M., Rakic J. M., Noel A. (2013) Laser-induced choroidal neovascularization model to study age-related macular degeneration in mice. Nat. Protoc. 8, 2197–2211 [DOI] [PubMed] [Google Scholar]
- 93.Chen J. (2014) Enhancing reliability of the laser-induced choroidal neovascularization mouse model: insights from a new study. Invest. Ophthalmol. Vis. Sci. 55, 6535 [DOI] [PubMed] [Google Scholar]
- 94.Poor S. H., Qiu Y., Fassbender E. S., Shen S., Woolfenden A., Delpero A., Kim Y., Buchanan N., Gebuhr T. C., Hanks S. M., Meredith E. L., Jaffee B. D., Dryja T. P. (2014) Reliability of the mouse model of choroidal neovascularization induced by laser photocoagulation. Invest. Ophthalmol. Vis. Sci. 55, 6525–6534 [DOI] [PubMed] [Google Scholar]
- 95.Krzystolik M. G., Afshari M. A., Adamis A. P., Gaudreault J., Gragoudas E. S., Michaud N. A., Li W., Connolly E., O’Neill C. A., Miller J. W. (2002) Prevention of experimental choroidal neovascularization with intravitreal anti-vascular endothelial growth factor antibody fragment. Arch. Ophthalmol. 120, 338–346 [DOI] [PubMed] [Google Scholar]
- 96.Li Y., Huang D., Xia X., Wang Z., Luo L., Wen R. (2011) CCR3 and choroidal neovascularization. PLoS One 6, e17106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Cao J., Zhao L., Li Y., Liu Y., Xiao W., Song Y., Luo L., Huang D., Yancopoulos G. D., Wiegand S. J., Wen R. (2010) A subretinal matrigel rat choroidal neovascularization (CNV) model and inhibition of CNV and associated inflammation and fibrosis by VEGF trap. Invest. Ophthalmol. Vis. Sci. 51, 6009–6017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Lafaut B. A., Aisenbrey S., Vanden Broecke C., Bartz-Schmidt K. U. (2000) Clinicopathological correlation of deep retinal vascular anomalous complex in age related macular degeneration. Br. J. Ophthalmol. 84, 1269–1274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Bermig J., Tylla H., Jochmann C., Nestler A., Wolf S. (2002) Angiographic findings in patients with exudative age-related macular degeneration. Graefes Arch. Clin. Exp. Ophthalmol. 240, 169–175 [DOI] [PubMed] [Google Scholar]
- 100.Bottoni F., Massacesi A., Cigada M., Viola F., Musicco I., Staurenghi G. (2005) Treatment of retinal angiomatous proliferation in age-related macular degeneration: a series of 104 cases of retinal angiomatous proliferation. Arch. Ophthalmol. 123, 1644–1650 [DOI] [PubMed] [Google Scholar]
- 101.Donati M. C., Carifi G., Virgili G., Menchini U. (2006) Retinal angiomatous proliferation: association with clinical and angiographic features. Ophthalmologica 220, 31–36 [DOI] [PubMed] [Google Scholar]
- 102.Heckenlively J. R., Hawes N. L., Friedlander M., Nusinowitz S., Hurd R., Davisson M., Chang B. (2003) Mouse model of subretinal neovascularization with choroidal anastomosis. Retina 23, 518–522 [DOI] [PubMed] [Google Scholar]
- 103.Li C., Huang Z., Kingsley R., Zhou X., Li F., Parke D. W. II, Cao W. (2007) Biochemical alterations in the retinas of very low-density lipoprotein receptor knockout mice: an animal model of retinal angiomatous proliferation. Arch. Ophthalmol. 125, 795–803 [DOI] [PubMed] [Google Scholar]
- 104.Okamoto N., Tobe T., Hackett S. F., Ozaki H., Vinores M. A., LaRochelle W., Zack D. J., Campochiaro P. A. (1997) Transgenic mice with increased expression of vascular endothelial growth factor in the retina: a new model of intraretinal and subretinal neovascularization. Am. J. Pathol. 151, 281–291 [PMC free article] [PubMed] [Google Scholar]
- 105.Tobe T., Okamoto N., Vinores M. A., Derevjanik N. L., Vinores S. A., Zack D. J., Campochiaro P. A. (1998) Evolution of neovascularization in mice with overexpression of vascular endothelial growth factor in photoreceptors. Invest. Ophthalmol. Vis. Sci. 39, 180–188 [PubMed] [Google Scholar]
- 106.Hu W., Jiang A., Liang J., Meng H., Chang B., Gao H., Qiao X. (2008) Expression of VLDLR in the retina and evolution of subretinal neovascularization in the knockout mouse model’s retinal angiomatous proliferation. Invest. Ophthalmol. Vis. Sci. 49, 407–415 [DOI] [PubMed] [Google Scholar]
- 107.Oka K., Ishimura-Oka K., Chu M. J., Sullivan M., Krushkal J., Li W. H., Chan L. (1994) Mouse very-low-density-lipoprotein receptor (VLDLR) cDNA cloning, tissue-specific expression and evolutionary relationship with the low-density-lipoprotein receptor. Eur. J. Biochem. 224, 975–982 [DOI] [PubMed] [Google Scholar]
- 108.Wyne K. L., Pathak K., Seabra M. C., Hobbs H. H. (1996) Expression of the VLDL receptor in endothelial cells. Arterioscler. Thromb. Vasc. Biol. 16, 407–415 [DOI] [PubMed] [Google Scholar]
- 109.Tiebel O., Oka K., Robinson K., Sullivan M., Martinez J., Nakamuta M., Ishimura-Oka K., Chan L. (1999) Mouse very low-density lipoprotein receptor (VLDLR): gene structure, tissue-specific expression and dietary and developmental regulation. Atherosclerosis 145, 239–251 [DOI] [PubMed] [Google Scholar]
- 110.Haines J. L., Schnetz-Boutaud N., Schmidt S., Scott W. K., Agarwal A., Postel E. A., Olson L., Kenealy S. J., Hauser M., Gilbert J. R., Pericak-Vance M. A. (2006) Functional candidate genes in age-related macular degeneration: significant association with VEGF, VLDLR, and LRP6. Invest. Ophthalmol. Vis. Sci. 47, 329–335 [DOI] [PubMed] [Google Scholar]
- 111.Frykman P. K., Brown M. S., Yamamoto T., Goldstein J. L., Herz J. (1995) Normal plasma lipoproteins and fertility in gene-targeted mice homozygous for a disruption in the gene encoding very low density lipoprotein receptor. Proc. Natl. Acad. Sci. USA 92, 8453–8457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Hua J., Guerin K. I., Chen J., Michán S., Stahl A., Krah N. M., Seaward M. R., Dennison R. J., Juan A. M., Hatton C. J., Sapieha P., Sinclair D. A., Smith L. E. (2011) Resveratrol inhibits pathologic retinal neovascularization in Vldlr-/- mice. Invest. Ophthalmol. Vis. Sci. 52, 2809–2816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Chen Y., Hu Y., Moiseyev G., Zhou K. K., Chen D., Ma J. X. (2009) Photoreceptor degeneration and retinal inflammation induced by very low-density lipoprotein receptor deficiency. Microvasc. Res. 78, 119–127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Dorrell M. I., Aguilar E., Jacobson R., Yanes O., Gariano R., Heckenlively J., Banin E., Ramirez G. A., Gasmi M., Bird A., Siuzdak G., Friedlander M. (2009) Antioxidant or neurotrophic factor treatment preserves function in a mouse model of neovascularization-associated oxidative stress. J. Clin. Invest. 119, 611–623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Kyosseva S. V., Chen L., Seal S., McGinnis J. F. (2013) Nanoceria inhibit expression of genes associated with inflammation and angiogenesis in the retina of Vldlr null mice. Exp. Eye Res. 116, 63–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Joyal J. S., Sun Y., Gantner M. L., Shao Z., Evans L. P., Saba N., Fredrick T., Burnim S., Kim J. S., Patel G., Juan A. M., Hurst C. G., Hatton C. J., Cui Z., Pierce K. A., Bherer P., Aguilar E., Powner M. B., Vevis K., Boisvert M., Fu Z., Levy E., Fruttiger M., Packard A., Rezende F. A., Maranda B., Sapieha P., Chen J., Friedlander M., Clish C. B., Smith L. E. (2016) Retinal lipid and glucose metabolism dictates angiogenesis through the lipid sensor Ffar1. Nat. Med. 22, 439–445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Westenskow P. D., Kurihara T., Aguilar E., Scheppke E. L., Moreno S. K., Wittgrove C., Marchetti V., Michael I. P., Anand S., Nagy A., Cheresh D., Friedlander M. (2013) Ras pathway inhibition prevents neovascularization by repressing endothelial cell sprouting. J. Clin. Invest. 123, 4900–4908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Sun Y., Liu C. H., SanGiovanni J. P., Evans L. P., Tian K. T., Zhang B., Stahl A., Pu W. T., Kamenecka T. M., Solt L. A., Chen J. (2015) Nuclear receptor RORα regulates pathologic retinal angiogenesis by modulating SOCS3-dependent inflammation. Proc. Natl. Acad. Sci. USA 112, 10401–10406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Sun Y., Lin Z., Liu C. H., Gong Y., Liegl R., Fredrick T. W., Meng S. S., Burnim S. B., Wang Z., Akula J. D., Pu W. T., Chen J., Smith L. E. H. (2017) Inflammatory signals from photoreceptor modulate pathological retinal angiogenesis via c-Fos. J. Exp. Med. 214, 1753–1767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Aiello L. P., Avery R. L., Arrigg P. G., Keyt B. A., Jampel H. D., Shah S. T., Pasquale L. R., Thieme H., Iwamoto M. A., Park J. E., Nguyen H. V., Aiello L. M., Ferrara N., King G. L. (1994) Vascular endothelial growth factor in ocular fluid of patients with diabetic retinopathy and other retinal disorders. N. Engl. J. Med. 331, 1480–1487 [DOI] [PubMed] [Google Scholar]
- 121.Sene A., Chin-Yee D., Apte R. S. (2015) Seeing through VEGF: innate and adaptive immunity in pathological angiogenesis in the eye. Trends Mol. Med. 21, 43–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Stalmans I., Ng Y. S., Rohan R., Fruttiger M., Bouché A., Yuce A., Fujisawa H., Hermans B., Shani M., Jansen S., Hicklin D., Anderson D. J., Gardiner T., Hammes H. P., Moons L., Dewerchin M., Collen D., Carmeliet P., D’Amore P. A. (2002) Arteriolar and venular patterning in retinas of mice selectively expressing VEGF isoforms. J. Clin. Invest. 109, 327–336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Neufeld G., Cohen T., Gengrinovitch S., Poltorak Z. (1999) Vascular endothelial growth factor (VEGF) and its receptors. FASEB J. 13, 9–22 [PubMed] [Google Scholar]
- 124.Mintz-Hittner H. A., Kennedy K. A., Chuang A. Z.; BEAT-ROP Cooperative Group (2011) Efficacy of intravitreal bevacizumab for stage 3+ retinopathy of prematurity. N. Engl. J. Med. 364, 603–615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Brown D. M., Kaiser P. K., Michels M., Soubrane G., Heier J. S., Kim R. Y., Sy J. P., Schneider S.; ANCHOR Study Group (2006) Ranibizumab versus verteporfin for neovascular age-related macular degeneration. N. Engl. J. Med. 355, 1432–1444 [DOI] [PubMed] [Google Scholar]
- 126.Brown D. M., Nguyen Q. D., Marcus D. M., Boyer D. S., Patel S., Feiner L., Schlottmann P. G., Rundle A. C., Zhang J., Rubio R. G., Adamis A. P., Ehrlich J. S., Hopkins J. J.; RIDE and RISE Research Group (2013) Long-term outcomes of ranibizumab therapy for diabetic macular edema: the 36-month results from two phase III trials: RISE and RIDE. Ophthalmology 120, 2013–2022 [DOI] [PubMed] [Google Scholar]
- 127.Robinson G. S., Ju M., Shih S. C., Xu X., McMahon G., Caldwell R. B., Smith L. E. (2001) Nonvascular role for VEGF: VEGFR-1, 2 activity is critical for neural retinal development. FASEB J. 15, 1215–1217 [DOI] [PubMed] [Google Scholar]
- 128.Kurihara T., Westenskow P. D., Bravo S., Aguilar E., Friedlander M. (2012) Targeted deletion of Vegfa in adult mice induces vision loss. J. Clin. Invest. 122, 4213–4217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Won J., Shi L. Y., Hicks W., Wang J., Hurd R., Naggert J. K., Chang B., Nishina P. M. (2011) Mouse model resources for vision research. J. Ophthalmol. 2011, 391384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Nagai N., Lundh von Leithner P., Izumi-Nagai K., Hosking B., Chang B., Hurd R., Adamson P., Adamis A. P., Foxton R. H., Ng Y. S., Shima D. T. (2014) Spontaneous CNV in a novel mutant mouse is associated with early VEGF-A-driven angiogenesis and late-stage focal edema, neural cell loss, and dysfunction. Invest. Ophthalmol. Vis. Sci. 55, 3709–3719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Hasegawa E., Sweigard H., Husain D., Olivares A. M., Chang B., Smith K. E., Birsner A. E., D’Amato R. J., Michaud N. A., Han Y., Vavvas D. G., Miller J. W., Haider N. B., Connor K. M. (2014) Characterization of a spontaneous retinal neovascular mouse model. PLoS One 9, e106507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Doyle S. L., López F. J., Celkova L., Brennan K., Mulfaul K., Ozaki E., Kenna P. F., Kurali E., Hudson N., Doggett T., Ferguson T. A., Humphries P., Adamson P., Campbell M. (2015) IL-18 immunotherapy for neovascular AMD: tolerability and efficacy in nonhuman primates. Invest. Ophthalmol. Vis. Sci. 56, 5424–5430 [DOI] [PubMed] [Google Scholar]
- 133.Paneghetti L., Ng Y. S. (2016) A novel endothelial-derived anti-inflammatory activity significantly inhibits spontaneous choroidal neovascularisation in a mouse model. Vasc. Cell 8, 2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Nagai N., Ju M., Izumi-Nagai K., Robbie S. J., Bainbridge J. W., Gale D. C., Pierre E., Krauss A. H., Adamson P., Shima D. T., Ng Y. S. (2015) Novel CCR3 antagonists are effective mono- and combination inhibitors of choroidal neovascular growth and vascular permeability. Am. J. Pathol. 185, 2534–2549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Foxton R., Osborne A., Martin K. R., Ng Y. S., Shima D. T. (2016) Distal retinal ganglion cell axon transport loss and activation of p38 MAPK stress pathway following VEGF-A antagonism. Cell Death Dis. 7, e2212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Pennesi M. E., Neuringer M., Courtney R. J. (2012) Animal models of age related macular degeneration. Mol. Aspects Med. 33, 487–509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Criswick V. G., Schepens C. L. (1969) Familial exudative vitreoretinopathy. Am. J. Ophthalmol. 68, 578–594 [DOI] [PubMed] [Google Scholar]
- 138.Warburg M. (1975) Norrie’s disease—differential diagnosis and treatment. Acta Ophthalmol. (Copenh.) 53, 217–236 [DOI] [PubMed] [Google Scholar]
- 139.Andersen S. R., Warburg M. (1961) Norrie’s disease: congenital bilateral pseudotumor of the retina with recessive X-chromosomal inheritance; preliminary report. Arch. Ophthalmol. 66, 614–618 [DOI] [PubMed] [Google Scholar]
- 140.Dejana E. (2010) The role of wnt signaling in physiological and pathological angiogenesis. Circ. Res. 107, 943–952 [DOI] [PubMed] [Google Scholar]
- 141.Toomes C., Bottomley H. M., Jackson R. M., Towns K. V., Scott S., Mackey D. A., Craig J. E., Jiang L., Yang Z., Trembath R., Woodruff G., Gregory-Evans C. Y., Gregory-Evans K., Parker M. J., Black G. C., Downey L. M., Zhang K., Inglehearn C. F. (2004) Mutations in LRP5 or FZD4 underlie the common familial exudative vitreoretinopathy locus on chromosome 11q. Am. J. Hum. Genet. 74, 721–730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Gal M., Levanon E. Y., Hujeirat Y., Khayat M., Pe’er J., Shalev S. (2014) Novel mutation in TSPAN12 leads to autosomal recessive inheritance of congenital vitreoretinal disease with intra-familial phenotypic variability. Am. J. Med. Genet. A. 164A, 2996–3002 [DOI] [PubMed] [Google Scholar]
- 143.Xu Q., Wang Y., Dabdoub A., Smallwood P. M., Williams J., Woods C., Kelley M. W., Jiang L., Tasman W., Zhang K., Nathans J. (2004) Vascular development in the retina and inner ear: control by Norrin and frizzled-4, a high-affinity ligand-receptor pair. Cell 116, 883–895 [DOI] [PubMed] [Google Scholar]
- 144.Kinnunen K., Ylä-Herttuala S. (2012) Vascular endothelial growth factors in retinal and choroidal neovascular diseases. Ann. Med. 44, 1–17 [DOI] [PubMed] [Google Scholar]
- 145.MacDonald B. T., Tamai K., He X. (2009) Wnt/beta-catenin signaling: components, mechanisms, and diseases. Dev. Cell 17, 9–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Kato M., Patel M. S., Levasseur R., Lobov I., Chang B. H., Glass D. A. II, Hartmann C., Li L., Hwang T. H., Brayton C. F., Lang R. A., Karsenty G., Chan L. (2002) Cbfa1-independent decrease in osteoblast proliferation, osteopenia, and persistent embryonic eye vascularization in mice deficient in Lrp5, a Wnt coreceptor. J. Cell Biol. 157, 303–314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Luhmann U. F., Lin J., Acar N., Lammel S., Feil S., Grimm C., Seeliger M. W., Hammes H. P., Berger W. (2005) Role of the Norrie disease pseudoglioma gene in sprouting angiogenesis during development of the retinal vasculature. Invest. Ophthalmol. Vis. Sci. 46, 3372–3382 [DOI] [PubMed] [Google Scholar]
- 148.Chen J., Stahl A., Krah N. M., Seaward M. R., Joyal J. S., Juan A. M., Hatton C. J., Aderman C. M., Dennison R. J., Willett K. L., Sapieha P., Smith L. E. (2012) Retinal expression of Wnt-pathway mediated genes in low-density lipoprotein receptor-related protein 5 (Lrp5) knockout mice. PLoS One 7, e30203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Wang Z., Liu C. H., Sun Y., Gong Y., Favazza T. L., Morss P. C., Saba N. J., Fredrick T. W., He X., Akula J. D., Chen J. (2016) Pharmacologic activation of Wnt signaling by lithium normalizes retinal vasculature in a murine model of familial exudative vitreoretinopathy. Am. J. Pathol. 186, 2588–2600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Stenman J. M., Rajagopal J., Carroll T. J., Ishibashi M., McMahon J., McMahon A. P. (2008) Canonical Wnt signaling regulates organ-specific assembly and differentiation of CNS vasculature. Science 322, 1247–1250 [DOI] [PubMed] [Google Scholar]
- 151.Junge H. J., Yang S., Burton J. B., Paes K., Shu X., French D. M., Costa M., Rice D. S., Ye W. (2009) TSPAN12 regulates retinal vascular development by promoting Norrin- but not Wnt-induced FZD4/beta-catenin signaling. Cell 139, 299–311 [DOI] [PubMed] [Google Scholar]
- 152.Chen J., Stahl A., Krah N. M., Seaward M. R., Dennison R. J., Sapieha P., Hua J., Hatton C. J., Juan A. M., Aderman C. M., Willett K. L., Guerin K. I., Mammoto A., Campbell M., Smith L. E. (2011) Wnt signaling mediates pathological vascular growth in proliferative retinopathy. Circulation 124, 1871–1881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Ye X., Wang Y., Cahill H., Yu M., Badea T. C., Smallwood P. M., Peachey N. S., Nathans J. (2009) Norrin, frizzled-4, and Lrp5 signaling in endothelial cells controls a genetic program for retinal vascularization. Cell 139, 285–298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Ye X., Wang Y., Nathans J. (2010) The Norrin/frizzled4 signaling pathway in retinal vascular development and disease. Trends Mol. Med. 16, 417–425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Rattner A., Wang Y., Zhou Y., Williams J., Nathans J. (2014) The role of the hypoxia response in shaping retinal vascular development in the absence of Norrin/frizzled4 signaling. Invest. Ophthalmol. Vis. Sci. 55, 8614–8625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Xia C. H., Yablonka-Reuveni Z., Gong X. (2010) LRP5 is required for vascular development in deeper layers of the retina. PLoS One 5, e11676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Zhou Y., Wang Y., Tischfield M., Williams J., Smallwood P. M., Rattner A., Taketo M. M., Nathans J. (2014) Canonical WNT signaling components in vascular development and barrier formation. J. Clin. Invest. 124, 3825–3846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Toivonen J. (1991) Plasma renin, catecholamines, vasopressin and aldosterone during hypotension induced by labetalol with isoflurane. Acta Anaesthesiol. Scand. 35, 496–501 [DOI] [PubMed] [Google Scholar]
- 159.Lobov I. B., Rao S., Carroll T. J., Vallance J. E., Ito M., Ondr J. K., Kurup S., Glass D. A., Patel M. S., Shu W., Morrisey E. E., McMahon A. P., Karsenty G., Lang R. A. (2005) WNT7b mediates macrophage-induced programmed cell death in patterning of the vasculature. Nature 437, 417–421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Ohlmann A., Scholz M., Goldwich A., Chauhan B. K., Hudl K., Ohlmann A. V., Zrenner E., Berger W., Cvekl A., Seeliger M. W., Tamm E. R. (2005) Ectopic norrin induces growth of ocular capillaries and restores normal retinal angiogenesis in Norrie disease mutant mice. J. Neurosci. 25, 1701–1710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Enomoto A., Murakami H., Asai N., Morone N., Watanabe T., Kawai K., Murakumo Y., Usukura J., Kaibuchi K., Takahashi M. (2005) Akt/PKB regulates actin organization and cell motility via Girdin/APE. Dev. Cell 9, 389–402 [DOI] [PubMed] [Google Scholar]
- 162.Anai M., Shojima N., Katagiri H., Ogihara T., Sakoda H., Onishi Y., Ono H., Fujishiro M., Fukushima Y., Horike N., Viana A., Kikuchi M., Noguchi N., Takahashi S., Takata K., Oka Y., Uchijima Y., Kurihara H., Asano T. (2005) A novel protein kinase B (PKB)/AKT-binding protein enhances PKB kinase activity and regulates DNA synthesis. J. Biol. Chem. 280, 18525–18535 [DOI] [PubMed] [Google Scholar]
- 163.Kitamura T., Asai N., Enomoto A., Maeda K., Kato T., Ishida M., Jiang P., Watanabe T., Usukura J., Kondo T., Costantini F., Murohara T., Takahashi M. (2008) Regulation of VEGF-mediated angiogenesis by the Akt/PKB substrate Girdin. Nat. Cell Biol. 10, 329–337 [DOI] [PubMed] [Google Scholar]
- 164.Ito T., Komeima K., Yasuma T., Enomoto A., Asai N., Asai M., Iwase S., Takahashi M., Terasaki H. (2013) Girdin and its phosphorylation dynamically regulate neonatal vascular development and pathological neovascularization in the retina. Am. J. Pathol. 182, 586–596 [DOI] [PubMed] [Google Scholar]
- 165.Gimbrone M. A. Jr, Cotran R. S., Leapman S. B., Folkman J. (1974) Tumor growth and neovascularization: an experimental model using the rabbit cornea. J. Natl. Cancer Inst. 52, 413–427 [DOI] [PubMed] [Google Scholar]
- 166.Bock F., Maruyama K., Regenfuss B., Hos D., Steven P., Heindl L. M., Cursiefen C. (2013) Novel anti(lymph)angiogenic treatment strategies for corneal and ocular surface diseases. Prog. Retin. Eye Res. 34, 89–124 [DOI] [PubMed] [Google Scholar]
- 167.Ellenberg D., Azar D. T., Hallak J. A., Tobaigy F., Han K. Y., Jain S., Zhou Z., Chang J. H. (2010) Novel aspects of corneal angiogenic and lymphangiogenic privilege. Prog. Retin. Eye Res. 29, 208–248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Azar D. T. (2006) Corneal angiogenic privilege: angiogenic and antiangiogenic factors in corneal avascularity, vasculogenesis, and wound healing (an American Ophthalmological Society thesis). Trans. Am. Ophthalmol. Soc. 104, 264–302 [PMC free article] [PubMed] [Google Scholar]
- 169.Chang J. H., Gabison E. E., Kato T., Azar D. T. (2001) Corneal neovascularization. Curr. Opin. Ophthalmol. 12, 242–249 [DOI] [PubMed] [Google Scholar]
- 170.Kendall R. L., Thomas K. A. (1993) Inhibition of vascular endothelial cell growth factor activity by an endogenously encoded soluble receptor. Proc. Natl. Acad. Sci. USA 90, 10705–10709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Ambati B. K., Nozaki M., Singh N., Takeda A., Jani P. D., Suthar T., Albuquerque R. J., Richter E., Sakurai E., Newcomb M. T., Kleinman M. E., Caldwell R. B., Lin Q., Ogura Y., Orecchia A., Samuelson D. A., Agnew D. W., St Leger J., Green W. R., Mahasreshti P. J., Curiel D. T., Kwan D., Marsh H., Ikeda S., Leiper L. J., Collinson J. M., Bogdanovich S., Khurana T. S., Shibuya M., Baldwin M. E., Ferrara N., Gerber H. P., De Falco S., Witta J., Baffi J. Z., Raisler B. J., Ambati J. (2006) Corneal avascularity is due to soluble VEGF receptor-1. Nature 443, 993–997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Cursiefen C., Chen L., Saint-Geniez M., Hamrah P., Jin Y., Rashid S., Pytowski B., Persaud K., Wu Y., Streilein J. W., Dana R. (2006) Nonvascular VEGF receptor 3 expression by corneal epithelium maintains avascularity and vision. Proc. Natl. Acad. Sci. USA 103, 11405–11410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Ksander B. R., Kolovou P. E., Wilson B. J., Saab K. R., Guo Q., Ma J., McGuire S. P., Gregory M. S., Vincent W. J., Perez V. L., Cruz-Guilloty F., Kao W. W., Call M. K., Tucker B. A., Zhan Q., Murphy G. F., Lathrop K. L., Alt C., Mortensen L. J., Lin C. P., Zieske J. D., Frank M. H., Frank N. Y. (2014) ABCB5 is a limbal stem cell gene required for corneal development and repair. Nature 511, 353–357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Ahmad S. (2012) Concise review: limbal stem cell deficiency, dysfunction, and distress. Stem Cells Transl. Med. 1, 110–115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Ouyang H., Xue Y., Lin Y., Zhang X., Xi L., Patel S., Cai H., Luo J., Zhang M., Zhang M., Yang Y., Li G., Li H., Jiang W., Yeh E., Lin J., Pei M., Zhu J., Cao G., Zhang L., Yu B., Chen S., Fu X. D., Liu Y., Zhang K. (2014) WNT7A and PAX6 define corneal epithelium homeostasis and pathogenesis. Nature 511, 358–361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Homer H., Chiang M. S., Hemmati H. D. H. (2013) Treatment of corneal neovascularization. Am. Acad. Opthalmol. 2013, 35–36 [Google Scholar]
- 177.Hsu C. C., Chang H. M., Lin T. C., Hung K. H., Chien K. H., Chen S. Y., Chen S. N., Chen Y. T. (2015) Corneal neovascularization and contemporary antiangiogenic therapeutics. J. Chin. Med. Assoc. 78, 323–330 [DOI] [PubMed] [Google Scholar]
- 178.Singh P., Tyagi M., Kumar Y., Gupta K. K., Sharma P. D. (2013) Ocular chemical injuries and their management. Oman J. Ophthalmol. 6, 83–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Wang Z., Zhao H., Ma J. X., Xu X. (2014) Inhibition of pathological corneal neovascularization by a small peptide derived from human apolipoprotein (a) Kringle V. Cornea 33, 405–413 [DOI] [PubMed] [Google Scholar]
- 180.Wang Z., Cheng R., Lee K., Tyagi P., Ding L., Kompella U. B., Chen J., Xu X., Ma J. X. (2015) Nanoparticle-mediated expression of a Wnt pathway inhibitor ameliorates ocular neovascularization. Arterioscler. Thromb. Vasc. Biol. 35, 855–864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Yoshida J., Wicks R. T., Zambrano A. I., Tyler B. M., Javaherian K., Grossman R., Daoud Y. J., Gehlbach P., Brem H., Stark W. J. (2015) Inhibition of corneal neovascularization by subconjunctival injection of Fc-endostatin, a novel inhibitor of angiogenesis. J. Ophthalmol. 2015, 137136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Ormerod L. D., Abelson M. B., Kenyon K. R. (1989) Standard models of corneal injury using alkali-immersed filter discs. Invest. Ophthalmol. Vis. Sci. 30, 2148–2153 [PubMed] [Google Scholar]
- 183.Giacomini C., Ferrari G., Bignami F., Rama P. (2014) Alkali burn versus suture-induced corneal neovascularization in C57BL/6 mice: an overview of two common animal models of corneal neovascularization. Exp. Eye Res. 121, 1–4 [DOI] [PubMed] [Google Scholar]
- 184.Hahn N., Dietz C. T., Kühl S., Vossmerbaeumer U., Kroll J. (2012) KLEIP deficiency in mice causes progressive corneal neovascular dystrophy. Invest. Ophthalmol. Vis. Sci. 53, 3260–3268 [DOI] [PubMed] [Google Scholar]
- 185.D’Amato R. J., Loughnan M. S., Flynn E., Folkman J. (1994) Thalidomide is an inhibitor of angiogenesis. Proc. Natl. Acad. Sci. USA 91, 4082–4085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Ferrari G., Giacomini C., Bignami F., Moi D., Ranghetti A., Doglioni C., Naldini L., Rama P., Mazzieri R. (2016) Angiopoietin 2 expression in the cornea and its control of corneal neovascularisation. Br. J. Ophthalmol. 100, 1005–1010 [DOI] [PubMed] [Google Scholar]
- 187.Kenyon B. M., Voest E. E., Chen C. C., Flynn E., Folkman J., D’Amato R. J. (1996) A model of angiogenesis in the mouse cornea. Invest. Ophthalmol. Vis. Sci. 37, 1625–1632 [PubMed] [Google Scholar]
- 188.Rogers M. S., Birsner A. E., D’Amato R. J. (2007) The mouse cornea micropocket angiogenesis assay. Nat. Protoc. 2, 2545–2550 [DOI] [PubMed] [Google Scholar]
- 189.Tang Z., Zhang F., Li Y., Arjunan P., Kumar A., Lee C., Li X. (2011) A mouse model of the cornea pocket assay for angiogenesis study. J. Vis. Exp. 18, 3077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Lee H., Schlereth S. L., Park E. Y., Emami-Naeini P., Chauhan S. K., Dana R. (2014) A novel pro-angiogenic function for interferon-γ-secreting natural killer cells. Invest. Ophthalmol. Vis. Sci. 55, 2885–2892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Sun Y., Su L., Wang Z., Xu Y., Xu X. (2013) H-RN, a peptide derived from hepatocyte growth factor, inhibits corneal neovascularization by inducing endothelial apoptosis and arresting the cell cycle. BMC Cell Biol. 14, 8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Niederkorn J. Y., Ubelaker J. E., Martin J. M. (1990) Vascularization of corneas of hairless mutant mice. Invest. Ophthalmol. Vis. Sci. 31, 948–953 [PubMed] [Google Scholar]
- 193.Kather J. N., Kroll J. (2014) Transgenic mouse models of corneal neovascularization: new perspectives for angiogenesis research. Invest. Ophthalmol. Vis. Sci. 55, 7637–7651 [DOI] [PubMed] [Google Scholar]
- 194.Cursiefen C., Ikeda S., Nishina P. M., Smith R. S., Ikeda A., Jackson D., Mo J. S., Chen L., Dana M. R., Pytowski B., Kruse F. E., Streilein J. W. (2005) Spontaneous corneal hem- and lymphangiogenesis in mice with destrin-mutation depend on VEGFR3 signaling. Am. J. Pathol. 166, 1367–1377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Mwaikambo B. R., Sennlaub F., Ong H., Chemtob S., Hardy P. (2008) Genetic ablation of CD36 induces age-related corneal neovascularization. Cornea 27, 1037–1041 [DOI] [PubMed] [Google Scholar]
- 196.Davis J., Piatigorsky J. (2011) Overexpression of Pax6 in mouse cornea directly alters corneal epithelial cells: changes in immune function, vascularization, and differentiation. Invest. Ophthalmol. Vis. Sci. 52, 4158–4168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Albuquerque R. J., Hayashi T., Cho W. G., Kleinman M. E., Dridi S., Takeda A., Baffi J. Z., Yamada K., Kaneko H., Green M. G., Chappell J., Wilting J., Weich H. A., Yamagami S., Amano S., Mizuki N., Alexander J. S., Peterson M. L., Brekken R. A., Hirashima M., Capoor S., Usui T., Ambati B. K., Ambati J. (2009) Alternatively spliced vascular endothelial growth factor receptor-2 is an essential endogenous inhibitor of lymphatic vessel growth. Nat. Med. 15, 1023–1030 [DOI] [PMC free article] [PubMed] [Google Scholar]