

Abstract
Intercellular adhesion molecule-1 (ICAM-1) mediates the firm adhesion of leukocytes to endothelial cells and initiates subsequent signaling that promotes their transendothelial migration (TEM). Vascular endothelial (VE)-cadherin plays a critical role in endothelial cell–cell adhesion, thereby controlling endothelial permeability and leukocyte transmigration. This study aimed to determine the molecular signaling events that originate from the ICAM-1–mediated firm adhesion of neutrophils that regulate VE-cadherin’s role as a negative regulator of leukocyte transmigration. We observed that ICAM-1 interacts with Src homology domain 2–containing phosphatase-2 (SHP-2), and SHP-2 down-regulation via silencing of small interfering RNA in endothelial cells enhanced neutrophil adhesion to endothelial cells but inhibited neutrophil transmigration. We also found that VE-cadherin associated with the ICAM-1–SHP-2 complex. Moreover, whereas the activation of ICAM-1 leads to VE-cadherin dissociation from ICAM-1 and VE-cadherin association with actin, SHP-2 down-regulation prevented ICAM-1–VE-cadherin association and promoted VE-cadherin–actin association. Furthermore, SHP-2 down-regulation in vivo promoted LPS-induced neutrophil recruitment in mouse lung but delayed neutrophil extravasation. These results suggest that SHP-2—via association with ICAM-1—mediates ICAM-1–induced Src activation and modulates VE-cadherin switching association with ICAM-1 or actin, thereby negatively regulating neutrophil adhesion to endothelial cells and enhancing their TEM.—Yan, M., Zhang, X., Chen, A., Gu, W., Liu, J., Ren, X., Zhang, J., Wu, X., Place, A. T., Minshall, R. D., Liu, G. Endothelial cell SHP-2 negatively regulates neutrophil adhesion and promotes transmigration by enhancing ICAM-1–VE-cadherin interaction.
Keywords: inflammation, lung injury, leukocyte, LPS
The endothelial barrier—by controlling the leakage of blood constituents and extravasation of leukocytes—plays a crucial role in regulating innate immunity. During pathologic inflammatory states, such as acute lung injury, the permeability of the endothelial barrier increases and trans-endothelial migration of leukocytes occurs (1, 2). Although these two events are related, they are differentially regulated (3, 4).
Endothelial barrier function is dependent on adherens junctions (AJs) between endothelial cells. AJs are comprised of vascular endothelial cell adhesion molecule, vascular endothelial cadherin (VE-cadherin), and associated catenins, and these are thought to be critical for the maintenance of endothelial barrier integrity (5). VE-cadherin is a Ca2+-dependent transmembrane adhesion glycoprotein (6, 7), and binding with p120 and β-catenin forms stable AJ complexes that are important for the maintenance of cell–cell adhesion (8). Stabilization of the VE-cadherin–β-catenin complex blocks vascular hyperpermeability and leukocyte extravasation in inflamed cremaster, lung, and skin tissues (9). Moreover, Rac-dependent enhancement of VE-cadherin trans-interactions also strengthens endothelial monolayer barrier and the recovery of endothelial monolayer integrity (10). Previous studies have revealed that VE-cadherin junctions disassemble in response to a variety of inflammatory mediators, such as thrombin (11), LPS, histamine, TNF (12), and VEGF (3, 13). Some mediators induce VE-cadherin phosphorylation, which leads to the destabilization of the VE-cadherin complex and disassembly of AJs. VE-cadherin internalization (14) or phosphorylation at Tyr731, Tyr658, Tyr685, or Ser665 has been shown to promote disassembly of junctions and disruption of endothelial cell contacts (4, 3, 15). In addition, it has been reported that VE-cadherin cleavage by neutrophil elastase and cathepsin G leads to junctional disruption and endothelial barrier breakdown (16, 17).
Leukocyte transendothelial migration (TEM) is mediated by several well-defined and orchestrated steps that re-associated with rolling, firm adhesion, and diapedesis-specific molecular interactions between endothelial cells and leukocytes are responsible for these actions. Intercellular adhesion molecule (ICAM-1) is the primary endothelial cell adhesion molecule that mediates the firm adhesion of leukocytes (18, 19). Proinflammatory cytokines increase ICAM-1 expression in endothelial cells in an NF-κB–dependent manner (20). Activation of ICAM-1 by integrin molecules on the leukocyte surface leads to a change in its distribution on the endothelial cell surface and initiates outside-in signaling (21). We have demonstrated that cytokines, such as TNF-α, can prime ICAM-1 in a phosphorylation-dependent manner, which leads to an increase in binding activity (22). ICAM-1 ligation induces the activation of Src tyrosine kinases, eNOS (23, 24), p38 MAPK, and Rho pathways (21, 25, 26). Recent studies have suggested that ICAM-1 activation-dependent signaling pathways also regulate the molecular events that are associated with diapedesis (23, 24).
Src homology domain 2–containing phosphatase-2 (SHP-2), a nonreceptor protein tyrosine phosphatase (PTP) that is encoded by PTPN-11 gene is known to be an ICAM-1–interacting protein (27). Normally, SHP-2 is self-inhibited by the interaction of its amino terminal, SH2 domain, with the PTP domain, thereby blocking the catalytic site (28). In endothelial cells, SHP-2 plays a critical role in several signal transduction pathways (29) and is also known to interact with the VE-cadherin–catenin complex (30). Recent studies demonstrated that SHP-2 is essential for the maintenance of endothelial barrier function both in cultured endothelial cells and in intact lungs by regulating the tyrosine phosphorylation of VE-cadherin, β-catenin, and RhoA (31, 32). Ablation of SHP-2 in endothelial cells results in a delay of the recovery of endothelial monolayer integrity (33). In addition to ICAM-1 and the VE-cadherin complex, SHP-2 also interacts with other membrane proteins that contain the unique ITIM motif, such as platelet endothelial cell adhesion molecule (PECAM), which is also important for leukocyte transmigration. SHP-2 regulates Src signaling (23) and also interacts with several molecules that play critical roles in leukocyte TEM; thus, identification of the specific roles of SHP-2 in the regulation of neutrophil transmigration may have important clinical implications.
In this study, we observed that ICAM-1 binding to SHP-2 is dependent on the ICAM-1 C-terminal tail tyrosine residue (27). Moreover, ICAM-1 and SHP-2 form a complex with VE-cadherin and β-catenin. Silencing endothelial SHP-2 interferes with ICAM-1–VE-cadherin complex formation and promotes VE-cadherin–actin interaction, thereby inhibiting neutrophil transmigration. We also show that silencing SHP-2 in vivo regulates the recruitment and infiltration of neutrophils into the alveolar space in an LPS-induced lung injury model. Our studies collectively suggest that SHP-2 plays an important role in regulating neutrophil recruitment and transmigration by regulating phosphorylation-dependent interactions between ICAM-1 and VE-cadherin.
MATERIALS AND METHODS
Reagents
SHP-2 small interfering RNA (siRNA; mouse) (GS19247) and Allstars Negative Control siRNA (SI03650318) were obtained from Qiagen (Dusseldorf, Germany). Anti–ICAM-1 (sc-1511), anti–SHP-2 (sc-7384), and anti–phospho-Src (Tyr419) (sc-139601) Abs; and SHP-2 siRNA (human; sc-36488), ICAM-1 siRNA (human; sc-29354), and control siRNA-A (sc-37007), as well as Protein A/G plus agarose (sc-2003) were obtained from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Anti-human VE-cadherin (BMS158) anti-mouse ICAM-1 mAb (YN1/1.7.4) and control rat IgG2b used in ICAM-1 crosslinking (XL) studies were purchased from eBioscience (San Diego, CA, USA). Anti–β-catenin (D10A8) (8480) was from Cell Signaling Technology (Danvers, MA, USA). Anti–ICAM-1 (phosphor Y512) (ab51033) was from Abcam (Cambridge, MA, USA). Anti–glyceraldehyde 3-phosphate dehydrogenase (60004-1-Ig), anti-Src (60315-1-Ig), and anti-actin (60008-1-Ig) Abs were from Proteintech (Wuhan, China), and Dylight 488 goat anti-rabbit IgG (A23220) was purchased from Abbkine (Wuhan, China). Lipofectamine, recombinant human TNF-α (10602HNAE50), and DAPI nucleic acid stain (D1306) were obtained from Thermo Fisher Scientific (Waltham, MA, USA). Amaxa Nucleofector kit (VPB-1002) was from Lonza (Walkersville, MD, USA). Calcein-AM (C3099) was from Thermo Fisher Scientific. Ficoll-Pacque Plus (17-1440-02) was purchased from GE Healthcare (Pittsburgh, PA, USA). LPS (L4524), dimethyldioctadecylammonium bromide (D2779), cholesterol (C8667), and glucose (G8270) were from Sigma-Aldrich (St. Louis, MO, USA). CHCl3 was purchased from Sinopharm Chemical Reagent (Sinopharm, Wuhan, China). A 24-well polycarbonate membrane insert with 3-μm pore size in Multidishes (140627) was obtained from Thermo Fisher Scientific.
Mice
Animal experiments were approved by the Animal Care Committee of Hubei Province, China, and performed in accordance with guidelines developed by the China Council on Animal Care and Protocol. Wild-type C57BL/6 female mice were purchased from the Animal Experiment Center of Wuhan University/Animal Biosafety Level III Laboratory. Wild-type C57BL/6 mice were used to generate SHP-2–deficient mice via i.v. tail injections and, after 48 h, mice received i.p. injections of LPS (8 mg/kg) (34, 35). ICAM-1−/− knockout mice were obtained from The Jackson Laboratory (Bar Harbor, ME, USA) (23). Mice age 8–10 wk and weighing 18–20 g were used for these studies.
Liposome preparation
Liposomes were prepared as described elsewhere (36, 37). In brief, dimethyldioctadecylammonium bromide and cholesterol were mixed in chloroform, then chloroform was evaporated by using a rotary evaporator under vacuum at 37°C until a thin lipid film formed. Liposomes that were dissolved in 5% glucose solution were mixed with control siRNA or SHP-2 siRNA (50 μg/mouse) (35) and injected into C57BL/6 mice by i.v. tail injection and transfection efficiency was assessed by Western blot.
Cell cultures
HUVECs purchased from Lonza were cultured in endothelial growth medium 2 that was supplemented with SingleQuots (Lonza). HUVECs were used between passages 6 and 9. Expression of mouse ICAM-1 in HUVECs was used to imitate ICAM-1 downstream signaling, as this method could keep Src kinase at baseline and only the ICAM-1 protein level is increased by transfection of the cDNA. EA.hy926 cells were maintained in DMEM that contained 10% heat-inactivated fetal bovine serum (FBS; Thermo Fisher Scientific). The EA.hy926 cell line was established by fusing primary HUVECs with A549—a thioguanine-resistant clone—by exposure to polyethylene glycol. EA.hy926 cells have characteristics of differentiated endothelial cell functions, so the cell line was used to perform immunoprecipitation analysis. The HL-60 cell line (GDC028) was purchased from the China Center for Type Culture Collection (Wuhan, China) and cultured in RPMI 1640 medium that contained 10% heat-inactivated FBS. Cell lines were transiently transfected with expression vectors according to manufacturer protocol by using Lipofectamine 2000 transfection reagent (Thermo Fisher Scientific). HUVEC transfection was performed by using the Lonza Nucleofector kit and the Nucleofector 2b Device according to manufacturer recommendations. Cells were routinely detected for mycoplasma contamination and cultured at 37°C in 5% CO2.
ICAM-1 XL
ICAM-1 was crosslinked as described elsewhere (23). In brief, monolayer cultures of HUVECs were washed with endothelial basal medium 2 and incubated with rat anti-mouse ICAM-1 Ab (15 µg/ml) for 20 min at 20–25°C. Control Ab was Rat IgG2b. Then, anti-rat IgG (50 μg/ml) was added to crosslink ICAM-1 for 20 min at 37°C.
Polymorphonuclear neutrophil isolation, adhesion, and TEM
Polymorphonuclear neutrophil (PMN) isolation was performed as described elsewhere (23). In brief, PMNs were isolated from mouse blood. The plasma layer was gathered to separate PMNs after 40 min. PMN isolation was proceeded by red blood cell removal and Ficoll-Pacque gradient centrifugation. Freshly isolated PMNs were labeled with calcein-AM according to manufacturer protocol and used immediately in PMN adhesion and TEM assays.
To determine PMN adhesion, transfected HUVECs were seeded onto 24-well plates in medium until confluent. Isolated PMNs were labeled with calcein-AM for 30 min at 37°C, then 5 × 106 PMNs per well were added to monolayers for 30 min (4). After washing with prewarmed HBSS, adherent PMNs were lysed for fluorescence measurement with PerkinElmer 2030 fluorometer at 485 and 535 nm (excitation and emission wavelengths, respectively).
For PMN TEM assay, HUVECs that were transfected with mouse ICAM-1 cDNA and control siRNA or SHP-2 siRNA were seeded onto 24-well inserts (3-μm pore size) until confluent (38). After overnight 0.1% FBS incubation, calcein-AM–labeled PMNs (2.5 × 105)—added to monolayers at a ratio of 10:1—were added and inserts were placed in medium that contained PMN activator fMLP (1 μM) for 3 h (23, 39). Transmigration PMNs were collected and lysed for fluorescence measurement as mentioned above.
Immunoprecipitation and Western blot analysis
Confluent cells in 100-mm dishes were washed with ice-cold PBS and lysed in NP-40 buffer that contained 25 mM Tris, 100 mM NaCl, 10 mM MgCl2, and 1% NP-40 (pH 7.4) that was supplemented with a phosphatase inhibitor cocktail and fresh protease inhibitor. In some experiments, cells were lysed in RIPA buffer (50 mM Tris, 150 mM NaCl, 10 mM MgCl2, 1% Triton X-100, 0.1% SDS, and 0.25% deoxycholic acid). After 10 min of incubation on ice, cell lysates were collected and centrifuged at 12,000 rpm for 10 min at 4°C. For coimmunoprecipitation experiments, supernatant was incubated with either a primary Ab or an equal amount of normal IgG, then with protein A/G plus agarose beads for 1 h at 4°C under continuous mixing. Beads were then centrifuged at 3000 rpm for 3 min at 4°C, washed 5 times with lysis buffer, and Western blot analysis was then performed as described previously (4).
Immunofluorescence
Endothelial cells were fixed in 4% paraformaldehyde in HBSS for 15 min at room temperature (RT) when cells reached confluence, then permeabilized with 0.1% Triton X-100 in HBSS for 10 min at RT (9). Cells were incubated with 5% bovine serum albumin in HBSS for 30 min at RT to block nonspecific binding, and were incubated in primary Ab for 1 h at RT, washed 3 times in HBSS, then incubated with Dylight 488 goat anti-rabbit IgG for 1 h at RT. Coverslips were washed 3 times in HBSS and mounted on antifade mounting medium. Images were captured by using a Zeiss LSM 510 laser scanning confocal microscope (Zeiss, Jena, Germany).
Myeloperoxidase activity assay
Myeloperoxidase (MPO) activity assay was performed as described elsewhere (23). In brief, after i.p. injection of LPS (8 mg/kg) or saline, bronchoalveolar lavage (BAL) fluid was obtained for protein concentration and MPO activity detection. Mouse lungs were then homogenized in 0.5% hexadecyl-trimethylammonium bromide. After centrifugation, aliquots of the supernatant were also used for total protein concentration and MPO activity detection. Supernatant was loaded onto a 96-well plate, then 0.0005% hydrogen peroxide and O-dianisidine dihydrochloride were added. Three minutes later, absorbance measurement was performed at 460 nm. MPO activity was expressed as the change in absorbance per minute per gram of tissue.
xCELLigence assay
HUVECs were seeded onto an E-Plate View 16 (Roche, Indianapolis, IL, USA) at a density of 2000 cells per well. Cells were transfected with SHP-2 or control siRNA and stimulated with ICAM-1 Ab once confluent. Impedance was monitored by using xCELLigence RTCA system (ACEA Biosciences, San Diego, CA, USA) as previously described (14, 40). Impedance—recorded as cell index—was measured every 15 min. After treatment, impedance was measured every 15 s for 6 h. All HUVEC experiments were performed using 2 different batches of HUVECs.
Experimental data and statistical analysis
All data are expressed as means ± sem. Comparison between groups was performed by using 2-way ANOVA, followed by Bonferroni post-tests. Pairwise comparisons were made by using 2-tailed Student’s t test. Results were considered significant when P < 0.05.
RESULTS
Activation of Src tyrosine kinases in response to ICAM-1 XL is mediated by SHP-2
It has been reported that ICAM-1 is able to interact with SHP-2 and form a complex in transfected cells (27, 41). Here, by using SHP-2 Ab, we immunoprecipitated SHP-2 from EA.hy926 cells that were transfected with mouse ICAM-1. These results indicate that expressed ICAM-1 interacts with endogenous SHP-2 (Fig. 1A). We also observed native endogenous ICAM-1 interaction with SHP-2 by coimmunoprecipitation of mouse lung lysates (Fig. 1B). These results demonstrate that ICAM-1 is able to interact with SHP-2 in vitro and in vivo.
Figure 1.
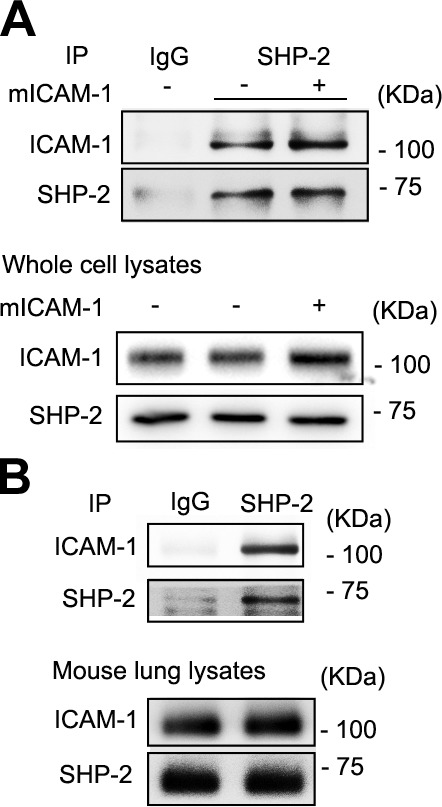
SHP-2 interacts with ICAM-1 in vitro and in vivo. A) EA.hy926 cells were transfected with mouse wild-type ICAM-1 cDNA and immunoprecipitation (IP) with Ab to SHP-2, followed by Western blot analysis with anti–SHP-2 or anti–ICAM-1. Total protein levels of ICAM-1 and SHP-2 were also determined by Western blot. B) Mouse lung was lysed, then IP was performed with Ab to SHP-2, followed by Western blot analysis with anti–SHP-2 and anti–ICAM-1. Whole-lung protein levels of ICAM-1 and SHP-2 were determined by Western blot. Three independent experiments were performed.
Previous studies demonstrated that ICAM-1 activation by Ab XL in HUVECs induces activation of Src (23, 25). To investigate whether SHP-2 is involved in ICAM-1–mediated Src activation, we used SHP-2–specific siRNA to down-regulate SHP-2 expression, then assessed Src phosphorylation at active site Y419, which is used as an index of Src activation after ICAM-1 Ab XL. Western blot showed that SHP-2 was down-regulated by approximately 90% in SHP-2 siRNA-transfected cells (Fig. 2A), and that in these cells, ICAM-1 XL-induced Src phosphorylation (Y419) was decreased by 40% in the presence of XL (Fig. 2B). Phosphorylation of ICAM-1 was also increased in SHP-2 siRNA transfected HUVECs (Fig. 2C). These observations suggest that SHP-2 mediates ICAM-1 XL-induced Src activation.
Figure 2.

SHP-2 mediates ICAM-1 activation-induced Src phosphorylation. HUVECs were cotransfected with mouse wild-type (WT) ICAM-1 cDNA and SHP-2 siRNA or control (Ctrl) siRNA for 48 h, then ICAM-1 XL was performed. A) Expression of SHP-2 in transfected cells was determined by Western blot analysis (Student’s unpaired t test, 2-tailed). B–E) Western blot analysis of phosphorylation of Src at Y419 (human; B), total protein levels of Src (C), phosphorylation of ICAM-1 at Y512 (D), and total protein levels of ICAM-1 (E) were determined. Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) served as loading control, and the blots of GAPDH were derived from the same samples. Data are presented as means ± sem of 3 independent experiments and data analysis was performed by Bonferroni post-tests after 2-way ANOVA. *P < 0.05; **P < 0.01; ***P < 0.001.
SHP-2 differentially regulates neutrophil adhesion and transmigration
To determine the functional significance of SHP-2–dependent Src activation, we assessed leukocyte adhesion and TEM via HUVEC monolayers. We first assessed the effect of SHP-2 siRNA on the adhesion of neutrophils to endothelial cells and observed that neutrophil adhesion to SHP-2 siRNA-transfected HUVECs was increased by 30% compared with cells that were transfected with control siRNA (Fig. 3A).
Figure 3.

SHP-2 differentially regulates neutrophil adhesion and transmigration. A) Neutrophils that were isolated from fresh mouse blood were labeled, activated, and added to HUVECs that were transfected with mouse ICAM-1 cDNA and control or SHP-2 siRNA. After 2–3 h, HUVECs were washed and lysed, then neutrophil fluorescence was measured. B) Cells were treated as in panel A, then plated on Transwell inserts, fMLP was added to the bottom chamber, and freshly isolated neutrophils that were labeled with fluorescent dye were added to the upper chamber. Neutrophils that transmigrated into the bottom chamber were collected and measured. C) Cotransfected HUVECs were grown on coverslips for 48 h, stimulated with rat anti-mouse ICAM-1 mAb, TNF-α, or left untreated, and then fixed and stained for VE-cadherin (green in merged image). Arrows indicate gap formation. Data are presented as means ± sem of 3 independent experiments and data analysis performed by 2-tailed Student’s unpaired t test. **P < 0.01. Scale bar, 20 μm.
We next investigated whether SHP-2 expression regulates neutrophil TEM. TEM assay was conducted after HUVECs were cotransfected with mouse ICAM-1 cDNA and SHP-2 siRNA and seeded in Transwells. Mouse neutrophils were added to the upper chamber of the insert and transmigration was induced by fMLP. We observed that neutrophil transmigration via HUVEC monolayers that have been transfected with SHP-2 siRNA was reduced by 20% compared with control siRNA-transfected cells (Fig. 3B).
To determine the effect of SHP-2 silencing on endothelial cell permeability, we first used confocal microscopy to examine that intercellular gap formation in HUVEC monolayers that were stimulated with TNF-α or by ICAM-1 Ab XL. As shown in Fig. 3C, SHP-2 down-regulation by siRNA did not affect intercellular gap formation in response to ICAM-1 Ab XL or stimulation with TNF-α. These results suggest that the gap formation was not impaired by SHP-2 down-regulation. To estimate the effect of SHP-2 down-regulation on endothelial barrier function, we monitored endothelial cell impedance to examine the effect of SHP-2 down-regulation on endothelial junctional integrity in the presence of ICAM-1 XL. As shown in Supplemental Fig. 1, SHP-2 down-regulation demonstrated no effects on monolayer formation, but the impedance of SHP-2 siRNA-transfected cells was slightly decreased after transfected HUVECs were activated by ICAM-1 Ab XL. These results suggest that SHP-2 might regulate endothelial barrier recovery.
SHP-2 regulates endothelial ICAM-1 interaction with the AJ complex
In addition to binding to ICAM-1, SHP-2 also specifically associates with β-catenin in endothelial cell AJs (30). To further understand the role of endothelial cell SHP-2 in neutrophil transmigration across endothelial monolayers and the relationship between SHP-2, ICAM-1, and VE-cadherin interaction in transmigration, we assessed these proteins in the immunoprecipitation complex of EA.hy926 cell lysates. By using SHP-2 Ab for the immunoprecipitation of EA.hy926 cell lysates, we observed VE-cadherin, β-catenin, and ICAM-1 in the immunoprecipitated complex (Fig. 4A). Activation of ICAM-1 by Ab XL decreased its association with SHP-2, but increased the association of VE-cadherin and β-catenin with SHP-2.
Figure 4.
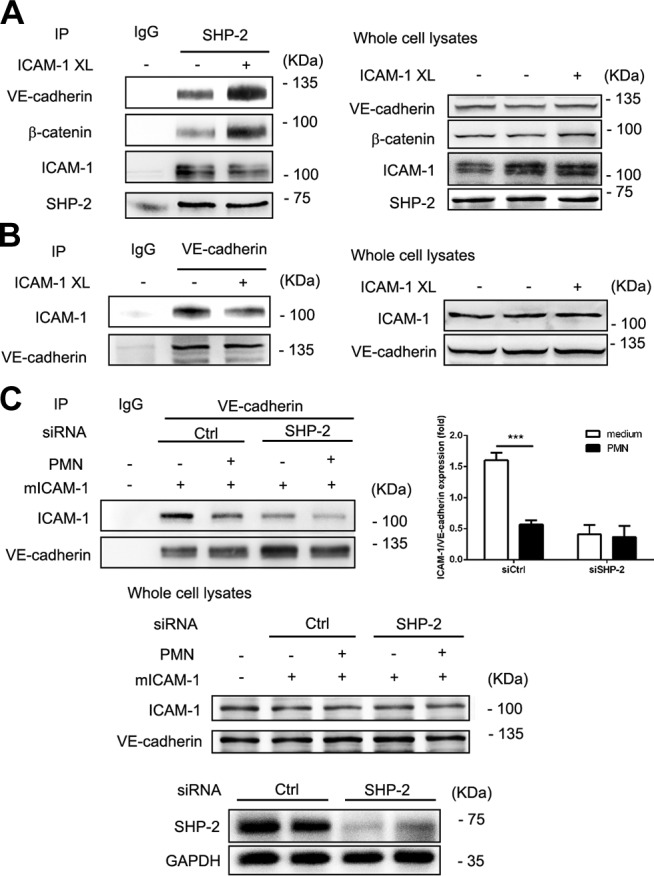
SHP-2 interacts with ICAM-1 and the VE-cadherin–β-catenin complex. A) EA.hy926 cells that were transfected with mouse ICAM-1 cDNA were crosslinked and lysed. Immunoprecipitation (IP) of proteins from cell lysates with Abs to SHP-2 was performed, followed by Western blot analysis with anti–SHP-2, anti–ICAM-1, anti–β-catenin, and anti–VE-cadherin Abs. Total protein levels of ICAM-1, SHP-2, β-catenin, and VE-cadherin were also determined by Western blot. B) EA.hy926 cell were treated as in panel A, followed by IP with Ab to VE-cadherin and Western blot analysis with anti–ICAM-1 and anti–VE-cadherin Abs. Total protein levels of ICAM-1 and VE-cadherin were determined by Western blot. C) EA.hy926 cell were cotransfected with mICAM-1 cDNA and SHP-2 siRNA or control (Ctrl) siRNA, and freshly isolated and activated neutrophils were added to EA.hy926 cell for 2 h, then cells were washed twice and lysed. Cell lysates were immunoprecipitated with Ab to VE-cadherin and Western blot analysis with anti–ICAM-1 and anti–VE-cadherin Abs. Then, ICAM-1–VE-cadherin expression was quantitated. Total protein levels of ICAM-1 and VE-cadherin were also determined by Western blot. In panels A–C, 3 independent experiments were performed. GAPDH, glyceraldehyde 3-phosphate dehydrogenase. Data are presented as means ± sem, and data analysis was performed by Bonferroni post-tests after 2-way ANOVA in panel C. ***P < 0.001.
To further understand the relationship between ICAM-1 and VE-cadherin in endothelial cells, we examined the association of ICAM-1 with VE-cadherin by using VE-cadherin Ab for immunoprecipitation. Results showed that activation of ICAM-1 by Ab XL decreased the association of ICAM-1 with VE-cadherin (Fig. 4B). Moreover, when freshly isolated and activated neutrophils were added to endothelial cells, the association of ICAM-1 with VE-cadherin was further decreased (Fig. 4C). These results suggest that during neutrophil adhesion and transmigration, ICAM-1 activation leads to VE-cadherin dissociation from ICAM-1 and SHP-2 association with ICAM-1. Tyrosine residue Y518 in the intracellular C terminus of ICAM-1 is important for interaction with SHP-2 (23, 27); therefore, we examined the effect of Y518 ICAM-1 mutants on the immunoprecipitated complex. Both Y518F and Y518D reduced the interaction between ICAM-1 and SHP-2 (Fig. 5A), which suggests that this tyrosine residue is important for their interaction. Another interesting observation was that β-catenin remained in the immunoprecipitated complex independent of ICAM-1 mutants. In contrast, VE-cadherin association was dependent on Y518 ICAM-1 (Fig. 5A). This observation further confirmed the observation that VE-cadherin is able to interact with ICAM-1 and suggested that SHP-2 may provide the linkage between these two molecules. To prove this hypothesis, siRNA that was specific to SHP-2 was transfected into EA.hy926 cells, then, using ICAM-1 Ab for immunoprecipitation, we found that SHP-2 down-regulation weakened the association between VE-cadherin and ICAM-1 (Fig. 5B). Conversely, overexpression of SHP-2 enhanced the interaction between ICAM-1 and VE-cadherin (Fig. 5B).
Figure 5.

SHP-2 mediates the interaction of ICAM-1 and the VE-cadherin–β-catenin complex. A) Immunoprecipitation (IP) with Ab to anti–SHP-2 Ab was performed by using EA.hy926 cell lysates after transfection with mouse ICAM-1 wild-type (WT), ICAM-1 Y518F, and ICAM-1 Y518D cDNA for 48 h, followed by Western blot analysis with anti–SHP-2, anti–ICAM-1, anti–VE-cadherin, and anti–β-catenin Abs. Whole-cell lysates (right). Whole protein levels of ICAM-1, SHP-2, VE-cadherin, and β-catenin were determined by Western blot. B) EA.hy926 cells were transfected with SHP-2 siRNA, control (Ctrl) siRNA, and SHP-2 cDNA, then IP of proteins from EA.hy926 cell lysates anti–ICAM-1 Ab was performed, followed by Western blot analysis with anti–SHP-2, anti–ICAM-1, anti–VE-cadherin, and anti–β-catenin Abs. Protein levels of ICAM-1, SHP-2, VE-cadherin, and β-catenin were determined by Western blot. C) EA.hy926 cells that were transfected with mouse ICAM-1 WT cDNA and control or SHP-2 siRNA for 48 h were crosslinked and lysed. IP of proteins from cell lysates with VE-cadherin Ab was performed, followed by Western blot analysis with anti-actin and anti–VE-cadherin Abs. Whole protein levels of actin and VE-cadherin were determined by Western blot. GAPDH, glyceraldehyde 3-phosphate dehydrogenase. In panels A–C, 3 independent experiments were performed.
We also examined the role of SHP-2 in regulating the interaction between ICAM-1 and VE-cadherin when transfected endothelial cells were exposed to freshly isolated and activated neutrophils. After SHP-2 down-regulation, the interaction of ICAM-1 with VE-cadherin decreased in the absence of neutrophils, and there were no changes in interaction after neutrophil adhesion. These results suggest that SHP-2 is able to regulate the interaction of ICAM-1 with VE-cadherin during neutrophil adhesion and transmigration. In addition to the interaction of ICAM-1 with VE-cadherin, VE-cadherin and β-catenin expressions were decreased at 0 h after LPS exposure in the absence of ICAM-1, and were further decreased after LPS exposure for 2 or 24 h (Supplemental Fig. 2).
Previously known, VE-cadherin associates with the actin cytoskeleton in endothelial cells. The interaction of VE-cadherin with actin increased after activation of ICAM-1 by Ab XL (Fig. 5C). In contrast, VE-cadherin–actin interaction increased in SHP-2 silencing endothelial cells at resting level, and there were no changes in interaction after ICAM-1 XL (Fig. 5C). These results suggest that SHP-2 is able to regulate the dynamic interaction of VE-cadherin with actin during neutrophil adhesion and transmigration.
SHP-2 regulates Src activity and neutrophil extravasation in vivo
To further investigate the role of SHP-2 in ICAM-1–mediated neutrophil transmigration in vivo, C57BL/6 mice were injected with either SHP-2 siRNA or control siRNA in liposomes and, after 48 h, mouse lung tissues were collected for analysis at 0, 2, 4, or 8 h after LPS injection. Expression of SHP-2 was examined by Western blot. SHP-2 siRNA in liposomes resulted in 80–90% down-regulation of SHP-2 in lung tissues (Fig. 6A) compared with control siRNA. LPS treatment induced down-regulation of SHP-2 within 2–8 h. Lung MPO activity of control siRNA-treated mice increased by 66% at 2 h after LPS injection, then returned to control levels by 4 h. In contrast, in SHP-2–depleted mice, MPO activity increased by 70% at 2 h after LPS injection and remained elevated through 8 h (Fig. 6B), which suggests that SHP-2 may regulate neutrophil recruitment to the lungs. MPO activity and protein concentration in BAL fluid were also examined. BAL MPO activity gradually increased after LPS injection in the control group, whereas basal MPO activity was initially elevated, then increased greatly at 8 h in mice that had been depleted of endothelial cell SHP-2 (Fig. 6C). BAL protein concentration was significantly increased at 4 h after LPS injection in the control group, whereas a delayed increase at 8 h was observed in the SHP-2 siRNA-treated group (Fig. 6D). These results suggest that SHP-2 regulates neutrophil extravasation in the lungs and affects the timing of endothelial barrier recovery.
Figure 6.
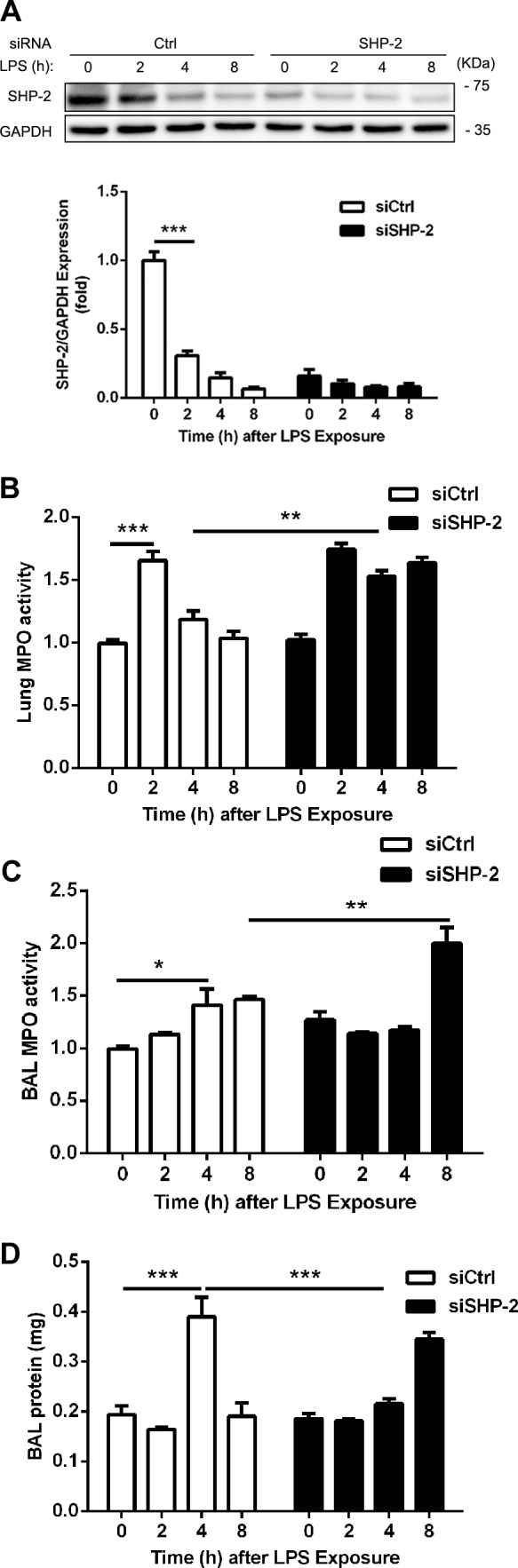
SHP-2 regulates neutrophil extravasation in vivo. Adult (age 8–10 wk) C57BL/6 mice were injected with control (Ctrl) or SHP-2 siRNA in liposomes. At 44 h after injection, mice were injected with LPS (8 mg/kg i.p.), and later lung tissue and BAL fluids were collected at 0, 2, 4, and 8 h. A) Down-regulation of SHP-2 by siRNA silencing was estimated by Western blot. B–D) Lung tissue MPO activity (B), BAL MPO activity (C), and protein contents in BAL were determined (D; n = 3 mice for each group). The blots of glyceraldehyde 3-phosphate dehydrogenase (GAPDH) were derived from the same samples. Data are presented as means ± sem of 3 independent experiments, and data analysis was performed by Bonferroni post-tests after 2-way ANOVA. *P < 0.05; **P < 0.01; ***P < 0.001.
To further understand the role of SHP-2 in neutrophil extravasation, we examined the expression of ICAM-1 and VE-cadherin and the phosphorylation of Src. There was no significant difference in basal ICAM-1 expression between control and SHP-2 siRNA-treated mice (Fig. 7A). LPS-induced ICAM-1 expression presented a different dynamic pattern; the level of Src pY419 was significantly increased by 3.4-fold in SHP-2–silenced mice (Fig. 7B). LPS-induced phosphorylation of Src Y419 peaked at 4 h after LPS injection in the control group, whereas phosphorylation of Src remained elevated in SHP-2–depleted mice (Fig. 7B). Expression of VE-cadherin also presented a different dynamic pattern. After SHP-2 down-regulation in mice, LPS induced the degradation of VE-cadherin (Fig. 7C). These results suggest that SHP-2 regulates the ICAM-1–VE-cadherin signaling pathway in mouse lungs.
Figure 7.
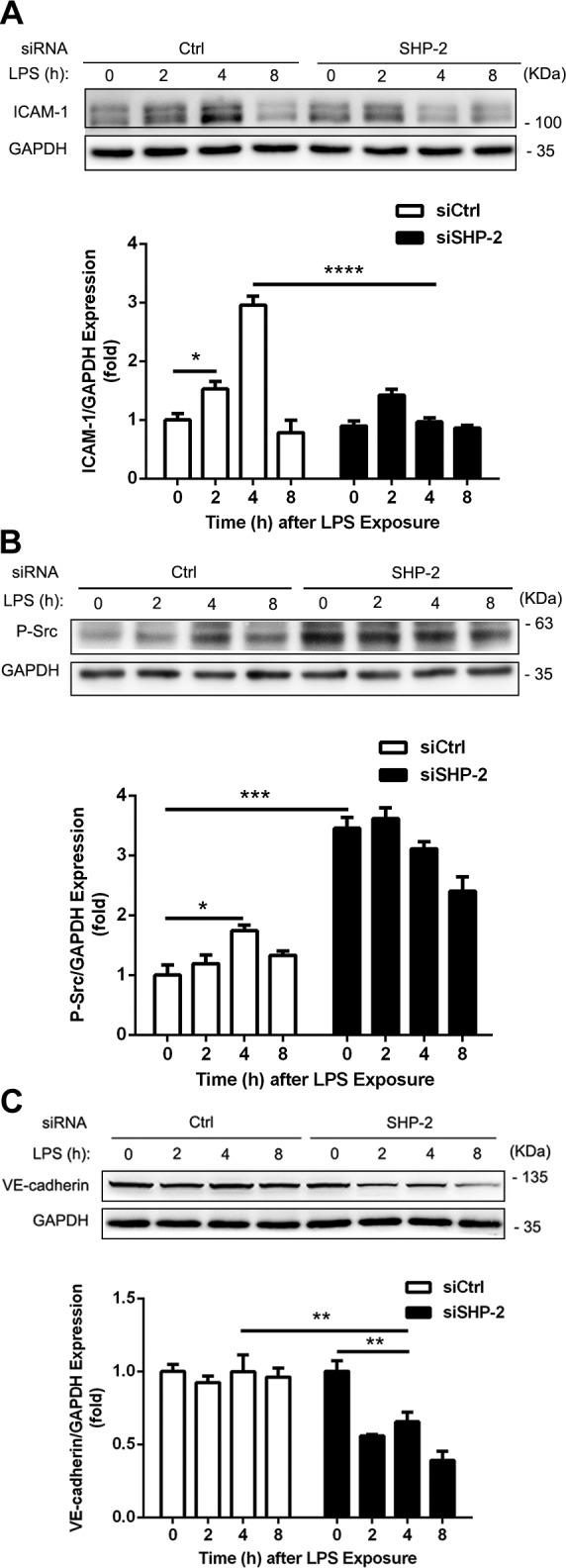
LPS-induced degradation of VE-cadherin in mouse lungs after SHP-2 down-regulation. Adult (age 8–10 wk) C57BL/6 mice were treated as in Fig. 6. Lungs were collected and lysed for total protein level by Western blot. Expression of ICAM-1 (A), phosphorylation levels of Src at Y419 (human; B), and protein levels of VE-cadherin (C) were determined by Western blot. Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) served as loading control and the blots of GAPDH were derived from the same samples (n = 3 mice for each group). Ctrl, control. Data are presented as means ± sem of 3 independent experiments and data analysis was performed by Bonferroni post-tests after 2-way ANOVA. *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001.
To exclude the possibility that increased neutrophil recruitment into lungs was a result of SHP-2 down-regulation in neutrophils, the neutrophil-like HL-60 cell line was used to detect the effect of SHP-2 down-regulation on neutrophil adhesion to endothelial cells and transmigration across endothelial cells. As shown in Supplemental Fig. 3, both adhesion and transmigration demonstrated no significant change when SHP-2 was silenced in HL-60 cells.
DISCUSSION
In this study, we explored the role of SHP-2 in ICAM-1–dependent neutrophil adhesion and subsequent TEM. Previously, studies have demonstrated that upon firm adhesion of neutrophils, ICAM-1 via Src and eNOS was able to initiate signaling involved in TEM via PECAM (23). Other molecules, including VE-cadherin, are involved in neutrophil TEM. The mechanism by which ICAM-1 signaling initiates TEM independent of PECAM has not been addressed; therefore, these studies were initiated to determine the ICAM-1–mediated signaling pathways that regulate AJ molecules during TEM.
The intracellular C-terminal domain of ICAM-1 contains the binding site for SHP-2 (27), and we have previously reported that SHP-2 is involved in ICAM-1–mediated Src activation in that SHP-2 phosphatase-defective mutant exhibited reduced Src phosphorylation at Y419 (23). In the current study, by using SHP-2 siRNA silencing, we observed that SHP-2 down-regulation blocks ICAM-1 activation-induced Src phosphorylation at Y419. This additional evidence further suggests that SHP-2 is involved in ICAM-1–induced Src activation. Another interesting observation was that basal Src phosphorylation was increased after SHP-2 down-regulation. Basal Src phosphorylation level also greatly increased in mouse lung tissues with SHP-2 siRNA silencing. The increase in basal Src phosphorylation suggests that SHP-2 is also required to keep Src in the inactivated state (42); therefore, it is possible that there are dual mechanisms of SHP-2–dependent regulation of Src activity. There are 2 phosphorylation sites on Src. Phosphorylation of Src Tyr530 is inhibitory, whereas phosphorylation of Src Tyr419 is associated with its activation. It is likely that SHP-2 is able to dephosphorylate both (42).
In the immunoprecipitation complex of ICAM-1 and SHP-2, we identified VE-cadherin and β-catenin. The interaction of SHP-2 with the VE-cadherin–β-catenin complex has been reported previously, and it is believed that SHP-2 associates with β-catenin in the AJ complex (30, 43). Immunoprecipitation results also demonstrated that SHP-2 is more likely to interact with β-catenin, as the association of VE-cadherin, but not β-catenin, was regulated by ICAM-1. Expression of ICAM-1 phosphomutants differentially associated with SHP-2, and the association of VE-cadherin with the complex was significantly affected by these mutants. It has recently been reported that SHP-2 interaction with VE-cadherin can be regulated by thioredoxin-interacting protein (44); therefore, the present findings suggest that the association of VE-cadherin with SHP-2 is more complicated than β-catenin binding to SHP-2. It is more likely that other proteins in the complex also affect their association.
In this study, we showed that ICAM-1 is able to regulate the interaction of SHP-2 with VE-cadherin. In addition, we observed VE-cadherin interaction with ICAM-1, which suggests that there may be direct interaction between ICAM-1 and VE-cadherin. In endothelial cells that were activated by ICAM-1 Ab XL or neutrophil adhesion, we observed that the interaction of VE-cadherin with ICAM-1 is dynamically regulated by the activation of ICAM-1. Thus, SHP-2 is able to regulate the interaction of ICAM-1 with VE-cadherin. Moreover, SHP-2 is also able to regulate the interaction of VE-cadherin with actin. Our results suggest that SHP-2 controls the switch of VE-cadherin interaction with ICAM-1 to interaction with actin. Taken together, these studies clearly suggest that the ICAM-1–SHP-2–VE-cadherin signaling pathway regulates TEM (Fig. 8). Other studies have addressed the role of SHP-2–VE-cadherin interaction in the phosphorylation of VE-cadherin (30). Our study suggests that ICAM-1 may mediate the signaling by direct association with VE-cadherin.
Figure 8.
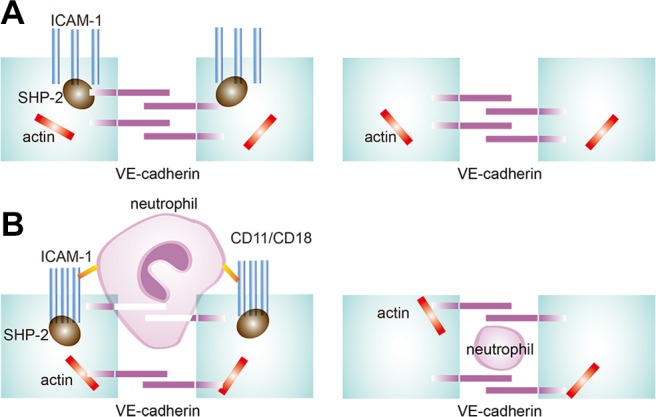
Schematic view of neutrophil–endothelial adhesion and transmigration. A) Under resting conditions, ICAM-1, SHP-2, and VE-cadherin form a complex in endothelial cells. ICAM-1 distributes uniformly on cells and VE-cadherin also localizes in sequence. B) In contrast to resting conditions, neutrophil–endothelial adhesion leads to ICAM-1–SHP-2–VE-cadherin complex disassembly, an increase in VE-cadherin–actin interaction, ICAM-1 cluster formation, and VE-cadherin behaving like a curtain.
SHP-2 down-regulation in endothelial cells results in more neutrophil adhesion and less TEM (Fig. 3). The increase in neutrophil adhesion may be a result of higher basal Src activation and the resultant ICAM-1 phosphorylation. We have previously shown that phosphorylation of ICAM-1 enhanced neutrophil binding (45). The inhibitory effects of SHP-2 down-regulation on neutrophil TEM is therefore not likely a result of reduced junction opening, as confocal studies demonstrated a normal endothelial cell response to ICAM-1 Ab XL and TNF-α treatment. Wessel et al. (4) showed that SHP-2 down-regulation exhibited similar results in TNF-α–treated End5 mouse brain endothelial cells. They proposed that VE-cadherin endocytosis may be responsible for reduced neutrophil TEM. In addition to endocytosis, other mechanisms may be at play. It has been reported that during neutrophil transmigration, VE-cadherin behaves like a curtain, first in an open and then a resealed configuration (46). Movement of VE-cadherin is supported by studies that have demonstrated VE-cadherin association with the actin cytoskeleton in endothelial cells (47–49). Our studies suggest that ICAM-1 activation by neutrophil adhesion triggers first the open and then resealed mode for VE-cadherin, and that SHP-2 down-regulation may inhibit this dynamic change.
We also examined the effect of SHP-2 down-regulation in vivo. We showed that more neutrophils are recruited to the mouse lung upon LPS stimulation in SHP-2–depleted lungs. This may be a result of the high basal level of Src activation; however, neutrophil transmigration into BAL fluid was less sensitive to LPS challenge at early time points, which suggests that neutrophil transmigration may be inhibited during the early phase of inflammation. At the 8-h time point after LPS challenge, more neutrophils were observed in BAL fluid, which suggests endothelial barrier dysfunction that may be a result of the failure of VE-cadherin recovery in SHP-2–depleted mouse lungs at that time (Fig. 7). Although the role of SHP-2 in neutrophil TEM in vivo is more complicated, our results suggest that SHP-2 is able to regulate neutrophil recruitment and transmigration in inflamed lungs.
In addition to the above roles of SHP-2 in regulating neutrophil adhesion, recruitment, and TEM, our studies also support the hypothesis that SHP-2 regulates endothelial permeability in vivo. A variety of inflammatory mediators may damage endothelial barrier function by VE-cadherin cleavage, endocytosis, phosphorylation, or internalization (17, 35, 50–52). When endothelial cells are subjected to these inflammatory mediators, Src family tyrosine kinases are activated, followed by increased phosphorylation of VE-cadherin and β-catenin and disassembly of AJs (4, 12). However, Src-mediated tyrosine phosphorylation of VE-cadherin is not sufficient to disrupt the VE-cadherin, p120-catenin, and β-catenin complexes or increase endothelial cell permeability. Recently, however, SHP-2 has been shown to support pulmonary endothelial barrier function as well as to mediate the recovery of impaired endothelial barrier function by stimulating AJ reassembly, as SHP-2 down-regulation in endothelial cells delayed the recovery of thrombin-induced hyperpermeability (31, 33).
In summary, SHP-2, via association with ICAM-1, mediates ICAM-1–induced Src activation and modulates ICAM-1 association with VE-cadherin, thereby negatively regulating neutrophil adhesion to endothelial cells and enhancing their TEM. SHP-2 may be a target for potential therapy in a sepsis or acute respiratory distress syndrome model in terms of potentially alleviating inflammation.
ACKNOWLEDGMENTS
This work was supported by National Natural Science Foundation of China (Grant 31372418; to G.L.), the Huazhong Agricultural University Scientific and Technology Self-innovation Foundation (program no. 2012RC011; to G.L.), the Fundamental Research Funds for the Central University (project no. 2662013PY054; to G.L.), the 948 Project of Chinese Ministry of Agriculture (Grant 2015-Z33; to G.L.), and U.S. National Institutes of Health National Heart, Lung, and Blood Institute Grants R01-HL71626 and P01-HL60678 (to R.D.M.). The authors thank Maricela Castellon (University of Illinois at Chicago) for excellent technical assistance. The authors declare no conflicts of interest.
Glossary
- AJ
adherens junction
- BAL
bronchoalveolar lavage
- FBS
fetal bovine serum
- ICAM-1
intercellular adhesion molecule-1
- MPO
myeloperoxidase
- PECAM
platelet endothelial cell adhesion molecule
- PMN
polymorphonuclear neutrophil
- PTP
protein tyrosine phosphatase
- RT
room temperature
- SHP-2
Src homology domain 2–containing phosphatase-2
- siRNA
small interfering RNA
- TEM
transendothelial migration
- VE-cadherin
vascular endothelial cadherin
- XL
crosslinking
Footnotes
This article includes supplemental data. Please visit http://www.fasebj.org to obtain this information.
AUTHOR CONTRIBUTIONS
M. Yan performed experiments, analyzed the data, prepared figures, participated in experimental design and in writing the manuscript; A. Chen and W. Gu performed some of the experiments and confocal microscopy; J. Liu and J. Zhang helped with in vivo experiments; X. Zhang and X. Ren performed some of the experiments with endothelial cells; A. T. Place and R. D. Minshall collaborated on the ICAM-1 knockout mouse studies and participated in experimental design and writing the manuscript; and G. Liu designed the experiments, supervised the work, analyzed the data, prepared the figures, and wrote the manuscript.
REFERENCES
- 1.Castillo R. L., Carrasco Loza R., Romero-Dapueto C. (2015) Pathophysiological approaches of acute respiratory distress syndrome: novel bases for study of lung injury. Open Respir. Med. J. 9, 83–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Grommes J., Soehnlein O. (2011) Contribution of neutrophils to acute lung injury. Mol. Med. 17, 293–307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wallez Y., Cand F., Cruzalegui F., Wernstedt C., Souchelnytskyi S., Vilgrain I., Huber P. (2007) Src kinase phosphorylates vascular endothelial-cadherin in response to vascular endothelial growth factor: identification of tyrosine 685 as the unique target site. Oncogene. 26, 1067–1077 [DOI] [PubMed] [Google Scholar]
- 4.Wessel F., Winderlich M., Holm M., Frye M., Rivera-Galdos R., Vockel M., Linnepe R., Ipe U., Stadtmann A., Zarbock A., Nottebaum A. F., Vestweber D. (2014) Leukocyte extravasation and vascular permeability are each controlled in vivo by different tyrosine residues of VE-cadherin. Nat. Immunol. 15, 223–230 [DOI] [PubMed] [Google Scholar]
- 5.Giannotta M., Trani M., Dejana E. (2013) VE-cadherin and endothelial adherens junctions: active guardians of vascular integrity. Dev. Cell 26, 441–454 [DOI] [PubMed] [Google Scholar]
- 6.Heimark R. L., Degner M., Schwartz S. M. (1990) Identification of a Ca2+-dependent cell-cell adhesion molecule in endothelial cells. J. Cell Biol. 110, 1745–1756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gooding J. M., Yap K. L., Ikura M. (2004) The cadherin-catenin complex as a focal point of cell adhesion and signalling: new insights from three-dimensional structures. BioEssays 26, 497–511 [DOI] [PubMed] [Google Scholar]
- 8.Ishiyama N., Lee S. H., Liu S., Li G. Y., Smith M. J., Reichardt L. F., Ikura M. (2010) Dynamic and static interactions between p120 catenin and E-cadherin regulate the stability of cell-cell adhesion. Cell 141, 117–128 [DOI] [PubMed] [Google Scholar]
- 9.Schulte D., Küppers V., Dartsch N., Broermann A., Li H., Zarbock A., Kamenyeva O., Kiefer F., Khandoga A., Massberg S., Vestweber D. (2011) Stabilizing the VE-cadherin-catenin complex blocks leukocyte extravasation and vascular permeability. EMBO J. 30, 4157–4170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Birukova A. A., Tian Y., Dubrovskyi O., Zebda N., Sarich N., Tian X., Wang Y., Birukov K. G. (2012) VE-cadherin trans-interactions modulate Rac activation and enhancement of lung endothelial barrier by iloprost. J. Cell. Physiol. 227, 3405–3416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rabiet M. J., Plantier J. L., Rival Y., Genoux Y., Lampugnani M. G., Dejana E. (1996) Thrombin-induced increase in endothelial permeability is associated with changes in cell-to-cell junction organization. Arterioscler. Thromb. Vasc. Biol. 16, 488–496 [DOI] [PubMed] [Google Scholar]
- 12.Angelini D. J., Hyun S. W., Grigoryev D. N., Garg P., Gong P., Singh I. S., Passaniti A., Hasday J. D., Goldblum S. E. (2006) TNF-alpha increases tyrosine phosphorylation of vascular endothelial cadherin and opens the paracellular pathway through fyn activation in human lung endothelia. Am. J. Physiol. Lung Cell. Mol. Physiol. 291, L1232–L1245 [DOI] [PubMed] [Google Scholar]
- 13.Oas R. G., Xiao K., Summers S., Wittich K. B., Chiasson C. M., Martin W. D., Grossniklaus H. E., Vincent P. A., Reynolds A. B., Kowalczyk A. P. (2010) p120-catenin is required for mouse vascular development. Circ. Res. 106, 941–951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gong H., Gao X., Feng S., Siddiqui M. R., Garcia A., Bonini M. G., Komarova Y., Vogel S. M., Mehta D., Malik A. B. (2014) Evidence of a common mechanism of disassembly of adherens junctions through Gα13 targeting of VE-cadherin. J. Exp. Med. 211, 579–591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Qian L. W., Greene W., Ye F., Gao S. J. (2008) Kaposi’s sarcoma-associated herpesvirus disrupts adherens junctions and increases endothelial permeability by inducing degradation of VE-cadherin. J. Virol. 82, 11902–11912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hermant B., Bibert S., Concord E., Dublet B., Weidenhaupt M., Vernet T., Gulino-Debrac D. (2003) Identification of proteases involved in the proteolysis of vascular endothelium cadherin during neutrophil transmigration. J. Biol. Chem. 278, 14002–14012 [DOI] [PubMed] [Google Scholar]
- 17.Golovkine G., Faudry E., Bouillot S., Voulhoux R., Attrée I., Huber P. (2014) VE-cadherin cleavage by LasB protease from Pseudomonas aeruginosa facilitates type III secretion system toxicity in endothelial cells. PLoS Pathog. 10, e1003939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yang L., Froio R. M., Sciuto T. E., Dvorak A. M., Alon R., Luscinskas F. W. (2005) ICAM-1 regulates neutrophil adhesion and transcellular migration of TNF-alpha-activated vascular endothelium under flow. Blood 106, 584–592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lawson C., Wolf S. (2009) ICAM-1 signaling in endothelial cells. Pharmacol. Rep. 61, 22–32. [DOI] [PubMed] [Google Scholar]
- 20.Sidibé A., Polena H., Pernet-Gallay K., Razanajatovo J., Mannic T., Chaumontel N., Bama S., Maréchal I., Huber P., Gulino-Debrac D., Bouillet L., Vilgrain I. (2014) VE-cadherin Y685F knock-in mouse is sensitive to vascular permeability in recurrent angiogenic organs. Am. J. Physiol. Heart Circ. Physiol. 307, H455–H463 [DOI] [PubMed] [Google Scholar]
- 21.Sano H., Nakagawa N., Chiba R., Kurasawa K., Saito Y., Iwamoto I. (1998) Cross-linking of intercellular adhesion molecule-1 induces interleukin-8 and RANTES production through the activation of MAP kinases in human vascular endothelial cells. Biochem. Biophys. Res. Commun. 250, 694–698 [DOI] [PubMed] [Google Scholar]
- 22.Liu G., Vogel S. M., Gao X., Javaid K., Hu G., Danilov S. M., Malik A. B., Minshall R. D. (2011) Src phosphorylation of endothelial cell surface intercellular adhesion molecule-1 mediates neutrophil adhesion and contributes to the mechanism of lung inflammation. Arterioscler. Thromb. Vasc. Biol. 31, 1342–1350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liu G., Place A. T., Chen Z., Brovkovych V. M., Vogel S. M., Muller W. A., Skidgel R. A., Malik A. B., Minshall R. D. (2012) ICAM-1-activated Src and eNOS signaling increase endothelial cell surface PECAM-1 adhesivity and neutrophil transmigration. Blood 120, 1942–1952 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Allingham M. J., van Buul J. D., Burridge K. (2007) ICAM-1-mediated, Src- and Pyk2-dependent vascular endothelial cadherin tyrosine phosphorylation is required for leukocyte transendothelial migration. J. Immunol. 179, 4053–4064 [DOI] [PubMed] [Google Scholar]
- 25.Wang Q., Pfeiffer G. R. II, Gaarde W. A. (2003) Activation of SRC tyrosine kinases in response to ICAM-1 ligation in pulmonary microvascular endothelial cells. J. Biol. Chem. 278, 47731–47743 [DOI] [PubMed] [Google Scholar]
- 26.Adamson P., Etienne S., Couraud P. O., Calder V., Greenwood J. (1999) Lymphocyte migration through brain endothelial cell monolayers involves signaling through endothelial ICAM-1 via a rho-dependent pathway. J. Immunol. 162, 2964–2973 [PubMed] [Google Scholar]
- 27.Pluskota E., Chen Y., D’Souza S. E. (2000) Src homology domain 2-containing tyrosine phosphatase 2 associates with intercellular adhesion molecule 1 to regulate cell survival. J. Biol. Chem. 275, 30029–30036 [DOI] [PubMed] [Google Scholar]
- 28.Hof P., Pluskey S., Dhe-Paganon S., Eck M. J., Shoelson S. E. (1998) Crystal structure of the tyrosine phosphatase SHP-2. Cell 92, 441–450 [DOI] [PubMed] [Google Scholar]
- 29.Qu C. K. (2000) The SHP-2 tyrosine phosphatase: signaling mechanisms and biological functions. Cell Res. 10, 279–288 [DOI] [PubMed] [Google Scholar]
- 30.Ukropec J. A., Hollinger M. K., Salva S. M., Woolkalis M. J. (2000) SHP2 association with VE-cadherin complexes in human endothelial cells is regulated by thrombin. J. Biol. Chem. 275, 5983–5986 [DOI] [PubMed] [Google Scholar]
- 31.Grinnell K. L., Casserly B., Harrington E. O. (2010) Role of protein tyrosine phosphatase SHP2 in barrier function of pulmonary endothelium. Am. J. Physiol. Lung Cell. Mol. Physiol. 298, L361–L370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hatanaka K., Lanahan A. A., Murakami M., Simons M. (2012) Fibroblast growth factor signaling potentiates VE-cadherin stability at adherens junctions by regulating SHP2. PLoS One 7, e37600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Timmerman I., Hoogenboezem M., Bennett A. M., Geerts D., Hordijk P. L., van Buul J. D. (2012) The tyrosine phosphatase SHP2 regulates recovery of endothelial adherens junctions through control of β-catenin phosphorylation. Mol. Biol. Cell 23, 4212–4225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Purswani M. U., Eckert S. J., Arora H. K., Noel G. J. (2002) Effect of ciprofloxacin on lethal and sublethal challenge with endotoxin and on early cytokine responses in a murine in vivo model. J. Antimicrob. Chemother. 50, 51–58 [DOI] [PubMed] [Google Scholar]
- 35.Chichger H., Duong H., Braza J., Harrington E. O. (2015) p18, a novel adaptor protein, regulates pulmonary endothelial barrier function via enhanced endocytic recycling of VE-cadherin. FASEB J. 29, 868–881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chichger H., Braza J., Duong H., Harrington E. O. (2015) SH2 domain-containing protein tyrosine phosphatase 2 and focal adhesion kinase protein interactions regulate pulmonary endothelium barrier function. Am. J. Respir. Cell Mol. Biol. 52, 695–707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hamborg M., Jorgensen L., Bojsen A. R., Christensen D., Foged C. (2013) Protein antigen adsorption to the DDA/TDB liposomal adjuvant: effect on protein structure, stability, and liposome physicochemical characteristics. Pharm. Res. 30, 140–155 [DOI] [PubMed] [Google Scholar]
- 38.Ludwig A, Sommer A, Uhlig S (2011) Assessment of endothelial permeability and leukocyte transmigration in human endothelial cell monolayers. Methods Mol. Biol. 763, 319–332 [DOI] [PubMed] [Google Scholar]
- 39.Bixel M. G., Petri B., Khandoga A. G., Khandoga A., Wolburg-Buchholz K., Wolburg H., März S., Krombach F., Vestweber D. (2007) A CD99-related antigen on endothelial cells mediates neutrophil but not lymphocyte extravasation in vivo. Blood 109, 5327–5336 [DOI] [PubMed] [Google Scholar]
- 40.Kustermann S., Manigold T., Ploix C., Skubatz M., Heckel T., Hinton H., Weiser T., Singer T., Suter L., Roth A. (2014) A real-time impedance-based screening assay for drug-induced vascular leakage. Toxicol. Sci. 138, 333–343 [DOI] [PubMed] [Google Scholar]
- 41.Miller I., Hatzivassiliou G., Cattoretti G., Mendelsohn C., Dalla-Favera R. (2002) IRTAs: a new family of immunoglobulin-like receptors differentially expressed in B cells. Blood 99, 2662–2669 [DOI] [PubMed] [Google Scholar]
- 42.Zhang S. Q., Yang W., Kontaridis M. I., Bivona T. G., Wen G., Araki T., Luo J., Thompson J. A., Schraven B. L., Philips M. R., Neel B. G. (2004) Shp2 regulates SRC family kinase activity and Ras/Erk activation by controlling Csk recruitment. Mol. Cell 13, 341–355 [DOI] [PubMed] [Google Scholar]
- 43.Muller W. A. (2015) The regulation of transendothelial migration: new knowledge and new questions. Cardiovasc. Res. 107, 310–320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Spindel O. N., Burke R. M., Yan C., Berk B. C. (2014) Thioredoxin-interacting protein is a biomechanical regulator of Src activity: key role in endothelial cell stress fiber formation. Circ. Res. 114, 1125–1132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Roebuck K. A., Finnegan A. (1999) Regulation of intercellular adhesion molecule-1 (CD54) gene expression. J. Leukoc. Biol. 66, 876–888 [DOI] [PubMed] [Google Scholar]
- 46.Su W. H., Chen H. I., Jen C. J. (2002) Differential movements of VE-cadherin and PECAM-1 during transmigration of polymorphonuclear leukocytes through human umbilical vein endothelium. Blood 100, 3597–3603 [DOI] [PubMed] [Google Scholar]
- 47.Mège R.-M., Gavard J., Lambert M. (2006) Regulation of cell-cell junctions by the cytoskeleton. Curr. Opin. Cell Biol. 18, 541–548 [DOI] [PubMed] [Google Scholar]
- 48.Yamada S., Pokutta S., Drees F., Weis W. I., Nelson W. J. (2005) Deconstructing the cadherin-catenin-actin complex. Cell 123, 889–901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Drees F., Pokutta S., Yamada S., Nelson W. J., Weis W. I. (2005) α-Catenin is a molecular switch that binds E-cadherin-β-catenin and regulates actin-filament assembly. Cell 123, 903–915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bannerman D. D., Sathyamoorthy M., Goldblum S. E. (1998) Bacterial lipopolysaccharide disrupts endothelial monolayer integrity and survival signaling events through caspase cleavage of adherens junction proteins. J. Biol. Chem. 273, 35371–35380 [DOI] [PubMed] [Google Scholar]
- 51.Di Lorenzo A., Lin M. I., Murata T., Landskroner-Eiger S., Schleicher M., Kothiya M., Iwakiri Y., Yu J., Huang P. L., Sessa W. C. (2013) eNOS-derived nitric oxide regulates endothelial barrier function through VE-cadherin and Rho GTPases. J. Cell Sci. 126, 5541–5552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yang J, Yao W, Qian G, Wei Z, Wu G, Wang G (2015) Rab5-mediated VE-cadherin internalization regulates the barrier function of the lung microvascular endothelium. Cell. Mol. Life Sci. 72, 4849–4866 [DOI] [PMC free article] [PubMed] [Google Scholar]